
Abstract
Among all three isoforms of the TGF-β ligand, TGF-β1 is the predominant isoform in the skin. In normal skin, canonical TGF-β signaling components, i.e., TGF-β receptors and signaling Smads, are broadly and highly expressed, whereas TGF-β ligands are expressed at very low levels. These expression patterns determine that the TGF-β signaling input to the skin is low under normal conditions but high once TGF-β ligands are upregulated under disease conditions. TGF-β1 is a potent growth inhibitor of epidermal keratinocytes, which dictates its tumor suppressive effect in early stages of skin cancer. However, cancer cells lose TGF-β-induced growth inhibition at late stages, and TGF-β-induced angiogenesis and skin inflammation create an environment favorable for skin cancer progression and metastasis. In fibrotic skin diseases, TGF-β plays a key role in activating fibroblast proliferation and stimulating the production of extracellular matrix proteins. Also, TGF-β affects many aspects of skin wound healing and in turn influences cutaneous scarring after skin damage. Fully understanding the mechanisms of TGF-β in the pathogenesis of skin cancer and fibrotic diseases will help design novel strategies in treating skin diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Transforming growth factor-β (TGF-β) is a ubiquitous and multifunctional cytokine that regulates a variety of events in cells and tissues, including cell proliferation, differentiation, migration, angiogenesis, inflammation, and immune surveillance. The function of TGF-β is largely dependent on its targeted cells and is influenced by other molecules and signaling. Thus, the in vivo role of TGF-β is complicated and context dependent. The skin is histologically divided into two parts: the epidermis and the dermis. Keratinocytes in the epidermis and fibroblasts in the dermis represent two major cell types showing opposite effects in response to TGF-β: TGF-β inhibits keratinocytes but promotes fibroblast proliferation (Anzano et al. 1982; Coffey et al. 1988; Shipley et al. 1986). Among the three TGF-β isoforms, TGF-β1 is the predominant isoform in most tissue types, including the skin (Frank et al. 1996; Quan et al. 2002; Wang 2001), and is expressed at a very low level in normal skin but elevated in skin diseases and cancer (Han et al. 2005; Quan et al. 2002; Querfeld et al. 1999). The role of TGF-β in the pathogenesis of skin cancer and fibrotic diseases has been extensively studied. In addition to the direct effect of TGF-β on keratinocytes and fibroblasts, TGF-β-mediated inflammation, angiogenesis, and regulation of immune cells have additional impacts on the development of skin cancer and fibrosis (Li et al. 2006; Martin 1997; Seifert and Mrowietz 2009; Wei et al. 2011). Particularly, recent studies on the receptors and Smads, components of TGF-β signaling in transgenic/knockout mouse models further advanced our understanding of the roles of TGF-β signaling in skin cancer and fibrotic diseases (Han and Wang 2011; Lakos et al. 2004; Sonnylal et al. 2007; Wei et al. 2011). This chapter will focus on the role of TGF-β signaling in the pathogenesis of non-melanoma skin cancer and skin fibrotic disorders, the latter including systemic sclerosis (SSc), hypertrophic scarring, and keloid scarring. Because hypertrophic and keloid scarring are consequences of wound healing affected by TGF-β during the healing process, the role of TGF-β signaling in cutaneous wound healing is also discussed.
2 TGF-β Signaling in Non-melanoma Skin Cancer
Non-melanoma skin squamous cell carcinoma (SCC) is the most common human cancer developed from epidermal keratinocytes. Genetic mutations and continuous proliferation of mutant keratinocytes are the prerequisites for SCC development. Studies indicate that angiogenesis, inflammation, and alterations in immune response in the stroma significantly influence the development of skin cancer and metastasis. Many molecules and signaling pathways have been implicated in the pathogenesis of SCC. The role of TGF-β and its downstream signaling components have been extensively investigated in SCC. The TGF-β signaling pathway consists of three components: (1) Ligands, i.e., TGF-β1, 2, and 3. (2) Secreted ligands function through two serine/threonine kinase receptors, TGF-β receptor type I (TGF-βRI) and type II (TGF-βRII), both of which are necessary for signal transduction. TGF-βRII receptor directly binds to ligands and interacts with TGF-βRI. (3) Intracellular downstream mediators, i.e., Smads, are phosphorylated and activated by TGF-βRI. To date, many components of TGF-β signaling have been studied for their differential roles in normal keratinocytes, SCC cells and tumor stromal cells. This chapter will give a brief review over these differential roles of TGF-β signaling in skin carcinogenesis.
2.1 TGF-β Inhibits Tumor Formation of ras Transformed Keratinocytes
TGF-β was initially identified as a growth factor stimulating the growth of normal rat kidney fibroblasts (Anzano et al. 1982; Sporn 1999, 2006). Soon, however, TGF-β was found to inhibit mink lung epithelial cell growth (Tucker et al. 1984). Thus, the diversity of TGF-β’s functions on different cell types was quickly recognized (Roberts et al. 1985). In particular, TGF-β inhibits the growth of normal human keratinocytes but can lose its growth inhibitory responses as it happens in the skin cancer cell line SCC-25 due to the possibility that tumor cells lack TGF-β receptors (Coffey et al. 1988; Shipley et al. 1986). However, tumor development in normal keratinocytes requires an initial oncogenic mutation. The Ras mutation in keratinocytes is considered an important initiating factor for papilloma development in mouse skin (Roop et al. 1986) and was identified in some human skin cancers (Dlugosz et al. 2002). Thus, Ras-transfected keratinocytes have been used to study the role of TGF-β signaling in skin carcinogenesis. In vitro studies showed that v-ras Ha transfected normal mouse keratinocytes increased basal expression and secretion of TGF-β1, and grafting v-ras Ha transfected normal keratinocytes onto nude mice formed well-differentiated papillomas with increased expression of TGF-β1 protein in the basal and spinous layers of papillomas (Glick et al. 1991, 1994). However, v-ras Ha transfected TGF-β1 null mouse keratinocytes grafted onto nude mice progressed rapidly to multifocal SCC. Tumor proliferation was also elevated in grafts initiated from v-ras Ha transfected TGF-β1 null keratinocytes compared to cells with v-ras Ha transfected wild-type keratinocytes. These studies indicated that TGF-β provides a tumor-suppressing function in skin carcinogenesis (Glick et al. 1991, 1994). Similarly, transient inactivation of TGF-β signaling by infecting with a dominant-negative TGF-βRII causes chromosome instability. These phenomena, observed in keratinocytes with TGF-β1 deletion or expressing dominant-negative TGF-βRII, could be suppressed by exogenous TGF-β1 (Glick et al. 1999). In the TGF-β signaling pathway, Smad3 is a critical factor mediating TGF-β signaling to its target genes (Millet and Zhang 2007). Smad3 null keratinocytes transduced with the v-ras Ha gene lost their growth inhibitory response to TGF-β. Nude mice with v-ras Ha-transduced Smad3 null keratinocyte grafts developed more papillomas and progressed to SCC with a higher frequency than nude mice with v-ras Ha-transduced Smad3 wild-type keratinocyte grafts (Vijayachandra et al. 2003). Therefore, data based on in vitro models of keratinocyte transformation studies demonstrated that TGF-β signaling inhibits keratinocyte proliferation and defects in TGF-β signaling accelerate tumor progression in multistage mouse carcinogenesis (Glick et al. 1999). HaCaT is a spontaneously immortalized normal human keratinocyte cell line (Boukamp et al. 1988). The cell line keeps most characteristics of normal keratinocytes and remains non-tumorigenic after long-term culture and passage. HaCaT cell became tumorigenic after transducing with a ras oncogene (Fusenig and Boukamp 1998; Gold et al. 2000), Within established ras-transfected HaCaT cells, benign and malignant clones have been characterized based on their behaviors in forming tumor after transplantation into nude mice (Fusenig and Boukamp 1998). HaCaT-ras cells with stable transduction of dominant-negative TGF-βRII lose differentiation and form metastatic SCC in vivo (Ganapathy et al. 2010). In contrast to the growth inhibitory effect of TGF-β on normal keratinocytes, HaCaT-ras cells with high TGF-β1 production in cultured conditions have increased tumorigenesis in vivo, and transducing TGF-β1 or TGF-β2 to the benign HaCaT-ras clone increases tumor formation, invasion, and metastasis in nude mice (Davies et al. 2012). Furthermore, the HaCaT-ras cells producing more TGF-β still retain the response to TGF-β-induced growth inhibition in vitro (Davies et al. 2012), suggesting that the growth inhibitory effect of TGF-β1 is insufficient for tumor suppression in vivo.
