
Abstract
The biological properties of the interleukin-1 (IL-1) family ligands and receptors are characteristically pro-inflammatory and act as adjuvants for specific immune responses to antigen. Thus, the IL-1 family of ligands and receptors is fundamental to innate immunity. Of the 11 members of the IL-1 family, IL-1β has emerged as a therapeutic target for an expanding number of systemic and local inflammatory conditions termed “auto-inflammatory” diseases. These diseases are distinct from autoimmune diseases and include several hereditary conditions. Howver, auto-inflammatory diseases are also common diseases such as heart failure, gouty arthritis, and type 2 diabetes. For these, neutralization of IL-1β results in a rapid and sustained reduction in disease severity. Another member of the IL-1 family, IL-1α, is also a mediator of inflammation but is classified as an “alarmin” because the cytokine is present in most cells and readily released upon cell death. Although treatment for autoimmune diseases often includes immunosuppressive drugs, blocking the IL-1 receptor is effective as an anti-inflammatory therapy for either IL-1α or IL-1β.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
As shown in Table 1.1, more than any other cytokine family, the interleukin-1 (IL-1) family plays a fundamental role in innate inflammation as well as reducing inflammation (Dinarello et al. 2012). It is this innate inflammation that facilitates specific immunological responses such as antibodies and cytotoxic T lymphocytes. In many ways, another interpretation of the innate response by IL-1 is its action as an adjuvant. Initially termed the nonspecific response to infection, a new name now used is “the innate immune response.” The cytosolic segment of each member of the IL-1-receptor family contains the Toll-IL-1-receptor (TIR) domain. This domain is also present in each Toll-like receptor (TLR), receptors that respond to microbial products, viruses, and nucleic acids. TIR is the functional domain for both the TLR and IL-1 receptor families, as mutations in this domain result in loss of response to IL-1 and TLR agonists.
With one exception, all members of the IL-1 family are initially translated as precursors lacking a signal peptide for secretion via the Golgi apparatus. The precursors are found in the cytosol and exit the cell following its death by necrosis, not apoptosis. For example, once released, IL-1α, IL-33, and IL-36 can be processed extracellularly by neutrophil proteases into active cytokines. Although IL-1β is primarily processed intracellularly by the cysteine protease caspase-1, the IL-1β precursor can also be cleaved extracellularly into an active cytokine by similar serine proteases of neutrophils (Joosten et al. 2009). The one member of the IL-1 family that is readily secreted is the IL-1-receptor antagonist (IL-1Ra). IL-1Ra is translated with a signal peptide (Fig. 1.1), although an intracellular form also exists (Arend 2002). IL-1Ra is produced in health and is found circulating in mice and humans where the antagonist serves as a brake on inflammation driven by endogenous IL-1α or IL-1β. IL-1Ra binds to IL-1RI and blocks the receptor from binding to either IL-1α or IL-1β (see Fig. 1.2c). Mice as well as humans born with a deficiency in functional IL-1Ra exhibit increased systemic and local inflammation; in humans a deficiency in IL-1Ra is lethal. The IL-36 receptor antagonist (IL-36Ra), another member of the IL-1 family, inhibits the activity of endogenous IL-36α, -β, and -γ. Although IL-36Ra is not readily secreted, individuals with a mutation in IL-36Ra develop a severe form of psoriasis. One may conclude that most members of the IL-1 family primarily promote inflammation and enhance specific acquired immune responses. However, there are also members that provide a brake on inflammation. The primary characteristics of each member of the IL-1 family are depicted in Table 1.1.

Organization of the interleukin (IL)-1 family into three subfamilies. The number of amino acids of the full length of each member is shown at the C-terminal end. The consensus sequence (AXD) is common to all IL-1 family members and serves to locate the N-terminus nine amino acids forward from this site (dark vertical bars). The N-terminus results in propieces of various lengths. The IL-1Ra has a bona fide signal peptide and is shown by comparison

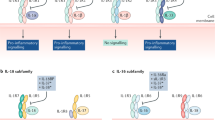
Interleukin (IL)-1 subfamily. (a) IL-1α or IL-1β binds to the IL-1RI and recruits the co-receptor IL-1RAcP. The heterodimeric IL-1 receptor complex results in a close approximation of the Toll-like receptor (TIR) domains on each receptor chain (arrows), resulting in the binding of intracellular MyD88 to the complex followed by phosphorylation of MyD88. Subsequent phosphorylations of IRAKs and IKKβ increase NFκB and IL-1R AcP-1 translocation to the nucleus, followed by expression of pro-inflammatory genes. (b) In the central nervous system, IL-1α or IL-1β binds IL-1RI, recruiting IL-1RAcP, but can also recruit the co-receptor IL-1RAcPb. IL-1RAcPb contains an altered TIR domain, which results in a reduced signal. (c) IL-1Ra binds to IL-1RI: there is no recruitment of the co-receptor IL-1RAcP, no approximation of the TIR domains, and there is no signal. (d) IL-33 binds to its specific receptor, ST2, recruits the co-receptor IL-1RAcP, and the TIR domains approximate: signal transduction is initiated, resulting in the induction of the pro-inflammatory gene profile
2 Interleukin-1 Family and Innate Responses
Independent of the type of organism or its products, the innate response is one of inflammation in which the host musters its defenses to increase the production and infiltration of phagocytic cells to the area of the invading microbe in an attempt limit infection and kill off the invader. Systemically, the liver increases the synthesis of acute-phase proteins, include anti-proteases. Even in humans, in most cases this process protects the subject without the use of antibiotics. For example, a break in the skin allows bacteria to gain access to the dermis and subsequent inflammation provides activation of complement, the release of preformed cytokines from keratinocytes, an increase in vascular wall adhesion molecules, and the extravasation of neutrophils. This response has functioned to battle against invaders for millions of years and can be traced back to fruit flies.
The skin, lung, and intestinal tract each provide a first line of defense against microbial invasion, and the lining cells, whether keratinocytes of the skin, the alveolar epithelial cells of the pulmonary tree, or the epithelial cells of the entire gastrointestinal tract, each contain preformed IL-1α, IL-18, and IL-33 as well as the members of the IL-36 subfamily. Because these members of the IL-1 family are each preformed in these cells, their release is a consequence of injury and is immediate. Therefore, they are termed “alarmins” as they alert the host to initiate the response. There are other alarmins from the lining cells that participate in defense, for example, defensins, which are directly antimicrobial. Each of the constitutively present IL-1 family members in lining cells is present as a precursor. In the case of IL-1α, the precursor is fully active; in the case of the other members, the precursors are weakly active at first but are converted to more active cytokines upon the infiltration of neutrophils and processing by extracellular neutrophil proteases. In the end, the infection is contained, the invading microorganism is eliminated, and the skin begins its process of repair.
Following the cloning of the mouse IL-1 receptor (Sims et al. 1988), the cytosolic domain of the IL-1 receptor was found to be homologous to Toll of the fruit fly (Gay and Keith 1991). Moreover, at the same time, the TIR domain for IL-1 signaling (see Fig. 1.2) was shown by Heguy to be required for IL-1 signaling (Heguy et al. 1992). Toll had been initially studied since its discovery in 1985 because of its central role in establishing dorsal ventral polarity in Drosophila. Only since 1996 was Toll linked to survival in fruit flies infected with fungi (Lemaitre et al. 1996). However, it had already been reported, back in 1988, that a member of the IL-1/TLR family, human IL-1β, protected mice from lethal Pseudomonas infection (van der Meer et al. 1988). As already noted, the TIR domain is essential for both IL-1-receptor family and TLR family signaling: a mutation in the TIR domain severely impairs responses to IL-1 family ligands as well to a large number of microbial products (O’Neill 2008).
The TIR domain binds MyD88 (Fig. 1.2), itself a TIR domain-containing protein, through TIR–TIR interactions triggering a cascade of kinases that propagate the IL-1 signal and result in transcription of a large number of genes, the majority of which code for other cytokines, chemokines, and a host of inflammatory mediators. Of these is IL-1 itself and other members of the IL-1 family such as IL-36 and IL-18.
The “innate immune response” regulates to the “acquired immune response.” The late Charles Janeway proposed that the innate response assists the host in mounting an acquired immune response. This relationship between a nonspecific cytokine providing help for a specific response to a microbial antigen is simply the adjuvant property of some cytokines. The adjuvant property of some cytokines functions by upregulating lymphocyte growth factors such as IL-2, IL-4, and IL-6 or lymphocyte receptors, resulting in expansion of lymphocyte clones, which will either rid the host of the invading microorganism with neutralizing antibodies or in generation of cytotoxic T cells to eliminate viral infections. In 1979, purified human IL-1β, a nonspecific macrophage product, was shown to augment the T-cell response to a specific antigen (Rosenwasser et al. 1979). It was nearly 20 years later that TLR were identified as inducing IL-1β from monocytes.
3 Organization of the IL-1 Family of Ligands and the Consensus Sequence
As depicted in Fig. 1.1, the IL-1 family can be divided into subfamilies according to the length of the precursor and the length of the propiece for each precursor. The IL-1 subfamily is composed of IL-1α, IL-1β, and IL-33. This subfamily has the longest proteins with the longest propieces. In the case of IL-1β, the propiece is cleaved intracellularly by caspase-1 and then the mature cytokine is secreted. In the case of IL-1α, cleavage appears to occur by the membrane protease calpain, but extracellular neutrophil proteases can also cleave the IL-1α precursor. Extracellular neutrophil proteases account for the cleavage of the propiece of IL-33. The exception in the IL-1 subfamily is the IL-1Ra, which contains a signal peptide.
The IL-18 subfamily is composed of IL-18 and IL-37. By comparison, this subfamily has a smaller propiece. IL-18 requires the cleavage of its propiece by caspase-1 to be active. IL-37 is part of the IL-18 subfamily because the cytokine binds to the IL-18Rα chain. It is unclear how the propiece of IL-37 is removed. The IL-36 subfamily is composed of IL-36α, -β, and -γ as well as IL-36 Ra. In addition, IL-38 likely belongs to this family because of its binding to the IL-36R. The IL-36 subfamily has the shortest propiece.
A consensus sequence in all members of the IL-1 family is A-X-D, where A is an aliphatic amino acid such as isoleucine, methionine, or leucine, X is any amino acid, and D is aspartic acid. The aspartic acid of the consensus sequence is not the aspartic acid of the caspase-1 cleavage recognition site. The A-X-D motif is conserved in the IL-1 family where it plays a role in the three-dimensional structure of the active cytokine. The actual N-terminus is often located nine amino acids before the A-X-D site. By eliminating the amino acids before the N-terminus, the first beta-sheet structure common to all members of the IL-1 family can form. For example, with the tenth amino acid before the A-X-D consensus site as the N-terminus, the specific activity of the IL-36 subfamily (IL-36α, IL-36β, IL-36γ, and IL-36Ra) is low. However, with the ninth amino acid as the N-terminus there was a marked increased in the activity (Towne et al. 2011). In the case of IL-1β, the ninth amino acid before the A-X-D site coincides exactly with the N-terminal alanine generated by the caspase-1 site.
4 Interleukin-1α
From an evolutionary point of view, IL-1α is the oldest member of the IL-1 family, and its primary amino acid sequence is closely related to that of the fibroblast growth factor (FGF) family. Similar to FGF, IL-1α does not have a signal peptide, binds to nuclear DNA, exits the cell upon death, and binds to its receptor as an unprocessed precursor. As shown in Fig. 1.2, IL-1α binds to the IL-1RI and recruits the IL-1R accessory protein (IL-1RAcP) to form a heterodimeric complex, which signals to induce inflammation. In health, primary cells contain constitutive levels of the IL-1α precursor but not IL-1β (Hacham et al. 2002). The IL-1α precursor is present in keratinocytes, thymic epithelium, hepatocytes, endothelial cells, the epithelial cells of mucous membranes, including the entire gastrointestinal tract, and fibroblasts regardless of their location. The propiece of IL-1α precursor can be cleaved extracellularly by neutrophil proteases, a step that increases its biological activity. However, IL-1α can also be active as a membrane-associated cytokine. Most cell lines including tumor cell lines contain constitutive levels of IL-1α (Hurgin et al. 2007; Lonnemann et al. 1995; Werman et al. 2004). Using an epithelial cell line, what were considered to be intrinsic interferon (IFN)-γ activities depended largely on constitutively expressed IL-1α. IFN-γ activities were inhibited by antibodies to IL-1α but not to IL-1β (Hurgin et al. 2007). The concept that IL-1α acts as an autocrine growth factor assumes that the intracellular IL-1α precursor regulates normal cellular differentiation, particularly in epithelial and ectodermal cells. In support of the concept, an antisense oligonucleotide to IL-1α reduces senescence in endothelial cells (Maier et al. 1990). In fibroblasts, the constitutive IL-1α precursor binds to HAX-1, a non-receptor substrate for tyrosine kinases in hematopoietic cells. In fibroblasts, the IL-1α HAX-1 complex translocates to the nucleus (Kawaguchi et al. 2006). Although the concept is that IL-1α acts as an autocrine growth factor in fibroblasts or endothelial cells in vitro, the data should be interpreted carefully because mice deficient in IL-1α show no demonstrable defects in growth and development, including skin, fur, epithelium, and gastrointestinal function (Horai et al. 1998). However, mice deficient in IL-1α still retain the N-terminal propiece, which functions as a nuclear factor (Werman et al. 2004). In fact, in another study, the N-terminal propiece of IL-1α was shown to bind HAX-1 (Yin et al. 2001).
Is there is a role for the intracellular precursor IL-1α in normal cell function? The IL-1α precursor is present in cells that also contain large amounts of the intracellular form of the IL-1Ra (icIL-1Ra), as reviewed by Arend (2002). This form of IL-1Ra also binds to the IL-1 receptor and prevents signal transduction. In fact, icIL-1Ra is thought to compete with the intracellular pool of precursor IL-1α for nuclear-binding sites.
4.1 Membrane-Associated IL-1α
Precursor IL-1α can be found on the surface of several cells, particularly on monocytes and B lymphocytes, where it is referred to as membrane IL-1α (Kurt-Jones et al. 1985). Membrane IL-1α is biologically active (Kaplanski et al. 1994); its biological activities are neutralized by antibodies to IL-1α but not those to IL-1β. Endothelial cells undergoing stress-induced apoptosis release membrane apoptotic body-like particles containing nuclear fragments and histones as well as the full-length IL-1α precursor and the processed mature form (Berda-Haddad et al. 2011). When injected into mice, apoptotic body-like particles containing the IL-1α precursor induce neutrophilic infiltration that was prevented by neutralization of IL-1α but not IL-1β (Berda-Haddad et al. 2011).
4.2 Processing and Secretion of IL-1α
Although the IL-1α precursor is biologically active, the processed form is more active. Furthermore, the binding of IL-1α to IL-1RI has been modeled using recombinant IL-1α with an N-terminus at 113. The processing of the IL-1α precursor is accomplished by calpain II, a membrane-associated, calcium-dependent cysteine protease (Miller et al. 1994). In macrophages treated with hydroquinone, calpain II levels fall and are associated with inhibition of IL-1α precursor processing (Miller et al. 1994). Not surprisingly, calcium influx induced IL-1α secretion of the processed form (Gross et al. 2012). The secretion of IL-1α requires the presence of IL-1β, because IL-1β-deficient mice do not secrete IL-1α (Fettelschoss et al. 2011). IL-1α binding to IL-1β has been reported in which IL-1β acts as a chaperone for the secretion mechanism via caspase-1 (Fettelschoss et al. 2011). In another study, IL-1β was shown to bind to, and enhance the activity of, HMGB1 (Sha et al. 2008). It is thus possible that both IL-1α exits the cell bound to IL-1β and HMGB1.
4.3 Biological Functions of Constitutive IL-1α: IL-1α and Sterile Inflammation
Large numbers of reports mention the use of bacterial and fungal products to induce cytokines as models of inflammatory disease; however, most inflammatory diseases are sterile. For example, the inflammation associated with atherosclerosis, myocardial infarction, stroke, cancer, renal, and liver failure is sterile. The hypoxic insult that takes place in ischemia results in local necrosis and release of cellular contents, including nucleic acids. Members of the IL-1 family contribute to sterile inflammation, and IL-1α plays a significant role in this regard. Upon cell death by necrosis, the IL-1α precursor is released (Carmi et al. 2009; Cohen et al. 2010) and binds to the IL-1 receptor on nearby tissue macrophages and epithelial cells, triggering a response (Luheshi et al. 2011; Rider et al. 2011). For example, infiltration of neutrophils occurs first and is followed by influx of monocytes (Rider et al. 2011). Extracts of tumor cells induce neutrophilic inflammation, which does not occur in mice deficient in IL-1RI and is prevented by neutralization of IL-1α, but not neutralization of IL-1β (Chen et al. 2007). Sterile inflammation is independent on TLR2 and TLR4 (Chen et al. 2007).
Thus, IL-1α, either the unprocessed precursor or the calpain cleavage form, is classified as an alarmin because the cytokine is preformed and triggers an inflammatory response rapidly. Endothelial cells subjected to nutritional stress release inflammatory apoptotic bodies, which contain both the precursor and processed forms of IL-1α (Berda-Haddad et al. 2011). Inflammatory apoptotic bodies induce chemokine and neutrophilic infiltration into the peritoneal cavity, both of which are IL-1α dependent (Berda-Haddad et al. 2011). Platelets also contain IL-1α as well as IL-1β (Hawrylowicz et al. 1989). Platelet-derived IL-1 induces chemokines such as IL-8 from endothelial cells (Kaplanski et al. 1993) and MCP-1 from monocytes (Hawrylowicz et al. 1991). Platelet-derived IL-1α is important in brain injury in stroke models (Thornton et al. 2010) and in atherosclerosis (Gawaz et al. 2000).
