
Abstract
Rod and cone photoreceptors initiate the visual process by capturing photons and transducing the information into chemical and electrical signals. These functionally specialized neurons have high metabolic activity and oxygen consumption, making them vulnerable to genetic insults and changes in microenvironment. Inherited retinal degenerative diseases are clinically and genetically heterogeneous, with more than 200 genes identified so far. In a majority of retinal degenerations, the genetic defects affect diverse functions in photoreceptors, such as phototransduction, gene regulation, splicing, or intracellular transport. To develop efficient therapies, it is critical to elucidate how genetic defects affect cellular functions and activate death pathways. Caspase-dependent and -independent apoptosis appear to be the major route for photoreceptor cell death in retinal diseases, although the importance of necrosis and autophagy has been demonstrated. Distinct molecules associated with oxidative or endoplasmic reticulum stress can activate these interconnected cell death pathways. Notably, in most inherited diseases, the first signs of photoreceptor dysfunction or loss are observed years after birth and late in life, indicating adaptive mechanisms that protect the cells and suggesting their breakdown may lead to cell death. Mitochondria are predicted to play a critical role in integrating cellular homeostasis to stress and initiation of death pathways. Investigations of adaptive behavior and pre-death molecules that modulate the response to genetic factors should provide attractive targets for the development of better therapeutic approaches.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
11.1 Introduction
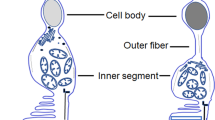
Vision is probably the most important of all senses in humans, allowing us to integrate the information from the surrounding world. From a functional perspective, the eye is sometimes compared to a camera with the ability to focus images with an optimal amount of light on the retina, which acts as the film. However, the light-sensitive retina is much more complicated and versatile than a camera, functioning as an extension of the central nervous system to detect, integrate, and process visual information before sending it to the cortex. Retinal photoreceptors capture light photons and initiate visual transduction cascade. The vertebrate retina contains two types of photoreceptors for light detection: rods optimally function under dim light conditions, whereas cones mediate color vision and high resolution in daylight conditions. The rod and cone photoreceptors are specialized neurons with distinct compartments to carry out distinct functions, as elaborated here.
-
The outer segment is made of membranous discs containing the phototransduction machinery (Hubbell and Bownds 1979), including visual pigment proteins, called opsins, which vary between rod and different types of cone photoreceptors. The outer segment discs are in contact with the retinal pigment epithelium (RPE), which serves as a selective barrier for transport of macromolecules between choroidal capillaries and photoreceptors and participates in the retinoid cycle necessary for phototransduction.
-
The inner segment encloses the metabolic machinery, including mitochondria, ribosomes, and other organelles associated with protein synthesis and transport to the outer segment and synaptic region. Massive amounts of protein are transported from the inner to the outer segment through a microtubule-based connecting cilium.
-
The nucleus is the control center localized in the outer nuclear layer, with discrete chromatin architecture depending on the type of the photoreceptor (Carter-Dawson and LaVail 1979; Solovei et al. 2009).
-
The synaptic terminal, called the “spherule” for rods and the “pedicle” for cone cells, is located in the outer plexiform layer and is connected to different types of interneurons, called bipolar cells, for signal transmission. Photoreceptors also make synapses with horizontal cells.
The photoreceptors have a high metabolic rate and carry out a complex phototransduction process (Fain et al. 2010; Hubbell and Bownds 1979; McBee et al. 2001). The unique characteristics of photoreceptors contribute to their high vulnerability to genetic defects and to changes in metabolism or microenvironment. Thus, photoreceptor dysfunction or death is commonly observed in many blinding diseases, including retinal and macular degeneration. Retinal degenerative diseases constitute a genetically and clinically heterogeneous group with almost 250 human genetic loci associated with Mendelian forms (such as retinitis pigmentosa and cone dystrophies) and more than 200 genes identified so far (Retnet; https://sph.uth.edu/retnet/sym-dis.htm ) (Ratnapriya and Swaroop 2013). In addition, dysfunction and death of retinal photoreceptors are observed in complex multifactorial diseases including age-related macular degeneration (AMD) (Jackson et al. 2002; Swaroop et al. 2009).
Genetic defects associated with photoreceptor degeneration can lead to different outcomes in terms of visual dysfunction:
-
Functional abnormalities, consequent to developmental defects or death of some/all of the photoreceptors.
-
Functional abnormalities associated with early cell death.
-
No or minimal functional abnormalities in early to mid-life, but late disease onset (including cell death).
Rod and cone photoreceptor function and survival can be differentially impacted in retinal diseases. Consequently, a clearer understanding of molecular mechanisms underlying photoreceptor cell death is crucial for developing effective treatments or cure to prevent vision loss or to preserve/restore vision. However, the clinical and genetic diversity associated with distinct forms of photoreceptor degeneration has complicated the elucidation of molecular mechanisms and hampered disease management (Goetz et al. 2012; Swaroop et al. 2007). Recent advances in gene therapy of Leber congenital amaurosis (LCA) caused by RPE65 mutations demonstrate the invaluable contribution of understanding fundamental cellular processes in designing therapies for blinding retinal diseases and other neurodegenerative disorders (Jacobson et al. 2012). Several excellent reviews have discussed the clinical and genetic aspects of retinal and macular degeneration (Ambati and Fowler 2012; Bramall et al. 2010; Fletcher 2010; Lim and Wong 2012; Ratnapriya and Swaroop 2013; Wright et al. 2010). In this chapter, we therefore focus on the discussion of cell death mechanisms involved in photoreceptor death. We also examine how the knowledge of affected cellular pathways can contribute to the development of therapeutics.
11.2 Retinal Diseases and Photoreceptor Degeneration
11.2.1 Retinitis Pigmentosa (RP)
RP is a genetically heterogeneous form of inherited blindness with prevalence ranging from 1 in 3,000 to 1 in 5,000 individuals worldwide. More than 50 genes have been identified for RP with autosomal dominant, autosomal recessive, or X-linked modes of inheritance. In almost half the patients it is difficult to assign the inheritance; these are called simplex RP cases. One can observe characteristic abnormal pigmentary deposits predominantly in the peripheral retina of RP patients. In some instances, nonocular phenotypes can be associated with RP in syndromic diseases.
In one form of RP, referred to as rod-cone dystrophy, patients initially exhibit progressive loss of night and peripheral vision because of the dysfunction or death of rod photoreceptors. Eventually, at later stages of the disease cone photoreceptors also die, resulting in loss of central vision and a severe restriction of the visual field.
Cone-rod dystrophy (CRD) has a prevalence of 1 in 40,000, with more than 15 genes identified so far. Even though CRD and RP overlap genetically and clinically, CRD often exhibits an initial loss of cone photoreceptors with subsequent or concomitant loss of rods.
11.2.2 Macular Degeneration (MD)
Macular degeneration initially affects the central part of the retina, called the macula, associated with high visual acuity. MD can be inherited in a Mendelian manner or manifest as a complex late-onset disease; the latter is termed age-related macular degeneration (AMD).
Mutations in several genes are associated with MD (Stone 2007). Among the monogenic disorders affecting the macula, Stargardt disease is the most common form of inherited juvenile MD with a prevalence of 1 in 8,000 to 1 in 10,000. In most cases, the disease is caused by mutations in the ABCA4 gene (Allikmets 1997), but ELOVL4 mutations have also been associated (Zhang et al. 2001). Stargardt patients display accumulation of yellowish-white deposits in the macular region and eventually exhibit photoreceptor and RPE cell death. Histological examination of donor eyes demonstrates the presence of oxidized cholesterol and insoluble aggregates in these deposits from incomplete degradation of shed photoreceptor outer segments (Molday and Zhang 2010). Another inherited MD, called Best disease or vitelliform macular dystrophy, is caused by mutations in the VDM2 gene that encodes the RPE protein Bestrophin. Best disease is also characterized by the accumulation of yellow, insoluble deposits within the subretinal space and in the RPE (Sun et al. 2002). Mutations in RDS, TIMP-3, and EFEMP1 (also known as Fibulin-3) genes also cause different forms of MD (Stone 2007).
In contrast to monogenic disease, AMD is a late-onset multifactorial disorder caused by interaction of environmental and genetic components (Priya et al. 2012; Swaroop et al. 2007; Chamberlain et al. 2006). In developed countries, AMD is a leading cause of blindness in the elderly. In the United States, almost 2 million people past the age of 50 years have some form of AMD, and the prevalence is more than 13% for people older than 85 years (Smith et al. 2001; Friedman et al. 2004); http://www.visionproblemsus.org/index.html). AMD is characterized by decreased central vision because of the loss of photoreceptors and RPE in the macular region of the retina. The Age-Related Eye Diseases Study (AREDS) classified AMD into four categories based on clinical features (Davis et al. 2005). Geographic atrophy is the major advanced form, characterized by the accumulation of large deposits, termed drusen, between the RPE and the Bruch’s membrane, and scattered areas of RPE atrophy and photoreceptor degeneration in the macula (Davis et al. 2005). Drusen are composed of numerous proteins and lipids, but their origin is still unclear (Rudolf et al. 2008). A more severe form of late AMD, called choroidal neovascularization (CNV), accounts for only about 10 % of cases and is characterized by the presence of new blood vessels in the macula. The nascent expanding blood vessels are leaky and lead to photoreceptor cell death. The patients usually experience sudden loss of central vision with distortion of straight lines and/or a dark spot in their central visual field (Lim et al. 2012).
Genome-wide association studies (GWAS) have identified a number of genetic risk variants linked to advanced stages of AMD (Priya et al. 2012). More recently, meta-analysis of GWAS from a large consortium of investigators has established as many as 19 AMD risk loci (Fritsche et al. 2013).
11.2.3 Cone Dystrophy
Cone dystrophies include a group of rare disorders that affect the function and survival of cone photoreceptors, resulting in high sensitivity to bright light, poor color vision, and low visual acuity (Simunovic and Moore 1998). Most cone dystrophy patients have better vision at dusk. Achromatopsia are usually autosomal recessive and represent a majority of stationary cone dystrophy. Individuals have impaired color discrimination, caused by mutations in one of the following five genes: CNGB3, CNGA3, GNAT2, PDE6C, PDE6H (Hamel 2007). In contrast, progressive cone dystrophy is inherited as autosomal dominant and overlaps with cone-rod dystrophy. For instance, mutations in GUCA1A encoding Guanylate Cyclase Activating Protein 1 (GUCAP1) are associated with autosomal dominant cone dystrophy, CRD or MD (Michaelides et al. 2005). Cone opsin gene mutations lead to X-linked cone dystrophy with varying severity from color blindness to progressive cone dystrophy (Gardner et al. 2010).
11.2.4 Leber Congenital Amaurosis (LCA)
LCA is a clinically and genetically heterogeneous congenital or early onset retinal disease (den Hollander et al. 2008) that is characterized by vision loss associated with other clinical symptoms including nystagmus and nonrecordable rod and cone electroretinogram (ERG) (Traboulsi 2010). LCA accounts for as much as 5% of all inherited retinopathies but 20% of the children attending schools for the blind and visually impaired (Koenekoop 2004). Seventeen genes have so far been associated with autosomal recessive LCA, and two, CRX and IMPDH1, can cause autosomal dominant disease (den Hollander et al. 2008; Freund et al. 1998; Bowne et al. 2002). LCA is the first neurodegenerative disease with effective gene-replacement therapy for patients with RPE65 mutations (Cideciyan et al. 2008).
11.3 Interdependence of Rod and Cone Photoreceptors for Survival
An interesting feature of retinal degeneration is that rod-specific mutations do not lead only to rod cell death. One would expect that mutations in genes exclusively expressed in rods would affect only night vision and have no major effect on daylight vision. However, patients with mutations in rod-specific genes also experience loss of cone photoreceptors. The kinetics of cone death is variable, depending on the affected gene (LaVail 1981; Punzo et al. 2009; Wright et al. 2004). Thus, in addition to cell-autonomous mechanisms mediating death pathways in rods, non-cell-autonomous factors appear to contribute to the death of normal and otherwise healthy cones. Chimera generated from a mixture of wild-type cells and those expressing mutant rhodopsin show homogenous retinal degeneration independent of the patch of normal or mutant cells (Huang et al. 1993). Similarly, female hemizygous-transgenic rds mutant mice also show uniform cell death in the outer nuclear layer, including those of genetically normal photoreceptors (Kedzierski et al. 1998). These studies reveal that photoreceptor death involves not only genetic defects but also cell interactions.
Even mutations that affect other cell types, such as those in RPE-specific genes, can cause non-cell-autonomous photoreceptor death. For instance, in mutant mice as well as human patients, mutations in RPE65, an enzyme expressed specifically in RPE, lead to absence of visual response and slow degeneration of rods (Redmond et al. 1998; Gu et al. 1997; Marlhens et al. 1997). Interestingly, rod photoreceptors are generally unaffected in cone dystrophies caused by cone-specific gene mutations.
11.4 Pathways of Photoreceptor Cell Death
Cell death is a part of normal development and can occur in different modes, such as apoptosis, necrosis and autophagy. Multiple interdependent pathways may participate sequentially or overlap to mediate photoreceptor cell death (Kunchithapautham and Rohrer 2007a, b; Metrailler et al. 2012; Lo et al. 2011; Lohr et al. 2006; Doonan et al. 2003).