2.2 In Vivo Expression of TGF-β1 in the Skin Inhibits Benign Tumor Formation and Promotes Malignant Conversion in Skin Carcinogenesis
The two-stage chemical carcinogenesis model is a well-established experimental system to study the mechanism of skin tumor development in vivo (Filler et al. 2007; Yuspa 1986). In this model, skin tumor is first initiated by exposing the skin to carcinogens such as the mutagen dimethylbenzanthracene (DMBA), which can cause mutation in the oncogene Ha-ras (Fujiki et al. 1989); second, tumor formation is promoted by treatments of a tumor promoter, such as 12-O-tetradecanoylphorbol-13-acetate (TPA). Some benign tumors (primarily papillomas) can convert to malignant SCCs after acquiring additional spontaneous genetic mutations (Akhurst and Balmain 1999). In wild-type mice, TGF-β1 mRNA expression is quickly induced by TPA in suprabasal cells of the epidermis (Akhurst et al. 1988). Chemically induced papillomas and carcinomas have elevated levels of TGF-β1 mRNA in the keratinocyte compartment (Fowlis et al. 1992; Patamalai et al. 1994). To investigate the role of TGF-β in skin homeostasis and carcinogenesis in vivo, keratinocyte-specific TGF-β transgenic mice were developed. Transgenic mouse models that target TGF-β1 to epidermal keratinocytes using different keratin promoters demonstrate that functions of TGF-β1 in skin development and epidermal keratinocyte proliferation depending on the location of TGF-β1 expression in different epidermal layers. When TGF-β1 was targeted to suprabasal layers of the epidermis driven by keratin 1 (K1) promoter (K1.TGF-β1), transgenic mice died at early neonatal stages (Sellheyer et al. 1993) due to the inhibition of keratinocyte proliferation. In contrast, mice overexpressing TGF-β1 driven by keratin 6 or keratin 10 promoters (K6.TGF-β1 or K10.TGF-β mice) lived to adulthood with no significant histological changes other than resistance to TPA-induced epidermal hyperplasia (Cui et al. 1995; Fowlis et al. 1996). However, study of long-term chemical carcinogenesis on K6.TGF-β1 and K10.TGF-β1 transgenic mice led to the conclusion that TGF-β1 has a biphasic effect on skin carcinogenesis: inhibiting tumorigenesis at an early stage but promoting malignant tumor conversion and rapid metastasis at a later stage (Cui et al. 1996). Using gene-switch-TGF-β1 transgenic mice to induce TGF-β 1 transgene expression in the skin at specific stages of chemical carcinogenesis, we found that TGF-β1 transgene induction at early stage inhibited tumor formation but TGF-β1 induction in papillomas significantly promoted tumor conversion from benign to malignant with increased metastasis (Weeks et al. 2001). Moreover, TGF-β transgenic papillomas exhibited down-regulation of TGF-β receptors and their signal transducer Smads, loss of the invasion suppressor E-cadherin/catenin complex in the cell membrane, elevated expression of matrix metalloproteinases and increased angiogenesis. Thus, although down regulation of TGF-β signaling components in tumor epithelia abolishes TGF-β-induced tumor cell growth inhibition, tumor stroma, which has intact TGF-β signaling components, respond to increased TGF-β ligand to promote tumor metastasis (Weeks et al. 2001).
2.3 In Vivo Abrogation of TGF-β Receptors in Skin Promotes Tumor Development and Metastasis
Mice with germline TGF-βRII deletion are embryonic lethal around 10.5 days of gestation (Oshima et al. 1996). Interruption of TGF-βRII function by either overexpressing dominant-negative TGF-βRII (ΔβRII) receptors or conditional deletion of TGF-βRII in specific epidermis has been utilized to study the role of TGF-β receptors in skin homeostasis and carcinogenesis (Amendt et al. 1998; Go et al. 1999; Guasch et al. 2007). Transgenic mice expressing ΔβRII in suprabasal keratinocytes driven by truncated mouse loricrin promoter (ML.ΔβRII) develop hyperproliferative skin conditions in the early neonatal stage but normalize in adulthood. In vitro cultured primary keratinocytes from these mice demonstrated resistance to exogenous TGF-β1-induced growth inhibition (Wang et al. 1997). When these transgenic mice were subjected to the two-stage chemical carcinogenesis study, they developed tumors earlier with a larger number of benign papillomas and higher frequencies of malignant cancer formation and metastasis compared to wild-type mice (Go et al. 1999). Analysis showed that ML.ΔβRII tumors had increased expressions of vascular endothelial growth factor (VEGF), a pro-angiogenesis factor, and decreased expression of thrombospondin-1, an angiogenesis inhibitor. Both factors are attributed to the increased angiogenesis found in tumors and malignant carcinoma progression and metastasis. Increased angiogenesis correlated with elevated endogenous TGF-β1 in ML.ΔβRII tumors (Go et al. 1999). Similarly, when ΔβRII was expressed in basal keratinocytes in mouse skin targeted by the keratin 5 promoter, the transgenic mice also have accelerated skin tumor development and malignancy transformation in chemical carcinogenesis (Amendt et al. 1998) compared to wild-type mice. Moreover, keratinocyte-specific TGF-βRII knockout mice in which deletion of TGF-βRII in stratified epithelia driven by the keratin 14 promoter results in spontaneous SCC formation in the transitional area between the stratified squamous epithelium of the skin and the mucosal epithelium, e.g. the anal and genital regions but not in mouse back skin (Guasch et al. 2007). Further analyses revealed that in comparison with mouse back skin, the epithelium of the transitional zone in the anus naturally showed enhanced Ras-MAPK signaling, locally increased inflammation and aberrant differentiation, which favor to tumorigenesis in the anal and genital regions. In aged mice with TGF-βRII deletion, reduced apoptosis and high epidermal proliferation due to blocking of TGF-β signaling eventually caused anal and genital SCC development. Tumor formation in these regions did not happen in young mice when enhanced proliferation, as a result of TGF-βRII deletion, was balanced with increased apoptosis (Guasch et al. 2007). Furthermore, when DMBA alone was applied to TGF-βRII knockout mice skin or grafting of Ha-Ras-infected TGF-βRII knockout keratinocytes onto nude mice, mice quickly developed invasive/metastatic SCC (Guasch et al. 2007). Therefore, the loss of TGF-β function in epidermal keratinocytes promotes skin carcinogenesis, but development of malignant tumors requires additional initiation factors (Amendt et al. 1998; Go et al. 1999; Guasch et al. 2007). Human malignant skin cancer frequently exhibits overexpression of TGF-β1 but reduced expression of TGF-βRII. To better understand how this combination affects cancer prognosis, we studied skin carcinogenesis using the chemical carcinogenesis protocol in inducible TGF-β transgenic mice with and without functional TGF-β receptors. In this study, TGF-β1/ΔβRII compound mice were generated; these mice stably express ΔβRII, which inhibits cell binding to the TGF-β ligand, but TGF-β1 can be induced at specific stages of carcinogenesis. When TGF-β1 expression was induced in the papilloma stage, mice developed malignant SCC at a high frequency, with more metastasis to the lymph nodes and lung. Overall, 30 % of tumors in mice overexpressing TGF-β1 exhibited spindle cell carcinoma (SPCC). TGF-β1 induction in the papilloma with inactivation of TGF-βRII function in the TGF-β/ΔβRII compound mice also developed more malignant SCC and metastasis. However, SPCC was rarely observed in TGF-β1/ΔβRII mice. Consistently, TGF-β1-induced SCCs exhibited loss of E-cadherin, an EMT marker. This phenomenon was not common in ΔβRII or TGF-β1/ΔβRII compound mice. Further analysis showed that TGF-β1-mediated SCCs had significant levels of Hey1 and Jag1, while upregulation of Erk1, JNK1, and RhoA/Rac1 was also observed in all TGF-β1 and ΔβRII mice. Therefore, TGF-β1-induced EMT and metastasis can be uncoupled. TGF-β1 mediates EMT via the activation of Notch signaling and requires intact TGF-β signaling components, whereas increased angiogenesis and matrix metalloproteinase (MMP) caused by TGF-β-induced MAPK and Rho/Rac activation via a Smad-independent mechanism largely contribute to cancer metastasis (Han et al. 2005). This study highlights an important fact that EMT cannot always be used to predict the metastatic potential.
2.4 Differential Roles of TGF-β Signaling Smads in Skin Cancer
The discovery and study of Smad family members mediating TGF-β signaling has significantly improved the understanding of the TGF-β signaling pathway’s multiple biological functions. Eight isoforms of Smads have been found in mammals and are generally classified into three subtypes: R-Smads (Smad1, 2, 3, 5, 8), Co-Smad (Smad4), and I-Smads (Smad6, 7) (Derynck and Zhang 1996; Massague 1996). Germline deletions of Smad1, Smad2, Smad4, Smad5, and Smad7 in mice result in embryonic death (Chang et al. 1999; Heyer et al. 1999; Kleiter et al. 2010; Nomura and Li 1998; Sirard et al. 1998; Tremblay et al. 2001; Waldrip et al. 1998; Weinstein et al. 1998; Yang et al. 1998). Smad3 knockout mice survive to adulthood (Datto et al. 1999; Yang et al. 1999; Zhu et al. 1998). To overcome embryonic lethality caused by knocking out individual Smads, conditional knockout mice targeting Smad deletion to keratinocytes were generated to study the role of Smads in skin carcinogenesis (Bornstein et al. 2007). So far, the potential roles of TGF-β signaling Smads, i.e., Smad2, Smad3, and Smad4, in skin carcinogenesis have been investigated, but the role of other Smads in skin tumors remains to be clarified. Thus, the current discussion will be focused on the role of TGF-β signaling Smads in skin cancer.