4.4 Studies in IL-1α-Deficient Mice
Mice deficient in IL-1α are born healthy and develop normally. In some models of local inflammatory responses, wild-type and IL-1α-deficient mice develop fever and acute-phase proteins, whereas IL-1β-deficient mice do not (Horai et al. 1998). In addition, although the inflammation-associated induction of glucocorticoids was suppressed in IL-1β-deficient mice, this suppression was not observed in IL-1α-deficient mice. However, expression of IL-1β mRNA in the brain decreased 1.5 fold in IL-1α-deficient mice whereas expression of IL-1α mRNA decreased more than 30 fold in IL-1β-deficient mice. These data suggest that IL-1β exerts greater control over production of IL-1α than does IL-1α over the production of IL-1β. In caspase-1-deficient mice, IL-1α production is also reduced (Kuida et al. 1995), further suggesting that production of IL-1α is under the control of IL-1β. It is important that caspase-1-deficient mice are also deficient in caspase-11 (Kenneth et al. 2012).
In mice fed a high-fat diet, serum amyloid A protein, a marker of inflammation in atherogenesis, was markedly lower in IL-1α-deficient mice compared to wild-type or IL-1β-deficient mice (Kamari et al. 2007). IL-1α-deficient mice had significantly higher levels of non-high density lipoprotein cholesterol. The beneficial effect of IL-1α deficiency was the result of hematopoietic cells transferred from the bone marrow of IL-1α-deficient mice, resulting in a reduction in aortic lesion size twice that observed in mice transplanted with IL-1β-deficient bone marrow cells. Therefore, IL-1α appears to play a greater role in the pathogenesis of lipid-mediated atherogenesis than IL-1β, and this may be the result of an effect of membrane IL-1α.
5 Interleukin-1β
5.1 IL-1β, the Master Cytokine in the IL-1 Family
More than any other member of the IL-1 family, IL-1β has been the focus of most studies. IL-1β is a highly inflammatory cytokine, particularly in humans, as reviewed by (Dinarello 2011a). As shown in Fig. 1.2a, IL-1β and IL-1α bind to the same IL-1RI and trigger a proinflammatory signal. The interest in IL-1β also results, in part, because it is a secreted cytokine from macrophages and from the importance of the macrophage in antigen presentation before the era of dendritic cells. The inactive IL-1β precursor is converted into an active cytokine by the intracellular cysteine protease caspase-1. In particular, persons with activating mutations in one of the key genes that control the activation of caspase-1 can develop life-threatening systemic inflammation, which is reversed by either blocking the IL-1 receptor or through the use of a neutralizing antibody to IL-1β. Other chronic inflammatory diseases are mediated by IL-1β, as neutralizing antibodies have been used to treat a broad spectrum of diseases.
The IL-1β-mediated illnesses fall into the category of “auto-inflammatory” diseases, which are to be distinguished from the classic “autoimmune” diseases. Although inflammation is common to both auto-inflammatory and autoimmune diseases, in the case of IL-1-mediated disease, there is no evidence for role of adaptive immunity in its induction.
5.2 IL-1β is an Inducible Cytokine
In contrast to IL-1α, the IL-1β precursor is not present in health. Also differing from IL-1α, IL-1β is primarily a product of monocytes, macrophages, and dendritic cells (DC) as well as B lymphocytes and natural killer (NK) cells. In health, circulating human blood monocytes or bone marrow cells do not constitutively express mRNA for IL-1β. Endothelial cells, skin keratinocytes, fibroblasts, and epithelial cells contain constitutive IL-1α and constitutive IL-33 as precursors as well as mRNA, but these cells do not express IL-1β mRNA, even upon stimulation with TLR ligands. Melanoma cells do express IL-1β as a precursor, and the more aggressive and metastatic the melanoma, the greater the likelihood of active caspase-1 and IL-1β secretion (Okamoto et al. 2009). In the bone marrow neutrophil precursors, IL-1β gene expression is inducible but mature neutrophils in the circulation no longer produce IL-1β. Neutrophil IL-1β plays a pathological role in the severe inflammation of mice with a mutant form of the phosphatase SHP1 (Croker et al. 2011). Several malignant tumors do express IL-1β as part of their neoplastic nature, particularly acute myelogenous leukemia, melanoma, multiple myeloma, and juvenile myelogenous leukemia, each of which exhibit constitutive expression of IL-1β. In contrast to most cytokine promoters, IL-1β regulatory regions are distributed more than several thousand base pairs upstream from the transcriptional start site. In addition to a cAMP response element, there are NF-κB-like and activating protein-1 (AP-1) sites. IL-1β gene regulation has been reviewed in detail (Unlu et al. 2007). Although steady-state mRNA levels for IL-1β may be present, there is distinct dissociation between transcription and translation of the IL-1β precursor. Non-TLR ligands such as the complement component C5a, hypoxia, adherence to surfaces, or clotting of blood induce the synthesis of large amounts of IL-1β mRNA in monocytic cells without significant translation into the IL-1β protein. In these cells, the IL-1β mRNA assembles into large polyribosomes, but there is no significant elongation of the peptide (Kaspar and Gehrke 1994). This failure to complete the translation into IL-1β protein may be caused by the instability element present in the coding region. This instability region is also found in IL-18 and IL-37 and appears to limit the mRNA of these cytokines (Bufler et al. 2004). However, completion of translation of the mRNA into the respective cytokines can be accomplished by adding low concentrations of TLR ligands or IL-1 itself to the “primed” monocytes (Schindler et al. 1990).
5.3 Processing and Secretion of IL-1β via Caspase-1
Nearly all microbial products induce IL-1β via TLR activation; in addition, IL-1 (either IL-1α or IL-1β) induces itself both in vivo and in monocytes in vitro (Dinarello et al. 1987). Other studies supporting this concept of IL-1-induced IL-1 have been reported (Boni-Schnetzler et al. 2008; Gattorno et al. 2007; Goldbach-Mansky et al. 2006; Greten et al. 2007). Regardless of the stimulus, processing and secretion of IL-1β require conversion of procaspase-1 to active caspase-1, although in some studies processing of the IL-1β precursor is caspase-1 independent (Wewers et al. 1997). The activation to active caspase-1 is dependent on a complex of intracellular proteins termed the “inflammasome” by the late Juerg Tschopp (Agostini et al. 2004; Martinon et al. 2009). The critical component of the inflammasome is NLRP3 (see Fig. 1.3). NLRP3 is also termed cryopyrin because the gene was initially discovered in patients with familial cold auto-inflammatory syndrome, a genetic disease characterized by constitutional symptoms, fevers, and elevated acute-phase proteins following exposure to cold (Hoffman et al. 2001).

Activation of caspase-1 by the NLRP3 inflammasome. 1 Activation of the cells following receptor binding for TLR or IL-1 receptors. 2 Transcription of the IL-1β gene. 3 Synthesis of the inactive IL-1β precursor. 4 Extracellular ATP binds P2X7 receptor. 5 Efflux of potassium. 6 Oligomerization of inflammasone components. 7 Activation of caspase-1. 8 Cleavage of the IL-1β precursor by caspase-1. 9 Maure IL-1β is released from the cell
As monocytes exit the bone marrow, they circulate in the bloodstream for approximately 3 days. In the absence of disease, it is likely that these cells do not enter tissues but are destroyed in the spleen or undergo apoptosis. There is no dearth of reports that circulating human blood monocytes release processed IL-1β upon stimulation starting 4 h after stimulation with TLR agonists and continue to release the cytokine during the following 20–40 h. Following lipopolysaccharide (LPS) stimulus, IL-1β mRNA levels rise rapidly within 15 min but begin to decline after 4 h because of the short half-life of their mRNA or the action of micro RNA. In contrast, using IL-1 itself as a stimulant, IL-1β mRNA levels are sustained for more than 24 h (Schindler et al. 1990). Raising intracellular cAMP levels with histamine enhances IL-1-induced IL-1 gene expression and protein synthesis. Monocytes of patients with auto-inflammatory diseases such as CAPS and HIDS release IL-1β even without TLR stimulation during a 24-h incubation (Drenth et al. 1996; Hoffman and Wanderer 2011).
When obtained from the venous blood of healthy subjects, human blood monocytes contain active caspase-1. Active caspase-1, as determined by its cleavage into the active dimer, is present even in the absence of stimulation (Netea et al. 2009). Active caspase-1 present in freshly obtained monocytes is nevertheless dependent on the presence of the key components of the inflammasome, namely, ASC and NLRP3 (Netea et al. 2009). However, during subsequent incubation, extracellular levels of ATP increase in the supernatant as IL-1β also increases and inhibition of ATP by oxidized ATP reduces the secretion of IL-1β (Netea et al. 2009). The inhibition of IL-1β secretion by oxidized ATP is consistent with the role of the P2X7 receptor, which binds ATP and opens the potassium channel for release of intracellular potassium. The presence of active caspase-1 in circulating blood monocytes suggests that the rate-limiting step in the processing and release of IL-1β is at the level of gene expression.
However, upon differentiation of the same blood monocytes into macrophages in vitro, TLR-induced IL-1β release requires activation of caspase-1 by exogenous ATP (Netea et al. 2009). The assembly of the inflammasome components with inactive pro-caspase-1 takes place following a fall in intracellular potassium triggered by ATP binding to the P2X7 receptor. ATP activation of the P2X7 receptor opens the potassium channel, and simultaneously, as potassium levels fall, caspase-1 is activated by the inflammasome (Andrei et al. 1999; Andrei et al. 2004; Elssner et al. 2004; Gardella et al. 2000; Perregaux et al. 2000). Without exogenous ATP, there is little or no processing of the IL-1β precursor in differentiated monocyte-derived macrophages. Alveolar macrophages obtained from the lungs of healthy human also do not release IL-1β with LPS stimulation unless exogenous ATP is added (Netea et al. 2009). In addition to ATP activation of P2X7, activation of IL-1β processing can also take place with a cathelicidin-derived peptide termed LL37, which is released from neutrophils (Elssner et al. 2004).
The cleavage of the IL-1β precursor by active caspase-1 can take place in the specialized secretory lysosomes or in the cytoplasm. However, more than one pathway seems available for processed IL-1β to exit the cell: these include by exocytosis of the secretory lysosomes (Andrei et al. 1999, 2004), shedding of plasma membrane microvesicles, direct release via transporters, or multivesicular bodies containing exosomes (Qu et al. 2007). In general, the release of processed IL-1β takes place before there is significant release of lactate dehydrogenase (Brough and Rothwell 2007), although in vitro cell death eventually takes place. Pyroptosis is a caspase-1-dependent form of cell death and is induced by certain bacteria using Ipaf, a member of the NLR family of intracellular receptors (Suzuki et al. 2007). An increase in intracellular calcium is also required for the mature IL-1β to exit the cell, and this is phopholipase C dependent (Andrei et al. 2004).
5.4 Gain-of-Function Mutation in Cryopyrin
Diseases associated with single amino acid-activating mutations in cryopyrin are termed cryopyrin-associated periodic syndromes (CAPS). In monocytes from patients with CAPS, activation of caspase-1 occurs without a requirement for a rapid fall in the level of intracellular potassium (Gattorno et al. 2007). Therefore, mutated cryopyrin allows for the assembly of the complex of interacting proteins in the presence of normal intracellular levels of potassium. Although LPS-induced synthesis of the IL-1β precursor is often studied (Kahlenberg et al. 2005), it is unlikely that LPS plays a role in auto-inflammatory diseases. On the other hand, spontaneous secretion of IL-1β from monocytes of patients is the result of endogenous IL-1β stimulation. In patients with CAPS, there is a decrease in steady-state levels of pro-caspase-1 mRNA with IL-1Ra treatment (Goldbach-Mansky et al. 2006), suggesting that IL-1β stimulates its own production and processing. Thus, in any disease process that includes an increase in the steady-state levels of pro-caspase-1 mRNA, components of the inflammasome or the IL-1β precursor explain the “auto-inflammatory” nature of the disease. Type 2 diabetes appears to be an example of an auto-inflammatory disease in which glucose induces IL-1β production from the insulin-producing beta cell and IL-1β induces the beta cell to produce its own IL-1β (Maedler et al. 2002).
5.5 Polymorphisms in P2X7 and the Activation of the Inflammasome
Patients with classic auto-inflammatory diseases such as FMF or CAPS have nearly identical clinical parameters, secrete more IL-1β, and respond dramatically to IL-1 receptor blockade, yet have no mutation in NALP3. It is therefore possible that mutations in P2X7 itself or regulation of the other genes controlling potassium channels (Pascual et al. 2005) may account for dysfunctional secretion of IL-1β. For example, monocytes from patients with rheumatoid arthritis are more sensitive to release of IL-1β following ATP activation of the P2X7 receptor compared to monocytes from healthy controls (Al-Shukaili et al. 2008). However, monocytes from subjects with a P2X7 Glu496Ala loss-of-function polymorphism secrete significantly less IL-1β (Sluyter et al. 2004b). Monocytes from subjects homozygous for this polymorphism also released significantly less IL-18 (Sluyter et al. 2004a). Another P2X7 receptor polymorphism is associated with increased mortality in patients undergoing allogeneic stem cell transplantation (Lee et al. 2007). Bacteremia was documented in 68 % of patients with this polymorphism compared to 18 % in wild-type control patients (Lee et al. 2007).
In mice deficient in P2X7 receptors, inflammation, pain, and IL-1β-mediated IL-6 production are markedly reduced (Chessell et al. 2005). In addition to a fall in intracellular potassium, ATP triggers formation of peroxynitrite, which is required for caspase-1 activation because peroxynitrite scavengers prevent IL-1β secretion (Hewinson et al. 2008). Pannexin-1, a mammalian protein that functions as a hemichannel for the uptake of dyes, is required for caspase-1 processing and release of IL-1β via the P2X7 receptor (Pelegrin and Surprenant 2006). Pannexin-1 can also function for LPS-induced IL-1β synthesis in the absence of TLR4 (Kanneganti et al. 2007). P2X7 receptor activity is also regulated by a “regeneration and tolerance factor” (Derks and Beaman 2004).
5.6 Polymorphism in NLRP1 and the Release of IL-1β
NLRP1 is genetically associated with risk of several autoimmune diseases including generalized vitiligo, Addison disease, type 1 diabetes, and rheumatoid arthritis. Predicted functional variations in NLRP1 reside in several common high-risk haplotypes, and the haplotypes that are high risk for disease share two substitutions, L155H and M1184V. Peripheral blood monocytes from healthy subjects homozygous for the predominant high-risk haplotype 2A released significantly greater amounts (P < 0.001) of the IL-1β precursor to mature bioactive IL-1β under basal (resting) conditions as well as in response to TLR2 and TLR4 agonists compared with monocytes from subjects homozygous for the reference haplotype 1 (Levandowski et al. 2013). The increase in basal release was 1.8 fold greater in haplotype 2A monocytes, and these differences between the two haplotypes were consistently observed three times during a 3-month period; no differences were observed for IL-1α or tumor necrosis factor (TNF)-α. NLRP1 RNA and protein levels were not altered by the predominant high-risk haplotype, indicating that altered function of the corresponding multivariant NLRP1 polypeptide predisposes to autoimmune diseases by activation of the NLRP1 inflammasome.
5.7 Non-caspase-1 Processing of IL-1β
Non-caspase-1 mechanisms also exist to generate active forms of IL-1β. For example, sterile inflammation induces fever, elevated IL-6, and increased production of hepatic acute-phase proteins. These responses are absent in mice deficient in IL-1β but present in mice deficient in caspase-1 (Fantuzzi et al. 1997a; Joosten et al. 2009). Sterile inflammation is often associated with neutrophilic infiltration, and the neutrophils produce IL-1β. Because neutrophils are short-lived cells, dying within hours upon emigration, release of the IL-1β precursor from intracellular stores is not unexpected. Processing of the IL-1β precursor extracellularly into an active cytokine has been reported for the common neutrophil protease, proteinase-3 (Coeshott et al. 1999; Joosten et al. 2009). Proteinase-3 also contributes to the processing of IL-18 (Sugawara et al. 2001). Other proteases such as elastase, matrix metalloprotease 9, and granzyme A process the IL-1β precursor extracellularly. In addition, a mast cell chymase generates active IL-1β.
Mice with a targeted IKK-β deletion in myeloid cells are more susceptible to LPS-induced shock than control mice (Greten et al. 2007) and markedly elevated levels of IL-1β are found in the circulation associated with a prominent neutrophilia (Greten et al. 2007). The elevated levels of IL-1β are lethal because blockade with IL-1Ra protects these mice from death. The source of the IL-1β in these mice is the neutrophil. When incubated with proteinase-3, cleavage of the IL-1β precursor is observed, yielding molecular weights of 25,000 and 15,000 Da (Greten et al. 2007). Because the cleavage of the IL-1β precursor by proteinase-3, elastase, and cathepsin G are within three amino acids of the caspase-1 cleavage site, the products of the non-caspase-1 cleavage are biologically active (Coeshott et al. 1999; Joosten et al. 2009). Therefore, in inflammatory conditions such as urate crystal arthritis, which is characterized by a prominent neutrophilic infiltration, proteinase-3 cleavage of extracellular IL-1β precursor likely takes place (Joosten et al. 2010). Mice deficient in caspase-1 are not protected against urate-induced inflammation. Although IL-1Ra is effective in treating gout, IL-1Ra would be equally effective in any disease with extracellular processing of the precursor (Schlesinger et al. 2011; So et al. 2007, 2010). The importance of extracellular processing of the IL-1β precursor by serine proteases may explain, in part, the anti-inflammatory properties of alpha-1-antitrypsin (Numanami et al. 2003).