11.4.1 Apoptosis
Apoptosis is the major cell death pathway (Portera-Cailliau et al. 1994), at times referred as programmed cell death (PCD). Apoptosis is a complex process leading to DNA fragmentation and generation of apoptotic bodies that can be phagocytized and quickly removed to prevent the surrounding cells from further damage by extrusion of cellular contents. Prompt removal of cellular debris avoids inflammation. The TUNEL assay that labels the fragmented DNA is extensively used to detect apoptosis, but other cell death pathways, such as necrosis and autolysis of the cells, can also exhibit DNA fragmentation (Grasl-Kraupp et al. 1995).
Apoptosis is a critical part of normal retinal development especially for photoreceptors that may be produced in excess (Vecino et al. 2004). In addition, apoptosis is a major cause of cell death under disease conditions, such as RP, retinal detachment, or light-induced apoptosis (Portera-Cailliau et al. 1994; Gregory and Bird 1995; Hafezi et al. 1997; Chang et al. 1995; Cook et al. 1995). Apoptosis is an active process involving several steps controlled by the cell itself (Fig. 11.1). The first step, called induction phase, triggers apoptosis by a wide variety of stimuli such as tumor necrosis factor (TNF), Fas-ligand, and other extracellular factors. During this phase, production of necessary effectors, called pro-apototic factors, is increased. Among them, c-Fos and c-Jun form a heterodimer complex, termed activator protein complex 1 (AP1) (Hafezi et al. 1997; Rich et al. 1997; Vuong et al. 2013; Wenzel et al. 2000). The subsequent executive phase leads to morphological changes including chromatin condensation, DNA fragmentation, cell shrinkage, and ultimately cell fragmentation into apoptotic bodies, in which organelles remain intact to prevent inflammation and damages to the surrounding cells. During the degradation phase, cleavage of proteins, further DNA fragmentation, and cellular shrinkage allow apoptotic bodies to be removed without inflammation by macrophages and activated microglia (Langmann 2007; Ng and Streilein 2001; Joly et al. 2009). Apoptosis can occur in a caspase-dependent or -independent manner.

Crosstalk between apoptotic cell death pathway and necrosis. Upon activation of death receptors, caspase-dependent apoptosis involves mitochondria and the release of cytochrome c, which activates APAF1 to cleave procaspases (2, 10, 9,8). The complex APAF1 and Caspase-9 then activate effector caspases such as caspase-3, -6, and -7, allowing the activation of caspase-activated DNAses that cleave genomic DNA and participate at the formation of apoptotic bodies. Caspase-independent apoptosis is also dependent on mitochondria and induced by cell death effectors such as AIF1, EndoG, and PARP1. AIF1 and EndoG are released from mitochondria under oxidative stress condition and translocate into the nucleus. PARP detects DNA strand breaks and transfers polymers of ADP-ribose to a wide range of nuclear proteins. High PARP activity leads to a high ATP consumption and accumulation of poly(ADP-ribosyl) polymers, which leads to the release of AIF1 from the mitochondria to trigger caspase-independent apoptosis. Caspase-dependent apoptosis and necrosis activation can overlap after cleavage of procaspase-8 by the release of RIP1 protein, the key protein to trigger necrosis. Phosphorylation of RIP1 and RIP3 leads to the formation of a pronecrotic complex initiating necrosis. This complex opens the mitochondrial permeability transition pores (mPTP) leading to an increase of intracellular Ca2+, reactive oxygen species (ROS), and the translocation of AIF1 into the nucleus. Necrosis activation ultimately causes the disruption of the plasma membrane and the release of HMGB1 into the extracellular environment to mediate inflammation
11.4.1.1 Caspase-Dependent Apoptosis
Caspases, a family of highly conserved cysteine proteases, play a central role in the executive phase. The initiator caspases (2, 9, 8, 10) activate the effector caspases (3, 6, 7) by cleavage. The role of activated caspases is to conduct proteolytic degradation of intracellular targets leading to the cell death program. During this phase, mitochondria play a critical role by releasing cytochrome c by permeabilization of the mitochondrial outer membrane (Green and Kroemer 2004). B-cell lymphoma 2 (Bcl-2) family members participate in the release of cytochrome c; some of these members are pro-apoptotic [e.g., Bcl-2 homologous antagonist/killer (Bak) and Bcl-2-associated X protein (Bax)], whereas others are anti-apoptotic (such as Bcl-2 and Bcl-xL). Cytochrome c activates the caspase cascade by binding to apoptotic peptidase activating factor 1 (Apaf1) and triggering the cleavage of initiator caspases, such as caspase-9, in an ATP-dependent manner (Brenner and Mak 2009; Liu et al. 1996). The cleavage of procaspase-9 to caspase-9 allows the cleavage and activation of the downstream effector caspases, such as caspase-3, which initiate the degradation phase by activating DNase. Caspase-dependent apoptosis has been demonstrated in rd1 (calcium overload) and rds (structural defects) mutant mice and in a light damage model (oxidative stress) (Jomary et al. 2001; Lohr et al. 2006; Perche et al. 2007).
11.4.1.2 Caspase-Independent Apoptosis
Although caspase-dependent apoptosis occurs in several retinal degenerative diseases, increasing evidence using a wide range of caspase inhibitors indicates that apoptosis can occur in a caspase-independent manner (Borner and Monney 1999; Bouchier-Hayes et al. 2005) in both in vitro and in vivo models of photoreceptor cell death (Carmody and Cotter 2000; Donovan and Cotter 2002) (Fig. 11.1). Caspase-independent apoptosis is also dependent on mitochondria and induced by cell death effectors, such as apoptosis-inducing factor 1 (AIF1), endonuclease G (EndoG), or poly(ADP-ribose) polymerase I (PARP1).
AIF1 is an oxido-reductase normally localized in the mitochondrial intermembrane space (Cande et al. 2002), and its activity is directly correlated to oxidative stress (Klein et al. 2002). After induction by appropriate apoptotic stimuli, AIF1 translocates to the nucleus and induces chromatin condensation and DNA fragmentation in cooperation with cyclophilin A to elicit apoptosis (Susin et al. 1999). In purified mitochondria, AIF1 triggers cytochrome C and caspase-9 release. AIF1 is involved in apoptosis triggered by retinal detachment in rodent models (Hisatomi et al. 2001), and its inhibition has a protective effect on photoreceptor degeneration (Hisatomi et al. 2008).
EndoG, similar to AIF1, is confined to the mitochondria under normal conditions but translocates to the nucleus upon apoptotic stimulation (Lemarie et al. 2004). In a mouse model of achromatopsia, these two factors are shown to participate in cone cell death caused by endoplasmic reticulum stress (Thapa et al. 2012).
PARP proteins also play a key role in mediating caspase-independent apoptosis. These factors detect DNA strand breaks and are involved in DNA repair (Barth et al. 2006). After activation, PARP enzymes use NAD+ as a substrate to transfer polymers of ADP-ribose onto itself as well as to many nuclear protein targets. Excessive DNA damage leads to enhanced activation of PARP1, with consequent accumulation of poly(ADP-ribosyl) polymers, which play a critical role in the release of AIF from mitochondria to trigger caspase-independent pathways (Yu et al. 2002; Yu et al. 2006). PARP activation has been shown in several models of retinal degeneration (Kaur et al. 2011; Paquet-Durand et al. 2007). Notably, the lack of PARP1 in the mouse retina does not alter retinal function and protects photoreceptors from cell death (Sahaboglu et al. 2010). Several studies suggest a possible clinical relevance of the pharmacological inhibition of PARP proteins (Jagtap and Szabo 2005; Paquet-Durand et al. 2007).
Photoreceptor cell death is shown to be caspase independent in a number of models of retinal degeneration, such as in rd mouse, N-methyl-N-nitrosourea (MNU)-treated mice, or in light-induced apoptosis (Donovan and Cotter 2002; Doonan et al. 2003; Donovan et al. 2001; Chahory et al. 2010). In these models, cytochrome c release from the mitochondria or activation of the caspase cascade could not be detected (Doonan et al. 2003).
It is widely accepted that caspase-dependent and -independent mechanisms often act in cooperation during apoptosis (Lohr et al. 2006; Kroemer et al. 2009). Hence, the success of therapeutics based on apoptosis inhibition would depend on targeting both caspase-dependent and caspase-independent mechanisms.
11.4.2 Autophagy
Autophagy is an alternative and equally critical death pathway that plays an important role during development and homeostasis of mature tissues. Growing evidence demonstrates that autophagy might be even more important under disease conditions (Kim et al. 2013; Mizushima et al. 2008). The primary function of this catabolic process is to maintain cellular integrity by targeting proteins (e.g., aggregates) and dysfunctional organelles (e.g., mitochondria) to the lysosome for degradation (Marino et al. 2011). A stringent regulation of autophagy is critical for cell homeostasis under normal conditions, and loss of control of degradation can be lethal. During starvation, the degradation of cellular components by autophagy provides resources for survival. Autophagy is an adaptive response under stress and disease conditions to promote cell survival; however, under other scenarios, it may promote cell death. The main autophagy pathway, called macroautophagy, involves organelle and protein degradation by engulfment in a double membrane and formation of an autophagosome, which is then targeted to and fuses with the lysosome for degradation of the enclosed material by lysosomal proteases. In contrast, microautophagy is characterized by direct invagination of the lysosomal membrane to completely enclose the cytoplasmic material. Chaperone-mediated autophagy (CMA) is a more complex and specific pathway in which cytosolic soluble proteins containing a recognition motif for chaperones are selectively transported to the lysosome. The translocation to the lysosome is triggered after binding of the chaperone to a lysosomal receptor, named lysosome-associated membrane protein type 2A (LAMP2A) (Cuervo 2010).
Macroautophagy is a nonspecific process often activated in response to nutrient deprivation and stress conditions. In contrast, CMA is usually activated after long periods of starvation leading to an increase of LAMP2A synthesis (Cuervo and Dice 2000; Marino et al. 2011; Massey et al. 2004). Crosstalk among different autophagic pathways can compensate for each other (Kaushik et al. 2008; Massey et al. 2008). Recent work on photoreceptor aging indicates that dysfunction of macroautophagy is correlated with significant increase of CMA markers in aged animals (Rodriguez-Muela et al. 2013). Such compensatory mechanisms can explain the delay between the start of expression of macroautophagy markers and apparent cell death occurring much later. Interestingly, inhibition of CMA in the retina does not lead to an increase in macroautophagy; instead, cells become more sensitive to stress.
In aging, AMD, and a majority of retinal dystrophies, rod photoreceptors die first, followed later by cones, which have a much slower rate of cell death (Kolesnikov et al. 2010; Punzo et al. 2009). Interestingly, macroautophagy is not activated in dying cones. On the contrary, it is inhibited with a concomitant increase of LAMP2A expression specifically in the lysosomal membrane of cones, potentially reflecting starvation of these cells. Microarray analysis of dying cone photoreceptors in mouse models of RP has identified several genes associated with insulin metabolism, suggesting that the level of intracellular glucose in cones may be altered (Punzo et al. 2009). Recently, glucose starvation of cones in vitro has also been shown to trigger autophagy (Balmer et al. 2013).
It is often difficult to determine whether autophagy is the cause or the consequence of photoreceptor stress and death. For instance, RPE accumulates proteins, lipofuscin, and damaged mitochondria during aging and in AMD (Kaarniranta et al. 2010; Kaarniranta et al. 2013; Krohne et al. 2010; Wang et al. 2009a, b). Accumulation of lipofuscin in the RPE can alter the fusion between the autophagosome and the lysosome. In addition, accumulation of A2E, the major lipofuscin fluorophore reportedly inhibits ATP-driven proton pump in the RPE cells, leading to increase of lysosomal pH. Increase in the autophagy flux and accumulation of toxic products may thus contribute to drusen formation, which are hallmarks of AMD (Bergmann et al. 2004). Autophagy is an important pathway for photoreceptor maintenance and homeostasis, especially during disc outer segment shedding (Kim et al. 2013; Long et al. 1986). Autophagy may also regulate opsin protein levels to adjust to lighting environment (Reme et al. 1999).
The ubiquitin proteasome system (UPS) is a major protein degradation pathway. Several lines of evidence indicate a crosstalk between autophagy and proteasomal degradation, especially in selective autophagy (Kraft et al. 2010). The UPS controls protein stability and degradation of the target proteins by posttranslational modifications, including covalent addition of several monomers of a small protein ubiquitin, transferred by sequential action of E1-, E2-, and E3-ligases. The proteasome complex then degrades poly-ubiquitinated proteins. This complex regulates a number of intracellular proteins associated with cell signaling, cell-cycle regulation, and synaptic plasticity. UPS also plays a critical role in regulating cell death (Tai and Schuman 2008; Vucic et al. 2011). Protein aggregation is a characteristic feature of a wide variety of neurological diseases. Parkinson’s disease, Alzheimer’s disease, and prion disease are among the most common neurodegenerative diseases, which reveal protein aggregation and dysfunction of the UPS-triggered apoptosis (Bence et al. 2001; Sherman and Goldberg 2001; Snyder et al. 2003). Several mutations associated with inherited retinal degeneration, such as those in rhodopsin, cause protein misfolding, mistargeting, or aggregation (Comitato et al. 2007; Surgucheva et al. 2005; Illing et al. 2002). In the case of formation of the transducin βγ-subunit complex, critical for phototransduction, lack of transducin γ-subunit (Gγ1) in mice leads to increase in production of its partner, transducin β1 (Gβ1), which in the absence of Gγ1 cannot be folded properly and is targeted to the proteasome (Lobanova et al. 2013). Consequently, large increases in Gβ1 overload the UPS, causing cellular stress and photoreceptor cell death. Defects in degrading misfolded proteins have also been observed in rodent models, including rds (PRPH2), Rho −/−, and Rho P23H. Such mechanisms are also described for extended polyglutamine proteins, such as SCA7 and Huntingtin, leading to neuronal cell death (Lobanova et al. 2013; Yvert et al. 2000; Abe et al. 2000; Helmlinger et al. 2002; Schipper-Krom et al. 2012). Mutations in components of the UPS have also been identified in autosomal dominant RP patients (Friedman et al. 2009).