2.4.1 The Role of Smad2 in Skin Carcinogenesis
Loss or reduction of Smad2 expression occurs in several human SCCs, including genital SCCs, head and neck SCCs and cervical SCCs (Han and Wang 2011). Immunostaining has shown Smad2 protein reduction/loss in 70 % of human skin cancers. Smad2 loss is especially high in poorly differentiated SCCs (Hoot et al. 2008). Although Smad2 mutation was not frequent in human SCCs (Han and Wang 2011), we found that approximately 67 % of poorly differentiated human skin SCCs have loss of heterozygosity (LOH) at the Smad2 locus, possibly due to accumulated ultraviolet radiation-induced genomic damage (Hoot et al. 2008). Since germline homozygous Smad2 deletion in mice results in embryonic lethality (Heyer et al. 1999; Nomura and Li 1998; Waldrip et al. 1998; Weinstein et al. 1998), the role of Smad2 in skin carcinogenesis was first investigated in Smad2 heterozygous mice (Smad2+/−) because these mice lived a normal lifespan without spontaneous tumor development. Smad2+/− mice developed a greater number of less-differentiated tumors compared to wild-type control mice when exposed to the two-stage chemical carcinogenesis protocol (Tannehill-Gregg et al. 2004). In our established conditional Smad2 knockout mice that target Smad2 deletions to keratinocytes using the keratin 14 (K14) promoter, we found Smad2 deletion did not develop spontaneous skin tumors throughout 18 months of observation (Hoot et al. 2008). Moreover, even in the presence of a DMBA-induced H-ras mutation, a genetic alteration mimicking early stage human skin cancer, mice with epidermal Smad2 deletion still failed to develop skin tumors without TPA promotion. Thus, Smad2 loss alone is insufficient to promote initiated cells to develop into cancer. Similar to the observation in Smad2+/− mice (Tannehill-Gregg et al. 2004), when the two-stage chemical carcinogenesis protocol was applied to mice with epidermal Smad2 loss, mice developed skin tumors early with increased numbers, and accelerated malignant conversion compared to wild-type mice (Hoot et al. 2008). Histological analysis showed that Smad2 loss induced papillomas to undergo early EMT, and about 25 % malignant tumors in conditional Smad2 knockout mice were classified as SPCC, which are rare in wild-type mice. Moreover, Smad2 deficient tumors have significantly increased angiogenesis in the stroma. Mechanism analyses revealed that levels of TGF-β1, TGF-β signaling, Smad3/Smad4, and VEGF were similar between Smad2 deletion tumors and wild-type tumors. Proliferation and apoptosis in tumors have a very limited effect from Smad2 deletion. However, Smad2 deletion significantly increased Snail and hepatocyte growth factor (HGF) expression in keratinocytes and tumor epithelial cells. Further analysis identified that Smad2 loss causes Snail and HGF upregulation via loss of Smad2-mediated transcriptional repression and enhanced Smad3/4-mediated transactivation for Snail and HGF in keratinocytes and tumor cells (Hoot et al. 2008; Hoot et al. 2010). These results provide potential new targets for treating skin cancer.
2.4.2 The Role of Smad3 in Skin Carcinogenesis
Smad3 acts as a major mediator for the TGF-β signaling pathway (Millet and Zhang 2007; Nakao et al. 1997). With respect to the role of Smad3 in SCC, Smad3 null keratinocytes transduced with v-ras Ha exhibited significant reduction of TGF-β-induced cell growth arrest and increased tumorigenesis after grafting onto nude mice. Overexpression of Smad3 in wild-type keratinocytes by infection with adenoviral-Smad3 induced keratinocyte growth arrest and senescence. All of the changes observed in Smad3 null keratinocytes cannot be amended by transfecting Smad2 and Smad4 (Bae et al. 2009; Vijayachandra et al. 2003). In contrast to the findings in Smad3 null keratinocytes and keratinocyte grafting bioassays, a carcinogenesis study in Smad3 knockout mice has shown that Smad3 is required for tumor formation in the two-stage carcinogenesis model. Germline Smad3 knockout mice did not develop spontaneous skin tumors after long-term observation (Datto et al. 1999; Yang et al. 1999; Zhu et al. 1998). Interestingly, both homozygous and heterozygous Smad3 knockout mice have shown resistance to chemically induced skin carcinogenesis (Li et al. 2004a; Tannehill-Gregg et al. 2004). Furthermore, Smad3 knockout tumors show reduced cell proliferation and inflammation, but increased apoptosis in response to TPA treatment (Li et al. 2004a). The observed phenomena may be attributed to Smad3 deletion-mediated blocking of TGF-β signaling, evidenced by the reduction of TGF-β-induced activator protein-1 family members and TGFα in TPA treated Smad3 null cells and tissues. Moreover, tumor tissues exhibited reduced leukocyte infiltration, particularly a reduction of tumor-associated macrophage infiltration. Consistently, the pro-inflammatory cytokine, IL-1β, and the monocyte/macrophage-attracting chemokine, MCP-1, are significantly reduced in Smad3 null tumors compared to wild-type tumors (Li et al. 2004a). Therefore, TGF-β mediated inflammation appears to require Smad3, which could explain the difference between Smad3-mediated tumor suppressive effects inside the keratinocytes and Smad3-mediated oncogenic effects on the tumor stroma.
2.4.3 The Role of Smad4 in Skin Carcinogenesis
To avoid embryonic lethality caused by germline deletion of Smad4 (Sirard et al. 1998; Yang et al. 1998), mouse models with Smad4 deletion specifically targeted at keratinocytes have been established (Bornstein et al. 2009; Li et al. 2003; Qiao et al. 2006; Yang et al. 2005). Smad4 deficient skin develops progressive alopecia with hair follicle degeneration without affecting keratinocyte differentiation (Owens et al. 2008; Yang et al. 2005). Molecular mechanisms for this pathological alteration are attributed to the loss of Smad4 that blocked Smad1/5 mediated desmoglein-4 transcription (Owens et al. 2008). When Smad4 was deleted in epidermal keratinocytes, mammary epithelium or head and neck epithelium, all mice developed spontaneous SCC (Bornstein et al. 2009; Li et al. 2003; Yang et al. 2005). These findings further confirmed that Smad4 mutation acts as an oncogenic factor (Hahn et al. 1996; Thiagalingam et al. 1996). Tumors in Smad4 knockout mice have reductions in phosphatase and tensin homolog (PTEN) and p21CIP/WAF, but upregulation of AKT, increased cell proliferation and nuclear accumulation of cyclin D1 (Qiao et al. 2006). In the established Smad4/PTEN double knockout mice, skin tumor formation was significantly accelerated compared to Smad4 deletion alone (Qiao et al. 2006; Yang et al. 2005). When Smad4 and PTEN genes were simultaneously deleted in epithelial cells of the upper digestive tract, forestomach tumor formation was also accelerated (Teng et al. 2006). Thus, Smad4 and PTEN act synergistically to regulate epidermal proliferation and differentiation. Loss of both genes may contribute to the initiation and promotion of tumorigenesis (Teng et al. 2006; Yang et al. 2005). The tumor suppressive role of Smad4 is also shown by the deletion of Smad4 in oral epithelia, which resulted in spontaneous head and neck SCC with downregulation of Fanc/Brca pathway genes, which increased genomic instability and inflammation (Bornstein et al. 2009). Thus, there are several critical differences between Smad2 and Smad4 deletion in the epithelium. Smad2 deletion in the epidermis promoted EMT at quite an early stage and caused significant increase in SPCC formation in chemical carcinogen-initiated skin cancer, but Smad2 loss itself is insufficient to initiate tumorigenesis. Increased angiogenesis observed in chemically induced Smad2−/−tumors was caused by directly upregulating HGF through recruiting Smad4 onto the SBE site of the HGF promoter (Hoot et al. 2010). Smad4 deletion in epithelial cells could initiate spontaneous carcinoma development by downregulation of PTEN or Fanc/Brca genes. EMT phenomena or SPCC was seldom observed in the Smad4 deletion tumors. Furthermore, Smad4 deleted but not Smad2 deleted tumors have increased expression of TGF-β1 and VEGF, which contributes to increased angiogenesis and inflammation (Bornstein et al. 2009; Owens et al. 2010; Qiao et al. 2006; Yang et al. 1998).
3 TGF-β and Systemic Sclerosis
SSc, also known as scleroderma, is a chronic and fatal autoimmune disease characterized by excessive extracellular matrix accumulation in connective tissues. SSc affects the skin and internal organs, including the lungs, gastrointestinal tract, and heart. Clinically, SSc is classified into two major types: diffuse SSc, characterized as cutaneous scleroderma with at least one internal organ involvement, and limited cutaneous scleroderma, characterized as limited skin scleroderma without internal organ involvement. The major pathologic feature of SSc is excess collagen deposition in the extracellular matrix. Although the mechanism of skin fibrosis remains unknown, abnormal dermal fibroblast activation and chronic inflammation have been implicated in the pathogenesis of SSc. TGF-β has been shown to promote fibroblast proliferation (Wynn 2007), stimulate the production of collagen by fibroblasts (Bettinger et al. 1996; Wu et al. 2012), and induce inflammation in the skin (Li et al. 2004b). Therefore, accumulated evidence indicates that TGF-β signaling plays a key pathogenic role in SSc.
3.1 Increased TGF-β Expression in SSc Lesions
Histologically, fibrosis is a cardinal feature of SSc, but vasculopathy and inflammatory infiltration may also be observed in the early stages of SSc (Beyer et al. 2012; Gabrielli et al. 2009; Varga and Abraham 2007). Fibrosis is characterized by excessive collagen formation in connective tissue, produced by activated fibroblasts. Since TGF-β is shown to promote fibroblast proliferation and enhance collagen production in normal human fibroblasts, TGF-β has been implicated in the pathogenesis of fibrotic disorders (Varga and Jimenez 1986). SSc patients have high levels of plasma TGF-β, and fibroblasts derived from SSc patients display elevated levels of TGF-β mRNA expression (Higley et al. 1994; Vuorio et al. 1991). Abnormal expression of TGF-β is shown to be involved in SSc skin lesion formation (Gay et al. 1992; Gruschwitz et al. 1990; Rudnicka et al. 1994). Increased TGF-β1 was also found in endothelial cells at the early stages of SSc skin (Gabrielli et al. 1993). For the three isoforms of TGF-β (1, 2, and 3) in mammals, the mRNA for each has been detected in inflamed areas of either localized or diffused SSc, but not in sclerotic or healthy skin. However, TGF-β1 and TGF-β2 proteins were confirmed in the inflammatory skin of patients, whereas TGF-β3 protein appears to be present in the dermis of both patients and healthy controls, indicating a reduced specificity of TGF-β3 in the pathogenesis of SSc (Querfeld et al. 1999). Furthermore, TGF-β2 mRNA was found to be co-localized with pro-α1(I) collagen expression around dermal blood vessels in the inflammatory stage of SSc, but no expression of TGF-β2 or pro-α1(I) collagen was found in the dermis of fibrotic stage patients, suggesting that TGF-β2 might be more important in the pathogenesis of early SSc (Kulozik et al. 1990). In several established SSc animal models, increased TGF-β acts as a key mediator in fibrosis formation, and the reduction of TGF-β expression by several therapeutic strategies shows antifibrotic effects (Artlett 2010; Batteux et al. 2011; Iwamoto et al. 2011; Rosenbloom et al. 2010; Yamamoto 2010). These experiments further verified the role of TGF-β in the pathogenesis of SSc.