5.8 Effects in Mice Deficient in IL-1β
After 10 years of continuous breeding, mice deficient in IL-1β exhibit no spontaneous disease. However, upon challenge, IL-1β-deficient mice exhibit specific differences from their wild-type controls. The most dramatic is the response to local inflammation induced by subcutaneous injection of an irritant. Within the first 24 h, IL-1β-deficient mice do not manifest an acute-phase response, do not develop anorexia, have no circulating IL-6, and no fever (Fantuzzi et al. 1997a; Zheng et al. 1995). These findings are consistent with those reported in the same model using anti-IL-1R type I antibodies in wild-type mice (Fantuzzi et al. 1997a; Zheng et al. 1995). IL-1β-deficient mice also have reduced inflammation because of zymosan-induced peritonitis (Fantuzzi et al. 1997a, b). In contrast, IL-1β-deficient mice have elevated febrile responses to LPS, IL-1β, or IL-1α compared to wild-type mice (Fantuzzi et al. 1996). Nevertheless, IL-1β-deficient mice injected with LPS have little or no expression of leptin mRNA or protein (Faggioni et al. 1998).
Mice deficient in IL-1β were compared to mice deficient in IL-1α after exposure to chemical carcinogens (Krelin et al. 2007). In IL-1β-deficient mice, tumors developed more slowly or did not develop in some mice. A deficiency in IL-1α, on the other hand, did not impair tumor development compared to wild-type mice injected with the same carcinogen. In IL-1Ra-deficient mice, tumor development was the most rapid. A leukocyte infiltrate was found at the site of carcinogen injection. The neutrophilic infiltrate was almost absent in IL-1β-deficient mice, whereas in IL-1Ra-deficient mice, a dense neutrophilic infiltrate was observed. In wild-type mice, the leukocytic infiltrate was sparse and the infiltrate that was observed in IL-1α-deficient mice was similar to that of control mice. These findings may reflect the fact that IL-1β is secreted into the microenvironment, resulting in the emigration of monocytes and neutrophils, whereas IL-1α, remaining cell associated, is less likely to affect the microenvironment.
5.9 IL-1β and Autophagy
Autophagy is an ancient process of recycling cellular components, such as cytosolic organelles and protein aggregates, through degradation mediated by lysosomes. Autophagy is activated in conditions of cell stress, hypoxia, starvation, or growth factor deprivation; it promotes cell survival by generating free metabolites and energy through degradation of the endogenous cellular components (Klionsky 2007). However, in addition to its role in the pathophysiology of cancer, neurodegenerative diseases, or aging, autophagy is also a modulator of inflammation (Schmid and Munz 2007). A role for autophagy in production of proinflammatory cytokines, particularly of IL-1β, has emerged with deletion of ATG16-L1. For example, macrophages from ATG16L1-deficient mice produce higher levels of IL-1β and IL-18 after stimulation with TLR4 ligands (Saitoh et al. 2008). The data suggest that higher activation of caspase-1 in the ATG16L1-deficient mice accounts for the higher production level (Saitoh et al. 2008). This observation was related to the specific degradation of the IL-1β precursor in autophagosomes in mouse macrophages (Harris et al. 2011). Additional studies in the ATG16L1-deficient mice point toward a regulatory effect of autophagy on caspase-1 activation through modulation of the NLRP3 inflammasome (Nakahira et al. 2011; Tschopp and Schroder 2010; Zhou et al. 2010).
This role of autophagy in the secretion of IL-1β was also observed in human primary monocytes, in which specific inhibition of autophagy leads to increased production of IL-1β (Crisan et al. 2011). However, in the same cells TNF-α production was decreased by autophagy inhibition. These data suggest divergent effects of autophagy on the production of these two important proinflammatory cytokines. In mice, the increase in IL-1β production is ascribed to increased activation of the inflammasome, but in human cells, it is IL-1β mRNA transcription that is elevated when autophagy was inhibited, whereas no effects were observed on caspase-1 activation (Crisan et al. 2011; Harris et al. 2011; Saitoh et al. 2008). Despite these differences between mouse and human cells, the inhibition of autophagy increases the production of IL-1β but not TNF-α.
The modulation of inflammation by autophagy in humans has been studied in Crohn’s disease. Genome-wide association studies in large cohorts of Crohn’s disease patients have revealed that genetic variants in two autophagy genes, ATG16L1 and IRGM, result in increased susceptibility to the disease. A nonsynonymous polymorphism in ATG16L1 on chromosome 2q37.1 and two polymorphisms in IRGM on chromosome 5q33.1 were significantly associated with Crohn’s disease risk (Hampe et al. 2007; Rioux et al. 2007). Another study revealed a significant association of Crohn’s disease susceptibility with an intronic polymorphism in the autophagy gene ULK1 (Henckaerts et al. 2011). Moreover, autophagy defects have been reported in individuals bearing NOD2 mutations and are consistent with the concept that impaired bacterial clearance and increased bacterial persistence are part of the pathogenesis of Crohn’s disease (Lapaquette et al. 2010).
The mechanism through which polymorphisms in autophagy genes influence susceptibility to Crohn’s disease appear to involve IL-1β production. The ATG16L1 300Ala risk allele was associated with elevated production of IL-1β and IL-6; however, this finding was only observed in cells stimulated with the NOD2 ligand muramyl dipeptide (MDP). In contrast, the expected levels of IL-1β and IL-6 were produced upon stimulation with TLR2 and TLR4 ligands (Plantinga et al. 2011). The increased production of IL-1β was associated with an increase in the steady-state levels of IL-1β mRNA rather than increased activation of the inflammasome (Plantinga et al. 2011). Studying the same polymorphism (ATG16L1 Thr300Ala) in human dendritic cells, Cooney et al. reported defective NOD2-induced, but not TLR-induced, autophagy and antigen presentation (Cooney et al. 2010). Furthermore, effects of this polymorphism on antibacterial autophagy in epithelial cells have been observed (Homer et al. 2010). The specific effect of the ATG16L1 polymorphism on the NOD2 pathway, and not on TLR-induced stimulation, is likely related to the fact that NOD2 and ATG16L1 form a protein complex that is essential for NOD2-induced autophagosome formation (Travassos et al. 2010). Because the ATG16L1 Thr300Ala polymorphism affects protein stability (Kuballa et al. 2008), defective induction of autophagy and therefore enhanced IL-1β mRNA transcription upon triggering of NOD2 may be caused by the presence of defective complex.
6 Interleukin-33
6.1 IL-33 as a Member of the IL-1 Subfamily
Formerly termed IL-1F11, IL-33 belongs to the IL-1 subfamily and has been studied for its role in the Th2 paradigm of immune responses. IL-1β is also linked to the Th2 response. The existence of IL-33 was predicted in 1994 following the discovery of a novel member of the IL-1 receptor family termed ST2 (Bergers et al. 1994). ST2 is the ligand-binding chain for IL-33 (see Table 1.2) and is structurally similar to the ligand-binding chain of IL-1α and IL-1β. In addition, the co-receptor for IL-33 is the IL-1RAcP, which is also the co-receptor for IL-1α and IL-1β. It was not until 2005 that IL-33 was reported as the ligand for ST2 (Schmitz et al. 2005). ST2 is regulated by the estrogen-inducible transcription factor Fos (Bergers et al. 1994), and this property of estrogens may be related to the large number of studies on the effect of estrogens to regulate IL-1 and inflammation.
Similar to most members of the IL-1 receptor family, ST2 is composed of three extracellular Ig domains and an intracellular TIR domain. Although the name ST2 is still used, the correct term is the IL-33 receptor α-chain (IL-33Rα). As shown in Fig. 1.2d, the IL-33Rα chain is similar to the IL-1R1 in that it is the ligand-binding chain for IL-33 but requires IL-1RAcP to signal (Ali et al. 2007; Chackerian et al. 2007).
Before the discovery of IL-33, several studies suggested that the putative ligand (IL-33) for the ST2 orphan receptor was playing a role in allergic-type diseases. It became clear that activation of ST2 was uniquely driving Th2 responses. Structurally, IL-33 is closer to IL-18 than IL-1β. Biologically, IL-33 is closest to IL-1α, as the precursors for IL-1α and IL-33 are constitutively present in all endothelial cells. As discussed below, similar to IL-1α, IL-33 functions as a DNA-binding molecule. The dominant property of IL-33 is the induction of IL-4, IL-5, and IL-13 as well as other properties anticipated for a Th2-type cytokine. Diseases thought to be caused by increased immunoglobulin production may also be related to IL-33. IL-33 induces the production of IL-6, IL-1β, and PGE2 from mast cells.
6.2 IL-33 and Th2 Responses
The properties of recombinant IL-33 recapitulate much of the existing data that ST2 promotes Th2-type responses. For example, before its discovery, a role for IL-33 in the Th2 response was observed using soluble extracellular forms of ST2 [reviewed in (Schmitz et al. 2005)]. However, IL-33 has properties that go beyond its role in the Th2 paradigm, because similar to IL-1α, IL-1β, and IL-36, IL-33 forms a heterodimeric complex with IL-1RAcP for signal transduction (Ali et al. 2007; Chackerian et al. 2007). Although the IL-1RAcP is expressed on most nucleated cells, ST2 is somewhat restricted to low expression on most cells with the notable exception of mast cells.
There are several mechanisms by which IL-33 favors the Th2 response. Similar to IL-1β, IL-33 induces IL-6, an adjuvant for antibody production. IL-33 induction of IL-6 is prevented by a blocking antibody to IL-1RAcP (Ali et al. 2007). IL-33 initiates signal transduction via activation of NF-κB, which is typical of IL-1α, IL-1β, and IL-18 (Schmitz et al. 2005), but other studies have shown that antibody cross-linking of ST2 does not result in activation of NF-κB but rather AP-1. IL-33 treatment also increased serum IgA and IgE, an expected response for a switch from Th1 to Th2.
6.3 Processing of the IL-33 Precursor
Initially, IL-33 was considered closely related to IL-1β and IL-18 because the IL-33 precursor contains a caspase-1 site, which upon activation would cleave the IL-33 precursor and release the active cytokine (Schmitz et al. 2005), similar to that for IL-1β and IL-18. Indeed, the first recombinant forms of IL-33 were produced with an N-terminus at the caspase-1 site (Schmitz et al. 2005). Although recombinant IL-33 was active, the concentrations required for activity were considerably higher than those of other members of the IL-1 family. Indeed, subsequent studies revealed that caspase-1 actually results in loss of IL-33 activity and that the full length IL-33 precursor binds to ST2 and is active (Cayrol and Girard 2009), similar to the ability of the IL-1α precursor to bind to IL-1RI. In addition, it was reported that the caspase-1 cleavage site at 178 is similar to the consensus sequence for caspase-3 and that intracellular IL-33 precursor is a substrate for caspase-3 (Cayrol and Girard 2009).
Using immobilized IL-33 precursor, neutrophil proteinase 3 (PR3) was isolated from human urinary proteins (Bae et al. 2012). Neutrophil PR3 is known to process the IL-1β precursor into an active cytokine (Joosten et al. 2009). PR3 converted human and mouse precursor IL-33 proteins to biological active forms; however, increasing the incubation time of PR3 abrogated IL-33 activities (Bae et al. 2012). Using the consensus amino acid sequence sites for PR3, six human and mouse recombinant IL-33 proteins were produced and assessed for biological activities; varying levels of activity were reported (Bae et al. 2012). Another study also demonstrated cleavage of the IL-33 precursor by neutrophil proteases such as PR3, neutrophil elastase, and cathepsin G (Lefrancais et al. 2012), resulting in the generation of IL-33 with different N-termini and varying levels of activity. These studies support the concept that extracellular IL-33 is released as a precursor, is rapidly processed by neutrophil enzymes, and generates active forms with varying levels of activity. The implications for generation of active IL-33 by neutrophil enzymes for Th2 polarization remain unclear. It may be more relevant to study the effect of proteases from eosinophils in the processing of the IL-33 precursor. Nevertheless, the IL-33 precursor binds to ST2 and recruits the accessory chain for signal transduction, but compared to IL-33 generated by neutrophil proteases, the activity of IL-33 precursor is weak (Bae et al. 2012; Lefrancais et al. 2012).
There is no dearth of studies on ST2 tissue-specific localization, regulation of its expression, and effects in transgenic mice overexpressing ST2 as well as deletion, neutralization, and antibody cross-linking of ST2. Elevated levels of the soluble form of ST2 were present in the circulation of patients with various inflammatory diseases, and exogenous administration of pharmacological doses of soluble ST2 neutralized endogenous levels of the then putative ligand IL-33 and reduced inflammation (Leung et al. 2004). IL-33 activates Th2 lymphocytes, mast cells, basophils, and eosinophils as well as NK T cells and blood monocytes. One of the best studied properties of IL-33 is the induction of IL-5 and IL-13 and their respective roles in lung inflammation, such as allergic-type asthma. For example, instillation of IL-33 into the airways triggers an immediate allergic response in the lung of naïve mice and worsens the response in mice sensitized to antigen peripherally but challenged by exposure of antigen in the lung (Louten et al. 2011).
Mice deficient in ST2 do not develop a Th2 response to Schistosoma egg antigen. Indeed, several studies have focused on the role of IL-33 in the pathogenesis of helminth worm infections. The Th2 response by the host contributes to the elimination of these worm infestations, which are worldwide and afflict hundreds of millions. The role of IL-33 in the induction of IL-4, IL-5, and IL-13 is of paramount importance in terms of pulmonary and intestinal complications that reduce lifespan. Using mice deficient in IL-33, a crucial role was demonstrated in mice to rid them of infection with Strongyloides venezuelensis (Yasuda et al. 2012). The infection induces a unique class of cells called natural helper cells or nuocytes, which upon activation by IL-33 produce IL-5 and IL-13, resulting in eosinophilic infiltration into the lungs. In this model, pulmonary inflammation causes damage via eosinophilic infiltration, which is IL-33 and IL-5 dependent (Yasuda et al. 2012).
Mice injected with human IL-33 exhibit impressive pathological changes in the arterial walls, lungs, and intestinal tissues (Schmitz et al. 2005). Of particular relevance to the concept that IL-33 drives a Th2 response, esosinophilic infiltration was a prominent finding in the lung and in allergic rhinitis as well as allergic conjunctivitis (Matsuba-Kitamura et al. 2010). These initial observations have been confirmed by other reports (Kim et al. 2012). Although the interpretation of in vivo effects following the administration of an exogenous cytokine should be conservative, the findings are clearly consistent with IL-33 being a pro-inflammatory ligand of the IL-1 receptor family. Even before the ability to test IL-33-mediated activation, others had reported that neutralization of the putative ST2 ligand using soluble ST2 markedly reduced joint inflammation, synovial hyperplasia, and joint erosion when given in the therapeutic phase of collagen-induced arthritis in mice (Leung et al. 2004).
6.4 IL-33 as an Anti-inflammatory Cytokine
Members of the IL-1 family of ligands bind to their specific cell-surface receptors and recruit an accessory chain. The IL-1RIAcP is used by IL-1α and IL-1β but also IL-36 and IL-33. The accessory chain for IL-18 is related to the IL-1RIAcP but is encoded by a distinct gene. We now recognize that other members of the IL-1 receptor family will bind more than one cytokine. The best example is IL-1α and IL-1β. Both bind with similar affinities to IL-1RI, but the three-dimensional structures of IL-1α and IL-1β are hardly identical (Wang et al. 2010). The IL-1β precursor binds to IL-1RII as well as a processed form with the first 112 amino acids cleaved from the precursor. IL-37 binds to the IL-18-receptor α-chain (Kumar et al. 2002), and both IL-36 and IL-38 bind to the IL-36 receptor (van de Veerdonk et al. 2012).
IL-33 forms a complex with ST2 IL-1RIAcP but also with SIGIRR (Bulek et al. 2009). This complex plays a role in the Th2 response by reducing IL-33 signaling (Bulek et al. 2009) and, consistent with these observations, Th2 responses are increased in mice deficient in SIGIRR. Furthermore, there is high expression of SIGIRR in Th2 polarized cells, and in models of Th2 antigen sensitization, SIGIRR-deficient mice exhibit a greater Th2 response (Bulek et al. 2009). The complex with SIGIRR and IL-33 may explain the anti-inflammatory properties of IL-33. ST2 can sequester TLR adaptor molecules such as MyD88 and Mal (Gadina and Jefferies 2007).
In mice deficient in ST2, there is myocardial hypertrophy, ventricle dilation, and fibrosis upon pressure overload, suggesting that IL-33 plays a protective role in the heart (Sanada et al. 2007). Furthermore, elevated levels of the extracellular domain of ST2 predict outcomes in patients with systolic heart failure or following a myocardial infarction (Sanada et al. 2007). In a model of cardiomyocyte hypertrophy induced by chronic administration of phenylephrine, administration of recombinant IL-33 inhibited the phosphorylation of IκB and reduced the hypertrophy and fibrosis (Sanada et al. 2007). One of the more challenging aspects of the properties of IL-33 to act as a Th2 cytokine is its role as an antagonist in the ApoE-deficient mouse model of artherosclerosis. In this model, arterial wall plaques of mice on a high-fat diet contain IL-33 and ST2. In mice treated with IL-33, the atherosclerotic plaques were markedly reduced (Miller et al. 2008). In mice treated with soluble ST2 to neutralize IL-33, the disease worsened (Miller et al. 2008).