11.4.3 Necrosis
Necrosis, also called necroptosis, is characterized by an increase in cell volume, swelling of the organelles, and rupture of the plasma membrane causing the release of intracellular components in the surrounding microenvironment and initiating an inflammatory response (Vandenabeele et al. 2010). If apoptosis is a regulated cellular process that can be beneficial and necessary for the organism, necrosis almost always has a negative impact. At first, necrosis was thought to be a nonspecific, uncontrolled cell death mechanism, but recent evidence indicates some level of regulation of this pathway (McCall 2010). Several regulators of necrosis have now been identified, including receptor-interacting protein (RIP) kinases. RIP1 is essential to trigger necrosis in response to tumor necrosis factor (TNF)-α, and FasL stimulation and promotes activation of nuclear factor κB (NF-κB) (Ea et al. 2006; Moquin and Chan 2010; Christofferson and Yuan 2010). In case of failure to activate caspase-dependant apoptosis, RIP1 interacts with RIP3 to form a pronecrotic complex, stabilized by phosphorylation that leads to necrosis pathway activation. Often, apoptosis and necrosis pathways overlap and crosstalk (Fig. 11.1) (Brenner and Mak 2009; Zhang et al. 2009). In necrosis, high-mobility group box 1 (HMGB1), a non-histone chromatin-binding protein, is usually released into the extracellular matrix and acts as a pro-inflammatory factor (Fang et al. 2012). However, little is known about the downstream effectors of necrosis, and additional studies are required.
The importance of RIP and necrotic pathway activation in the secondary death of cone photoreceptors has recently been demonstrated in models of rod-cone dystrophy (Murakami et al. 2012). In the rd10 mouse, a model of rod-cone dystrophy caused by a mutation in the rod-specific gene, cGMP phosphodiesterase β-subunit (Pde6β), an increase in RIP proteins has been observed in the late phase of retinal degeneration during the period of cone death but not in earlier stages, when most of the rods die. A detailed morphological analysis of dying photoreceptors shows that necrosis is an important cell death pathway for cones in contrast to rods, where apoptosis appears to be the preferred cell death pathway. In concordance, lack of RIP3 expression does not prevent rod cell death but leads to preservation of cones. The importance of RIP proteins in mediating necrosis is also shown recently in a mouse model of retinal degeneration induced by subretinal injection of dsRNA, a component of the drusen, causing RPE cell death and inflammation in AMD patients (Murakami et al. 2013). In such a model, lack of RIP3 expression has a protective effect by preventing necrosis and inflammation and suppressing the release of HMGB1 (Fang et al. 2012; Murakami et al. 2013). Recent studies have highlighted the contribution of necrosis in several models of photoreceptor degeneration, such as retinal detachment, light damage, and retinal ischemia (Shahinfar et al. 1991; Trichonas et al. 2010; Rosenbaum et al. 2010).
11.5 Mediators of Cell Death Pathways
11.5.1 Calcium Signaling and Photoreceptor Cell Death
Under dark conditions, rod photoreceptors are depolarized because of an inward flow of sodium and calcium through cyclic nucleotide-gated channels (cGMP-dependent) located in the outer segment. Upon light stimulation, 11-cis-retinal bound to rhodopsin is isomerized into all-trans-retinal, triggering the release of α-subunit of the G-protein transducin, which in turn activates a photoreceptor-specific cyclic nucleotide phosphodiesterase (PDE). Activated PDE hydrolyzes cGMP to GMP, reducing its intracellular concentration, and thereby closing cyclic nucleotide-gated channels and triggering an outward flux of potassium leading to hyperpolarization of the photoreceptor membrane. Channel closure also leads to a decrease in calcium influx and a decrease of glutamate release at the synaptic terminal. cGMP concentration is restored by activation of guanylate cyclase upon reduction in intracellular calcium. Enzyme dysregulation, altered energy utilization, and changes in signaling molecule concentration are predicted to cause photoreceptor cell death when the visual transduction cascade is continuously active. Thus, calcium may play a crucial role in cell death.
Overload of intracellular calcium is a major contributor to cell death, including in photoreceptors (Wenzel et al. 2005; Zhivotovsky and Orrenius 2011; Fox et al. 1999; Takano et al. 2004; Read et al. 2002; Fox et al. 2003; Sharma and Rohrer 2004). However, it is still unclear whether the additional Ca2+ enters from the extracellular space or is released from intracellular storage in mitochondria or endoplasmic reticulum (Orrenius et al. 2003). The importance of Ca2+ in photoreceptor cell death has been investigated in the rd1 mouse retina, where a strong activation of Ca2+-dependent enzymes occurs (Paquet-Durand et al. 2006; Hauck et al. 2006). Here, photoreceptors go under severe and rapid degeneration caused by a mutation in PDE-β, which, when mutated, also causes photoreceptor death in humans (Bowes et al. 1990; McLaughlin et al. 1993). The mutated PDE6β is not functional, potentially leading to cGMP accumulation and constitutive opening of cGMP-gated channels, resulting in an influx of Ca2+ into the cells. The deleterious effect of Ca2+ has been further demonstrated using rd1 mice that also lack a major L-type voltage-dependent Ca2+ channel at the photoreceptor synapse. In the double mutant mice, photoreceptor cell death is slower compared to the rd1 mice (Read et al. 2002). Calpains are a group of 15 Ca2+-activated cysteine proteases that have been implicated in degeneration of neuronal tissues (Paquet-Durand et al. 2006; Smith and Schnellmann 2012; Nakazawa 2011). Association of calpains with calpastatins in the endoplasmic reticulum specifically inhibits their activity, and overexpression of calpastatins has neuroprotective effects (Suzuki et al. 2004; Schoch et al. 2013; Wingrave et al. 2004; Cao et al. 2007). In the retina, calpains also play a critical role in photoreceptor cell death, and their inhibition can delay photoreceptor demise (Paquet-Durand et al. 2010; Mahajan et al. 2012; Ozaki et al. 2013). In the rd1 mutant, calpains are strongly activated during photoreceptor cell death with a concomitant downregulation of calpastatin and cyclic AMP response element-binding protein (CREB)-1 (Paquet-Durand et al. 2006). CREB1, a transcription factor critical for neuronal survival (Finkbeiner 2000; Mantamadiotis et al. 2002), is critical for integrating Ca2+ and cAMP signals. It can be activated by the AC/cAMP/PKA pathway, triggered by high intracellular Ca2+, or by Ca2+/calmodulin-dependent protein kinases (CAMK).
11.5.2 Oxidative Stress and Photoreceptor Cell Death
Oxidative stress is closely linked to a number of neurodegenerative diseases (Andersen 2004; Komeima et al. 2006; Shen et al. 2005; Anderson et al. 1999) and is produced by increase in free radicals that cause damage to proteins, lipids, and DNA. Reactive oxygen species (ROS) are highly reactive molecules produced by normal oxygen metabolism from the mitochondria or by exogenous sources such as ionizing radiation. Varying rates of ROS production have been proposed to underlie differential rates of cell death in neurodegenerative diseases sharing the same mutation (Wright et al. 2004). Neuronal cells have very high metabolism and their membranes are rich in lipids and unsaturated fatty acids, which are ideal targets of lipid peroxidation, thus making them highly susceptible to damage by ROS.
During aging, the scavenging activities of endogenous antioxidants decrease and trigger oxidative stress (Klein and Ackerman 2003). However, it is still difficult to discriminate whether oxidative stress is the cause or the consequence of neuronal cell death (Andersen 2004). Additional investigations are required to identify signaling pathways activated by ROS and to define their role in death pathways.
Similar to other neuronal cells, photoreceptors are under constant environmental and intrinsic stress. Their function and location in the retina and the presence of high concentration of lipids in the outer segments make photoreceptors specially vulnerable as they are exposed to light, and ultraviolet radiations in particular, which can lead to the production of free radicals (Kagan et al. 1973; Oguni et al. 1996; Yang et al. 2003). These deleterious effects are amplified by high oxygen tension caused by proximity of the choroidal blood vessels that also participate in the production of ROS (Nickla and Wallman 2010). Interestingly, impairment of choroidal blood circulation in light-exposed albino rats is shown to delay photoreceptor cell death (Tanito et al. 2007b).
Mitochondrial metabolism is a major source of ROS. In photoreceptors, mitochondria are located in the inner segments. Mouse cone photoreceptors contain twofold more mitochondria than rods, and the ratio reaches tenfold in humans and primates (Hoang et al. 2002; Perkins et al. 2003). If rod and cone mitochondrial membranes have the same width, the total membrane surface is greater in cones compared to rods, suggesting high-energy demand and higher cytochrome c oxidase activity in cones (Hoang et al. 2002; Perkins et al. 2003). Oxidative stress and photoreceptor degeneration are tightly linked (Costa et al. 2008; Shen et al. 2005), as has been described in models of retinal degeneration (Cao and Phillis 1995; Costa et al. 2008; Komeima et al. 2006; Kowluru and Odenbach 2004). As a by-product of energy production, mitochondria produce free radicals, including superoxide (O2 −) radicals. Mitochondria are also equipped with antioxidant mechanisms to preserve cell homeostasis. Among them are superoxide dismutase (SOD), in charge of converting O2 − to hydrogen peroxide (H2O2), and glutathione peroxidase and related enzymes, which convert H2O2 to H2O, making glutathione an important cellular antioxidant. Transgenic mice with gain-of- function mutation in SOD1, associated with familial amyotrophic lateral sclerosis in human, show specific light-induced photoreceptor cell death (Mittag et al. 1999). In addition, decreased expression of SOD2 leads to photoreceptor cell death (Justilien et al. 2007). Such alterations in antioxidant properties of mitochondria may also be linked to AMD (Khandhadia and Lotery 2010). Because rod photoreceptors outnumber cones, it has been proposed that rod death leads to a great increase of oxygen tension on the remaining cone photoreceptors, thereby increasing their oxidative stress (Yu et al. 2000). Such mechanisms might participate in the non-cell-autonomous death occurring in rod-specific inherited retinal degeneration and explain the initially slow death rate in cones. Oxidative damage is reported in a transgenic pig model with rhodopsin mutation, in which cone photoreceptors progressively accumulate oxidative damage over time in parallel to their death (Shen et al. 2005). A similar pattern of oxidative damage and cell death is observed in rd1 mice (Komeima et al. 2006). In contrast, reduction of superoxide radical production can reduce cone cell death (Komeima et al. 2008). Thus, loss of rod photoreceptors in RP may lead to hyperoxia in the outer nuclear layer, causing oxidative damage and slow death of cone photoreceptors.
The presence of free radicals in photoreceptor outer segments can also result in peroxidation of lipids, which account for about 15% of the total weight of photoreceptors compared to about 1% in most other cells (Bramall et al. 2010). Photoreceptors have a high amount of decosahexaenoic acid (DHA), a very long chain polyunsaturated fatty acid, which can be oxidized to highly reactive molecules such as molondialdehde (MDA) and 4-hydroxy-2-neoenal (4-HNE), two by-products that indicate the level of oxidative stress in cells (Moreira et al. 2010). Furthermore, oxidized phosphatidylcholine increases in human photoreceptors and RPE located in the macula during aging and at even higher levels in AMD patients (Suzuki et al. 2007). Oxidative stress is also shown to damage mitochondrial DNA in human RPE (Liang and Godley 2003). The decreased antioxidant capacity during aging, together with a concomitant damage to mitochondrial DNA, can contribute to AMD pathophysiology. Our recent studies in cone-only Nrl −/− mouse retina have shown an association between rapid and transient death of cones and transient increase of oxidative stress that are followed by long-term preservation of the remaining cone photoreceptors after lipid peroxidation suppresses (Roger et al. 2012).
Daily shedding and renewal of photoreceptor outer segments by the RPE can also be considered an antioxidant mechanism, designed to remove oxidized lipids and their by-products (Winkler 2008). Additional antioxidant mechanisms can protect photoreceptors; for example, thioredoxins are small proteins playing an important role in the redox signaling cascade (Tanaka et al. 2000). Their expression is upregulated in response to oxidative stress and shown to have neuroprotective effects, especially in the retina (Tanito et al. 2002a, b, 2005, 2007a; Wang et al. 2008; Kong et al. 2010). Thioredoxin-like protein rod-derived cone survival factor (RdCVF) has been identified based on its property to sustain the viability of cone photoreceptors (Leveillard et al. 2004). RdCVF proteins are localized in the cone extracellular matrix, and lack of their expression in mice affects photoreceptor function and increases oxidative stress, suggesting a relationship between RdCVF and redox signaling (Cronin et al. 2010).