3.2 Upregulation of TGF-β Receptors in SSc Fibroblasts
It was also reported that alteration of TGF-β receptors occurred in SSc fibroblasts. Increased TGF-βRI and TGF-βRII mRNAs were found in cultured fibroblasts derived from SSc patients compared to healthy control fibroblasts. Consistently, overexpression of either TGF-βRI or TGF-βRII significantly increases α2 (I) collagen promoter activity in transient transfection assays in dermal fibroblasts (Kawakami et al. 1998). Furthermore, both TGF-βRI and TGF-βRII increased in dermal fibroblasts and inflammatory cells around vascular blood in localized scleroderma and SSc lesional skin in comparison with healthy controls (Kubo et al. 2001, 2002). These studies suggest that overexpression of TGF-βRI and TGF-βRII in dermal fibroblasts results in autocrine TGF-β signaling, which plays a pathological role in SSc development. However, these findings have not been fully verified by other studies. One study reported that SSc derived fibroblasts have high levels of TGF-β RI expression, but 30 % decreased TGF-βRII expression compared to normal healthy skin derived fibroblasts (Pannu et al. 2004). Furthermore, normal fibroblasts infected with adenoviral-TGF-βRI but not adenoviral-TGF-βRII demonstrated increased basal level of type-I collagen expression. They concluded that increasing the ratio of TGF-βRI to TGF-βRII contributed to collagen formation involved in the pathogenesis of SSc (Pannu et al. 2004). Perturbation of TGF-βRI and TGF-βRII in the pathogenesis of fibrosis has also been seen in animal models of SSc. In the induced TGF-βRI (βRICA/Cre-ER) mouse models, postnatal induction of TGF-βRI in fibroblasts resulted in multiple organ fibrosis mimicking features of SSc in humans (Sonnylal et al. 2007). When constitutively active TGF-βRI was induced in fibroblasts in βRICA/Cre-ER transgenic mice, the mice developed generalized dermal fibrosis in their skin and a small-vessel vasculopathy with both smooth muscle and endothelial abnormalities in the lungs and kidneys (Sonnylal et al. 2007). Additionally, overexpression of dominant-negative TGF-βRII in fibroblasts in TGF-βRIIΔk-fib mice also caused lung and skin fibrosis as endogenous TGF-βRI was activated in transgenic fibroblasts (Denton et al. 2003, 2005; Hoyles et al. 2008).
3.3 Abnormal Expression of Smads in SSc
Type I collagen proteins, which consist of glycine- and proline-rich two α1(I) chains (COL1A1) and one α2(I) chain (COL1A2), have been recognized as major excess protein in SSc lesion (Jinnin 2010). COL1A2 genes contain Smad binding sites at their promoters. Transient overexpression of Smad3 and Smad4, but not Smad1 or Smad2, causes trans-activation of the human COL1A2 promoter in normal human dermal fibroblasts (Chen et al. 1999; Dennler et al. 1998; Zhang et al. 2000). In vitro studies demonstrated that scleroderma-derived fibroblasts had increased COL1A2 promoter activity with high levels of Smad3 binding to the promoter compared to normal control fibroblasts (Jinnin et al. 2006). Compared to fibroblasts derived from healthy adult volunteers, increased Smad3 expression is found in fibroblasts from scleroderma patients (Mori et al. 2003). Since subcutaneous injection of bleomycin into mouse skin for 4 weeks results in fibrosis in the skin and lung, bleomycin-induced fibrosis has been extensively used to study the pathogenesis of SSc (Yamamoto 2010). When the bleomycin-inducing sclerosis protocol was applied to Smad3 knockout mice, inflammatory infiltration and TGF-β expression in the dermis at an early time point were similar in Smad3 knockout and wild-type mice. However, by day 28 after injection, lesional skin from Smad3 knockout mice exhibited attenuated fibrosis, lower synthesis and accumulation of collagen, and reduced collagen gene transcription in situ, compared to wild-type mice (Lakos et al. 2004). These data support the idea that Smad3 mediates TGF-β-induced fibrosis in SSc thus targeting Smad3 signaling could at least in part ameliorate scleroderma. Smad7, as an antagonist of the TGF-β signaling pathway, has been reported to inhibit α1 (I) collagen production in normal human fibroblasts and decrease α-smooth muscle actin expression in hypertrophic scar-derived fibroblasts (Kopp et al. 2005). Reduced Smad7 functions, due to either reduced expression or functional defect, have been observed in SSc. In one study, SSc lesions or SSc derived fibroblasts have reduced Smad7 expression but increased Smad3 expression in comparison with that of normal skin or fibroblasts, indicating that Smad7 deficiency and Smad3 upregulation attribute to upregulated TGF-β signaling in scleroderma (Dong et al. 2002). In a separate study, SSc lesions and SSc derived fibroblasts were found to express high levels of Smad7 and TGF-βRI, and TGF-β signaling Smads remained unchanged compared to normal controls (Asano et al. 2004). Further analysis showed that the inhibitory effect of Smad7 on human α2(I) collagen promoter activity was completely impaired in SSc fibroblasts, suggesting that increased Smad7 in SSc fibroblasts may be caused by autocrine TGF-β signaling (Asano et al. 2004). The discrepancy of Smad7 expression levels in SSc skin and fibroblasts has also been reported by other studies (Kreuter et al. 2006; Zhu et al. 2012) and may be due to different sample sources or different stages of SSc (Asano et al. 2004). Recent reports indicate that the Smad1 pathway may also be involved in the pathogenesis of SSc. The first evidence came from a study on forced TGF-βRI expression-induced fibrosis, which upregulates collagen production and CCN2 (CTGF) via the TGF-βRI/Smad1 and ERK1/2 pathways (Pannu et al. 2007). The same group later showed that total and phosphorylated Smad1 levels were significantly elevated in SSc skin biopsy samples and cultured SSc fibroblasts and correlated with elevated CCN2 protein levels. Furthermore, this group identified that Smad1 was a direct activator of the CCN2 gene through binding the GC-motif in the CCN2 promoter. SSc fibroblasts exhibited elevated CCN2 promoter activity, which is correlated with the level of increased Smad1. Knocking down Smad1 in SSc fibroblasts reduced production of CCN2 and collagen (Pannu et al. 2008). Therefore, it is likely that Smad1 and Smad3 play complementary roles in the pathogenesis of SSc.
3.4 TGF-β Regulates Other Fibrotic Factors Implicated in SSc
In addition to TGF-β, many cytokines, chemokines, and growth factors have been implicated in the pathogenesis of SSc. However, TGF-β seems to still be a master regulator in SSc as evidenced by its crosstalk with other molecules during SSc fibrosis. The platelet derived growth factor (PDGF) pathway has long been recognized as another key player in fibrotic diseases. PDGF regulates many aspects of fibrotic diseases including promoting myofibroblast proliferation and stimulating production of inflammatory cytokines and ECM. In SSc, TGF-β enhances the function of PDGF by upregulating expression of PDGF receptors in fibroblasts (Bonner 2004). c-Abelson (c-Abl), a non-receptor tyrosine kinase associated with chronic myelogenous leukemia (CML), is produced by TGF-β-activated fibroblasts and is also involved in TGF-β-induced fibrotic processes (Bhattacharyya et al. 2009; Rosenbloom et al. 2010). Inhibition of c-Abl by dasatinib or nilotinib has shown antifibrotic effects (Beyer et al. 2012). CCN2 (CTGF), early growth response gene I (EGR1), and Endothelin 1 (ET-1) are all induced by TGF-β and upregulated in SSc skin. All have been shown to mediate TGF-β-induced fibrosis. Targeting CCN2, EGR1 (e.g., imatinib) or ET-1 (e.g., Bosentan) may become new therapeutic strategies for treating fibrotic diseases (Bhattacharyya et al. 2012).
4 TGF-β and Cutaneous Hypertrophic and Keloid Scarring
Hypertrophic scarring and keloid scarring are frequently observed in clinical practice. Hypertrophic scarring occurs when the raised scar is limited to within the margin of the original injury, while keloid scarring is defined as a continually growing scar extending beyond the original injury. Scarring is usually the outcome of normal wound healing and can regress spontaneously after injury; however, hypertrophic scars caused by acute injuries such as severe burns usually result in life-long physical disability. Keloid scarring has been shown to have a strong genetic predisposition and rarely regresses (Tuan and Nichter 1998). Clinically, hypertrophic scarring and keloid scarring cause the patient physical and psychological distress and can significantly affect the patient’s quality of life (Shih and Bayat 2010). Causally, both result from overgrowth of fibrous tissue characterized by excess production of ECM and collagen formation after skin damage due to injury or surgery (Seifert and Mrowietz 2009; Tziotzios et al. 2012). Many cytokines and growth factors are proposed to be involved in scar formation; TGF-β and its signaling pathway, which influences all aspects of wound healing, play key roles in regulating scar development (Seifert and Mrowietz 2009).