6.5 IL-33 as a Transcription Factor
Similar to IL-1α, there is another side to IL-33. Although IL-33 binds to its specific surface receptor, IL-33 is identical to a nuclear factor dominantly expressed in high endothelial venules (HEV) (Carriere et al. 2007). This nuclear factor is termed NF-HEV. In addition to endothelial cells, constitutive nuclear localization of IL-33 has been reported in several cell types such as type II lung epithelial cells (Yasuda et al. 2012), epithelial cells (Moussion et al. 2008), and pancreatic stellate cells (Masamune et al. 2010). In fact, IL-33 binding to DNA and acting as a nuclear factor is similar to IL-1α binding to chromatin and functioning as a nuclear factor (Cohen et al. 2010; Stevenson et al. 1997; Werman et al. 2004). A short IL-33 peptide similar to a sequence in Kaposi sarcoma virus binds chromatin (Roussel et al. 2008). The full-length IL-33 precursor, but not mature IL-33, binds to the N-terminal Rel homology domain of NF-κB p65 (Ali et al. 2011). In cells overexpressing the IL-33 precursor, there was a reduction in IL-1β-induced TNF-α (Ali et al. 2011). These data are consistent with other data that IL-33 possesses anti-inflammatory properties (see foregoing), and the mechanism for this property of IL-33 appears to be nuclear sequestration similar to that of IL-1α (Cohen et al. 2010).
7 IL-18 and IL-37 Subfamily
7.1 IL-18
7.1.1 Background
IL-18 was first described in 1989 as “IFN-γ-inducing factor” isolated in the serum of mice following an injection of endotoxin. The mice had been pretreated with Proprionibacterium acnes, which stimulates the reticuloendothelial system, particularly the Kupffer cells of the liver. Many investigators concluded that the serum factor was IL-12. With molecular cloning of “IFN-γ-inducing factor” in 1995 (Okamura et al. 1995), the name was changed to IL-18. Surprisingly, the new cytokine was related to IL-1 and particularly to IL-1β. Similar to IL-1β, IL-18 lacks a signal peptide, is first synthesized as an inactive precursor, and remains as an intracellular cytokine. The tertiary structure of the mature form of IL-18 closely resembles that of IL-1β (Okamura et al. 1995), although the IL-18 precursor is closely related to the IL-37 precursor. Since 1995, many studies have used neutralization of endogenous IL-18- or IL-18-deficient mice to demonstrate the role for this cytokine in promoting inflammation and immune responses [reviewed by (Dinarello 2007)]. However, the biology of IL-18 is hardly the recapitulation of IL-1β. There are several unique and specific differences between IL-18 and IL-1β. For example, in healthy human subjects and also in healthy mice, gene expression for IL-1β in blood mononuclear cells and hematopoietic cells is absent and there is no evidence that the IL-1β precursor is constitutively present in epithelial cells (Puren et al. 1999). In contrast, in the same blood cells large amounts of the IL-18 precursor are present. Peritoneal macrophages and mouse spleen contain the IL-18 precursor in the absence of disease (Puren et al. 1999). The IL-18 precursor is also present in keratinocytes and nearly all epithelial cells. In this regard, IL-18 is similar to IL-1α and IL-33.
7.1.2 Processing of the IL-18 Precursor
The IL-18 precursor has a molecular weight of 24,000 and is processed by caspase-1 cleavage into a mature molecule of 18,000. Compared to wild-type mice, following an injection of endotoxin into caspase-1-deficient mice, circulating IFN-γ is absent. IL-12-induced IFN-γ is also absent in caspase-1-deficient mice (Fantuzzi et al. 1999). Importantly, any phenotypic characteristic of capsase-1-deficient mice must be studied as whether the deficiency is caused by reduced IL-1β or IL-18 activity. For example, the caspase-1-deficient mouse is resistant to colitis (Siegmund et al. 2001b) but the IL-1β-deficient mouse is susceptible in the same disease. Because neutralizing antibodies to IL-18 are protective in the colitis model, caspase-1 deficiency appears to prevent processing of IL-18 (Siegmund et al. 2001a, b). On the other hand, there are examples where caspase-1 processing of IL-18 is not required. For example, Fas ligand stimulation results in release of biologically active IL-18 in caspase-1-deficient murine macrophages (Tsutsui et al. 2000). Similar to IL-1β processing, proteinase-3 appears to activate processing to mature IL-18 (Sugawara et al. 2001).
Similar to IL-1α and IL-33, the IL-18 precursor is constitutively expressed in endothelial cells, keratinocytes, and intestinal epithelial cells throughout the gastrointestinal tract. Macrophages and dendritic cells are the primary sources for the release of active IL-18, whereas the inactive precursor remains in the intracellular compartment of mesenchymal cells. Also, similar to IL-1α and IL-33, the IL-18 precursor is released from dying cells and processed extracellularly, most likely by neutrophil proteases such as proteinase-3.
7.1.3 Signal Transduction by IL-18
As shown in Fig. 1.4a, IL-18 forms a signaling complex by binding to the IL-18 α-chain (IL-18Rα), which is the ligand-binding chain for mature IL-18; however, this binding is of low affinity. In cells that express the co-receptor, termed IL-18-receptor β-chain (IL-18Rβ), a high-affinity complex is formed, which then signals. The complex of IL-18 with the IL-18Rα and IL-18Rβ chains is similar to that formed by other members of the IL-1 family with the co-receptor, the IL-1R accessory chain IL-1RAcP. Following the formation of the heterodimer, the TIR domains approximate, and it appears that the cascade of sequential recruitment of MyD88, the four IRAKs, and TRAF-6 followed by the degradation of IκB and release of NFκB are nearly identical as that for IL-1 (Weber et al. 2010). There are differences between IL-1 and IL-18 signaling that remain unexplained. With few exceptions, IL-1α or IL-1βis active on cells in the low nanogram/ml range and often in the picogram/ml range. In contrast, IL-18 activation of cells expressing the two IL-18-receptor chains requires 10–20 ng/ml and sometime higher levels (Lee et al. 2004; Morel et al. 2001).

IL-18 subfamily. (a) IL-18 binds to the IL-18Rα chain and recruits the co-receptor IL-18Rβ. The signaling cascade of the IL-18 receptor complex is nearly the same as that of IL-1α and IL-1β, resulting in the expression of pro-inflammatory genes. (b) The naturally occurring IL-18BP binds IL-18, thus neutralizing the activity of the cytokine. (c) IL-37 also binds to the IL-18Rα but with an affinity lower than that of IL-18 binding to the same receptor. Furthermore, the binding of IL-37 to IL-18Rα does not recruit the co-receptor, IL-18Rβ, and therefore there is no pro-inflammatory signal. The anti-inflammatory properties of IL-37 require SIGIRR, which may act as a “decoy” for MyD88. (d) IL-18BP also binds to IL-37, thus preventing binding of IL-37 to IL-18Rα. (e) IL-37 binds to IL-18BP, forming a complex, which then binds to IL-18Rα, enhancing the anti-inflammatory property of IL-18BP
Although nearly all cells express IL-1RI, not all cells express IL-1RAcP. Similarly, most cells express IL-18Rα but not all cell express IL-18Rβ. IL-8Rβ is expressed on T cells and dendritic cells but is not commonly expressed in mesenchymal cells. The best example is the A549 cell. This cell line, derived from a lung carcinoma epithelial cell, does not express IL-18Rβ (Kim et al. 2005), and there is no signal unless IL-12 is added to induce IL-18Rβ (Nakanishi et al. 2001b). In the absence of IL-18Rβ, IL-18 binds to IL-18Rα without a pro-inflammatory signal. In A549 cells transfected with IL-18Rβ, IL-18 induces IL-8 and a large number of genes. One of these genes is the former IL-2-induced gene termed NK4 (Dahl et al. 1992), now termed IL-32 (Kim et al. 2005). IL-32 is not a member of the IL-1 family but plays an important role in the regulation of cytokines such as IL-1β and TNF-α.
7.1.4 IL-18 as an Immunoregulatory Cytokine
Together with IL-12, IL-18 participates in the Th1 paradigm. This property of IL-18 is the result of its ability to induce IFN-γ with either IL-12 or IL-15. Without IL-12 or IL-15, IL-18 does not induce IFN-γ. IL-12 or IL-15 increases IL-18Rβ, which is essential for IL-18 signal transduction. Without IL-12 or IL-15, IL-18 plays a role in Th2 diseases (Nakanishi et al. 2001a). The importance of IL-18 as an immunoregulatory cytokine is derived from its prominent biological property of inducing IFN-γ from NK cells. Macrophage colony-stimulating factor (M-CSF) induces human blood monocytes to develop into a subset of macrophages; these cells express a membrane-bound form of IL-18 (Bellora et al. 2012). Membrane IL-18 is expressed in 30–40 % of M-CSF-primed macrophages. In contrast, monocytes, dendritic cells, and monocytes differentiated into M1 macrophages did not express membrane IL-18. Although the expression of membrane IL-18 is caspase-1 dependent (Bellora et al. 2012), LPS treatment was necessary for the release of membrane IL-18 (Bellora et al. 2012). A major immunoregulating role for IL-18 is on the NK cell. Upon shedding of membrane IL-18 into a soluble form, NK cells expressed CCR7 and produced high levels of IFN-γ. As expected, IFN-γ production was prevented by neutralization of IL-18. This mechanism may account for the role of IL-18 as a major IFN-γ-inducing factor from NK cells and the role of NK cells in the pathogenesis of autoimmune diseases.
The induction of IFN-γ by IL-18 has been studied with co-inducer IL-12. For example, mice injected with the combination of IL-18 plus IL-12 develop high levels of IFN-γ and die of hypoglycemia, intestinal inflammation, and inanition (Nakamura et al. 2000). In leptin-deficient mice, IL-18 plus IL-12 induce acute pancreatitis (Sennello et al. 2008). Several human autoimmune diseases are associated with elevated production of IFN-γ and IL-18. Diseases such as systemic lupus erythematosus, rheumatoid arthritis, type 1 diabetes, Crohn’s disease, psoriasis, and graft-versus-host disease are thought to be mediated, in part, by IL-18.
7.1.5 Pro-inflammatory Properties of IL-18
IL-18 exhibits characteristics of other pro-inflammatory cytokines, such as increases in cell adhesion molecules, nitric oxide synthesis, and chemokine production. Blocking IL-18 activity reduces metastasis in a mouse model of melanoma, caused by a reduction in IL-18-induced expression of vascular cell adhesion molecule-1 (Vidal-Vanaclocha et al. 2000). A unique property of IL-18 is the induction of Fas ligand (FasL), which may account for the hepatic damage that takes place in macrophage activation syndrome (Mazodier et al. 2005; Tsutsui et al. 2000). The induction of fever, a well-studied property of IL-1α and IL-1β as well as IL-6, is not a property of IL-18. Injection of IL-18 into mice, rabbits, or humans does not produce fever (Gatti et al. 2002; Li et al. 2003). In contrast to IL-1 and TNF-α, IL-18 does not induce cyclooxygenase-2 and hence there is no production of prostaglandin E2 (Lee et al. 2004; Reznikov et al. 2000). IL-18 has been administered to humans for the treatment of cancer to increase the activity and expansion of cytotoxic T cells. Not unexpectedly, and similar to several cytokines, the therapeutic focus on IL-18 has shifted from its use as an immune stimulant to inhibition of its activity (Dinarello 2007; Tak et al. 2006).
Because IL-18 can increase IFN-γ production, blocking IL-18 activity in autoimmune diseases is an attractive therapeutic target as anti-IL-12/23 reduces the severity of Crohn’s disease as well as that of psoriasis. As discussed next, there appears to be a role for blocking IL-18 in Crohn’s disease. However, there are several activities of IL-18 that are independent of IFN-γ. For example, IL-18 inhibits proteoglycan synthesis in chondrocytes (Joosten et al. 2000), and proteoglycan synthesis is essential for maintaining healthy cartilage. IL-18 also increases VCAM-1 expression in endothelial cells independently of IFN-γ. VCAM-1 plays a major role in multiple sclerosis and other autoimmune diseases as well as in the metastatic process (Carrascal et al. 2003).
7.1.6 IL-18, Hyperphagia, and the Metabolic Syndrome
Although there is no constitutive gene expression for IL-1β in freshly obtained human peripheral blood mononuclear cells (PBMC), the same cells express constitutive mRNA for IL-18 (Puren et al. 1999). In Western blot analysis from the same cells, the IL-18 precursor was present but not the IL-1β precursor. Similar observations were also made in mice (Puren et al. 1999). These findings suggest that IL-18 may act as regulator of homeostasis. Starting at 16 weeks of age, IL-18-deficient mice start to overeat, become obese, and exhibit lipid abnormalities; there is increased atherosclerosis, insulin resistance, and diabetes mellitus, reminiscent of the metabolic syndrome (Netea et al. 2006). IL-18Rα-deficient mice also develop a similar phenotype. The higher body weight is attributed to enhanced food intake, in which the IL-18-deficient mice begin to diverge from wild-type animals at a relatively early age, and to reach values 30–40 % higher than those of wild-type mice. Others have observed similar findings (Zorrilla et al. 2007). A striking finding was an increase of more than 100 % in the percent of adipose tissue in the IL-18-deficient animals, which was accompanied by fat deposition in the arterial walls. The insulin resistance in these mice is corrected by exogenous recombinant IL-18. Mice deficient in IL-18 respond normally to a challenge with exogenous leptin, suggesting that expression of the leptin receptor is unaffected. The unexpected and unique mechanism responsible for the higher food intake in the IL-18-deficient animals appears to be caused by a central nervous system loss of appetite control. IL-18 deficient-mice eat throughout the day whereas wild-type mice eat once, nocturnally.
7.1.7 IL-18 as a Protected Cytokine
As already stated, mice deficient in caspase-1 experience increased disease severity when subjected to dextran sulfate sodium (DSS)-induced colitis and that administration of exogenous IL-18 restored mucosal healing in these mice (Dupaul-Chicoine et al. 2010). In addition, mice deficient in NLRP3 were more susceptible to DSS colitis, which is thought to be caused by decreased IL-18 (Hirota et al. 2011). Mice deficient in NLRP6 are also more vulnerable to DSS (Chen et al. 2007; Elinav et al. 2011), and the susceptibility appears to be lack of sufficient IL-18. Thus, a growing number of studies support a protective role for IL-18. The fact that mice deficient in IL-18 develop a metabolic syndrome-like phenotype is consistent with a role for IL-18 in homeostasis. A study in age-related macular degeneration is also consistent with a protective role for IL-18. In that study, drusen, which is a mixture of complement-derived apolipoproteins and lipids, was shown to activate NLRP3 and induce the production of mature IL-1β and IL-18 (Doyle et al. 2012). In a mouse model of “wet” age-related macular degeneration, the disease was worse in mice deficient in NLRP3 but not in IL-1RI-deficient mice (Doyle et al. 2012). Therefore, IL-18 rather than IL-1α or IL-1β was protective and, upon administration of IL-IL-18, the disease severity improved. Taken together, there is a case for IL-18 being a protective rather than inflammatory cytokine.
7.1.8 IL-18-Binding Protein
The discovery of IL-18BP took place during the search for the soluble receptors for IL-18 (Novick et al. 1999). IL-18BP is a constitutively secreted protein with an exceptionally high affinity for IL-18 (400 pM) (Fig. 1.4b). Present in the serum of healthy humans at a 20-fold molar excess compared to IL-18 (Novick et al. 2001), IL-18BP may contribute to a default mechanism by which a Th1 response to foreign organisms is blunted to reduce triggering an autoimmune response to a routine infection. Although IL-18BP is readily secreted, it falls into the functional category of being a shed soluble receptor. As shown in Fig. 1.4b, IL-18BP contains only one IgG domain whereas the type II IL-1 receptor contains three domains. In this regard, the single IgG domain of IL-18BP is similar to SIGIRR, which also has a single IgG domain and also functions as a decoy receptor. The salient property of IL-18BP in immune responses is in downregulating Th1 responses by binding to IL-18 and thus reducing the induction of IFN-γ (Nakanishi et al. 2001a). Because IL-18 also affects Th2 responses, IL-18BP also has properties controlling a Th2 cytokine response (Nakanishi et al. 2001a). IL-18BP has a classic signal peptide and therefore is readily secreted. Serum levels in healthy subjects are in the range of 2,000–3,000 pg/ml compared to the levels of IL-18 in the same sera of 80–120 pg/ml. Moreover, IL-18BP binds IL-18 with an affinity of 3–5 nM (Novick et al. 1999), an affinity significantly higher than that of IL-18Rα. Because a single IL-18BP molecule binds a single IL-18 molecule, one can calculate bound versus free IL-18 in a mixture of both molecules (Novick et al. 2001).
If one examines immunologically mediated diseases where IFN-γ plays a pathological role such as Wegener’s granulomatosis and systemic lupus erythematosus, one must consider the level of free IL-18 compared to IL-18 bound to IL-18BP. In fact, in these diseases both IL-18BP and IL-18 are high (Novick et al. 2009, 2011) but the level of IL-18BP is not sufficiently high enough to neutralize IL-18 and, therefore, the level of free IL-18 is higher than in healthy subjects. In macrophage activation syndrome where IFN-γ plays a pathological role, both IL-18BP and IL-18 are also high, but the clinical and hematological abnormalities correlate with elevated free IL-18 (Mazodier et al. 2005).