11.5.3 Purine and Photoreceptor Death
Intracellular ATP is a major source of cellular energy. When released into the extracellular space, it can also be used as a purinergic neurotransmitter in the central and peripheral nervous system. If the intracellular concentration of ATP is in the millimolar range, the extracellular concentration ranging from nanomolar to micromolar must result from the balance between release and degradation (Schwiebert 2000). Enzymes located at the cellular membrane, called ectonucleotidases, which produce ADP or AMP, control ATP concentration in the extracellular space. ATP can be released by damaged membranes of dying cells, by ATP transporters, channels, or osmotic transporters, or by exocytosis from synaptic vesicles. Two families of receptors, called P2X (seven subtypes) and P2Y receptors (eight subtypes), mediate ATP signaling. P2X receptors assemble into ATP-activated cation channels, selectively permeable to sodium and calcium ions, leading to an increase of intracellular calcium concentration and depolarization of the cell membrane (Abbracchio et al. 2009). P2X receptors have low affinity for ATP and have a fast response because they do not require the diffusion of a second messenger (Volonte et al. 2003). In contrast, P2Y receptors are metabotropic receptors belonging to the family of seven-transmembrane domain G protein-coupled receptors that generate a slower response because they have a higher affinity for their ligand and are coupled to potassium or calcium channels (Weisman et al. 2012).
ATP is demonstrated to regulate retinal function, especially as a neuromodulator released by Müller glia cells (Ward et al. 2010; Neal and Cunningham 1994; Newman 2003). P2X7 and P2Y6 receptors are expressed in the plexiform layers of rodent retinas, especially in the photoreceptor synaptic terminals (Puthussery and Fletcher 2004; Puthussery et al. 2006; Zhang et al. 2012). In addition to their role in modulating neurotransmission, P2 receptors are shown to be involved in cell homeostasis in the central nervous system (Franke and Illes 2006). The release of ATP into the extracellular environment can also participate in apoptosis-induced cell death after injury (Wang et al. 2004). In the retina, intravitreal injections of ATP, but not its breakdown products (i.e., ADP or AMP), induce extensive and selective photoreceptor apoptosis through activation of P2 purinoreceptors in a dose-dependent manner and lead to severe photoreceptor degeneration and impaired ERG (Puthussery and Fletcher 2009). Notably, injection of the purinergic antagonist pyridoxal-phosphate-6-azophenyl-2',4'-disulfonic acid (PPADS) prevents ATP-induced apoptosis and reduces photoreceptor cell death in rd1 mouse retina. The critical role of P2RX7 in ATP-mediated apoptosis has been reported in cells under physiological stress such as nutrient starvation (Notomi et al. 2011). Injection of a P2RX7 agonist triggered photoreceptor cell death in wild-type animals but has no effect in mice lacking P2X7 receptor. Apoptosis can be mediated by activation of caspase-8 and -9, leading to the translocation of AIF1 from mitochondria to the nuclei (Notomi et al. 2011). Such a mechanism has also been identified in AMD patients with subretinal hemorrhage caused by choroidal neovascularization (CNV), in which increased levels of ATP are observed (Notomi et al. 2013). In vitro, extravascular blood initiates apoptotic pathways after binding to P2X7 receptors, suggesting an important role for ATP in exacerbating photoreceptor cell apoptosis in AMD patients with CNV. The preceding discussion suggests that dying photoreceptors may release ATP into the extracellular space, and released ATP can bind to P2 receptors on neighboring photoreceptors and contribute to cell death by excessive stimulation.
11.6 Complexities Associated with Photoreceptor Degeneration
In addition to the large number of genes associated with photoreceptor degeneration, different mutations in the same gene can have diverse outcomes in terms of disease type, progression, and severity. For instance, mutations in CRX, a critical transcription factor for photoreceptor development, can cause dominant LCA, cone-rod dystrophy, or RP (Huang et al. 2012). In addition to allelic differences, environmental factors and genetic modifiers can also cause phenotypic variability. Modifier genes can protect or have additional deleterious effects, as described for RPGR/RPGRIP1L, RPE65, or CEP290 (Cideciyan et al. 2013; Coppieters et al. 2010; Fahim et al. 2011; Khanna et al. 2009). Furthermore, clear heterogeneity exists in disease progression and cell death location within the retina. Nonuniform photoreceptor cell death illustrates the importance and differential sensitivity of photoreceptors expressing the exact same mutation (Jacobson et al. 2000). In mice, photoreceptor degeneration can follow in either way around a central/peripheral axis, suggesting that a gradient of molecules makes the photoreceptor cells more or less sensitive to cellular stress. One can also hypothesize the existence of distinct rod subtypes in the retina with differential sensitivity to cell death. Consequently, gene-profiling analysis from macula versus peripheral retina may reveal factors influencing photoreceptor homeostasis (Sharon et al. 2002). The heterogeneity of retinal photoreceptors is illustrated with different distribution of cones among vertebrates and especially by a dorsal–ventral gradient of short and medium/long wavelength opsins in the mouse retina (Applebury et al. 2000; Ng et al. 2001). However, very few molecules displaying gradient have so far been identified in the retina. Alternatively, a gradient of neurotrophic factors may create variability in the neuroprotective effects, generating resistance of photoreceptors to cellular stress (Cornish et al. 2005).
A large number of mutations leading to photoreceptor cell death converge to common death pathways such as apoptosis, necrosis, and autophagy (Fig. 11.2). Consequently, all these mutations must share common death effectors (Swaroop et al. 2010). Many genes associated with photoreceptor degeneration are ubiquitously expressed and exhibit extensive functional diversity. In addition, mutations in disease-causing genes do not always lead directly to nonfunctional photoreceptors. Slow progression of disease in many mutations supports a model involving the accumulation of toxic molecules and damage before cell death (Clarke et al. 2001). LCA and AMD represent two extremes, with LCA-causing genes leading to early childhood blindness, whereas in AMD the first signs of the disease appear after many decades.

Convergence of cell death signals to trigger autonomous and nonautonomous cell death during photoreceptor degeneration. The large number of mutations in genes causing photoreceptor degeneration can alter any subcellular compartments of the photoreceptor cells. Mitochondria play an important role in the integration of pre-death factors generated by the cellular stress caused by mutated genes over time. Exchange of signals from the nucleus to the mitochondria (anterograde signaling) and vice versa (retrograde signaling) is an important part of this integration of life and death signal by the mitochondria to maintain cell homeostasis. Accumulation above a certain threshold of pre-death signal and cellular stress, such as ROS, activates photoreceptor cell death. In rod-cone dystrophies, rod photoreceptor cell death by apoptosis, necrosis, or autophagy leads to nonautonomous cone cell death by the release of toxic signals or a decrease of neurotrophic factors produced
It is critical to identify pre-cell death molecules shared by different types of mutations because it is unlikely that each mutated gene triggers its own pre-apoptotic pathway (Swaroop et al. 2010). In such a model, the pre-death signal must reach a certain level to trigger cell death. The pre-death signal may also participate in death of neighboring photoreceptors because of the accumulation of toxic products extruded by dying cells into the microenvironment or the depletion of pro-survival factors [such as rod-derived cone viability factor (Rdcvf) and ciliary neurotrophic factor (Cntf)] (Fig. 11.2). The presence of diffusible toxic factors released by dying photoreceptors is supported by studies using chimeric animals in which the retina contains both normal and mutant photoreceptors and the cell death rate is proportional to the number of mutant cells (Burns et al. 2002; Huang et al. 1993). In such a model, the pre-death signal may converge and be integrated by an organelle in the cell to trigger cell death. One organelle where such integration takes place is the mitochondria, as the production of ROS appears to be correlated with the rate of photoreceptor cell death (Perez-Campo et al. 1998; Sastre et al. 2000; Wright et al. 2004). Overexpression of antioxidant defenses, such as SOD, which has neuroprotective effects, can extend cellular lifespan (Sampayo et al. 2003). However, the correlation between oxidative stress and neurodegeneration is still unclear, because most genes do not alter or directly induce ROS production. Mitochondrial DNA is more vulnerable to mutations compared to nuclear DNA, and such accumulation of genomic abnormalities would occur independently in each photoreceptor (Wright et al. 2009). The consequence could be altered mitochondrial bioenergetic functions, which then act as redox damage sensors. Mitochondria and the nucleus are suggested to have anterograde (nucleus to mitochondria) and retrograde signaling to maintain homeostasis and prevent the triggering of cell death pathways (Galganska et al. 2010). Oversaturation of ROS can modify proteins involved in cell death pathways and damage key mitochondrial proteins (Luce et al. 2010). The other consequence of ROS would be the modification of lipids in the mitochondria, which may influence the release of cytochrome c (Ott et al. 2002). In the retina, P53 and HDAC4 are two important components regulating photoreceptor cell death partly because of their sensitivity to oxidative stress (Ali et al. 1998; Chen and Cepko 2009).
11.7 Concluding Remarks and Therapeutic Perspective
Photoreceptor degeneration has often been associated with classical caspase-dependent apoptotic pathways. However, as discussed here, caspase-independent apoptosis and nonapoptotic pathways, such as necrosis or autophagy, can also play important roles. In addition, crosstalk among different death pathways would make therapeutic approaches for photoreceptor degeneration challenging (Doonan et al. 2005; Lohr et al. 2006). One target can be mitochondria, which are one of the main hubs to integrate cellular stress signal and activate downstream cell death pathways (Lezi and Swerdlow 2012; Newmeyer and Ferguson-Miller 2003).
Retinal dystrophies offer great therapeutic potential because of easy anatomical access of the eye, an immunologically confined environment, and availability of noninvasive tools for assessing treatment outcomes in clinical trials. Such tools include direct vision assessment, the electroretinogram to test retinal function, and optical coherence tomography, which provides live imaging of the retina.
To prevent loss of vision in individuals with predisposing mutations, several approaches must be attempted concurrently: gene therapy, cell therapy, prosthesis implantation, and more recently optogenetic methods. Neuroprotection can also be used to preserve photoreceptors by blocking cell death or to strengthen endogenous pro-survival pathways. Such methods present the advantage of being mutation independent. However, studies using neurotrophic factors to delay degeneration showed significant variation in efficiency depending on the type of animal model studied, reflecting the complexity of cell death pathways (Trifunovic et al. 2012). The heterogeneous effects observed with neurotrophic factors suggest that multiple and combined pharmacological approaches may be required to thwart photoreceptor cell death.
Neurotrophic factors (i.e., bFGF, NGF, BDNF, PEDF, CNTF) have shown different degrees of neuroprotection in mouse models. Among these neurotrophic factors, ciliary neurotrophic factor (CNTF) has shown efficiency in more than ten different animal models (Wen et al. 2012; MacDonald et al. 2007). The use of encapsulated CNTF-expressing cells implanted in the eye of RP patients has shown promising results, especially for cone preservation, although the protective mechanism is still unclear (Sieving et al. 2006; Talcott et al. 2011). However, sustained CNTF expression leads to morphological and functional alterations (Bok et al. 2002; Rhee et al. 2007). It is still unclear if CNTF directly protects photoreceptors or indirectly by activating secretion from Müller glia cells of extrinsic factors having neuroprotective effects on photoreceptors.
As discussed here, oxidative stress and photoreceptor cell death are linked, and endogenous antioxidants play critical roles in redox homeostasis and photoreceptor survival (i.e., RdCVF, glutathione-S-transferase), which have been tested for different type of retinal dystrophies. For instance, in a clinical trial involving patients with RP, the combination of vitamin E, diltiazem, and taurin showed protective effects by delaying the progressive visual field reduction (Pasantes-Morales et al. 2002). Still, therapeutic success appears to be dependent on the genetic background and the nature of the mutation, as described for neurotrophic factors. In regard to the great variety of genetic modifications leading to photoreceptor cell death, it is critical to identify downstream death effectors commonly used in most mutant photoreceptors. The identification of a drug inhibiting such common death effectors will be of interest for further pharmaceutical development as it could have broader neuroprotective effects in a large number of retinal diseases. All these approaches require a detailed understanding of death pathways activated during photoreceptor degeneration and of the intracellular effectors mediating neuroprotection. The technological advances made during the past few years in high-throughput genomic sequencing and in proteomics will certainly further our understanding of cellular mechanisms mediating photoreceptor homeostasis and assist in the development of therapeutics.