4.1 TGF-β and Scar Free Wounds
Previous studies have demonstrated that fetal wounds heal without scar formation in humans and various experimental animal models (Bullard et al. 2003). Compared to adult wounds, fetal wounds are characterized by fine and reticular collagen deposition with less cross-linking, less inflammation, higher hyaluronic acid concentration, and a greater ratio of collagen type III to type I (Namazi et al. 2011). Multiple differences in molecules and gene expression have been identified between fetal wounds and adult wounds (Larson et al. 2010; Namazi et al. 2011). Among them, the role of TGF-β has been extensively studied. The expression of TGF-β1 and TGF-β2 is increased in adult wounds while levels of TGF-β1 and TGF-β2 are low in fetal wounding mouse models (Nath et al. 1994; Sullivan et al. 1995; Whitby and Ferguson 1991). Lack of TGF-β1 has also been observed in fetal platelets (Olutoye et al. 1996). To verify the pathological role of TGF-β1 and TGF-β2 in scar formation, TGF-β1 and TGF-β2 neutralizing antibodies were used in adult rat wounds, which resulted in reduced monocyte and macrophage profile, neovascularization, fibronectin, collagen III and collagen I deposition in the early stages of wound healing. Only given a single antibody to either TGF-β1 or TGF-β2 did the wound show less or no relieved scarring (Shah et al. 1995). In addition, some studies indicated that scar-less wounds have significantly increased levels of TGF-β3, while TGF-β1 levels remained unchanged (Eslami et al. 2009; Hsu et al. 2001). Administration of the TGF-β3 peptide to the wounded area exhibited an antifibrotic effect (Shah et al. 1995). Those studies clearly demonstrate isoform-specific differences of TGF-β playing different roles in wound healing and cutaneous scarring. Although several other reports have shown increased TGF-β1 and TGF-β2 in fetal wounds after acute wounding, these increases clear rapidly. This transient induction of TGF-β1 and TGF-β2 in fetal wounds might not be enough to induce inflammation (Cowin et al. 2001; Martin et al. 1993; Soo et al. 2003). Moreover, recent studies indicate that fetal fibroblasts do not exhibit TGF-β1-induced collagen production when compared with their mature counterparts (Rolfe et al. 2007). Thus, the difference in response to TGF-β stimuli between fetal fibroblasts and mature fibroblasts may also contribute to scar-free fetal wounds.
4.2 TGF-β and Cutaneous Scarring
The mechanism of keloid formation is still poorly understood, and no effective therapy for keloids exists. As studies have shown that TGF-β has a potent role in stimulating the proliferation of fibroblasts and causing inflammation, activation of TGF-β signaling has been postulated to play a central role in the pathogenesis of keloids. Compared to normal skin, keloid-derived fibroblasts have high expression of TGF-β1 and TGF-β2 proteins, but expression of TGF-β3 remains unchanged (Lee et al. 1999). Both TGF-β1 and VEGF expressions were increased in keloid fibroblasts and the upregulation of VEGF expression was dependent on TGF-β (Fujiwara et al. 2005). Furthermore, upregulation of TGF-β1, TGF-βRI, and TGF-βRII has been shown in burn-induced hypertrophic scar tissue in vivo (Ghahary et al. 1993; Schmid et al. 1998; Zhang et al. 1995). In addition, high levels of TGF-β signaling Smads including Smad2, 3, 4, TGF-βRI and TGF-βRII proteins were reported in keloid and hypertrophic scar tissue, and keloid-derived fibroblasts (Phan et al. 2005; Tsujita-Kyutoku et al. 2005). In contrast, inhibitory Smads including Smad6 and Smad7 have been shown to decrease in keloid tissue (Yu et al. 2006). In vitro studies showed that TGF-β1 increases collagen production by keloid fibroblasts and promotes proliferation and migration of keloid fibroblasts (Bettinger et al. 1996; Wu et al. 2012). These results indicated the pathogenic role of TGF-β signaling in cutaneous scar formation (Mrowietz and Seifert 2009; Seifert and Mrowietz 2009). Ideally, accelerated wound healing without scars or with minimal scarring is the aim for perfect regeneration in adult skin injuries. TGF-β is produced by many cellular components involved in wound healing, including keratinocytes, platelets, and macrophages, and therefore production of TGF-β increases after injury (Singer and Clark 1999). However, the controversy surrounding the role of TGF-β in wound healing has existed for a long time and is still not resolved. Analysis identified that TGF-β3 in serum plays a crucial role in mediating keratinocyte migration and inhibiting dermal fibroblast migration, implying TGF-β3 is key factor initiating re-epithelialization at early stages of wound healing as keratinocytes are exposed to serum due to skin damage (Bandyopadhyay et al. 2006; Henry et al. 2003). In contrast, transgenic mice overexpressing the TGF-β1 transgene in keratinocytes exhibited delayed healing after burn injury or excisional wounding due to delayed re-epithelialization and significant skin inflammation (Chan et al. 2002; Wang et al. 2006; Yang et al. 2001). Consistently, TGF-β1 knockout mice showed accelerated re-epithelialization during incisional wound repair in comparison with wild-type mice (Koch et al. 2000), and transgenic mice overexpressing a dominant-negative TGF-βRII receptor or keratinocyte-specific deletion of TGF-βRII receptor also exhibited accelerated re-epithelialization in skin wounds (Amendt et al. 2002; Guasch et al. 2007). Smad3 knockout mice, in which TGF-β signaling is partially abolished, exhibited accelerated wound healing, featuring increased keratinocyte proliferation and migration, and reduced monocyte infiltration (Ashcroft et al. 1999). Overexpression of Smad2 in basal epidermal keratinocytes delayed wound healing due to a defect in basal keratinocyte migration (Hosokawa et al. 2005). These studies indicate that both ectopically and endogenously expressed TGF-β1 inhibits re-epithelialization and activation of TGF-β signaling in skin wounds might promote scar formation. Thus, a better wound healing outcome may be achieved by selectively blocking the negative effects of TGF-β1 (Massague 1999). Recently, we reported that transgenic mice undergoing temporal induction of Smad7, an antagonist of the TGF-β signaling pathway, in the epidermis during wound healing exhibited accelerated wound healing characterized by increased re-epithelialization and reduced inflammation, angiogenesis and the production of type I collagen in wound stroma. The mechanism involved in blocking TGF-β-induced epithelial proliferation arrest and inhibiting NFκB signaling in the epidermis by Smad7 (Han et al. 2011; Hong et al. 2007). These findings highlight the importance of future studies to evaluate whether temporal Smad7 application in skin wounds can be used as a therapeutic strategy for impaired wound healing related to defects in keratinocyte migration, excessive inflammation, and scarring.
5 Concluding Remarks
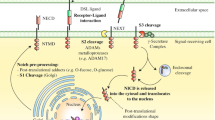
Skin cancer and fibrotic disease are common skin diseases lacking efficient therapies. TGF-β has been implicated in the pathogenesis of both due to its differential regulation of epidermal keratinocytes, fibroblasts, and other dermal cell components. The role of TGF-β signaling in skin carcinogenesis is schematically summarized in Fig. 9.1. TGF-β first acts as a tumor suppressor in carcinogenesis as TGF-β inhibits normal keratinocyte proliferation and maintains genomic stability. However, TGF-β has also been demonstrated to play a crucial role in promoting malignant skin cancer conversion and cancer metastasis. Dominant-negative TGF-βRII transgenic mice and Smad4 deletion tumors have also increased TGF-β expression associated with tumor development and metastasis. Although SCC tumor cells express high levels of TGF-β, cancer cells lost their response to TGF-β-induced growth inhibition due to the deficient component of TGF-β signaling in tumor cells. However, TGF-β-induced angiogenesis and inflammation in tumor stroma further accelerate tumor development and metastasis. Therefore, TGF-β contributes to either tumor inhibition or promotion depending on tumor stages. Figure 9.2 summarizes the role of TGF-β signaling in fibrotic diseases and cutaneous wound healing. In skin fibrotic diseases, it is well established that TGF-β plays a crucial role in fibrosis formation due to its ability to activate fibroblasts and stimulate collagen production in the extracellular matrix. Activation of TGF-β receptors, especially TGF-βRI, and abnormal expression of Smads have been documented in skin fibrosis. Based on wound healing studies in the fetus, the isoforms of TGF-β subtypes may have different roles in cutaneous scar formation. TGF-β1 is a negative factor in cutaneous wound healing and promotes skin scarring due to its action in fibroblast activation and induction of inflammation. Blocking TGF-β signaling may lead to novel strategies for treating impaired wound and skin fibrotic diseases in humans.