A unique property of IL-18BP is that the molecule also binds IL-37 (Bufler et al. 2002) and, in so doing, enhances the ability of IL-18BP to inhibit the induction of IFN-γ by IL-18. IL-37 binds to the IL-18Rα with a very low affinity, but in mice expressing human IL-37, a profound anti-inflammatory effect is observed (Nold et al. 2010), particularly of LPS-induced cytokines and dendritic cell maturation (Nold et al. 2010). Human IL-37-expressing mice are also resistant to colitis (McNamee et al. 2011). Thus, the anti-inflammatory property of IL-37 can be affected by the concentration of IL-18BP. As the concentration of IL-18BP increases and binds IL-37, there is the possibility that IL-37 becomes less available as an anti-inflammatory cytokine. Indeed, this has been observed in mice injected with IL-18BP. At low dosing of IL-18BP, there is reduced inflammation in a model of rheumatoid arthritis, but as the doing of IL-18BP increases, the anti-inflammatory properties of IL-18BP are lost (Banda et al. 2003).
IL-18BP is highly regulated at the level of gene expression and, unexpectedly, IFN-γ increases gene expression and synthesis of IL-18BP (Hurgin et al. 2002; Muhl et al. 2000). Therefore, IFN-γ driving an increase in the natural and potent inhibitor of IL-18 falls into the category of a negative feedback loop. The concept is supported by clinical data showing that patients being treated with IFN-α for hepatitis have elevated levels of IL-18BP (Kaser et al. 2002; Ludwiczek et al. 2002). IL-27, similar to IFN-γ, functions as both a pro- and an anti-inflammatory cytokine, and both may accomplish their roles as anti-inflammatory cytokines at the level of increased production of IL-18BP. In the skin, IL-27 also acts through a negative feedback loop for inflammation. IL-27 is acting, as is IFN-γ, by induction of IL-18BP gene expression and synthesis (Wittmann et al. 2012).
7.1.9 Viral IL-18BP
Natural neutralization of human IL-18 by IL-18BP takes place during a common viral infection. In molluscum contagiosum infection, characterized by raised but bland eruptions, there are large numbers of viral particles in the epithelial cells of the skin but histologically there are few inflammatory or immunologically active cells in or near the lesions. Clearly, the virus fails to elicit an inflammatory or immunological response. Amino acid similarity exists between human IL-18BP and a gene found in various members of the poxviruses; the greatest degree of homology is found to be expressed by the molluscum contagiosum gene (Xiang and Moss 2001). The ability of viral IL-18BP to reduce the activity of mammalian IL-18 likely explains the lack of inflammatory and immune cells in the infected tissues and provides further evidence for the natural ability of IL-18BP to interfere with IL-18 activity.
7.2 IL-37
7.2.1 IL-37
IL-37 was formerly termed IL-1F7. IL-37 lacks a signal peptide, has a caspase-1 site, but the secretion of IL-37 has not been documented with any certainty. It is likely, however, that similar to IL-1α and IL-33, with loss of membrane integrity upon cell death, the IL-37 precursor exits from the cell. The recombinant form of the IL-37 precursor suppresses LPS-induced IL-1β, IL-6, and TNF-α. However, this is observed primarily in macrophages that have been differentiated into the M1 phenotype by 5 days in the presence of GM-CSF. There are two consensus sequences (A-X-D) in the N-terminal domain of IL-37, IHD, and LED. A recombinant form of IL-37 with an N-terminus nine amino acids from the IHD site is active in suppressing LPS-induced TNF-α and IL-6. Whether this short form of recombinant IL-37 exists in nature is unclear.
7.2.2 IL-37 Reduces IL-1β- and LPS-Induced Inflammation In Vivo
A mouse homologue for human IL-37 has not been identified. Therefore, to define the in vivo functional role of IL-37, a strain of transgenic mice was generated (Nold et al. 2010). The full-length IL-37 cDNA was inserted into a vector using the standard CMV promoter for constitutive expression of the transgene in all cells. Both heterozygous and homozygous IL-37 transgenic mice (IL-37 tg) mice breed normally and exhibit no obvious phenotype. Despite the presence of the CMV promoter, the IL-37 transcript is not constitutively expressed in the tissues of the IL-37 transgenic mice. The failure to express IL-37 is likely caused by a functional instability sequence found in IL-37, which limits the half-life of IL-37 mRNA (Bufler et al. 2004). Nevertheless, upon stimulation with LPS or IL-1β, levels of IL-37 increase after 4–24 h. Once the transcript is present, the IL-37 precursor can be found in peripheral blood cells taken from the transgenic mice (Nold et al. 2003).
IL-37 transgenic mice are protected against LPS challenge compared to similarly challenged wild-type mice. IL-37 transgenic mice exhibit significantly less hypothermia, acidosis, hyperkalemia, hepatitis, and dehydration (Nold et al. 2010). In addition, circulating cytokines are significantly reduced as well as cytokines induced in whole blood cultures and in lung and spleen cell homogenates. In addition to LPS-induced cytokines, whole blood cultures from IL-37 transgenic mice produce significantly less IL-6 and TNF-α when stimulated by IL-1β or the combination of IL-12 plus IL-18. The anti-inflammatory activity of IL-37 was not limited to a reduction of the cytokines and chemokines of innate immunity. Dendritic cells isolated from the spleen of IL-37 transgenic mice upon LPS stimulation revealed a marked reduction (75 % and 89 %) in expression of CD86 and MHC II, respectively (Nold et al. 2010). The total numbers of dendritic cells, macrophages, natural killer cells, and CD4+ T cells were similar in all strains and experimental conditions.
7.2.3 A Role for IL-37 During Experimental Colitis
IL-37 transgenic mice have been subjected to dextran sulfate sodium (DSS)-induced colitis. Despite the presence of a CMV promoter to drive expression of IL-37, mRNA transcripts were not present in colons in the resting state (McNamee et al. 2011). Expression was observed only upon disruption of the epithelial barrier, with a 6- to 7-fold increase on days 3 and 5 after continuous exposure to DSS. During the development of colitis, clinical disease scores were reduced by 50 % and histological indices of colitis were one-third less in IL-37 transgenic mice compared with wild-type counterparts. Reduced inflammation was associated with decreased leukocyte recruitment into the colonic lamina propria. In addition, release of IL-1β and TNF-α from ex vivo colonic explant tissue was decreased 5- and 13 fold, respectively, compared with wild-type mice, whereas IL-10 was increased 6 fold. However, IL-10 was not required for the anti-inflammatory effects of IL-7 because IL-10 receptor antibody blockade did not reverse IL-37-mediated protection. Mechanistically, IL-37 originating from hematopoietic cells was sufficient to exert anti-inflammatory effects because wild-type mice reconstituted with bone marrow from IL-37 transgenic mice were protected from colitis.
7.2.4 A Nuclear Role for IL-37
In stable transfectants of human IL-37 in RAW macrophages stimulated with LPS, levels of TNF-α, IL-1α, IL-6, and the chemokine MIP-2 were substantially reduced (72–98 %) compared with LPS-stimulated cells transfected with the empty plasmid (Sharma et al. 2008). Similar to IL-1α and IL-33, IL-37 translocates to the nucleus following stimulation (Sharma et al. 2008). In mouse RAW macrophages stably expressing IL-37, the mature carboxyl terminal was detected in the nucleus. Furthermore, a specific caspase-1 inhibitor markedly reduced nuclear entry of IL-37 (Sharma et al. 2008). The data demonstrate that IL-37 translocates to the nucleus after caspase-1 processing and may act as a transcriptional modulator reducing the production of LPS-stimulated pro-inflammatory cytokines, consistent with IL-37 being an anti-inflammatory member of the IL-1 family.
IL-37 was identified in a proteomics-based search for proteins that interacted with Smad3 (Grimsby et al. 2004). To test for a functional interaction of Smad3 with IL-37, IL-37 was transfected into A549 cells. IL-37 colocalized with phospho-Smad3 was found in perinuclear and cytosolic regions and a IL-37–Smad3 complex was observed (Nold et al. 2010). A specific inhibitor of Smad3 reversed the inhibition of IL-6 and IL-1β expression in RAW cells stably transfected with IL-37. In stable human macrophage lines expressing IL-37, depletion of Smad3 by lentiviral introduction of short hairpin (sh)RNA that inhibits Smad3 expression prevented the ability of IL-37 to reduce IL-1β- or LPS-induced production of IL-8, IL-6, and TNF. These in vitro findings were confirmed in vivo. IL-37 transgenic mice were pretreated intranasally with a Smad3-specific small interfering (si)RNA and then challenged with intranasal LPS. The reduction of lung cytokines in IL-37 transgenic mice was reversed in transgenic mice with a lung knockdown of Smad3 (Nold et al. 2010).
7.2.5 Role of IL-18Rα for IL-37
From the first reports on IL-37, it was observed that the recombinant forms bound to the IL-18Rα (Kumar et al. 2002; Pan et al. 2001). The binding of IL-37 to IL-18Rα has also been observed in cells from IL-37 transgenic mice using immunofluoresence, immunoprecipitation, and FRET analysis (Nold et al. 2011). IL-37 specifically binds to the third domain of the IL-18Rα (Bufler et al. 2002). Despite these studies showing binding of IL-37 to the IL-18Rα-chain, IL-37 does not act as a classical receptor antagonist for IL-18 in that the ability of recombinant IL-18 to induce IFN-γ is not inhibited by high concentrations of IL-37. However, in the presence of low concentrations of IL-18BP, recombinant IL-37 modestly reduces IL-18-induced IFN-γ (Bufler et al. 2002). The concept that IL-37 binds to the IL-18Rα and reduces cytokine production is supported, in part, with the finding that embryonic fibroblasts from mice deficient in IL-18Rα produce tenfold more IL-6 in response to IL1β than do wild-type embryonic fibroblasts (Nold-Petry et al. 2009). In addition, silencing of IL-18Rα in primary human blood monocytes results in a fourfold increase in the secretion of LPS-induced IL-1β, IL-6, IFN-γ, and CD40 ligand (Nold-Petry et al. 2009). Thus, the seemingly paradoxical hyperresponsive state in cells deficient in IL-18Rα supports the concept that IL-18Rα participates in both pro- and anti-inflammatory responses and that the endogenous ligand IL-37 engages the IL-18Rα to deliver an inhibitory signal.
7.2.6 Role of SIGIRR in the Anti-inflammatory Property of IL-37
The mechanism by which an IL-1β or an LPS signal is suppressed by IL-37 requires an understanding of SIGIRR. The IL-1RAcP serves as the co-receptor for IL-1α, IL-1β, IL-36α, IL-36β, IL-36γ, and IL-33, each a pro-inflammatory cytokine. However, in the IL-1 family of receptors, three co-receptors contain unusually long intracellular domains: these are SIGIRR, a variant of the IL-1RAcP termed IL-1RAcPb and receptors termed “three Ig IL-1 related receptor” (TIGIRR). There are two TIGIRRs, TIGIRR-1 and TIGIRR-2. IL-1RAcPb is expressed only in the brain and TIGIRR has limited expression. However, SIGIRR is expressed in most cells. The TIR domain of the three co-receptors is also different from that of other members of the IL-1 co-receptor family in that the TIR domain contains an amino acid sequence different from that of wild-type TIR (Smith et al. 2009). IL-1RAcPb forms the expected complex with IL-1 and IL-1R1 but does not recruit MyD88 or phosphorylate IRAK4 (Smith et al. 2009). Therefore, most IL-1 signaling is arrested. However, as some genes are increased in response to the formation of the IL-1RI/IL–1RAcPb complex, partial IL-1 signaling must take place. Nevertheless, IL-1RAcPb functions as an inhibitory receptor chain but only in the brain. Mice deficient in IL-1RAcPb exhibit a normal inflammatory response in the periphery but greater neurodegeneration in the brain. As such, IL-1RAcPb could play a role in chronic inflammatory responses in the brain by “buffering” IL-1-mediated neurodegeneration.
Similar to IL-1RAcPb, SIGIRR contains the same amino acid differences in its TIR domain, termed TIRb. Compared to wild-type TIR, TIRb likely has reduced binding of MyD88 (Smith et al. 2009). In addition to an altered TIR domain, SIGIRR has a carboxyl extension of 140 amino acids. Carboxyl extensions are also present in IL-1RAcPb as well as the two TIGIRRs. TIGIRR-2, which is associated with an X-linked cognitive deficiency, is apparently independent of IL-1 function. Little is known whether these C-terminal segments contribute to the inhibitory properties of these receptors. Nevertheless, it seems likely that the alternative sequence in the TIRb domain of SIGIRR may act as a partial decoy for MyD88. MyD88 is phosphorylated upon TLR4 as well as IL-1β and IL-18 signaling and results in downstream phosphorylation of IRAK-4. In cells expressing SIGIRR and activated by IL-37 binding to the IL-18Rα, the signal from either IL-1 or LPS initiates phosphorylation of MyD88. However, the decoy effect by the mutated TIRb of SIGIRR reduces the degree of phosphorylation of MyD88 and thus the phosphorylation of IRAK-4. The reduction, however, is partial. Indeed, the suppression by IL-37 added to blood macrophages is in the range of 20 % to 50 % and is unlike the total loss of activation by a deficiency in MyD88.
Upon binding to the IL-18Rα, the IL-37 precursor may activate SIGIRR and provide a negative signal. An inhibitory signal from IL-37 is enhanced by a low concentration of IL-18BP (Bufler et al. 2002). As shown in Fig. 1.4c,d, IL-18BP binds IL-37 (Bufler et al. 2002) and likely presents the complex of the cytokine with the binding protein to the IL-18Rα. Because A549 cells express SIGIRR, it is likely that the inhibitory signal from IL-37 activates SIGIRR or alternatively IL-37 recruits SIGIRR as the accessory chain. The inhibitory signal of SIGIRR is established in several mouse models of inflammation in which SIGIRR deficient mice exhibit more inflammation compared to wild-type control mice (Garlanda et al. 2009). In differentiated human blood M1 macrophages, recombinant IL-37 suppresses LPS-induced TNF-α and IL-6 production by 50–70 %. A source of IL-18BP in this culture may be fetal calf serum. During the differentiation of macrophages into the M1 subset by GM-CSF, it is possible that SIGIRR expression increases whereas the level of IL-18Rβ decreases (see Fig. 1.4a). In the absence of IL-18Rβ, a proinflammatory complex is not formed with IL-18Rα and thus IL-37 binding to IL-18Rα may recruit or activate SIGIRR. Thus, expression of SIGIRR and the absence of IL-18Rβ would best explain the inhibitory properties of recombinant IL-37 reducing the response to LPS induction of IL-6 and TNFα.
8 IL-36 Subfamily
The IL-1 family members IL-1F5, IL-1F6, IL-1F8, and IL-1F9 are now termed IL-36Ra, IL-36α, IL-36β, and IL-36γ, respectively (Dinarello et al. 2010). Each member of the IL-36 subfamily binds to the IL-1Rpr2, now termed IL-36R (Towne et al. 2004). As IL-38 also binds to the IL-36R (van de Veerdonk et al. 2012), IL-38 is included in the IL-36R subfamily. The IL-36 subfamily is closely related to the IL-1 subfamily because, similar to IL-1α and IL-1β and IL-33, IL-36R forms a signaling complex with IL-1RAcP (Ali et al. 2007; Towne et al. 2004). Thus, of the 11 members of the entire IL-1 family, 6 members use IL-1RAcP as the co-receptor for signal transduction.
8.1 IL-36
IL-36R is the ligand-binding chain and therefore is comparable to the ligand-binding IL-1R1 and IL-18Rα. However, two members of the IL-36 subfamily bind to IL-36R but do not signal: the IL-36 receptor antagonist (IL-36Ra) and IL-38. As such, these function as receptor antagonists (Towne et al. 2011; van de Veerdonk et al. 2012). An unusual property of IL-38 is that low concentrations (1–10 ng/ml) are able to reduce the activity of endogenous IL-36 (van de Veerdonk et al. 2012), whereas in the case of IL-1Ra, higher concentrations are required to prevent the activation of endogenous IL-1α or IL-1β.
None of the members of the IL-36 subfamily has a signal peptide indicating the generation of the N-terminus and secretion via the Golgi. In addition, each member of the IL-36 subfamily has an unusually short propiece compared to those of IL-1α, IL-1β, and IL-33 (see Fig. 1.1). Similar to IL-1β and IL-18, there is no true caspase-1 cleavage site for generation of an N-terminus in the IL-36 subfamily. It remains unknown which specific proteases generate the various N-termini of the IL-36 subfamily; nevertheless, each member has a unique N-terminus with a different levels of biological activity (Towne et al. 2011). What determines the N-terminus with optimal biological activity in the IL-36 subfamily? Each member of the IL-36 subfamily contains the IL-1 family consensus sequence of A-X-D. The aspartic acid is not the recognition amino acid for caspase-1 or caspase-3, but rather participates in the stabilization of the first beta-sheet of the three-dimensional structure that characterizes the entire IL-1 family.
The “A” of the consensus sequence is for any aliphatic amino acid, for example, leucine or isoleucine. Nine amino acids before the “A” of the consensus sequence is the N-terminal site, which results in the cytokine with the greatest activity (Towne et al. 2011). For example, the biological activity of IL-36-γ increases by a factor of 1,000 when the N-terminus is at the site nine amino acids before the consensus sequence and, in the case of the IL-36β, there is a 10,000 fold increase (Towne et al. 2011). The site nine amino acids forward from the consensus sequence is not only the N-terminus for the agonist members of the IL-36 family but also the IL-36 receptor antagonist (IL-36Ra) (Towne et al. 2011), which increases from a low level of blocking of IL-36 family ligands to a high degree of blockade. It is unclear what specific protease cleaves at this site as the amino acid is different for each member of the IL-36 subfamily. Moreover, the site for the N-terminus of the IL-36Ra (valine) is but one amino acid from the N-terminal precursor methionine, and yet IL-36Ra with an N-terminus at the valine site is 10,000 fold more potent than the IL-36 precursor. It is also an unusual situation that proteases that usually are inflammatory in processing members of the IL-1 family in that case of IL-36Ra generate an anti-inflammatory molecule.