References
Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H (2009) Purinergic signalling in the nervous system: an overview. Trends Neurosci 32(1):19–29. doi:10.1016/j.tins.2008.10.001
Abe T, Tsuda T, Yoshida M, Wada Y, Kano T, Itoyama Y, Tamai M (2000) Macular degeneration associated with aberrant expansion of trinucleotide repeat of the SCA7 gene in 2 Japanese families. Arch Ophthalmol 118(10):1415–1421
Ali RR, Reichel MB, Kanuga N, Munro PM, Alexander RA, Clarke AR, Luthert PJ, Bhattacharya SS, Hunt DM (1998) Absence of p53 delays apoptotic photoreceptor cell death in the rds mouse. Curr Eye Res 17(9):917–923
Allikmets R (1997) A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Gen 17(1):122. doi:10.1038/ng0997-122a
Ambati J, Fowler BJ (2012) Mechanisms of age-related macular degeneration. Neuron 75(1):26–39. doi:10.1016/j.neuron.2012.06.018
Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10(Suppl):S18–S25. doi:10.1038/nrn1434
Anderson KM, Seed T, Ou D, Harris JE (1999) Free radicals and reactive oxygen species in programmed cell death. Med Hypotheses 52(5):451–463. doi:10.1054/mehy.1997.0521
Applebury ML, Antoch MP, Baxter LC, Chun LL, Falk JD, Farhangfar F, Kage K, Krzystolik MG, Lyass LA, Robbins JT (2000) The murine cone photoreceptor: a single cone type expresses both S and M opsins with retinal spatial patterning. Neuron 27(3):513–523
Balmer D, Emery M, Andreux P, Auwerx J, Ginet V, Puyal J, Schorderet DF, Roduit R (2013) Autophagy defect is associated with low glucose-induced apoptosis in 661W photoreceptor Cells. PloS One 8(9):e74162. doi:10.1371/journal.pone.0074162
Barth E, Radermacher P, Szabo C (2006) The world according to poly(ADP-ribose) polymerase (PARP)–update 2006. Intensive Care Med 32(10):1470–1474. doi:10.1007/s00134-006-0336-x
Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292(5521):1552–1555. doi:10.1126/science.292.5521.1552
Bergmann M, Schutt F, Holz FG, Kopitz J (2004) Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J 18(3):562–564. doi:10.1096/fj.03-0289fje
Bok D, Yasumura D, Matthes MT, Ruiz A, Duncan JL, Chappelow AV, Zolutukhin S, Hauswirth W, LaVail MM (2002) Effects of adeno-associated virus-vectored ciliary neurotrophic factor on retinal structure and function in mice with a P216L rds/peripherin mutation. Exp Eye Res 74(6):719–735
Borner C, Monney L (1999) Apoptosis without caspases: an inefficient molecular guillotine? Cell Death Differ 6(6):497–507. doi:10.1038/sj.cdd.4400525
Bouchier-Hayes L, Lartigue L, Newmeyer DD (2005) Mitochondria: pharmacological manipulation of cell death. J Clin Invest 115(10):2640–2647. doi:10.1172/JCI26274
Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB (1990) Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature (Lond) 347(6294):677–680. doi:10.1038/347677a0
Bowne SJ, Sullivan LS, Blanton SH, Cepko CL, Blackshaw S, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP (2002) Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet 11(5):559–568
Bramall AN, Wright AF, Jacobson SG, McInnes RR (2010) The genomic, biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu Rev Neurosci 33:441–472. doi:10.1146/annurev-neuro-060909-153227
Brenner D, Mak TW (2009) Mitochondrial cell death effectors. Curr Opin Cell Biol 21(6):871–877. doi:10.1016/j.ceb.2009.09.004
Burns J, Clarke G, Lumsden CJ (2002) Photoreceptor death: spatiotemporal patterns arising from one-hit death kinetics and a diffusible cell death factor. Bull Math Biol 64(6):1117–1145. doi:10.1006/bulm.2002.0320
Cande C, Cecconi F, Dessen P, Kroemer G (2002) Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci 115(pt 24):4727–4734
Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J (2007) Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci 27(35):9278–9293. doi:10.1523/JNEUROSCI.2826-07.2007
Cao X, Phillis JW (1995) The free radical scavenger, alpha-lipoic acid, protects against cerebral ischemia-reperfusion injury in gerbils. Free Radic Res 23(4):365–370
Carmody RJ, Cotter TG (2000) Oxidative stress induces caspase-independent retinal apoptosis in vitro. Cell Death Differ 7(3):282–291. doi:10.1038/sj.cdd.4400646
Carter-Dawson LD, LaVail MM (1979) Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol 188(2):245–262. doi:10.1002/cne.901880204
Chahory S, Keller N, Martin E, Omri B, Crisanti P, Torriglia A (2010) Light induced retinal degeneration activates a caspase-independent pathway involving cathepsin D. Neurochem Int 57(3):278–287. doi:10.1016/j.neuint.2010.06.006
Chamberlain M, Baird P, Dirani M, Guymer R (2006) Unraveling a complex genetic disease: age-related macular degeneration. Surv Ophthalmol 51(6):576–586. doi:10.1016/j.survophthal.2006.08.003
Chang CJ, Lai WW, Edward DP, Tso MO (1995) Apoptotic photoreceptor cell death after traumatic retinal detachment in humans. Arch Ophthalmol 113(7):880–886
Chen B, Cepko CL (2009) HDAC4 regulates neuronal survival in normal and diseased retinas. Science 323(5911):256–259. doi:10.1126/science.1166226
Christofferson DE, Yuan J (2010) Cyclophilin A release as a biomarker of necrotic cell death. Cell Death Differ 17(12):1942–1943. doi:10.1038/cdd.2010.123
Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, Pang JJ, Sumaroka A, Windsor EA, Wilson JM, Flotte TR, Fishman GA, Heon E, Stone EM, Byrne BJ, Jacobson SG, Hauswirth WW (2008) Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA 105(39): 15112–15117. doi:10.1073/pnas.0807027105
Cideciyan AV, Jacobson SG, Beltran WA, Hauswirth WW, Aguirre GD (2013) Reply to Townes-Anderson: RPE65 gene therapy does not alter the natural history of retinal degeneration. Proc Natl Acad Sci USA 110(19):E1706
Clarke G, Lumsden CJ, McInnes RR (2001) Inherited neurodegenerative diseases: the one-hit model of neurodegeneration. Hum Mol Gen 10(20):2269–2275
Comitato A, Spampanato C, Chakarova C, Sanges D, Bhattacharya SS, Marigo V (2007) Mutations in splicing factor PRPF3, causing retinal degeneration, form detrimental aggregates in photoreceptor cells. Hum Mol Genet 16(14):1699–1707. doi:10.1093/hmg/ddm118
Cook B, Lewis GP, Fisher SK, Adler R (1995) Apoptotic photoreceptor degeneration in experimental retinal detachment. Invest Ophthalmol Vis Sci 36(6):990–996
Coppieters F, Casteels I, Meire F, De Jaegere S, Hooghe S, van Regemorter N, Van Esch H, Matuleviciene A, Nunes L, Meersschaut V, Walraedt S, Standaert L, Coucke P, Hoeben H, Kroes HY, Vande Walle J, de Ravel T, Leroy BP, De Baere E (2010) Genetic screening of LCA in Belgium: predominance of CEP290 and identification of potential modifier alleles in AHI1 of CEP290-related phenotypes. Hum Mut 31(10):E1709–E1766. doi:10.1002/humu.21336
Cornish EE, Madigan MC, Natoli R, Hales A, Hendrickson AE, Provis JM (2005) Gradients of cone differentiation and FGF expression during development of the foveal depression in macaque retina. Vis Neurosci 22(4):447–459. doi:10.1017/S0952523805224069
Costa BL, Fawcett R, Li GY, Safa R, Osborne NN (2008) Orally administered epigallocatechin gallate attenuates light-induced photoreceptor damage. Brain Res Bull 76(4):412–423. doi:10.1016/j.brainresbull.2008.01.022
Cronin T, Raffelsberger W, Lee-Rivera I, Jaillard C, Niepon ML, Kinzel B, Clerin E, Petrosian A, Picaud S, Poch O, Sahel JA, Leveillard T (2010) The disruption of the rod-derived cone viability gene leads to photoreceptor dysfunction and susceptibility to oxidative stress. Cell Death Differ 17(7):1199–1210. doi:10.1038/cdd.2010.2
Cuervo AM (2010) Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab 21(3):142–150. doi:10.1016/j.tem.2009.10.003
Cuervo AM, Dice JF (2000) Regulation of lamp2a levels in the lysosomal membrane. Traffic 1(7):570–583
Davis MD, Gangnon RE, Lee LY, Hubbard LD, Klein BE, Klein R, Ferris FL, Bressler SB, Milton RC, Age-Related Eye Disease Study Group (2005) The Age-Related Eye Disease Study severity scale for age-related macular degeneration: AREDS Report No. 17. Arch Ophthalmol 123(11):1484–1498. doi:10.1001/archopht.123.11.1484
den Hollander AI, Roepman R, Koenekoop RK, Cremers FP (2008) Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res 27(4):391–419. doi:10.1016/j.preteyeres.2008.05.003
Donovan M, Carmody RJ, Cotter TG (2001) Light-induced photoreceptor apoptosis in vivo requires neuronal nitric-oxide synthase and guanylate cyclase activity and is caspase-3-independent. J Biol Chem 276(25):23000–23008. doi:10.1074/jbc.M005359200
Donovan M, Cotter TG (2002) Caspase-independent photoreceptor apoptosis in vivo and differential expression of apoptotic protease activating factor-1 and caspase-3 during retinal development. Cell Death Differ 9(11):1220–1231. doi:10.1038/sj.cdd.4401105
Doonan F, Donovan M, Cotter TG (2003) Caspase-independent photoreceptor apoptosis in mouse models of retinal degeneration. J Neurosci 23(13):5723–5731
Doonan F, Donovan M, Cotter TG (2005) Activation of multiple pathways during photoreceptor apoptosis in the rd mouse. Invest Ophthalmol Vis Sci 46(10):3530–3538. doi:10.1167/iovs.05-0248
Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ (2006) Activation of IKK by TNF-alpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 22(2):245–257. doi:10.1016/j.molcel.2006.03.026
Fahim AT, Bowne SJ, Sullivan LS, Webb KD, Williams JT, Wheaton DK, Birch DG, Daiger SP (2011) Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to RPGR mutations. PloS One 6(8):e23021. doi:10.1371/journal.pone.0023021
Fain GL, Hardie R, Laughlin SB (2010) Phototransduction and the evolution of photoreceptors. Curr Biol 20(3):R114–R124. doi:10.1016/j.cub.2009.12.006
Fang P, Schachner M, Shen YQ (2012) HMGB1 in development and diseases of the central nervous system. Mol Neurobiol 45(3):499–506. doi:10.1007/s12035-012-8264-y
Finkbeiner S (2000) CREB couples neurotrophin signals to survival messages. Neuron 25(1):11–14
Fletcher EL (2010) Mechanisms of photoreceptor death during retinal degeneration. Optom Vis Sci 87(4):269–275. doi:10.1097/OPX.0b013e3181c9132b
Fox DA, Poblenz AT, He L (1999) Calcium overload triggers rod photoreceptor apoptotic cell death in chemical-induced and inherited retinal degenerations. Ann NY Acad Sci 893:282–285
Fox DA, Poblenz AT, He L, Harris JB, Medrano CJ (2003) Pharmacological strategies to block rod photoreceptor apoptosis caused by calcium overload: a mechanistic target-site approach to neuroprotection. Eur J Ophthalmol 13(suppl 3):S44–S56
Franke H, Illes P (2006) Involvement of P2 receptors in the growth and survival of neurons in the CNS. Pharmacol Ther 109(3):297–324. doi:10.1016/j.pharmthera.2005.06.002
Freund CL, Wang QL, Chen S, Muskat BL, Wiles CD, Sheffield VC, Jacobson SG, McInnes RR, Zack DJ, Stone EM (1998) De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Gen 18(4):311–312. doi:10.1038/ng0498-311
Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J, Eye Diseases Prevalence Research Group (2004) Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 122(4):564–572. doi:10.1001/archopht.122.4.564
Friedman JS, Ray JW, Waseem N, Johnson K, Brooks MJ, Hugosson T, Breuer D, Branham KE, Krauth DS, Bowne SJ, Sullivan LS, Ponjavic V, Granse L, Khanna R, Trager EH, Gieser LM, Hughbanks-Wheaton D, Cojocaru RI, Ghiasvand NM, Chakarova CF, Abrahamson M, Goring HH, Webster AR, Birch DG, Abecasis GR, Fann Y, Bhattacharya SS, Daiger SP, Heckenlively JR, Andreasson S, Swaroop A (2009) Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am J Hum Genet 84(6):792–800. doi:10.1016/j.ajhg.2009.05.007
Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, Igo RP, Jr., Buitendijk GH, Sim X, Weeks DE, Guymer RH, Merriam JE, Francis PJ, Hannum G, Agarwal A, Armbrecht AM, Audo I, Aung T, Barile GR, Benchaboune M, Bird AC, Bishop PN, Branham KE, Brooks M, Brucker AJ, Cade WH, Cain MS, Campochiaro PA, Chan CC, Cheng CY, Chew EY, Chin KA, Chowers I, Clayton DG, Cojocaru R, Conley YP, Cornes BK, Daly MJ, Dhillon B, Edwards AO, Evangelou E, Fagerness J, Ferreyra HA, Friedman JS, Geirsdottir A, George RJ, Gieger C, Gupta N, Hagstrom SA, Harding SP, Haritoglou C, Heckenlively JR, Holz FG, Hughes G, Ioannidis JP, Ishibashi T, Joseph P, Jun G, Kamatani Y, Katsanis N, C NK, Khan JC, Kim IK, Kiyohara Y, Klein BE, Klein R, Kovach JL, Kozak I, Lee CJ, Lee KE, Lichtner P, Lotery AJ, Meitinger T, Mitchell P, Mohand-Said S, Moore AT, Morgan DJ, Morrison MA, Myers CE, Naj AC, Nakamura Y, Okada Y, Orlin A, Ortube MC, Othman MI, Pappas C, Park KH, Pauer GJ, Peachey NS, Poch O, Priya RR, Reynolds R, Richardson AJ, Ripp R, Rudolph G, Ryu E, Sahel JA, Schaumberg DA, Scholl HP, Schwartz SG, Scott WK, Shahid H, Sigurdsson H, Silvestri G, Sivakumaran TA, Smith RT, Sobrin L, Souied EH, Stambolian DE, Stefansson H, Sturgill-Short GM, Takahashi A, Tosakulwong N, Truitt BJ, Tsironi EE, Uitterlinden AG, van Duijn CM, Vijaya L, Vingerling JR, Vithana EN, Webster AR, Wichmann HE, Winkler TW, Wong TY, Wright AF, Zelenika D, Zhang M, Zhao L, Zhang K, Klein ML, Hageman GS, Lathrop GM, Stefansson K, Allikmets R, Baird PN, Gorin MB, Wang JJ, Klaver CC, Seddon JM, Pericak-Vance MA, Iyengar SK, Yates JR, Swaroop A, Weber BH, Kubo M, Deangelis MM, Leveillard T, Thorsteinsdottir U, Haines JL, Farrer LA, Heid IM, Abecasis GR, Consortium AMDG (2013) Seven new loci associated with age-related macular degeneration. Nat Genet 45(4):433–439, 439e, 431–432. doi:10.1038/ng.2578
Galganska H, Karachitos A, Wojtkowska M, Stobienia O, Budzinska M, Kmita H (2010) Communication between mitochondria and nucleus: putative role for VDAC in reduction/oxidation mechanism. Biochim Biophys Acta 1797(6-7):1276–1280. doi:10.1016/j.bbabio.2010.02.004
Gardner JC, Webb TR, Kanuga N, Robson AG, Holder GE, Stockman A, Ripamonti C, Ebenezer ND, Ogun O, Devery S, Wright GA, Maher ER, Cheetham ME, Moore AT, Michaelides M, Hardcastle AJ (2010) X-linked cone dystrophy caused by mutation of the red and green cone opsins. Am J Hum Genet 87(1):26–39. doi:10.1016/j.ajhg.2010.05.019
Goetz KE, Reeves MJ, Tumminia SJ, Brooks BP (2012) eyeGENE(R): a novel approach to combine clinical testing and researching genetic ocular disease. Curr Opin Ophthalmol 23(5):355–363. doi:10.1097/ICU.0b013e32835715c9
Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R (1995) In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 21(5):1465–1468
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305(5684):626–629. doi:10.1126/science.1099320
Gregory CY, Bird AC (1995) Cell loss in retinal dystrophies by apoptosis–death by informed consent! Br J Ophthalmol 79(2):186–190
Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A (1997) Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet 17(2):194–197. doi:10.1038/ng1097-194
Hafezi F, Steinbach JP, Marti A, Munz K, Wang ZQ, Wagner EF, Aguzzi A, Reme CE (1997) The absence of c-fos prevents light-induced apoptotic cell death of photoreceptors in retinal degeneration in vivo. Nat Med 3(3):346–349
Hamel CP (2007) Cone rod dystrophies. Orphanet J Rare Dis 2:7. doi:10.1186/1750-1172-2-7
Hauck SM, Ekstrom PA, Ahuja-Jensen P, Suppmann S, Paquet-Durand F, van Veen T, Ueffing M (2006) Differential modification of phosducin protein in degenerating rd1 retina is associated with constitutively active Ca2+/calmodulin kinase II in rod outer segments. Mol Cell Proteomics 5(2):324–336. doi:10.1074/mcp.M500217-MCP200
Helmlinger D, Yvert G, Picaud S, Merienne K, Sahel J, Mandel JL, Devys D (2002) Progressive retinal degeneration and dysfunction in R6 Huntington’s disease mice. Hum Mol Gen 11(26):3351–3359
Hisatomi T, Nakazawa T, Noda K, Almulki L, Miyahara S, Nakao S, Ito Y, She H, Kohno R, Michaud N, Ishibashi T, Hafezi-Moghadam A, Badley AD, Kroemer G, Miller JW (2008) HIV protease inhibitors provide neuroprotection through inhibition of mitochondrial apoptosis in mice. J Clin Invest 118(6):2025–2038. doi:10.1172/JCI34267
Hisatomi T, Sakamoto T, Murata T, Yamanaka I, Oshima Y, Hata Y, Ishibashi T, Inomata H, Susin SA, Kroemer G (2001) Relocalization of apoptosis-inducing factor in photoreceptor apoptosis induced by retinal detachment in vivo. Am J Pathol 158(4):1271–1278. doi:10.1016/S0002-9440(10)64078-3
Hoang QV, Linsenmeier RA, Chung CK, Curcio CA (2002) Photoreceptor inner segments in monkey and human retina: mitochondrial density, optics, and regional variation. Vis Neurosci 19(4):395–407
Huang L, Xiao X, Li S, Jia X, Wang P, Guo X, Zhang Q (2012) CRX variants in cone-rod dystrophy and mutation overview. Biochem Biophys Res Commun 426(4):498–503. doi:10.1016/j.bbrc.2012.08.110
Huang PC, Gaitan AE, Hao Y, Petters RM, Wong F (1993) Cellular interactions implicated in the mechanism of photoreceptor degeneration in transgenic mice expressing a mutant rhodopsin gene. Proc Natl Acad Sci USA 90(18):8484–8488
Hubbell WL, Bownds MD (1979) Visual transduction in vertebrate photoreceptors. Annu Rev Neurosci 2:17–34. doi:10.1146/annurev.ne.02.030179.000313
Illing ME, Rajan RS, Bence NF, Kopito RR (2002) A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem 277(37):34150–34160. doi:10.1074/jbc.M204955200
Jackson GR, Owsley C, Curcio CA (2002) Photoreceptor degeneration and dysfunction in aging and age-related maculopathy. Ageing Res Rev 1(3):381–396
Jacobson SG, Cideciyan AV, Iannaccone A, Weleber RG, Fishman GA, Maguire AM, Affatigato LM, Bennett J, Pierce EA, Danciger M, Farber DB, Stone EM (2000) Disease expression of RP1 mutations causing autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 41(7):1898–1908
Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, Peden MC, Aleman TS, Boye SL, Sumaroka A, Conlon TJ, Calcedo R, Pang JJ, Erger KE, Olivares MB, Mullins CL, Swider M, Kaushal S, Feuer WJ, Iannaccone A, Fishman GA, Stone EM, Byrne BJ, Hauswirth WW (2012) Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 130(1):9–24. doi:10.1001/archophthalmol.2011.298
Jagtap P, Szabo C (2005) Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 4(5):421–440. doi:10.1038/nrd1718
Joly S, Francke M, Ulbricht E, Beck S, Seeliger M, Hirrlinger P, Hirrlinger J, Lang KS, Zinkernagel M, Odermatt B, Samardzija M, Reichenbach A, Grimm C, Reme CE (2009) Cooperative phagocytes: resident microglia and bone marrow immigrants remove dead photoreceptors in retinal lesions. Am J Pathol 174(6):2310–2323. doi:10.2353/ajpath.2009.090023
Jomary C, Neal MJ, Jones SE (2001) Characterization of cell death pathways in murine retinal neurodegeneration implicates cytochrome c release, caspase activation, and bid cleavage. Mol Cell Neurosci 18(4):335–346. doi:10.1006/mcne.2001.1036
Justilien V, Pang JJ, Renganathan K, Zhan X, Crabb JW, Kim SR, Sparrow JR, Hauswirth WW, Lewin AS (2007) SOD2 knockdown mouse model of early AMD. Invest Ophthalmol Vis Sci 48(10):4407–4420. doi:10.1167/iovs.07-0432
Kaarniranta K, Hyttinen J, Ryhanen T, Viiri J, Paimela T, Toropainen E, Sorri I, Salminen A (2010) Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front Biosci (Elite Ed) 2:1374–1384
Kaarniranta K, Sinha D, Blasiak J, Kauppinen A, Vereb Z, Salminen A, Boulton ME, Petrovski G (2013) Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 9(7):973–984
Kagan VE, Shvedova AA, Novikov KN, Kozlov YP (1973) Light-induced free radical oxidation of membrane lipids in photoreceptors of frog retina. Biochim Biophys Acta 330(1):76–79
Kaur J, Mencl S, Sahaboglu A, Farinelli P, van Veen T, Zrenner E, Ekstrom P, Paquet-Durand F, Arango-Gonzalez B (2011) Calpain and PARP activation during photoreceptor cell death in P23H and S334ter rhodopsin mutant rats. PloS One 6(7):e22181. doi:10.1371/journal.pone.0022181
Kaushik S, Massey AC, Mizushima N, Cuervo AM (2008) Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 19(5):2179–2192. doi:10.1091/mbc.E07-11-1155
Kedzierski W, Bok D, Travis GH (1998) Non-cell-autonomous photoreceptor degeneration in rds mutant mice mosaic for expression of a rescue transgene. J Neurosci 18(11):4076–4082
Khandhadia S, Lotery A (2010) Oxidation and age-related macular degeneration: insights from molecular biology. Expert Rev Mol Med 12:e34. doi:10.1017/S146239941000164X
Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, Maubaret C, Diaz-Font A, MacDonald I, Muzny DM, Wheeler DA, Morgan M, Lewis LR, Logan CV, Tan PL, Beer MA, Inglehearn CF, Lewis RA, Jacobson SG, Bergmann C, Beales PL, Attie-Bitach T, Johnson CA, Otto EA, Bhattacharya SS, Hildebrandt F, Gibbs RA, Koenekoop RK, Swaroop A, Katsanis N (2009) A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet 41(6):739–745. doi:10.1038/ng.366
Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, Ferguson TA (2013) Noncanonical autophagy promotes the visual cycle. Cell 154(2):365–376. doi:10.1016/j.cell.2013.06.012
Klein JA, Ackerman SL (2003) Oxidative stress, cell cycle, and neurodegeneration. J Clin Invest 111(6):785–793. doi:10.1172/JCI18182
Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL (2002) The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature (Lond) 419(6905):367–374. doi:10.1038/nature01034
Koenekoop RK (2004) An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol 49(4):379–398. doi:10.1016/j.survophthal.2004.04.003
Kolesnikov AV, Fan J, Crouch RK, Kefalov VJ (2010) Age-related deterioration of rod vision in mice. J Neurosci 30(33):11222–11231. doi:10.1523/JNEUROSCI.4239-09.2010
Komeima K, Rogers BS, Lu L, Campochiaro PA (2006) Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci USA 103(30):11300–11305. doi:10.1073/pnas.0604056103
Komeima K, Usui S, Shen J, Rogers BS, Campochiaro PA (2008) Blockade of neuronal nitric oxide synthase reduces cone cell death in a model of retinitis pigmentosa. Free Radic Biol Med 45(6):905–912. doi:10.1016/j.freeradbiomed.2008.06.020
Kong L, Zhou X, Li F, Yodoi J, McGinnis J, Cao W (2010) Neuroprotective effect of overexpression of thioredoxin on photoreceptor degeneration in Tubby mice. Neurobiol Dis 38(3):446–455. doi:10.1016/j.nbd.2010.03.005
Kowluru RA, Odenbach S (2004) Role of interleukin-1beta in the development of retinopathy in rats: effect of antioxidants. Invest Ophthalmol Vis Sci 45(11):4161–4166. doi:10.1167/iovs.04-0633
Kraft C, Peter M, Hofmann K (2010) Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 12(9):836–841. doi:10.1038/ncb0910-836
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G, Nomenclature Committee on Cell Death (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16(1):3–11. doi:10.1038/cdd.2008.150
Krohne TU, Stratmann NK, Kopitz J, Holz FG (2010) Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp Eye Res 90(3):465–471. doi:10.1016/j.exer.2009.12.011
Kunchithapautham K, Rohrer B (2007a) Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy 3(5):433–441
Kunchithapautham K, Rohrer B (2007b) Autophagy is one of the multiple mechanisms active in photoreceptor degeneration. Autophagy 3(1):65–66
Langmann T (2007) Microglia activation in retinal degeneration. J Leukoc Biol 81(6):1345–1351. doi:10.1189/jlb.0207114
LaVail MM (1981) Analysis of neurological mutants with inherited retinal degeneration. Friedenwald Lecture. Invest Ophthalmol Vis Sci 21(5):638–657
Lemarie A, Lagadic-Gossmann D, Morzadec C, Allain N, Fardel O, Vernhet L (2004) Cadmium induces caspase-independent apoptosis in liver Hep3B cells: role for calcium in signaling oxidative stress-related impairment of mitochondria and relocation of endonuclease G and apoptosis-inducing factor. Free Radic Biol Med 36(12):1517–1531. doi:10.1016/j.freeradbiomed.2004.03.020
Leveillard T, Mohand-Said S, Lorentz O, Hicks D, Fintz AC, Clerin E, Simonutti M, Forster V, Cavusoglu N, Chalmel F, Dolle P, Poch O, Lambrou G, Sahel JA (2004) Identification and characterization of rod-derived cone viability factor. Nat Genet 36(7):755–759. doi:10.1038/ng1386
Lezi E, Swerdlow RH (2012) Mitochondria in neurodegeneration. Adv Exp Med Biol 942:269–286. doi:10.1007/978-94-007-2869-1_12
Liang FQ, Godley BF (2003) Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res 76(4):397–403
Lim JH, Wickremasinghe SS, Xie J, Chauhan DS, Baird PN, Robman LD, Hageman G, Guymer RH (2012) Delay to treatment and visual outcomes in patients treated with anti-vascular endothelial growth factor for age-related macular degeneration. Am J Ophthalmol 153(4):678–686. doi:10.1016/j.ajo.2011.09.013