Schematic summarizing the role of TGF-β signaling in skin carcinogenesis. TGF-β expression in skin suppresses tumor formation associated with inhibiting proliferation and maintaining genomic stability of keratinocytes. During tumor development, TGF-β-induced inflammation and angiogenesis accelerate malignant SCC conversion and metastasis. Smad2 loss in epithelia causes increased Snail and HGF by recruiting Smad4 to the promoter of Snail and HGF in tumor cells. These molecular changes lead to EMT and increasing angiogenesis, which promote SCC formation. TGF-βRII or Smad4-deficient cells express high level of TGF-β but cell itself loses the response to TGF-β-induced tumor suppression, and additional TGF-β increase stromal angiogenesis and inflammation, all of which accelerate SCC development and metastasis. Smad3 loss in skin abrogates TGF-β-induced inflammation during skin carcinogenesis

Schematic summarizing the role of TGF-β signaling in fibrotic diseases. TGF-β acts a key player in mediating fibroblast activation to differentiate into myofibroblasts and stimulates the production of collagen via Smad3 and Smad1/Erk pathways. TGF-β also synergizes with other fibrotic factors such as CCN2, c-Ab1, Et-1, EGR1 in promoting collagen production. Excess collagen production and TGF-β-induced inflammation and angiogenesis contribute to skin fibrosis. In response to skin damage, TGF-β directly stimulates inflammation and angiogenesis in the stroma and inhibits keratinocyte migration via Smad2/3 signaling, which result in delayed wound healing and skin scarring. Overexpression of Smad7 or deletion of Smad3 reduces skin fibrosis/skin scarring and promotes wound healing through anti-inflammation, accelerating keratinocyte migration, and reducing collagen production
References
Akhurst RJ, Balmain A (1999) Genetic events and the role of TGF β in epithelial tumour progression. J Pathol 187:82–90
Akhurst RJ, Fee F, Balmain A (1988) Localized production of TGF-β mRNA in tumour promoter-stimulated mouse epidermis. Nature 331:363–365
Amendt C, Schirmacher P, Weber H, Blessing M (1998) Expression of a dominant negative type II TGF-β receptor in mouse skin results in an increase in carcinoma incidence and an acceleration of carcinoma development. Oncogene 17:25–34
Amendt C, Mann A, Schirmacher P, Blessing M (2002) Resistance of keratinocytes to TGFβ-mediated growth restriction and apoptosis induction accelerates re-epithelialization in skin wounds. J Cell Sci 115:2189–2198
Anzano MA, Roberts AB, Meyers CA, Komoriya A, Lamb LC, Smith JM, Sporn MB (1982) Synergistic interaction of two classes of transforming growth factors from murine sarcoma cells. Cancer Res 42:4776–4778
Artlett CM (2010) Animal models of scleroderma: fresh insights. Curr Opin Rheumatol 22:677–682
Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K (2004) Impaired Smad7-Smurf-mediated negative regulation of TGF-β signaling in scleroderma fibroblasts. J Clin Invest 113:253–264
Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, Roberts AB (1999) Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol 1:260–266
Bae DS, Blazanin N, Licata M, Lee J, Glick AB (2009) Tumor suppressor and oncogene actions of TGFβ1 occur early in skin carcinogenesis and are mediated by Smad3. Mol Carcinog 48:441–453
Bandyopadhyay B, Fan J, Guan S, Li Y, Chen M, Woodley DT, Li W (2006) A “traffic control” role for TGFβ3: orchestrating dermal and epidermal cell motility during wound healing. J Cell Biol 172:1093–1105
Batteux F, Kavian N, Servettaz A (2011) New insights on chemically induced animal models of systemic sclerosis. Curr Opin Rheumatol 23:511–518
Bettinger DA, Yager DR, Diegelmann RF, Cohen IK (1996) The effect of TGF-β on keloid fibroblast proliferation and collagen synthesis. Plast Reconstr Surg 98:827–833
Beyer C, Distler O, Distler JH (2012) Innovative antifibrotic therapies in systemic sclerosis. Curr Opin Rheumatol 24:274–280
Bhattacharyya S, Ishida W, Wu M, Wilkes M, Mori Y, Hinchcliff M, Leof E, Varga J (2009) A non-Smad mechanism of fibroblast activation by transforming growth factor-β via c-Abl and Egr-1: selective modulation by imatinib mesylate. Oncogene 28:1285–1297
Bhattacharyya S, Wei J, Varga J (2012) Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol 8:42–54
Bonner JC (2004) Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev 15:255–273
Bornstein S, Hoot K, Han GW, Lu SL, Wang XJ (2007) Distinct roles of individual Smads in skin carcinogenesis. Mol Carcinog 46:660–664
Bornstein S, White R, Malkoski S, Oka M, Han G, Cleaver T, Reh D, Andersen P, Gross N, Olson S, Deng C, Lu SL, Wang XJ (2009) Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest 119:3408–3419
Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE (1988) Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106:761–771
Bullard KM, Longaker MT, Lorenz HP (2003) Fetal wound healing: current biology. World J Surg 27:54–61
Chan T, Ghahary A, Demare J, Yang L, Iwashina T, Scott PG, Tredget EE (2002) Development, characterization, and wound healing of the keratin 14 promoted transforming growth factor-β1 transgenic mouse. Wound Repair Regen 10:177–187
Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A (1999) Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 126:1631–1642
Chen SJ, Yuan W, Mori Y, Levenson A, Trojanowska M, Varga J (1999) Stimulation of type I collagen transcription in human skin fibroblasts by TGF-β: involvement of Smad 3. J Invest Dermatol 112:49–57
Coffey RJ Jr, Sipes NJ, Bascom CC, Graves-Deal R, Pennington CY, Weissman BE, Moses HL (1988) Growth modulation of mouse keratinocytes by transforming growth factors. Cancer Res 48:1596–1602
Cowin AJ, Holmes TM, Brosnan P, Ferguson MW (2001) Expression of TGF-β and its receptors in murine fetal and adult dermal wounds. Eur J Dermatol 11:424–431
Cui W, Fowlis DJ, Cousins FM, Duffie E, Bryson S, Balmain A, Akhurst RJ (1995) Concerted action of TGF-β 1 and its type II receptor in control of epidermal homeostasis in transgenic mice. Genes Dev 9:945–955
Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, Akhurst RJ (1996) TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 86:531–542
Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF (1999) Targeted disruption of Smad3 reveals an essential role in transforming growth factor β-mediated signal transduction. Mol Cell Biol 19:2495–2504
Davies M, Prime SS, Eveson JW, Price N, Ganapathy A, D’Mello A, Paterson IC (2012) Transforming growth factor-β enhances invasion and metastasis in Ras-transfected human malignant epidermal keratinocytes. Int J Exp Pathol 93:148–156
Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM (1998) Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 17:3091–3100
Denton CP, Zheng B, Evans LA, Shi-wen X, Ong VH, Fisher I, Lazaridis K, Abraham DJ, Black CM, de Crombrugghe B (2003) Fibroblast-specific expression of a kinase-deficient type II transforming growth factor β (TGFβ) receptor leads to paradoxical activation of TGFβ signaling pathways with fibrosis in transgenic mice. J Biol Chem 278:25109–25119
Denton CP, Lindahl GE, Khan K, Shiwen X, Ong VH, Gaspar NJ, Lazaridis K, Edwards DR, Leask A, Eastwood M, Leoni P, Renzoni EA, Bou Gharios G, Abraham DJ, Black CM (2005) Activation of key profibrotic mechanisms in transgenic fibroblasts expressing kinase-deficient type II transforming growth factor-{β} receptor (T{β}RII{Δ}k). J Biol Chem 280:16053–16065
Derynck R, Zhang Y (1996) Intracellular signalling: the mad way to do it. Curr Biol 6:1226–1229
Dlugosz A, Merlino G, Yuspa SH (2002) Progress in cutaneous cancer research. J Investig Dermatol Symp Proc 7:17–26
Dong C, Zhu S, Wang T, Yoon W, Li Z, Alvarez RJ, ten Dijke P, White B, Wigley FM, Goldschmidt-Clermont PJ (2002) Deficient Smad7 expression: a putative molecular defect in scleroderma. Proc Natl Acad Sci USA 99:3908–3913
Eslami A, Gallant-Behm CL, Hart DA, Wiebe C, Honardoust D, Gardner H, Hakkinen L, Larjava HS (2009) Expression of integrin αvβ6 and TGF-β in scarless vs scar-forming wound healing. J Histochem Cytochem 57:543–557
Filler RB, Roberts SJ, Girardi M (2007) Cutaneous two-stage chemical carcinogenesis. CSH Protoc 2007: pdb prot4837
Fowlis DJ, Flanders KC, Duffie E, Balmain A, Akhurst RJ (1992) Discordant transforming growth factor β 1 RNA and protein localization during chemical carcinogenesis of the skin. Cell Growth Differ 3:81–91
Fowlis DJ, Cui W, Johnson SA, Balmain A, Akhurst RJ (1996) Altered epidermal cell growth control in vivo by inducible expression of transforming growth factor β 1 in the skin of transgenic mice. Cell Growth Differ 7:679–687
Frank S, Madlener M, Werner S (1996) Transforming growth factors β1, β2, and β3 and their receptors are differentially regulated during normal and impaired wound healing. J Biol Chem 271:10188–10193
Fujiki H, Suganuma M, Yoshizawa S, Kanazawa H, Sugimura T, Manam S, Kahn SM, Jiang W, Hoshina S, Weinstein IB (1989) Codon 61 mutations in the c-Harvey-ras gene in mouse skin tumors induced by 7,12-dimethylbenz[a]anthracene plus okadaic acid class tumor promoters. Mol Carcinog 2:184–187