8.1.1 IL-36α, -β, and -γ, Proinflammatory Members of the IL-36 Subfamily
IL-36α was highly expressed in the murine model of glomerulonephritis (Ichii et al. 2010), where the cytokine was localized to the kidney epithelium, and also in CD3 T cells surrounding the tubules. IL-36α is also found in the kidneys of the MRL/lupus, nephritic syndrome, and streptozotocin-induced diabetic models (Ichii et al. 2010). IL-36γ increases IL-8, CXCL3, and the Th17 chemokine CCL20 in human lung fibroblasts (Chustz et al. 2011) and thus may account for acute neutrophilic lung inflammation. In addition to CD4+ T cells, human articular chondrocytes and synovial fibroblasts express the IL-36R (Magne et al. 2006). In chondrocytes, there is also constitutive gene expression of IL-36β. Following stimulation with IL-1β or TNF-α, levels of the IL-36β precursor rise intracellularly but the cytokine is not secreted. Although IL-36β levels were detected in the joint fluids of patients with rheumatoid arthritis as well as in serum samples, there was no correlation with disease severity (Magne et al. 2006). It is likely that IL-36 ligands are functional only when released from dying cells and can be processed extracellularly by enzymes present in inflammatory conditions such as the joint of patients with rheumatoid arthritis. It unclear to what extent IL-36β plays a role in joint disease, although constitutive expression in primary chondrocytes may indicate a role for the cytokine in osteoarthritis (Magne et al. 2006).
High levels of this cytokine are found in mouse embryonic tissues rich in epithelial cells (Debets et al. 2001). In humans, IL-36α is observed in keratinocytes, not fibroblasts, and is thought to contribute to the inflammation of psoriasis. Upon forming the heterodimer with IL-36R and IL-1RAcP, IL-36α activates NF-κB similar to that of IL-1β (Towne et al. 2004). In addition to NF-κB activation, IL-36α also activates MAPK, JNK, and ERK1/2 (Towne et al. 2004). In the mouse, bone marrow-derived dendritic cells and CD4+ T cells express IL-36 receptors in health. In a comparison with IL-1 as a stimulant, the three IL-36 ligands are more active in inducing IL-1β, IL-6, IL-12, TNF-α, and IL-23 (Vigne et al. 2011). In addition, IL-36 ligands induced the production of IFN-γ, IL-4, and IL-17 from CD4+ T cells. Not unexpectedly, cytokines induced by IL-36 ligands were prevented by 100- to 1,000-fold excess IL-36Ra (Vigne et al. 2011).
8.1.2 IL-36 in Psoriasis
Several studies implicate IL-36 ligands in the pathogenesis of psoriasis (Blumberg et al. 2007; Johnston et al. 2011; Muhr et al. 2011). IL-36γ is highly expressed in keratinocytes from healthy human skin and increases upon stimulation with TLR polyI:C (Lian et al. 2012). Furthermore, polyI:C induced caspase-3, which resulted in cell death and the release of IL-36γ. Unexpectedly, stimulation of IL-36γ gene expression was dependent on caspase-1 (Lian et al. 2012). The caspase-1 dependency may be caused by IL-18 as this cytokine is constitutively present in keratinocytes as is IL-1α. With the release of IL-36γ by polyI:C and the subsequent death of the cell, IL-36γ falls into the category of being an alarmin in the skin, particularly because of infection (Lian et al. 2012). There is also a role for IL-36 in the production of IL-17: studies suggest that each of the IL-36 ligands is expressed in the skin and dependent on IL-22 (Carrier et al. 2011). Furthermore, the expression of IL-36 ligands in the psoriatic skin correlated with IL-17 (Carrier et al. 2011). Similar to other models in the IL-1 family, auto- and co-induction accounts for a role in a pathological process.
Overexpression of IL-36 in mice results in inflammatory skin lesions that resemble psoriasis in humans, as reviewed by (Towne and Sims 2012).Similarly, mice deficient in endogenous IL-36Ra exhibit a severe lesion similar to that of humans with pustular psoriasis. The role of IL-36 in pustular psoriasis may include IL-1α, as humans with pustular psoriasis respond to an antibody that neutralizes IL-1α. Both IL-36 and IL-1α are found in the keratinocytes in healthy skin.
8.1.3 A Role for IL-36R in Metabolic Regulation
Obesity is characterized by chronic low-grade inflammation originating from expanding adipose tissue. Human adipogenic tissue levels of IL-36α are primarily present in adipose tissue-resident macrophages and are induced by inflammation; however, IL-36β is absent (van Asseldonk et al. 2010). IL-36α, but not IL-36β, reduces adipocyte differentiation, as shown by a significant decrease in PPAR-γ gene expression. Both IL-36α and IL-36β induce inflammatory gene expression in mature adipocytes (van Asseldonk et al. 2010). Therefore, IL-36α and IL-36β are present in adipose tissue and are involved in the regulation of adipose tissue gene expression. Importantly, IL-36α inhibits PPAR-γ expression, which may lead to reduced adipocyte differentiation, suggesting metabolic effects of this cytokine.
Although IL-36Ra is known to occupy the IL-36R and act as a receptor antagonist, earlier studies revealed that IL-36Ra inhibited the induction of IL-1β by LPS. The role of IL-36Ra was also examined in the brain. IL-36Ra injected into the rat brain induced IL-4 and also in glial cells in vitro (Costelloe et al. 2008). Moreover, the reduction in LPS-induced IL-1β was not observed in cells deficient in IL-4 and also not observed in cells deficient in SIGIRR (Costelloe et al. 2008). However, these unique properties of IL-36Ra were not observed in peripheral monocytes or dendritic cells but only in the brain.
8.1.4 Role of IL-36 in Human Disease
The importance of any cytokine in human biology can be found in mutations that result in a profound clinical picture. In IL-1, a mutation in the natural IL-1Ra results in severe systemic inflammation with erosive bone lesions, sterile meningitis, and death; the syndrome is called deficiency of IL-1Ra (Aksentijevich et al. 2009; Reddy et al. 2009). In case of the IL-36 family, persons with a mutation in the naturally occurring IL-36Ra suffer with a severe form of pustular psoriasis (Marrakchi et al. 2011; Onoufriadis et al. 2011). These human studies are consistent with the data from transgenic mice overexpressing IL-36α in the skin and the ability of IL-36Ra to suppress the severity of the inflammation (Blumberg et al. 2007). In mice overexpressing IL-36α in the skin, crossing the mice to generate a strain of mice heterozygous for natural IL-36Ra knockout results in worsening of the skin lesions(Blumberg et al. 2007).
9 IL-38
IL-8 is the name for the IL-1 family member 10. During the nomenclature revision of the IL-1 family in 2010 (Dinarello et al. 2010), the term IL-38 was assigned to IL-1F10 without any known biological function. Since then, IL-38 has been shown to bind to the IL-36 receptor (formerly IL-1Rrp2) (van de Veerdonk et al. 2012). To find the receptor for IL-38, each member of the IL-1 receptor family was immobilized, recombinant IL-38 precursor containing 152 amino acids was added, and binding was assessed using an antibody to the ligand. IL-38 bound only to the IL-36 receptor, as did IL-36Ra (van de Veerdonk et al. 2012). To assess the biological function of IL-38, heat-killed Candida albicans was used to stimulate memory T-lymphocyte cytokine production in freshly obtained human peripheral blood mononuclear cells from healthy subjects. The addition of recombinant IL-38 inhibited the production of T-cell cytokines IL-22 and IL-17. The dose–response suppression of IL-38 as well as that of IL-36Ra of Candida-induced IL-22 and IL-17 was not that of the classic IL-1 receptor antagonist, because low concentrations were optimal for inhibiting IL-22 production (van de Veerdonk et al. 2012). These data provide evidence that IL-38 binds to the IL-36R, as does IL-36Ra, and that IL-38 and IL-36Ra have similar biological effects on immune cells by engaging the IL-36 receptor.
10 The Influence of the IL-1 Family on Th17 Responses
The IL-1 family plays a significant role in IFN-γ production, which is essential for the defense against intracellular pathogens. On the other hand, Th2 cells are characterized by the production of IL-4 and are important in the host defense against parasitic infections. For more than one decade, the dichotomy between Th1 and Th2 has been the focus of studies on differentiation of CD4+ T lymphocytes. More recently, Th17 helper cells have been described and are characterized by their production of IL-17. IL-17 plays a major role in neutrophil recruitment and host defense against extracellular bacteria and fungi. Th17 cells produce a distinct cytokine profile, namely, IL-17A, IL-17F, IL-21, and IL-22. The cytokines produced by Th17 cells, in addition to activating neutrophils, are also crucial for nonimmune cells, for example, induction of defensins by IL-22 in epithelial cells and keratinocytes, which are part of mucosal and skin defenses. It has become apparent that Th17 responses are associated with chronic inflammation and autoimmune diseases such as multiple sclerosis, type 1 diabetes, Crohn’s disease, and psoriasis. Furthermore, Th17 responses are fundamental for host defense against many microorganisms, although they also contribute to the inflammation during infection.
Although IL-4 and IL-12 were the first cytokines described as influencing Th cell differentiation, cytokines of the IL-1 family also influence cytokine differentiation. IL-18 was initially described as an IFN-γ-inducing factor because of its strong stimulatory effect on Th1/IFN-γ responses (Nakanishi et al. 2001b). It is now known that IL-18 is, in fact, a crucial cytokine directing the development of Th1 cells, and one role of IL-12 is the induction of the expression of IL-18 receptors. In contrast, binding of IL-33, another member of the IL-1 family, to its ST2 receptor plays a role in inducing Th2 responses (Schmitz et al. 2005), and it thus appeared as if distinct members of the IL-1 family of cytokines directed Th1 versus Th2 differentiation. Considering these effects of IL-18 and IL-33, it came as no surprise that IL-1, the most well known member of the family, participates in the function of Th cells.
It has been known for more than 30 years that IL-1 enhances T-cell activation and recognition of antigen: one of the early names of IL-1 was lymphocyte activation factor (LAF). The specificity of this response was, however, not known. Although initially only IL-23, IL-6, IL-21, and TGF-β were suggested to play a role in the development of Th17 responses in mice, there is no dearth of data that a more complex picture exists. Thus, IL-1β, IL-6, and TGF-β have been reported to induce the development of Th17 cells, whereas IL-23 has been reported to be important for the maintenance of Th17 cells. The combination of IL-23 and IL-1β induces the development of human Th17 cells expressing IL-17A, IL-17F, IL-22, IL-26, the chemokine CCL20, and transcription factor RORγt (Wilson et al. 2007). Interestingly, these cells also released IFN-γ, displaying a phenotype common to both Th17 and Th1 cells (Wilson et al. 2007). The strong capacity of IL-1 to induce Th17 differentiation has been also linked to its well-known capacity to induce the release of prostaglandins, as reviewed by Dinarello (2011b). PGE2 induced by COX-2 is a stimulator of Th17 induction and inhibitors of cyclooxyugenase decrease IL-17 production (Chizzolini et al. 2008). On the other hand, engagement of the aryl-hydrocarbon receptor, a pathway demonstrated to be crucial for the generation of Th17 cells, has been shown to strongly induce IL-1β (Henley et al. 2004). In addition to inducing IL-17 production from the Th17 subset of lymphocytes, IL-1β is required for the production of IL-17 by NKT cells (Moreira et al. 2011) and of IL-22 from NK cells (Hughes et al. 2010).
Thus, cytokines of the IL-1 family have an important role in the differentiation of the Th subsets, with IL-1β strongly inducing Th17 responses, IL-18 being crucial for the generation of Th1 cells, and IL-33 being important in Th2 responses. Interestingly, reciprocal regulation has been demonstrated between the various Th subsets, with cytokines released by Th2 cells inhibiting Th1 responses, whereas IFN-γ release from Th1 cells impairs both Th2 and Th17 responses.