Lim LS, Wong TY (2012) Lipids and diabetic retinopathy. Expert Opin Biol Ther 12(1):93–105. doi:10.1517/14712598.2012.641531
Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86(1):147–157
Lo AC, Woo TT, Wong RL, Wong D (2011) Apoptosis and other cell death mechanisms after retinal detachment: implications for photoreceptor rescue. Ophthalmologica 226(suppl 1):10–17. doi:10.1159/000328206
Lobanova ES, Finkelstein S, Skiba NP, Arshavsky VY (2013) Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc Natl Acad Sci USA 110(24):9986–9991. doi:10.1073/pnas.1305521110
Lohr HR, Kuntchithapautham K, Sharma AK, Rohrer B (2006) Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp Eye Res 83(2):380–389. doi:10.1016/j.exer.2006.01.014
Long KO, Fisher SK, Fariss RN, Anderson DH (1986) Disc shedding and autophagy in the cone-dominant ground squirrel retina. Exp Eye Res 43(2):193–205
Luce K, Weil AC, Osiewacz HD (2010) Mitochondrial protein quality control systems in aging and disease. Adv Exp Med Biol 694:108–125
MacDonald IM, Sauve Y, Sieving PA (2007) Preventing blindness in retinal disease: ciliary neurotrophic factor intraocular implants. Can J Ophthalmol 42(3):399–402. doi:10.3129/can j ophthalmol.i07-039
Mahajan VB, Skeie JM, Bassuk AG, Fingert JH, Braun TA, Daggett HT, Folk JC, Sheffield VC, Stone EM (2012) Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet 8(10):e1003001. doi:10.1371/journal.pgen.1003001
Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schutz G (2002) Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 31(1):47–54. doi:10.1038/ng882
Marino G, Madeo F, Kroemer G (2011) Autophagy for tissue homeostasis and neuroprotection. Curr Opin Cell Biol 23(2):198–206. doi:10.1016/j.ceb.2010.10.001
Marlhens F, Bareil C, Griffoin JM, Zrenner E, Amalric P, Eliaou C, Liu SY, Harris E, Redmond TM, Arnaud B, Claustres M, Hamel CP (1997) Mutations in RPE65 cause Leber’s congenital amaurosis. Nat Genet 17(2):139–141. doi:10.1038/ng1097-139
Massey A, Kiffin R, Cuervo AM (2004) Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell Biol 36(12):2420–2434. doi:10.1016/j.biocel.2004.04.010
Massey AC, Follenzi A, Kiffin R, Zhang C, Cuervo AM (2008) Early cellular changes after blockage of chaperone-mediated autophagy. Autophagy 4(4):442–456
McBee JK, Palczewski K, Baehr W, Pepperberg DR (2001) Confronting complexity: the interlink of phototransduction and retinoid metabolism in the vertebrate retina. Prog Retin Eye Res 20(4):469–529
McCall K (2010) Genetic control of necrosis: another type of programmed cell death. Curr Opin Cell Biol 22(6):882–888. doi:10.1016/j.ceb.2010.09.002
McLaughlin ME, Sandberg MA, Berson EL, Dryja TP (1993) Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet 4(2):130–134. doi:10.1038/ng0693-130
Metrailler S, Schorderet DF, Cottet S (2012) Early apoptosis of rod photoreceptors in Rpe65(−/−) mice is associated with the upregulated expression of lysosomal-mediated autophagic genes. Exp Eye Res 96(1):70–81. doi:10.1016/j.exer.2011.12.019
Michaelides M, Wilkie SE, Jenkins S, Holder GE, Hunt DM, Moore AT, Webster AR (2005) Mutation in the gene GUCA1A, encoding guanylate cyclase-activating protein 1, causes cone, cone-rod, and macular dystrophy. Ophthalmology 112(8):1442–1447. doi:10.1016/j.ophtha.2005.02.024
Mittag TW, Bayer AU, La VM (1999) Light-induced retinal damage in mice carrying a mutated SOD I gene. Exp Eye Res 69(6):677–683. doi:10.1006/exer.1999.0748
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature (Lond) 451(7182):1069–1075. doi:10.1038/nature06639
Molday RS, Zhang K (2010) Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog Lipid Res 49(4):476–492. doi:10.1016/j.plipres.2010.07.002
Moquin D, Chan FK (2010) The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci 35(8):434–441. doi:10.1016/j.tibs.2010.03.001
Moreira PI, Sayre LM, Zhu X, Nunomura A, Smith MA, Perry G (2010) Detection and localization of markers of oxidative stress by in situ methods: application in the study of Alzheimer disease. Methods Mol Biol 610:419–434. doi:10.1007/978-1-60327-029-8_25
Murakami Y, Matsumoto H, Roh M, Giani A, Kataoka K, Morizane Y, Kayama M, Thanos A, Nakatake S, Notomi S, Hisatomi T, Ikeda Y, Ishibashi T, Connor KM, Miller JW, Vavvas DG (2013) Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. doi:10.1038/cdd.2013.109
Murakami Y, Matsumoto H, Roh M, Suzuki J, Hisatomi T, Ikeda Y, Miller JW, Vavvas DG (2012) Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc Natl Acad Sci USA 109(36):14598–14603. doi:10.1073/pnas.1206937109
Nakazawa M (2011) Effects of calcium ion, calpains, and calcium channel blockers on retinitis pigmentosa. J Ophthalmol 2011:292040. doi:10.1155/2011/292040
Neal M, Cunningham J (1994) Modulation by endogenous ATP of the light-evoked release of ACh from retinal cholinergic neurones. Br J Pharmacol 113(4):1085–1087
Newman EA (2003) Glial cell inhibition of neurons by release of ATP. J Neurosci 23(5):1659–1666
Newmeyer DD, Ferguson-Miller S (2003) Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112(4):481–490
Ng L, Hurley JB, Dierks B, Srinivas M, Salto C, Vennstrom B, Reh TA, Forrest D (2001) A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet 27(1):94–98. doi:10.1038/83829
Ng TF, Streilein JW (2001) Light-induced migration of retinal microglia into the subretinal space. Invest Ophthalmol Vis Sci 42(13):3301–3310
Nickla DL, Wallman J (2010) The multifunctional choroid. Prog Retin Eye Res 29(2):144–168. doi:10.1016/j.preteyeres.2009.12.002
Notomi S, Hisatomi T, Kanemaru T, Takeda A, Ikeda Y, Enaida H, Kroemer G, Ishibashi T (2011) Critical involvement of extracellular ATP acting on P2RX7 purinergic receptors in photoreceptor cell death. Am J Pathol 179(6):2798–2809. doi:10.1016/j.ajpath.2011.08.035
Notomi S, Hisatomi T, Murakami Y, Terasaki H, Sonoda S, Asato R, Takeda A, Ikeda Y, Enaida H, Sakamoto T, Ishibashi T (2013) Dynamic increase in extracellular ATP accelerates photoreceptor cell apoptosis via ligation of P2RX7 in subretinal hemorrhage. PloS One 8(1):e53338. doi:10.1371/journal.pone.0053338
Oguni M, Tamura H, Kato K, Setogawa T (1996) Chronic retinal effects by ultraviolet irradiation, with special reference to superoxide dismutases. Histol Histopathol 11(3):695–702
Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol 4(7):552–565. doi:10.1038/nrm1150
Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S (2002) Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 99(3):1259–1263. doi:10.1073/pnas.241655498
Ozaki T, Ishiguro S, Hirano S, Baba A, Yamashita T, Tomita H, Nakazawa M (2013) Inhibitory peptide of mitochondrial mu-calpain protects against photoreceptor degeneration in rhodopsin transgenic S334ter and P23H rats. PloS One 8(8):e71650. doi:10.1371/journal.pone.0071650
Paquet-Durand F, Azadi S, Hauck SM, Ueffing M, van Veen T, Ekstrom P (2006) Calpain is activated in degenerating photoreceptors in the rd1 mouse. J Neurochem 96(3):802–814. doi:10.1111/j.1471-4159.2005.03628.x
Paquet-Durand F, Sanges D, McCall J, Silva J, van Veen T, Marigo V, Ekstrom P (2010) Photoreceptor rescue and toxicity induced by different calpain inhibitors. J Neurochem 115(4):930–940. doi:10.1111/j.1471-4159.2010.06983.x
Paquet-Durand F, Silva J, Talukdar T, Johnson LE, Azadi S, van Veen T, Ueffing M, Hauck SM, Ekstrom PA (2007) Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J Neurosci 27(38):10311–10319. doi:10.1523/JNEUROSCI.1514-07.2007
Pasantes-Morales H, Quiroz H, Quesada O (2002) Treatment with taurine, diltiazem, and vitamin E retards the progressive visual field reduction in retinitis pigmentosa: a 3-year follow-up study. Metab Brain Dis 17(3):183–197
Perche O, Doly M, Ranchon-Cole I (2007) Caspase-dependent apoptosis in light-induced retinal degeneration. Invest Ophthalmol Vis Sci 48(6):2753–2759. doi:10.1167/iovs.06-1258
Perez-Campo R, Lopez-Torres M, Cadenas S, Rojas C, Barja G (1998) The rate of free radical production as a determinant of the rate of aging: evidence from the comparative approach. J Comp Physiol B Biochem System Environ Physiol 168(3):149–158
Perkins GA, Ellisman MH, Fox DA (2003) Three-dimensional analysis of mouse rod and cone mitochondrial cristae architecture: bioenergetic and functional implications. Mol Vis 9:60–73
Portera-Cailliau C, Sung CH, Nathans J, Adler R (1994) Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci USA 91(3):974–978
Priya RR, Chew EY, Swaroop A (2012) Genetic studies of age-related macular degeneration: lessons, challenges, and opportunities for disease management. Ophthalmology 119(12):2526–2536. doi:10.1016/j.ophtha.2012.06.042
Punzo C, Kornacker K, Cepko CL (2009) Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci 12(1):44–52. doi:10.1038/nn.2234
Puthussery T, Fletcher E (2009) Extracellular ATP induces retinal photoreceptor apoptosis through activation of purinoceptors in rodents. J Comp Neurol 513(4):430–440. doi:10.1002/cne.21964
Puthussery T, Fletcher EL (2004) Synaptic localization of P2X7 receptors in the rat retina. J Comp Neurol 472(1):13–23. doi:10.1002/cne.20045
Puthussery T, Yee P, Vingrys AJ, Fletcher EL (2006) Evidence for the involvement of purinergic P2X receptors in outer retinal processing. Eur J Neurosci 24(1):7–19. doi:10.1111/j.1460-9568.2006.04895.x
Ratnapriya R, Swaroop A (2013) Genetic architecture of retinal and macular degenerative diseases: the promise and challenges of next-generation sequencing. Genome Med 5(9):84. doi:10.1186/gm488
Read DS, McCall MA, Gregg RG (2002) Absence of voltage-dependent calcium channels delays photoreceptor degeneration in rd mice. Exp Eye Res 75(4):415–420
Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K (1998) Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet 20(4):344–351. doi:10.1038/3813
Reme CE, Wolfrum U, Imsand C, Hafezi F, Williams TP (1999) Photoreceptor autophagy: effects of light history on number and opsin content of degradative vacuoles. Invest Ophthalmol Vis Sci 40(10):2398–2404
Rhee KD, Ruiz A, Duncan JL, Hauswirth WW, Lavail MM, Bok D, Yang XJ (2007) Molecular and cellular alterations induced by sustained expression of ciliary neurotrophic factor in a mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 48(3):1389–1400. doi:10.1167/iovs.06-0677
Rich KA, Zhan Y, Blanks JC (1997) Aberrant expression of c-Fos accompanies photoreceptor cell death in the rd mouse. J Neurobiol 32(6):593–612
Rodriguez-Muela N, Koga H, Garcia-Ledo L, de la Villa P, de la Rosa EJ, Cuervo AM, Boya P (2013) Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 12(3):478–488. doi:10.1111/acel.12072
Roger JE, Ranganath K, Zhao L, Cojocaru RI, Brooks M, Gotoh N, Veleri S, Hiriyanna A, Rachel RA, Campos MM, Fariss RN, Wong WT, Swaroop A (2012) Preservation of cone photoreceptors after a rapid yet transient degeneration and remodeling in cone-only Nrl−/− mouse retina. J Neurosci 32(2):528–541. doi:10.1523/JNEUROSCI.3591-11.2012
Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI (2010) Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res 88(7):1569–1576. doi:10.1002/jnr.22314
Rudolf M, Malek G, Messinger JD, Clark ME, Wang L, Curcio CA (2008) Sub-retinal drusenoid deposits in human retina: organization and composition. Exp Eye Res 87(5):402–408. doi:10.1016/j.exer.2008.07.010
Sahaboglu A, Tanimoto N, Kaur J, Sancho-Pelluz J, Huber G, Fahl E, Arango-Gonzalez B, Zrenner E, Ekstrom P, Lowenheim H, Seeliger M, Paquet-Durand F (2010) PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function. PloS One 5(11):e15495. doi:10.1371/journal.pone.0015495
Sampayo JN, Olsen A, Lithgow GJ (2003) Oxidative stress in Caenorhabditis elegans: protective effects of superoxide dismutase/catalase mimetics. Aging Cell 2(6):319–326
Sastre J, Pallardo FV, Vina J (2000) Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life 49(5):427–435. doi:10.1080/152165400410281
Schipper-Krom S, Juenemann K, Reits EA (2012) The ubiquitin-proteasome system in Huntington’s disease: are proteasomes impaired, initiators of disease, or coming to the rescue? Biochem Res Int 2012:837015. doi:10.1155/2012/837015