Fujiwara M, Muragaki Y, Ooshima A (2005) Upregulation of transforming growth factor-β1 and vascular endothelial growth factor in cultured keloid fibroblasts: relevance to angiogenic activity. Arch Dermatol Res 297:161–169
Fusenig NE, Boukamp P (1998) Multiple stages and genetic alterations in immortalization, malignant transformation, and tumor progression of human skin keratinocytes. Mol Carcinog 23: 144–158
Gabrielli A, Di Loreto C, Taborro R, Candela M, Sambo P, Nitti C, Danieli MG, DeLustro F, Dasch JR, Danieli G (1993) Immunohistochemical localization of intracellular and extracellular associated TGF β in the skin of patients with systemic sclerosis (scleroderma) and primary Raynaud’s phenomenon. Clin Immunol Immunopathol 68:340–349
Gabrielli A, Avvedimento EV, Krieg T (2009) Scleroderma. N Engl J Med 360:1989–2003
Ganapathy A, Paterson IC, Prime SS, Eveson JW, Pring M, Price N, Threadgold SP, Davies M (2010) TGF-β inhibits metastasis in late stage human squamous cell carcinoma of the skin by a mechanism that does not involve Id1. Cancer Lett 298:107–118
Gay S, Trabandt A, Moreland LW, Gay RE (1992) Growth factors, extracellular matrix, and oncogenes in scleroderma. Arthritis Rheum 35:304–310
Ghahary A, Shen YJ, Scott PG, Gong Y, Tredget EE (1993) Enhanced expression of mRNA for transforming growth factor-β, type I and type III procollagen in human post-burn hypertrophic scar tissues. J Lab Clin Med 122:465–473
Glick AB, Sporn MB, Yuspa SH (1991) Altered regulation of TGF-β 1 and TGF-α in primary keratinocytes and papillomas expressing v-Ha-ras. Mol Carcinog 4:210–219
Glick AB, Lee MM, Darwiche N, Kulkarni AB, Karlsson S, Yuspa SH (1994) Targeted deletion of the TGF-β 1 gene causes rapid progression to squamous cell carcinoma. Genes Dev 8: 2429–2440
Glick A, Popescu N, Alexander V, Ueno H, Bottinger E, Yuspa SH (1999) Defects in transforming growth factor-β signaling cooperate with a Ras oncogene to cause rapid aneuploidy and malignant transformation of mouse keratinocytes. Proc Natl Acad Sci USA 96:14949–14954
Go C, Li P, Wang XJ (1999) Blocking transforming growth factor β signaling in transgenic epidermis accelerates chemical carcinogenesis: a mechanism associated with increased angiogenesis. Cancer Res 59:2861–2868
Gold LI, Jussila T, Fusenig NE, Stenback F (2000) TGF-β isoforms are differentially expressed in increasing malignant grades of HaCaT keratinocytes, suggesting separate roles in skin carcinogenesis. J Pathol 190:579–588
Gruschwitz M, Muller PU, Sepp N, Hofer E, Fontana A, Wick G (1990) Transcription and expression of transforming growth factor type β in the skin of progressive systemic sclerosis: a mediator of fibrosis? J Invest Dermatol 94:197–203
Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E (2007) Loss of TGFβ signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell 12:313–327
Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE (1996) DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271:350–353
Han G, Wang XJ (2011) Roles of TGFβ signaling Smads in squamous cell carcinoma. Cell Biosci 1:41
Han G, Lu SL, Li AG, He W, Corless CL, Kulesz-Martin M, Wang XJ (2005) Distinct mechanisms of TGF-β1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest 115:1714–1723
Han G, Li F, Ten Dijke P, Wang XJ (2011) Temporal smad7 transgene induction in mouse epidermis accelerates skin wound healing. Am J Pathol 179:1768–1779
Henry G, Li W, Garner W, Woodley DT (2003) Migration of human keratinocytes in plasma and serum and wound re-epithelialisation. Lancet 361:574–576
Heyer J, Escalante-Alcalde D, Lia M, Boettinger E, Edelmann W, Stewart CL, Kucherlapati R (1999) Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proc Natl Acad Sci USA 96:12595–12600
Higley H, Persichitte K, Chu S, Waegell W, Vancheeswaran R, Black C (1994) Immunocytochemical localization and serologic detection of transforming growth factor β 1. Association with type I procollagen and inflammatory cell markers in diffuse and limited systemic sclerosis, morphea, and Raynaud’s phenomenon. Arthritis Rheum 37:278–288
Hong S, Lim S, Li AG, Lee C, Lee YS, Lee EK, Park SH, Wang XJ, Kim SJ (2007) Smad7 binds to the adaptors TAB2 and TAB3 to block recruitment of the kinase TAK1 to the adaptor TRAF2. Nat Immunol 8:504–513
Hoot KE, Lighthall J, Han G, Lu SL, Li A, Ju W, Kulesz-Martin M, Bottinger E, Wang XJ (2008) Keratinocyte-specific Smad2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. J Clin Invest 118:2722–2732
Hoot KE, Oka M, Han G, Bottinger E, Zhang Q, Wang XJ (2010) HGF upregulation contributes to angiogenesis in mice with keratinocyte-specific Smad2 deletion. J Clin Invest 120:3606–3616
Hosokawa R, Urata MM, Ito Y, Bringas P Jr, Chai Y (2005) Functional significance of Smad2 in regulating basal keratinocyte migration during wound healing. J Invest Dermatol 125: 1302–1309
Hoyles RK, Khan K, Shiwen X, Howat SL, Lindahl GE, Leoni P, du Bois RM, Wells AU, Black CM, Abraham DJ, Denton CP (2008) Fibroblast-specific perturbation of transforming growth factor β signaling provides insight into potential pathogenic mechanisms of scleroderma-associated lung fibrosis: exaggerated response to alveolar epithelial injury in a novel mouse model. Arthritis Rheum 58:1175–1188
Hsu M, Peled ZM, Chin GS, Liu W, Longaker MT (2001) Ontogeny of expression of transforming growth factor-β 1 (TGF-β 1), TGF-β 3, and TGF-β receptors I and II in fetal rat fibroblasts and skin. Plast Reconstr Surg 107:1787–1794; discussion 1795–1786
Iwamoto N, Distler JH, Distler O (2011) Tyrosine kinase inhibitors in the treatment of systemic sclerosis: from animal models to clinical trials. Curr Rheumatol Rep 13:21–27
Jinnin M (2010) Mechanisms of skin fibrosis in systemic sclerosis. J Dermatol 37:11–25
Jinnin M, Ihn H, Mimura Y, Asano Y, Tamaki K (2006) Potential regulatory elements of the constitutive up-regulated α2(I) collagen gene in scleroderma dermal fibroblasts. Biochem Biophys Res Commun 343:904–909
Kawakami T, Ihn H, Xu W, Smith E, LeRoy C, Trojanowska M (1998) Increased expression of TGF-β receptors by scleroderma fibroblasts: evidence for contribution of autocrine TGF-β signaling to scleroderma phenotype. J Invest Dermatol 110:47–51
Kleiter I, Song J, Lukas D, Hasan M, Neumann B, Croxford AL, Pedre X, Hovelmeyer N, Yogev N, Mildner A, Prinz M, Wiese E, Reifenberg K, Bittner S, Wiendl H, Steinman L, Becker C, Bogdahn U, Neurath MF, Steinbrecher A, Waisman A (2010) Smad7 in T cells drives T helper 1 responses in multiple sclerosis and experimental autoimmune encephalomyelitis. Brain 133:1067–1081
Koch RM, Roche NS, Parks WT, Ashcroft GS, Letterio JJ, Roberts AB (2000) Incisional wound healing in transforming growth factor-β1 null mice. Wound Repair Regen 8:179–191
Kopp J, Preis E, Said H, Hafemann B, Wickert L, Gressner AM, Pallua N, Dooley S (2005) Abrogation of transforming growth factor-β signaling by SMAD7 inhibits collagen gel contraction of human dermal fibroblasts. J Biol Chem 280:21570–21576
Kreuter A, Hyun J, Skrygan M, Sommer A, Tomi NS, Breuckmann F, Altmeyer P, Gambichler T (2006) Ultraviolet A1 phototherapy decreases inhibitory SMAD7 gene expression in localized scleroderma. Arch Dermatol Res 298:265–272
Kubo M, Ihn H, Yamane K, Tamaki K (2001) Up-regulated expression of transforming growth factor β receptors in dermal fibroblasts in skin sections from patients with localized scleroderma. Arthritis Rheum 44:731–734
Kubo M, Ihn H, Yamane K, Tamaki K (2002) Upregulated expression of transforming growth factor-β receptors in dermal fibroblasts of skin sections from patients with systemic sclerosis. J Rheumatol 29:2558–2564
Kulozik M, Hogg A, Lankat-Buttgereit B, Krieg T (1990) Co-localization of transforming growth factor β 2 with α 1(I) procollagen mRNA in tissue sections of patients with systemic sclerosis. J Clin Invest 86:917–922
Lakos G, Takagawa S, Chen SJ, Ferreira AM, Han G, Masuda K, Wang XJ, DiPietro LA, Varga J (2004) Targeted disruption of TGF-β/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am J Pathol 165:203–217
Larson BJ, Longaker MT, Lorenz HP (2010) Scarless fetal wound healing: a basic science review. Plast Reconstr Surg 126:1172–1180
Lee TY, Chin GS, Kim WJ, Chau D, Gittes GK, Longaker MT (1999) Expression of transforming growth factor β 1, 2, and 3 proteins in keloids. Ann Plast Surg 43:179–184
Li W, Qiao W, Chen L, Xu X, Yang X, Li D, Li C, Brodie SG, Meguid MM, Hennighausen L, Deng CX (2003) Squamous cell carcinoma and mammary abscess formation through squamous metaplasia in Smad4/Dpc4 conditional knockout mice. Development 130:6143–6153
Li AG, Lu SL, Zhang MX, Deng C, Wang XJ (2004a) Smad3 knockout mice exhibit a resistance to skin chemical carcinogenesis. Cancer Res 64:7836–7845
Li AG, Wang D, Feng XH, Wang XJ (2004b) Latent TGFβ1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. EMBO J 23:1770–1781
Li AG, Lu SL, Han G, Hoot KE, Wang XJ (2006) Role of TGFβ in skin inflammation and carcinogenesis. Mol Carcinog 45:389–396
Martin P (1997) Wound healing—aiming for perfect skin regeneration. Science 276:75–81