References
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J (2004) NALP3 forms an IL-1beta processing inflammasome with increased activity in Muckle–Wells auto-inflammatory disorder. Immunity 20:319–325
Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, Laxer R, Tedgard U, Cowen EW, Pham TH, Booty M, Estes JD, Sandler NG, Plass N, Stone DL, Turner ML, Hill S, Butman JA, Schneider R, Babyn P, El-Shanti HI, Pope E, Barron K, Bing X, Laurence A, Lee CC, Chapelle D, Clarke GI, Ohson K, Nicholson M, Gadina M, Yang B, Korman BD, Gregersen PK, van Hagen PM, Hak AE, Huizing M, Rahman P, Douek DC, Remmers EF, Kastner DL, Goldbach-Mansky R (2009) An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 360:2426–2437
Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU (2007) IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci USA 104:18660–18665
Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, Martin MU (2011) The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J Immunol 187:1609–1616
Al-Shukaili A, Al-Kaabi J, Hassan B (2008) A comparative study of interleukin-1beta production and p2x7 expression after ATP stimulation by peripheral blood mononuclear cells isolated from rheumatoid arthritis patients and normal healthy controls. Inflammation 31:84–90
Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A (1999) The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell 10:1463–1475
Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A (2004) Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: implications for inflammatory processes. Proc Natl Acad Sci USA 101:9745–9750
Arend WP (2002) The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev 13:323–340
Bae S, Kang T, Hong J, Lee S, Choi J, Jhun H, Kwak A, Hong K, Kim E, Jo S, Kim S (2012) Contradictory functions (activation/termination) of neutrophil proteinase 3 enzyme (PR3) in interleukin-33 biological activity. J Biol Chem 287:8205–8213
Banda NK, Vondracek A, Kraus D, Dinarello CA, Kim SH, Bendele A, Senaldi G, Arend WP (2003) Mechanisms of inhibition of collagen-induced arthritis by murine IL-18 binding protein. J Immunol 170:2100–2105
Bellora F, Castriconi R, Doni A, Cantoni C, Moretta L, Mantovani A, Moretta A, Bottino C (2012) M-CSF induces the expression of a membrane-bound form of IL-18 in a subset of human monocytes differentiating in vitro toward macrophages. Eur J Immunol 42:1618–1626
Berda-Haddad Y, Robert S, Salers P, Zekraoui L, Farnarier C, Dinarello CA, Dignat-George F, Kaplanski G (2011) Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1alpha. Proc Natl Acad Sci USA 108:20684–20689
Bergers G, Reikerstorfer A, Braselmann S, Graninger P, Busslinger M (1994) Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. EMBO J 13:1176–1188
Blumberg H, Dinh H, Trueblood ES, Pretorius J, Kugler D, Weng N, Kanaly ST, Towne JE, Willis CR, Kuechle MK, Sims JE, Peschon JJ (2007) Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med 204:2603–2614
Boni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, Kerr-Conte J, Pattou F, Halban PA, Weir GC, Donath MY (2008) Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta-cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab 93:4065–4074
Brough D, Rothwell NJ (2007) Caspase-1-dependent processing of pro-interleukin-1beta is cytosolic and precedes cell death. J Cell Sci 120:772–781
Bufler P, Azam T, Gamboni-Robertson F, Reznikov LL, Kumar S, Dinarello CA, Kim SH (2002) A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc Natl Acad Sci USA 99:13723–13728
Bufler P, Gamboni-Robertson F, Azam T, Kim SH, Dinarello CA (2004) Interleukin-1 homologues IL-1F7b and IL-18 contain functional mRNA instability elements within the coding region responsive to lipopolysaccharide. Biochem J 381:503–510
Bulek K, Swaidani S, Qin J, Lu Y, Gulen MF, Herjan T, Min B, Kastelein RA, Aronica M, Kosz-Vnenchak M, Li X (2009) The essential role of single Ig IL-1 receptor-related molecule/Toll IL-1R8 in regulation of Th2 immune response. J Immunol 182:2601–2609
Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M, Huszar M, White MR, Dinarello CA, Apte RN (2009) The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J Immunol 183:4705–4714
Carrascal MT, Mendoza L, Valcarcel M, Salado C, Egilegor E, Telleria N, Vidal-Vanaclocha F, Dinarello CA (2003) Interleukin-18 binding protein reduces b16 melanoma hepatic metastasis by neutralizing adhesiveness and growth factors of sinusoidal endothelium. Cancer Res 63:491–497
Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O’Toole M, Young DA, Fouser LA, Nickerson-Nutter C, Collins M, Dunussi-Joannopoulos K, Medley QG (2011) Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol 131:2428–2437
Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP (2007) IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA 104:282–287
Cayrol C, Girard JP (2009) The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA 106:9021–9026
Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA (2007) IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol 179:2551–2555
Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL (2007) Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 13:851–856
Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CB, Casula MA, Yiangou Y, Birch R, Anand P, Buell GN (2005) Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain 114:386–396
Chizzolini C, Chicheportiche R, Alvarez M, de Rham C, Roux-Lombard P, Ferrari-Lacraz S, Dayer JM (2008) Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood 112:3696–3703
Chustz RT, Nagarkar DR, Poposki JA, Favoreto S Jr, Avila PC, Schleimer RP, Kato A (2011) Regulation and function of the IL-1 family cytokine IL-1F9 in human bronchial epithelial cells. Am J Respir Cell Mol Biol 45:145–153
Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, Leimer AH, Cheronis J (1999) Converting enzyme-independent release of TNFα and IL-1β from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase-3. Proc Natl Acad Sci USA 96:6261–6266
Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN (2010) Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci USA 107:2574–2579
Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16:90–97
Costelloe C, Watson M, Murphy A, McQuillan K, Loscher C, Armstrong ME, Garlanda C, Mantovani A, O’Neill LA, Mills KH, Lynch MA (2008) IL-1F5 mediates anti-inflammatory activity in the brain through induction of IL-4 following interaction with SIGIRR/TIR8. J Neurochem 105:1960–1969
Crisan TO, Plantinga TS, van de Veerdonk FL, Farcas MF, Stoffels M, Kullberg BJ, van der Meer JW, Joosten LA, Netea MG (2011) Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One 6:e18666
Croker BA, Lewis RS, Babon JJ, Mintern JD, Jenne DE, Metcalf D, Zhang JG, Cengia LH, O’Donnell JA, Roberts AW (2011) Neutrophils require SHP1 to regulate IL-1beta production and prevent inflammatory skin disease. J Immunol 186:1131–1139
Dahl CA, Schall RP, He HL, Cairns JS (1992) Identification of a novel gene expressed in activated natural killer cells and T cells. J Immunol 148:597–603
Debets R, Timans JC, Homey B, Zurawski S, Sana TR, Lo S, Wagner J, Edwards G, Clifford T, Menon S, Bazan JF, Kastelein RA (2001) Two novel IL-1 family members, IL-1 delta and IL-1 epsilon, function as an antagonist and agonist of NF-kappa B activation through the orphan IL-1 receptor-related protein 2. J Immunol 167:1440–1446
Derks R, Beaman K (2004) Regeneration and tolerance factor modulates the effect of adenosine triphosphate-induced interleukin 1 beta secretion in human macrophages. Hum Immunol 65:676–682
Dinarello CA (2007) Interleukin-18 and the pathogenesis of inflammatory diseases. Semin Nephrol 27:98–114
Dinarello CA (2011a) A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol 41:1203–1217
Dinarello CA (2011b) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117:3720–3732
Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG, Libby P (1987) Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol 139:1902–1910
Dinarello C, Arend W, Sims J, Smith D, Blumberg H, O’Neill L, Goldbach-Mansky R, Pizarro T, Hoffman H, Bufler P, Nold M, Ghezzi P, Mantovani A, Garlanda C, Boraschi D, Rubartelli A, Netea M, van der Meer J, Joosten L, Mandrup-Poulsen T, Donath M, Lewis E, Pfeilschifter J, Martin M, Kracht M, Muehl H, Novick D, Lukic M, Conti B, Solinger A, Peyman K, van de Veerdonk F, Gabel C (2010) IL-1 family nomenclature. Nat Immunol 11:973
Dinarello CA, Simon A, van der Meer JW (2012) Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 11:633–652
Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, Farrar GJ, Kiang AS, Humphries MM, Lavelle EC, O’Neill LA, Hollyfield JG, Humphries P (2012) NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med 18:791–798
Drenth JP, van der Meer JW, Kushner I (1996) Unstimulated peripheral blood mononuclear cells from patients with the hyper-IgD syndrome produce cytokines capable of potent induction of C-reactive protein and serum amyloid A in Hep3B cells. J Immunol 157:400–404
Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, Meunier C, Turbide C, Gros P, Beauchemin N, Vallance BA, Saleh M (2010) Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32:367–378
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145:745–757
Elssner A, Duncan M, Gavrilin M, Wewers MD (2004) A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 beta processing and release. J Immunol 172:4987–4994
Faggioni R, Fantuzzi G, Fuller J, Dinarello CA, Feingold KR, Grunfeld C (1998) IL-1b mediates leptin induction during inflammation. Am J Physiol 274:R204–R208
Fantuzzi G, Zheng H, Faggioni R, Benigni F, Ghezzi P, Sipe JD, Shaw AR, Dinarello CA (1996) Effect of endotoxin in IL-1b-deficient mice. J Immunol 157:291–296
Fantuzzi G, Ku G, Harding MW, Livingston DL, Sipe JD, Kuida K, Flavell RA, Dinarello CA (1997a) Response to local inflammation of IL-1b converting enzyme-deficient mice. J Immunol 158:1818–1824
Fantuzzi G, Sacco S, Ghezzi P, Dinarello CA (1997b) Physiological and cytokine responses in interleukin-1b-deficient mice after zymosan-induced inflammation. Am J Physiol 273:R400–R406
Fantuzzi G, Reed DA, Dinarello CA (1999) IL-12-induced IFNγ is dependent on caspase-1 processing of the IL-18 precursor. J Clin Invest 104:761–767
Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, Contassot E, Bachmann MF, French LE, Oxenius A, Kundig TM (2011) Inflammasome activation and IL-1beta target IL-1alpha for secretion as opposed to surface expression. Proc Natl Acad Sci USA 108:18055–18060
Gadina M, Jefferies CA (2007) IL-33: a sheep in wolf’s clothing? Sci STKE 2007:pe31
Gardella S, Andrei C, Costigliolo S, Olcese L, Zocchi MR, Rubartelli A (2000) Secretion of bioactive interleukin-1beta by dendritic cells is modulated by interaction with antigen specific T cells. Blood 95:3809–3815
Garlanda C, Anders HJ, Mantovani A (2009) TIR8/SIGIRR: an IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol 30:439–446
Gatti S, Beck J, Fantuzzi G, Bartfai T, Dinarello CA (2002) Effect of interleukin-18 on mouse core body temperature. Am J Physiol Regul Integr Comp Physiol 282:R702–R709
Gattorno M, Tassi S, Carta S, Delfino L, Ferlito F, Pelagatti MA, D'Osualdo A, Buoncompagni A, Alpigiani MG, Alessio M, Martini A, Rubartelli A (2007) Pattern of interleukin-1beta secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum 56:3138–3148
Gawaz M, Brand K, Dickfeld T, Pogatsa-Murray G, Page S, Bogner C, Koch W, Schomig A, Neumann F (2000) Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin-1-dependent mechanism. Implications for atherogenesis. Atherosclerosis 148:75–85
Gay NJ, Keith FJ (1991) Drosophila Toll and IL-1 receptor. Nature (Lond) 351:355–356
Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, Kim HJ, Brewer C, Zalewski C, Wiggs E, Hill S, Turner ML, Karp BI, Aksentijevich I, Pucino F, Penzak SR, Haverkamp MH, Stein L, Adams BS, Moore TL, Fuhlbrigge RC, Shaham B, Jarvis JN, O’Neil K, Vehe RK, Beitz LO, Gardner G, Hannan WP, Warren RW, Horn W, Cole JL, Paul SM, Hawkins PN, Pham TH, Snyder C, Wesley RA, Hoffmann SC, Holland SM, Butman JA, Kastner DL (2006) Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med 355:581–592
Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Goktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O’Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M (2007) NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130:918–931
Grimsby S, Jaensson H, Dubrovska A, Lomnytska M, Hellman U, Souchelnytskyi S (2004) Proteomics-based identification of proteins interacting with Smad3: SREBP-2 forms a complex with Smad3 and inhibits its transcriptional activity. FEBS Lett 577:93–100
Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, Quadroni M, Drexler SK, Tschopp J (2012) Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36:388–400
Hacham M, Argov S, White RM, Segal S, Apte RN (2002) Different patterns of interleukin-1alpha and interleukin-1beta expression in organs of normal young and old mice. Eur Cytokine Netw 13:55–65
Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Gunther S, Prescott NJ, Onnie CM, Hasler R, Sipos B, Folsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, Schreiber S (2007) A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 39:207–211
Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC (2011) Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 286:9587–9597
Hawrylowicz CM, Santoro SA, Platt FM, Unanue ER (1989) Activated platelets express IL-1 activity. J Immunol 143:4015–4018
Hawrylowicz CM, Howells GL, Feldmann M (1991) Platelet-derived interleukin 1 induces human endothelial adhesion molecule expression and cytokine production. J Exp Med 174:785–790
Heguy A, Baldari CT, Macchia G, Telford JL, Melli M (1992) Amino acids conserved in interleukin-1 receptors (IL-1Rs) and the Drosophila Toll protein are essential for IL-1R signal transduction. J Biol Chem 267:2605–2609
Henckaerts L, Cleynen I, Brinar M, John JM, Van Steen K, Rutgeerts P, Vermeire S (2011) Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm Bowel Dis 17:1392–1397
Henley DV, Bellone CJ, Williams DA, Ruh TS, Ruh MF (2004) Aryl hydrocarbon receptor-mediated posttranscriptional regulation of IL-1beta. Arch Biochem Biophys 422:42–51
Hewinson J, Moore SF, Glover C, Watts AG, MacKenzie AB (2008) A key role for redox signaling in rapid P2X7 receptor-induced IL-1 beta processing in human monocytes. J Immunol 180:8410–8420
Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, Lam V, Potentier MS, Ng K, Bawa M, McCafferty DM, Rioux KP, Ghosh S, Xavier RJ, Colgan SP, Tschopp J, Muruve D, MacDonald JA, Beck PL (2011) NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis 17:1359–1372
Hoffman HM, Wanderer AA (2011) Inflammasome and IL-1beta-mediated disorders. Curr Allergy Asthma Rep 10:229–235
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat Genet 29:301–305
Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C (2010) ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 139:1630–1641, 1641 e1631–e1632
Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, Takahashi M, Iwakura Y (1998) Production of mice deficient in genes for interleukin (IL)-1a, IL- 1b, IL-1a/b, and IL-1 receptor antagonist shows that IL-1b is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med 187:1463–1475
Hughes T, Becknell B, Freud AG, McClory S, Briercheck E, Yu J, Mao C, Giovenzana C, Nuovo G, Wei L, Zhang X, Gavrilin MA, Wewers MD, Caligiuri MA (2010) Interleukin-1beta selectively expands and sustains interleukin-22+ immature human natural killer cells in secondary lymphoid tissue. Immunity 32:803–814
Hurgin V, Novick D, Rubinstein M (2002) The promoter of IL-18 binding protein: activation by an IFN-gamma-induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein beta. Proc Natl Acad Sci USA 99:16957–16962
Hurgin V, Novick D, Werman A, Dinarello CA, Rubinstein M (2007) Antiviral and immunoregulatory activities of IFN-gamma depend on constitutively expressed IL-1alpha. Proc Natl Acad Sci USA 104:5044–5049
Ichii O, Otsuka S, Sasaki N, Yabuki A, Ohta H, Takiguchi M, Hashimoto Y, Endoh D, Kon Y (2010) Local overexpression of interleukin-1 family, member 6 relates to the development of tubulointerstitial lesions. Lab Invest 90:459–475
Johnston A, Xing X, Guzman AM, Riblett M, Loyd CM, Ward NL, Wohn C, Prens EP, Wang F, Maier LE, Kang S, Voorhees JJ, Elder JT, Gudjonsson JE (2011) IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol 186:2613–2622
Joosten LA, van De Loo FA, Lubberts E, Helsen MM, Netea MG, van Der Meer JW, Dinarello CA, van Den Berg WB (2000) An IFN-gamma-independent proinflammatory role of IL-18 in murine streptococcal cell wall arthritis. J Immunol 165:6553–6558
Joosten LA, Netea MG, Fantuzzi G, Koenders MI, Helsen MM, Sparrer H, Pham CT, van der Meer JW, Dinarello CA, van den Berg WB (2009) Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum 60:3651–3662
Joosten LA, Netea MG, Mylona E, Koenders MI, Malireddi RK, Oosting M, Stienstra R, van de Veerdonk FL, Stalenhoef AF, Giamarellos-Bourboulis EJ, Kanneganti TD, van der Meer JW (2010) Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1beta production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum 62:3237–3248
Kahlenberg JM, Lundberg KC, Kertesy SB, Qu Y, Dubyak GR (2005) Potentiation of caspase-1 activation by the P2X7 receptor is dependent on TLR signals and requires NF-kappaB-driven protein synthesis. J Immunol 175:7611–7622
Kamari Y, Werman-Venkert R, Shaish A, Werman A, Harari A, Gonen A, Voronov E, Grosskopf I, Sharabi Y, Grossman E, Iwakura Y, Dinarello CA, Apte RN, Harats D (2007) Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis 195:31–38
Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P, Nunez G (2007) Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26:433–443
Kaplanski G, Porat R, Aiura K, Erban JK, Gelfand JA, Dinarello CA (1993) Activated platelets induce endothelial secretion of interleukin-8 in vitro via an interleukin-1-mediated event. Blood 81:2492–2495
Kaplanski G, Farnarier C, Kaplanski S, Porat R, Shapiro L, Bongrand P, Dinarello CA (1994) Interleukin-1 induces interleukin-8 from endothelial cells by a juxacrine mechanism. Blood 84:4242–4248
Kaser A, Novick D, Rubinstein M, Siegmund B, Enrich B, Koch RO, Vogel W, Kim SH, Dinarello CA, Tilg H (2002) Interferon-alpha induces interleukin-18 binding protein in chronic hepatitis C patients. Clin Exp Immunol 129:332–338
Kaspar RL, Gehrke L (1994) Peripheral blood mononuclear cells stimulated with C5a or lipopolysaccharide to synthesize equivalent levels of IL-1b mRNA show unequal IL-1b protein accumulation but similar polyribosome profiles. J Immunol 153:277–286
Kawaguchi Y, Nishimagi E, Tochimoto A, Kawamoto M, Katsumata Y, Soejima M, Kanno T, Kamatani N, Hara M (2006) Intracellular IL-1alpha-binding proteins contribute to biological functions of endogenous IL-1alpha in systemic sclerosis fibroblasts. Proc Natl Acad Sci USA 103:14501–14506
Kenneth NS, Younger JM, Hughes ED, Marcotte D, Barker PA, Saunders TL, Duckett CS (2012) An inactivating caspase 11 passenger mutation originating from the 129 murine strain in mice targeted for c-IAP1. Biochem J 443:355–359
Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA (2005) Interleukin-32: a cytokine and inducer of TNF-alpha. Immunity 22:131–142
Kim YH, Yang TY, Park CS, Ahn SH, Son BK, Kim JH, Lim DH, Jang TY (2012) Anti-IL-33 antibody has a therapeutic effect in a murine model of allergic rhinitis. Allergy 67:183–190
Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931–937
Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, Huszar M, Iwakura Y, Segal S, Dinarello CA, Apte RN (2007) Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res 67:1062–1071
Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ (2008) Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One 3:e3391
Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS-S, Flavell RA (1995) Altered cytokine export and apoptosis in mice deficient in interleukin-1b converting enzyme. Science 267:2000–2003
Kumar S, Hanning CR, Brigham-Burke MR, Rieman DJ, Lehr R, Khandekar S, Kirkpatrick RB, Scott GF, Lee JC, Lynch FJ, Gao W, Gambotto A, Lotze MT (2002) Interleukin-1F7B (IL-1H4/IL-1F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine 18:61–71