Schoch KM, von Reyn CR, Bian J, Telling GC, Meaney DF, Saatman KE (2013) Brain injury-induced proteolysis is reduced in a novel calpastatin-overexpressing transgenic mouse. J Neurochem 125(6):909–920. doi:10.1111/jnc.12144
Schwiebert EM (2000) Extracellular ATP-mediated propagation of Ca(2+) waves. Focus on mechanical strain-induced Ca(2+) waves are propagated via ATP release and purinergic receptor activation. Am J Physiol Cell Physiol 279(2):C281–283
Shahinfar S, Edward DP, Tso MO (1991) A pathologic study of photoreceptor cell death in retinal photic injury. Curr Eye Res 10(1):47–59
Sharma AK, Rohrer B (2004) Calcium-induced calpain mediates apoptosis via caspase-3 in a mouse photoreceptor cell line. J Biol Chem 279(34):35564–35572. doi:10.1074/jbc.M401037200
Sharon D, Blackshaw S, Cepko CL, Dryja TP (2002) Profile of the genes expressed in the human peripheral retina, macula, and retinal pigment epithelium determined through serial analysis of gene expression (SAGE). Proc Natl Acad Sci USA 99(1):315–320. doi:10.1073/pnas.012582799
Shen J, Yang X, Dong A, Petters RM, Peng YW, Wong F, Campochiaro PA (2005) Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J Cell Physiol 203(3):457–464. doi:10.1002/jcp.20346
Sherman MY, Goldberg AL (2001) Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29(1):15–32
Sieving PA, Caruso RC, Tao W, Coleman HR, Thompson DJ, Fullmer KR, Bush RA (2006) Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci USA 103(10):3896–3901. doi:10.1073/pnas.0600236103
Simunovic MP, Moore AT (1998) The cone dystrophies. Eye 12(pt 3b):553–565. doi:10.1038/eye.1998.145
Smith MA, Schnellmann RG (2012) Calpains, mitochondria, and apoptosis. Cardiovasc Res 96(1):32–37. doi:10.1093/cvr/cvs163
Smith W, Assink J, Klein R, Mitchell P, Klaver CC, Klein BE, Hofman A, Jensen S, Wang JJ, de Jong PT (2001) Risk factors for age-related macular degeneration: pooled findings from three continents. Ophthalmology 108(4):697–704
Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B (2003) Aggregated and monomeric alpha-synuclein bind to the S6' proteasomal protein and inhibit proteasomal function. J Biol Chem 278(14):11753–11759. doi:10.1074/jbc.M208641200
Solovei I, Kreysing M, Lanctot C, Kosem S, Peichl L, Cremer T, Guck J, Joffe B (2009) Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137(2):356–368. doi:10.1016/j.cell.2009.01.052
Stone EM (2007) Macular degeneration. Annu Rev Med 58:477–490. doi:10.1146/annurev.med.58.111405.133335
Sun H, Tsunenari T, Yau KW, Nathans J (2002) The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci USA 99(6):4008–4013. doi:10.1073/pnas.052692999
Surgucheva I, Ninkina N, Buchman VL, Grasing K, Surguchov A (2005) Protein aggregation in retinal cells and approaches to cell protection. Cell Mol Neurobiol 25(6):1051–1066. doi:10.1007/s10571-005-8474-1
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature (Lond) 397(6718):441–446. doi:10.1038/17135
Suzuki K, Hata S, Kawabata Y, Sorimachi H (2004) Structure, activation, and biology of calpain. Diabetes 53(suppl 1):S12–S18
Suzuki M, Kamei M, Itabe H, Yoneda K, Bando H, Kume N, Tano Y (2007) Oxidized phospholipids in the macula increase with age and in eyes with age-related macular degeneration. Mol Vis 13:772–778
Swaroop A, Branham KE, Chen W, Abecasis G (2007) Genetic susceptibility to age-related macular degeneration: a paradigm for dissecting complex disease traits. Human Mol Genet 16(Spec No. 2):R174–R182. doi:10.1093/hmg/ddm212
Swaroop A, Chew EY, Rickman CB, Abecasis GR (2009) Unraveling a multifactorial late-onset disease: from genetic susceptibility to disease mechanisms for age-related macular degeneration. Annu Rev Genomics Hum Genet 10:19–43. doi:10.1146/annurev.genom.9.081307.164350
Swaroop A, Kim D, Forrest D (2010) Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat Rev Neurosci 11(8):563–576. doi:10.1038/nrn2880
Tai HC, Schuman EM (2008) Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci 9(11):826–838. doi:10.1038/nrn2499
Takano Y, Ohguro H, Dezawa M, Ishikawa H, Yamazaki H, Ohguro I, Mamiya K, Metoki T, Ishikawa F, Nakazawa M (2004) Study of drug effects of calcium channel blockers on retinal degeneration of rd mouse. Biochem Biophys Res Commun 313(4):1015–1022
Talcott KE, Ratnam K, Sundquist SM, Lucero AS, Lujan BJ, Tao W, Porco TC, Roorda A, Duncan JL (2011) Longitudinal study of cone photoreceptors during retinal degeneration and in response to ciliary neurotrophic factor treatment. Invest Ophthalmol Vis Sci 52(5):2219–2226. doi:10.1167/iovs.10-6479
Tanaka T, Nakamura H, Nishiyama A, Hosoi F, Masutani H, Wada H, Yodoi J (2000) Redox regulation by thioredoxin superfamily: protection against oxidative stress and aging. Free Radic Res 33(6):851–855
Tanito M, Agbaga MP, Anderson RE (2007a) Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic Biol Med 42(12):1838–1850. doi:10.1016/j.freeradbiomed.2007.03.018
Tanito M, Kaidzu S, Anderson RE (2007b) Delayed loss of cone and remaining rod photoreceptor cells due to impairment of choroidal circulation after acute light exposure in rats. Invest Ophthalmol Vis Sci 48(4):1864–1872. doi:10.1167/iovs.06-1065
Tanito M, Masutani H, Kim YC, Nishikawa M, Ohira A, Yodoi J (2005) Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in mice. Invest Ophthalmol Vis Sci 46(3):979–987. doi:10.1167/iovs.04-1120
Tanito M, Masutani H, Nakamura H, Ohira A, Yodoi J (2002a) Cytoprotective effect of thioredoxin against retinal photic injury in mice. Invest Ophthalmol Vis Sci 43(4):1162–1167
Tanito M, Masutani H, Nakamura H, Oka S, Ohira A, Yodoi J (2002b) Attenuation of retinal photooxidative damage in thioredoxin transgenic mice. Neurosci Lett 326(2):142–146
Thapa A, Morris L, Xu J, Ma H, Michalakis S, Biel M, Ding XQ (2012) Endoplasmic reticulum stress-associated cone photoreceptor degeneration in cyclic nucleotide-gated channel deficiency. J Biol Chem 287(22):18018–18029. doi:10.1074/jbc.M112.342220
Traboulsi EI (2010) The Marshall M. Parks Memorial Lecture. Making sense of early-onset childhood retinal dystrophies: the clinical phenotype of Leber congenital amaurosis. Br J Ophthalmol 94(10):1281–1287. doi:10.1136/bjo.2009.165654
Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG (2010) Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA 107(50):21695–21700. doi:10.1073/pnas.1009179107
Trifunovic D, Sahaboglu A, Kaur J, Mencl S, Zrenner E, Ueffing M, Arango-Gonzalez B, Paquet-Durand F (2012) Neuroprotective strategies for the treatment of inherited photoreceptor degeneration. Curr Mol Med 12(5):598–612
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11(10):700–714. doi:10.1038/nrm2970
Vecino E, Hernandez M, Garcia M (2004) Cell death in the developing vertebrate retina. Int J Dev Biol 48(8-9):965–974. doi:10.1387/ijdb.041891ev
Volonte C, Amadio S, Cavaliere F, D’Ambrosi N, Vacca F, Bernardi G (2003) Extracellular ATP and neurodegeneration. Curr Drug Targets CNS Neurol Disord 2(6):403–412
Vucic D, Dixit VM, Wertz IE (2011) Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol 12(7):439–452. doi:10.1038/nrm3143
Vuong L, Brobst DE, Ivanovic I, Sherry DM, Al-Ubaidi MR (2013) p53 selectively regulates developmental apoptosis of rod photoreceptors. PloS One 8(6):e67381. doi:10.1371/journal.pone.0067381
Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH (2009a) Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PloS One 4(1):e4160. doi:10.1371/journal.pone.0004160
Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH (2009b) Autophagy, exosomes and drusen formation in age-related macular degeneration. Autophagy 5(4):563–564
Wang X, Arcuino G, Takano T, Lin J, Peng WG, Wan P, Li P, Xu Q, Liu QS, Goldman SA, Nedergaard M (2004) P2X7 receptor inhibition improves recovery after spinal cord injury. Nat Med 10(8):821–827. doi:10.1038/nm1082
Wang XW, Tan BZ, Sun M, Ho B, Ding JL (2008) Thioredoxin-like 6 protects retinal cell line from photooxidative damage by upregulating NF-kappaB activity. Free Radic Biol Med 45(3):336–344. doi:10.1016/j.freeradbiomed.2008.04.028
Ward MM, Puthussery T, Vessey KA, Fletcher EL (2010) The role of purinergic receptors in retinal function and disease. Adv Exp Med Biol 664:385–391. doi:10.1007/978-1-4419-1399-9_44
Weisman GA, Woods LT, Erb L, Seye CI (2012) P2Y receptors in the mammalian nervous system: pharmacology, ligands and therapeutic potential. CNS Neurol Disord Drug Targets 11(6):722–738
Wen R, Tao W, Li Y, Sieving PA (2012) CNTF and retina. Prog Retin Eye Res 31(2):136–151. doi:10.1016/j.preteyeres.2011.11.005
Wenzel A, Grimm C, Marti A, Kueng-Hitz N, Hafezi F, Niemeyer G, Reme CE (2000) c-fos controls the “private pathway” of light-induced apoptosis of retinal photoreceptors. J Neurosci 20(1):81–88
Wenzel A, Grimm C, Samardzija M, Reme CE (2005) Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res 24(2):275–306. doi:10.1016/j.preteyeres.2004.08.002
Wingrave JM, Sribnick EA, Wilford GG, Matzelle DD, Mou JA, Ray SK, Hogan EL, Banik NL (2004) Higher calpastatin levels correlate with resistance to calpain-mediated proteolysis and neuronal apoptosis in juvenile rats after spinal cord injury. J Neurotrauma 21(9):1240–1254. doi:10.1089/neu.2004.21.1240
Winkler BS (2008) An hypothesis to account for the renewal of outer segments in rod and cone photoreceptor cells: renewal as a surrogate antioxidant. Invest Ophthalmol Vis Sci 49(8):3259–3261. doi:10.1167/iovs.08-1785
Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS (2010) Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet 11(4):273–284. doi:10.1038/nrg2717
Wright AF, Jacobson SG, Cideciyan AV, Roman AJ, Shu X, Vlachantoni D, McInnes RR, Riemersma RA (2004) Lifespan and mitochondrial control of neurodegeneration. Nat Genet 36(11):1153–1158. doi:10.1038/ng1448
Wright AF, Murphy MP, Turnbull DM (2009) Do organellar genomes function as long-term redox damage sensors? Trends Genet 25(6):253–261. doi:10.1016/j.tig.2009.04.006
Yang JH, Basinger SF, Gross RL, Wu SM (2003) Blue light-induced generation of reactive oxygen species in photoreceptor ellipsoids requires mitochondrial electron transport. Invest Ophthalmol Vis Sci 44(3):1312–1319
Yu DY, Cringle SJ, Su EN, Yu PK (2000) Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Invest Ophthalmol Vis Sci 41(12):3999–4006
Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL (2006) Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA 103(48):18314–18319. doi:10.1073/pnas.0606528103
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297(5579):259–263. doi:10.1126/science.1072221
Yvert G, Lindenberg KS, Picaud S, Landwehrmeyer GB, Sahel JA, Mandel JL (2000) Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum Mol Genet 9(17):2491–2506
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325(5938):332–336. doi:10.1126/science.1172308
Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML, Allikmets R, Zack DJ, Kakuk LE, Lagali PS, Wong PW, MacDonald IM, Sieving PA, Figueroa DJ, Austin CP, Gould RJ, Ayyagari R, Petrukhin K (2001) A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet 27(1):89–93. doi:10.1038/83817
Zhang PP, Yang XL, Zhong YM (2012) Cellular localization of P2Y(6) receptor in rat retina. Neuroscience 220:62–69. doi:10.1016/j.neuroscience.2012.06.032
Zhivotovsky B, Orrenius S (2011) Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium 50(3):211–221. doi:10.1016/j.ceca.2011.03.003
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Japan (outside the USA)
About this chapter
Cite this chapter
Roger, J.E., Swaroop, A. (2014). Photoreceptor Degeneration: Molecular Mechanisms of Photoreceptor Degeneration. In: Furukawa, T., Hurley, J., Kawamura, S. (eds) Vertebrate Photoreceptors. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54880-5_11
Download citation
DOI: https://doi.org/10.1007/978-4-431-54880-5_11
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54879-9
Online ISBN: 978-4-431-54880-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)