Kurt-Jones EA, Beller DI, Mizel SB, Unanue ER (1985) Identification of a membrane-associated interleukin-1 in macrophages. Proc Natl Acad Sci USA 82:1204–1208
Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A (2010) Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol 12:99–113
Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL, Dinarello CA (2004) Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci USA 101:8815–8820
Lee KH, Park SS, Kim I, Kim JH, Ra EK, Yoon SS, Hong YC, Park S, Kim BK (2007) P2X7 receptor polymorphism and clinical outcomes in HLA-matched sibling allogeneic hematopoietic stem cell transplantation. Haematologica 92:651–657
Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, Cayrol C (2012) IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA 109:1673–1678
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA (1996) The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86:973–983
Leung BP, Xu D, Culshaw S, McInnes IB, Liew FY (2004) A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J Immunol 173:145–150
Levandowski CB, Mailloux CM, Ferrara TM, Gowan K, Ben S, Jin Y, McFann KK, Holland PJ, Fain PR, Dinarello CA, Spritz RA (2013) NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1beta processing via the NLRP1 inflammasome. Proc Natl Acad Sci USA 110(8):2952–2956
Li S, Goorha S, Ballou LR, Blatteis CM (2003) Intracerebroventricular interleukin-6, macrophage inflammatory protein-1 beta and IL-18: pyrogenic and PGE(2)-mediated? Brain Res 992:76–84
Lian LH, Milora KA, Manupipatpong KK, Jensen LE (2012) The double-stranded RNA analogue polyinosinic-polycytidylic acid induces keratinocyte pyroptosis and release of IL-36gamma. J Invest Dermatol 132(5):1346–1353
Lonnemann G, Engler-Blum G, Müller GA, Koch KM, Dinarello CA (1995) Cytokines in human renal interstitial fibrosis. II. Intrinsic Interleukin (IL)-1 synthesis and IL-1-dependent production of IL-6 and IL-8 by cultured kidney fibroblasts. Kidney Int 47:845–854
Louten J, Rankin AL, Li Y, Murphy EE, Beaumont M, Moon C, Bourne P, McClanahan TK, Pflanz S, de Waal MR (2011) Endogenous IL-33 enhances Th2 cytokine production and T-cell responses during allergic airway inflammation. Int Immunol 23:307–315
Ludwiczek O, Kaser A, Novick D, Dinarello CA, Rubinstein M, Vogel W, Tilg H (2002) Plasma levels of interleukin-18 and interleukin-18 binding protein are elevated in patients with chronic liver disease. J Clin Immunol 22:331–337
Luheshi NM, Kovacs KJ, Lopez-Castejon G, Brough D, Denes A (2011) Interleukin-1alpha expression precedes IL-1beta after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J Neuroinflammation 8:186
Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Magne D, Palmer G, Barton JL, Mezin F, Talabot-Ayer D, Bas S, Duffy T, Noger M, Guerne PA, Nicklin MJ, Gabay C (2006) The new IL-1 family member IL-1F8 stimulates production of inflammatory mediators by synovial fibroblasts and articular chondrocytes. Arthritis Res Ther 8:R80
Maier JAM, Voulalas P, Roeder D, Maciag T (1990) Extension of the life span of human endothelial cells by an interleukin-1α antisense oligomer. Science 249:1570–1574
Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, Zribi J, Bal E, Cluzeau C, Chrabieh M, Towne JE, Douangpanya J, Pons C, Mansour S, Serre V, Makni H, Mahfoudh N, Fakhfakh F, Bodemer C, Feingold J, Hadj-Rabia S, Favre M, Genin E, Sahbatou M, Munnich A, Casanova JL, Sims JE, Turki H, Bachelez H, Smahi A (2011) Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med 365:620–628
Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annu Rev Immunol 27:229–265
Masamune A, Watanabe T, Kikuta K, Satoh K, Kanno A, Shimosegawa T (2010) Nuclear expression of interleukin-33 in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol 299:G821–G832
Matsuba-Kitamura S, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Taki Y, Muto T, Ikeda T, Mimura O, Nakanishi K (2010) Contribution of IL-33 to induction and augmentation of experimental allergic conjunctivitis. Int Immunol 22:479–489
Mazodier K, Marin V, Novick D, Farnarier C, Robitail S, Schleinitz N, Veit V, Paul P, Rubinstein M, Dinarello CA, Harle JR, Kaplanski G (2005) Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood 106:3483–3489
McNamee EN, Masterson JC, Jedlicka P, McManus M, Grenz A, Collins CB, Nold MF, Nold-Petry C, Bufler P, Dinarello CA, Rivera-Nieves J (2011) Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci USA 108:16711–16716
Miller AC, Schattenberg DG, Malkinson AM, Ross D (1994) Decreased content of the IL1 alpha processing enzyme calpain in murine bone marrow-derived macrophages after treatment with the benzene metabolite hydroquinone. Toxicol Lett 74:177–184
Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, Baker AH, McInnes IB, Liew FY (2008) IL-33 reduces the development of atherosclerosis. J Exp Med 205:339–346
Moreira AP, Cavassani KA, Ismailoglu UB, Hullinger R, Dunleavy MP, Knight DA, Kunkel SL, Uematsu S, Akira S, Hogaboam CM (2011) The protective role of TLR6 in a mouse model of asthma is mediated by IL-23 and IL-17A. J Clin Invest 121:4420–4432
Morel JC, Park CC, Woods JM, Koch AE (2001) A novel role for interleukin-18 in adhesion molecule induction through NFkappa B and phosphatidylinositol (PI) 3-kinase-dependent signal transduction pathways. J Biol Chem 276:37069–37075
Moussion C, Ortega N, Girard JP (2008) The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel “alarmin”? PLoS One 3:e3331
Muhl H, Kampfer H, Bosmann M, Frank S, Radeke H, Pfeilschifter J (2000) Interferon-gamma mediates gene expression of IL-18 binding protein in nonleukocytic cells. Biochem Biophys Res Commun 267:960–963
Muhr P, Zeitvogel J, Heitland I, Werfel T, Wittmann M (2011) Expression of interleukin (IL)-1 family members upon stimulation with IL-17 differs in keratinocytes derived from patients with psoriasis and healthy donors. Br J Dermatol 165:189–193
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–230
Nakamura S, Otani T, Ijiri Y, Motoda R, Kurimoto M, Orita K (2000) IFN-gamma-dependent and -independent mechanisms in adverse effects caused by concomitant administration of IL-18 and IL-12. J Immunol 164:3330–3336
Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H (2001a) Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev 12:53–72
Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H (2001b) Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol 19:423–474
Netea MG, Joosten LA, Lewis E, Jensen DR, Voshol PJ, Kullberg BJ, Tack CJ, van Krieken H, Kim SH, Stalenhoef AF, van de Loo FA, Verschueren I, Pulawa L, Akira S, Eckel RH, Dinarello CA, van den Berg W, van der Meer JW (2006) Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med 12:650–656
Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, van de Veerdonk FL, Ferwerda G, Heinhuis B, Devesa I, Funk CJ, Mason RJ, Kullberg BJ, Rubartelli A, van der Meer JW, Dinarello CA (2009) Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113:2324–2335
Nold M, Hauser IA, Hofler S, Goede A, Eberhardt W, Ditting T, Geiger H, Pfeilschifter J, Muhl H (2003) IL-18BPa:Fc cooperates with immunosuppressive drugs in human whole blood. Biochem Pharmacol 66:505–510
Nold MF, Nold-Petry CA, Zepp JA, Palmer BE, Bufler P, Dinarello CA (2010) IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol 11:1014–1022
Nold MF, Nold-Petry CA, Zepp JA, Lo C, Garlanda C, Mantovani A, Bufler P, Dinarello CA (2011) Interleukin 37 exerts its anti-inflammatory functions by associating with IL-18R alpha and SIGIRR. Cytokine 56:12 (Abstract)
Nold-Petry CA, Nold MF, Nielsen JW, Bustamante A, Zepp JA, Storm KA, Hong JW, Kim SH, Dinarello CA (2009) Increased cytokine production in interleukin-18 receptor alpha-deficient cells is associated with dysregulation of suppressors of cytokine signaling. J Biol Chem 284:25900–25911
Novick D, Kim S-H, Fantuzzi G, Reznikov L, Dinarello CA, Rubinstein M (1999) Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity 10:127–136
Novick D, Schwartsburd B, Pinkus R, Suissa D, Belzer I, Sthoeger Z, Keane WF, Chvatchko Y, Kim SH, Fantuzzi G, Dinarello CA, Rubinstein M (2001) A novel IL-18BP ELISA shows elevated serum IL-18BP in sepsis and extensive decrease of free IL-18. Cytokine 14:334–342
Novick D, Elbirt D, Dinarello CA, Rubinstein M, Sthoeger ZM (2009) Interleukin-18 binding protein in the sera of patients with Wegener’s granulomatosis. J Clin Immunol 29:38–45
Novick D, Elbirt D, Miller G, Dinarello CA, Rubinstein M, Sthoeger ZM (2011) High circulating levels of free interleukin-18 in patients with active SLE in the presence of elevated levels of interleukin-18 binding protein. J Autoimmun 34:121–126
Numanami H, Koyama S, Sato E, Haniuda M, Nelson DK, Hoyt JC, Freels JL, Habib MP, Robbins RA (2003) Serine protease inhibitors modulate chemotactic cytokine production by human lung fibroblasts in vitro. Am J Physiol Lung Cell Mol Physiol 284:L882–L890
Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, Dinarello CA, Fujita M (2009) Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J Biol Chem 285:6477–6488
Okamura H, Nagata K, Komatsu T, Tanimoto T, Nukata Y, Tanabe F, Akita K, Torigoe K, Okura T, Fukuda S, Kurimoto M (1995) A novel costimulatory factor for gamma interferon induction found in the livers of mice causes endotoxic shock. Infect Immun 63:3966–3972
O'Neill LA (2008) The interleukin-1 receptor/toll-like receptor superfamily: 10 years of progress. Immunol Rev 226:10–18
Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, Knight J, Spain SL, Nestle FO, Burden AD, Capon F, Trembath RC, Barker JN (2011) Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet 89:432–437
Pan G, Risser P, Mao W, Baldwin DT, Zhong AW, Filvaroff E, Yansura D, Lewis L, Eigenbrot C, Henzel WJ, Vandlen R (2001) IL-1H, an interleukin 1-related protein that binds IL-18 receptor/IL-1Rrp. Cytokine 13:1–7
Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J (2005) Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med 201:1479–1486
Pelegrin P, Surprenant A (2006) Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J 25:5071–5082
Perregaux DG, McNiff P, Laliberte R, Conklyn M, Gabel CA (2000) ATP acts as an agonist to promote stimulus-induced secretion of IL-1 beta and IL-18 in human blood. J Immunol 165:4615–4623
Plantinga TS, Crisan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, van der Meer JW, Girardin SE, Joosten LA, Netea MG (2011) Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 60:1229–1235
Puren AJ, Fantuzzi G, Dinarello CA (1999) Gene expression, synthesis and secretion of IL-1b and IL-18 are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci USA 96:2256–2261
Qu Y, Franchi L, Nunez G, Dubyak GR (2007) Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol 179:1913–1925
Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, Hessner MJ, Verbsky J (2009) An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 360:2438–2444
Reznikov LL, Kim SH, Westcott JY, Frishman J, Fantuzzi G, Novick D, Rubinstein M, Dinarello CA (2000) IL-18 binding protein increases spontaneous and IL-1-induced prostaglandin production via inhibition of IFN-gamma. Proc Natl Acad Sci U S A 97:2174–2179
Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN (2011) IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 187:4835–4843
Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ, Brant SR (2007) Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39:596–604
Rosenwasser LJ, Dinarello CA, Rosenthal AS (1979) Adherent cell function in murine T-lymphocyte antigen recognition IV. Enhancement of murine T-cell antigen recognition by human leukocytic pyrogen. J Exp Med 150:709–714
Roussel L, Erard M, Cayrol C, Girard JP (2008) Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep 9:1006–1012
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (Lond) 456:264–268
Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT (2007) IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest 117:1538–1549
Schindler R, Clark BD, Dinarello CA (1990) Dissociation between interleukin-1b mRNA and protein synthesis in human peripheral blood mononuclear cells. J Biol Chem 265:10232–10237
Schlesinger N, Mysler E, Lin HY, De Meulemeester M, Rovensky J, Arulmani U, Balfour A, Krammer G, Sallstig P, So A (2011) Canakinumab reduces the risk of acute gouty arthritis flares during initiation of allopurinol treatment: results of a double-blind, randomised study. Ann Rheum Dis 70:1264–1271
Schmid D, Munz C (2007) Innate and adaptive immunity through autophagy. Immunity 27:11–21
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA (2005) IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23:479–490
Sennello JA, Fayad R, Pini M, Gove ME, Ponemone V, Cabay RJ, Siegmund B, Dinarello CA, Fantuzzi G (2008) Interleukin-18, together with interleukin-12, induces severe acute pancreatitis in obese but not in nonobese leptin-deficient mice. Proc Natl Acad Sci USA 105:8085–8090
Sha Y, Zmijewski J, Xu Z, Abraham E (2008) HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol 180:2531–2537
Sharma S, Kulk N, Nold MF, Graf R, Kim SH, Reinhardt D, Dinarello CA, Bufler P (2008) The IL-1 family member 7b translocates to the nucleus and down-regulates proinflammatory cytokines. J Immunol 180:5477–5482
Siegmund B, Fantuzzi G, Rieder F, Gamboni-Robertson F, Lehr HA, Hartmann G, Dinarello CA, Endres S, Eigler A (2001a) Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-γ and TNF-α production. Am J Physiol Regul Integr Comp Physiol 281:R1264–R1273
Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA (2001b) IL-1beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A 98:13249–13254
Sims JE, March CJ, Cosman D, Widmer MB, MacDonald HR, McMahan CJ, Grubin CE, Wignall JM, Jackson JL, Call SM (1988) cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science 241:585–589
Sluyter R, Dalitz JG, Wiley JS (2004a) P2X7 receptor polymorphism impairs extracellular adenosine 5′-triphosphate-induced interleukin-18 release from human monocytes. Genes Immun 5:588–591
Sluyter R, Shemon AN, Wiley JS (2004b) Glu496 to Ala polymorphism in the P2X7 receptor impairs ATP-induced IL-1 beta release from human monocytes. J Immunol 172:3399–3405
Smith DE, Lipsky BP, Russell C, Ketchem RR, Kirchner J, Hensley K, Huang Y, Friedman WJ, Boissonneault V, Plante MM, Rivest S, Sims JE (2009) A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity 30:817–831
So A, De Smedt T, Revaz S, Tschopp J (2007) A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 9:R28
So A, De Meulemeester M, Pikhlak A, Yucel AE, Richard D, Murphy V, Arulmani U, Sallstig P, Schlesinger N (2010) Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: results of a multicenter, phase II, dose-ranging study. Arthritis Rheum 62:3064–3076
Stevenson FT, Turck J, Locksley RM, Lovett DH (1997) The N-terminal propiece of interleukin 1 alpha is a transforming nuclear oncoprotein. Proc Natl Acad Sci USA 94:508–513
Sugawara S, Uehara A, Nochi T, Yamaguchi T, Ueda H, Sugiyama A, Hanzawa K, Kumagai K, Okamura H, Takada H (2001) Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol 167:6568–6575
Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nunez G (2007) Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 3:e111
Tak PP, Bacchi M, Bertolino M (2006) Pharmacokinetics of IL-18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur J Drug Metab Pharmacokinet 31:109–116
Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ (2010) Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood 115:3632–3639
Towne J, Sims J (2012) IL-36 in psoriasis. Curr Opin Pharmacol 12(4):486–490
Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE (2004) Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem 279:13677–13688
Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, Sims JE (2011) Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J Biol Chem 286:42594–42602
Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, Philpott DJ (2010) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11:55–62
Tschopp J, Schroder K (2010) NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10:210–215
Tsutsui H, Matsui K, Okamura H, Nakanishi K (2000) Pathophysiological roles of interleukin-18 in inflammatory liver diseases. Immunol Rev 174:192–209
Unlu S, Kumar A, Waterman WR, Tsukada J, Wang KZ, Galson DL, Auron PE (2007) Phosphorylation of IRF8 in a pre-associated complex with Spi-1/PU.1 and non-phosphorylated Stat1 is critical for LPS induction of the IL1B gene. Mol Immunol 44:3364–3379
van Asseldonk EJ, Stienstra R, Koenen TB, van Tits LJ, Joosten LA, Tack CJ, Netea MG (2010) The effect of the interleukin-1 cytokine family members IL-1F6 and IL-1F8 on adipocyte differentiation. Obesity 18:2234–2236
van de Veerdonk FL, Stoeckman AK, Wu G, Boeckermann AN, Azam T, Netea MG, Joosten LA, van der Meer JW, Hao R, Kalabokis V, Dinarello CA (2012) IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc Natl Acad Sci USA 109:3001–3005
van der Meer JWM, Barza M, Wolff SM, Dinarello CA (1988) A low dose of recombinant interleukin 1 protects granulocytopenic mice from lethal gram-negative infection. Proc Natl Acad Sci USA 85:1620–1623
Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, Fuentes AM, Anasagasti MJ, Martin J, Carrascal T, Walsh P, Reznikov LL, Kim SH, Novick D, Rubinstein M, Dinarello CA (2000) IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci USA 97:734–739
Vigne S, Palmer G, Lamacchia C, Martin P, Talabot-Ayer D, Rodriguez E, Ronchi F, Sallusto F, Dinh H, Sims JE, Gabay C (2011) IL-36R ligands are potent regulators of dendritic and T cells. Blood 118:5813–5823
Wang D, Zhang S, Li L, Liu X, Mei K, Wang X (2010) Structural insights into the assembly and activation of IL-1beta with its receptors. Nat Immunol 11:905–911
Weber A, Wasiliew P, Kracht M (2010) Interleukin-1 (IL-1) pathway. Sci Signal 3:cm1
Werman A, Werman-Venkert R, White R, Lee JK, Werman B, Krelin Y, Voronov E, Dinarello CA, Apte RN (2004) The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci USA 101:2434–2439
Wewers MD, Dare HA, Winnard AV, Parker JM, Miller DK (1997) IL-1b-converting enzyme (ICE) is present and functional in human alveolar macrophages: macrophage IL-1b release limitation is ICE independent. J Immunol 159:5964–5972
Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal MR (2007) Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 8:950–957
Wittmann M, Bachmann M, Doble R, Pfeilschifter J, Werfel T, Mühl H (2012) IL-27 regulates IL-18 binding protein in skin resident cells. PLoS One 7(6):e38751
Xiang Y, Moss B (2001) Correspondence of the functional epitopes of poxvirus and human interleukin-18-binding proteins. J Virol 75:9947–9954
Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, Matsushita K, Taki Y, Futatsugi-Yumikura S, Tsutsui H, Ishii KJ, Yoshimoto T, Akira S, Nakanishi K (2012) Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci USA 109:3451–3456
Yin H, Morioka H, Towle CA, Vidal M, Watanabe T, Weissbach L (2001) Evidence that HAX-1 is an interleukin-1 alpha N-terminal binding protein. Cytokine 15:122–137
Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann K, Conn CC, Siszynski D, Grabiec C, Trumbauer MA, Shaw AR, Kostura MJ, Stevens K, Rosen H, North RJ, Chen HY, Tocci MJ, Kluger MJ, Van der Ploeg LHT (1995) Resistance to fever induction and impaired acute-phase response in interleukin-1b deficient mice. Immunity 3:9–19
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11:136–140
Zorrilla EP, Sanchez-Alavez M, Sugama S, Brennan M, Fernandez R, Bartfai T, Conti B (2007) Interleukin-18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc Natl Acad Sci USA 104:11097–11102
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Japan
About this chapter
Cite this chapter
Dinarello, C.A., Netea, M.G. (2014). The Interleukin-1 Family. In: Yoshimoto, T., Yoshimoto, T. (eds) Cytokine Frontiers. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54442-5_1
Download citation
DOI: https://doi.org/10.1007/978-4-431-54442-5_1
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54441-8
Online ISBN: 978-4-431-54442-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)