Martin P, Dickson MC, Millan FA, Akhurst RJ (1993) Rapid induction and clearance of TGFβ 1 is an early response to wounding in the mouse embryo. Dev Genet 14:225–238
Massague J (1996) TGFβ signaling: receptors, transducers, and Mad proteins. Cell 85:947–950
Massague J (1999) Wounding Smad. Nat Cell Biol 1:E117–E119
Millet C, Zhang YE (2007) Roles of Smad3 in TGF-β signaling during carcinogenesis. Crit Rev Eukaryot Gene Expr 17:281–293
Mori Y, Chen SJ, Varga J (2003) Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum 48:1964–1978
Mrowietz U, Seifert O (2009) Keloid scarring: new treatments ahead. Actas Dermosifiliogr 100(Suppl 2):75–83
Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin CH, Miyazono K, ten Dijke P (1997) TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J 16:5353–5362
Namazi MR, Fallahzadeh MK, Schwartz RA (2011) Strategies for prevention of scars: what can we learn from fetal skin? Int J Dermatol 50:85–93
Nath RK, LaRegina M, Markham H, Ksander GA, Weeks PM (1994) The expression of transforming growth factor type β in fetal and adult rabbit skin wounds. J Pediatr Surg 29:416–421
Nomura M, Li E (1998) Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 393:786–790
Olutoye OO, Yager DR, Cohen IK, Diegelmann RF (1996) Lower cytokine release by fetal porcine platelets: a possible explanation for reduced inflammation after fetal wounding. J Pediatr Surg 31:91–95
Oshima M, Oshima H, Taketo MM (1996) TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol 179:297–302
Owens P, Bazzi H, Engelking E, Han G, Christiano AM, Wang XJ (2008) Smad4-dependent desmoglein-4 expression contributes to hair follicle integrity. Dev Biol 322:156–166
Owens P, Engelking E, Han G, Haeger SM, Wang XJ (2010) Epidermal Smad4 deletion results in aberrant wound healing. Am J Pathol 176:122–133
Pannu J, Gore-Hyer E, Yamanaka M, Smith EA, Rubinchik S, Dong JY, Jablonska S, Blaszczyk M, Trojanowska M (2004) An increased transforming growth factor β receptor type I:type II ratio contributes to elevated collagen protein synthesis that is resistant to inhibition via a kinase-deficient transforming growth factor β receptor type II in scleroderma. Arthritis Rheum 50:1566–1577
Pannu J, Nakerakanti S, Smith E, ten Dijke P, Trojanowska M (2007) Transforming growth factor-β receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J Biol Chem 282:10405–10413
Pannu J, Asano Y, Nakerakanti S, Smith E, Jablonska S, Blaszczyk M, ten Dijke P, Trojanowska M (2008) Smad1 pathway is activated in systemic sclerosis fibroblasts and is targeted by imatinib mesylate. Arthritis Rheum 58:2528–2537
Patamalai B, Burow DL, Gimenez-Conti I, Zenklusen JC, Conti CJ, Klein-Szanto AJ, Fischer SM (1994) Altered expression of transforming growth factor-β 1 mRNA and protein in mouse skin carcinogenesis. Mol Carcinog 9:220–229
Phan TT, Lim IJ, Aalami O, Lorget F, Khoo A, Tan EK, Mukhopadhyay A, Longaker MT (2005) Smad3 signalling plays an important role in keloid pathogenesis via epithelial-mesenchymal interactions. J Pathol 207:232–242
Qiao W, Li AG, Owens P, Xu X, Wang XJ, Deng CX (2006) Hair follicle defects and squamous cell carcinoma formation in Smad4 conditional knockout mouse skin. Oncogene 25:207–217
Quan T, He T, Kang S, Voorhees JJ, Fisher GJ (2002) Ultraviolet irradiation alters transforming growth factor β/smad pathway in human skin in vivo. J Invest Dermatol 119:499–506
Querfeld C, Eckes B, Huerkamp C, Krieg T, Sollberg S (1999) Expression of TGF-β 1, -β 2 and -β 3 in localized and systemic scleroderma. J Dermatol Sci 21:13–22
Roberts AB, Anzano MA, Wakefield LM, Roche NS, Stern DF, Sporn MB (1985) Type β transforming growth factor: a bifunctional regulator of cellular growth. Proc Natl Acad Sci USA 82:119–123
Rolfe KJ, Richardson J, Vigor C, Irvine LM, Grobbelaar AO, Linge C (2007) A role for TGF-β1-induced cellular responses during wound healing of the non-scarring early human fetus? J Invest Dermatol 127:2656–2667
Roop DR, Lowy DR, Tambourin PE, Strickland J, Harper JR, Balaschak M, Spangler EF, Yuspa SH (1986) An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature 323:822–824
Rosenbloom J, Castro SV, Jimenez SA (2010) Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Ann Intern Med 152:159–166
Rudnicka L, Varga J, Christiano AM, Iozzo RV, Jimenez SA, Uitto J (1994) Elevated expression of type VII collagen in the skin of patients with systemic sclerosis. Regulation by transforming growth factor-β. J Clin Invest 93:1709–1715
Schmid P, Itin P, Cherry G, Bi C, Cox DA (1998) Enhanced expression of transforming growth factor-β type I and type II receptors in wound granulation tissue and hypertrophic scar. Am J Pathol 152:485–493
Seifert O, Mrowietz U (2009) Keloid scarring: bench and bedside. Arch Dermatol Res 301:259–272
Sellheyer K, Bickenbach JR, Rothnagel JA, Bundman D, Longley MA, Krieg T, Roche NS, Roberts AB, Roop DR (1993) Inhibition of skin development by overexpression of transforming growth factor β 1 in the epidermis of transgenic mice. Proc Natl Acad Sci USA 90:5237–5241
Shah M, Foreman DM, Ferguson MW (1995) Neutralisation of TGF-β 1 and TGF-β 2 or exogenous addition of TGF-β 3 to cutaneous rat wounds reduces scarring. J Cell Sci 108(Pt 3):985–1002
Shih B, Bayat A (2010) Genetics of keloid scarring. Arch Dermatol Res 302:319–339
Shipley GD, Pittelkow MR, Wille JJ Jr, Scott RE, Moses HL (1986) Reversible inhibition of normal human prokeratinocyte proliferation by type β transforming growth factor-growth inhibitor in serum-free medium. Cancer Res 46:2068–2071
Singer AJ, Clark RA (1999) Cutaneous wound healing. N Engl J Med 341:738–746
Sirard C, de la Pompa JL, Elia A, Itie A, Mirtsos C, Cheung A, Hahn S, Wakeham A, Schwartz L, Kern SE, Rossant J, Mak TW (1998) The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev 12:107–119
Sonnylal S, Denton CP, Zheng B, Keene DR, He R, Adams HP, Vanpelt CS, Geng YJ, Deng JM, Behringer RR, de Crombrugghe B (2007) Postnatal induction of transforming growth factor β signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum 56:334–344
Soo C, Beanes SR, Hu FY, Zhang X, Dang C, Chang G, Wang Y, Nishimura I, Freymiller E, Longaker MT, Lorenz HP, Ting K (2003) Ontogenetic transition in fetal wound transforming growth factor-β regulation correlates with collagen organization. Am J Pathol 163:2459–2476
Sporn MB (1999) TGF-β: 20 years and counting. Microbes Infect 1:1251–1253
Sporn MB (2006) The early history of TGF-β, and a brief glimpse of its future. Cytokine Growth Factor Rev 17:3–7
Sullivan KM, Lorenz HP, Meuli M, Lin RY, Adzick NS (1995) A model of scarless human fetal wound repair is deficient in transforming growth factor β. J Pediatr Surg 30:198–202; discussion 202–193
Tannehill-Gregg SH, Kusewitt DF, Rosol TJ, Weinstein M (2004) The roles of Smad2 and Smad3 in the development of chemically induced skin tumors in mice. Vet Pathol 41:278–282
Teng Y, Sun AN, Pan XC, Yang G, Yang LL, Wang MR, Yang X (2006) Synergistic function of Smad4 and PTEN in suppressing forestomach squamous cell carcinoma in the mouse. Cancer Res 66:6972–6981
Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, Kinzler KW, Vogelstein B (1996) Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet 13:343–346
Tremblay KD, Dunn NR, Robertson EJ (2001) Mouse embryos lacking Smad1 signals display defects in extra-embryonic tissues and germ cell formation. Development 128:3609–3621
Tsujita-Kyutoku M, Uehara N, Matsuoka Y, Kyutoku S, Ogawa Y, Tsubura A (2005) Comparison of transforming growth factor-β/Smad signaling between normal dermal fibroblasts and fibroblasts derived from central and peripheral areas of keloid lesions. In Vivo 19:959–963
Tuan TL, Nichter LS (1998) The molecular basis of keloid and hypertrophic scar formation. Mol Med Today 4:19–24
Tucker RF, Shipley GD, Moses HL, Holley RW (1984) Growth inhibitor from BSC-1 cells closely related to platelet type β transforming growth factor. Science 226:705–707
Tziotzios C, Profyris C, Sterling J (2012) Cutaneous scarring: pathophysiology, molecular mechanisms, and scar reduction therapeutics Part II. Strategies to reduce scar formation after dermatologic procedures. J Am Acad Dermatol 66:13–24
Varga J, Abraham D (2007) Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 117:557–567
Varga J, Jimenez SA (1986) Stimulation of normal human fibroblast collagen production and processing by transforming growth factor-β. Biochem Biophys Res Commun 138:974–980
Vijayachandra K, Lee J, Glick AB (2003) Smad3 regulates senescence and malignant conversion in a mouse multistage skin carcinogenesis model. Cancer Res 63:3447–3452
Vuorio T, Kahari VM, Black C, Vuorio E (1991) Expression of osteonectin, decorin, and transforming growth factor-β 1 genes in fibroblasts cultured from patients with systemic sclerosis and morphea. J Rheumatol 18:247–251
Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ (1998) Smad2 signaling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo. Cell 92:797–808
Wang XJ (2001) Role of TGFβ signaling in skin carcinogenesis. Microsc Res Tech 52:420–429
Wang XJ, Greenhalgh DA, Bickenbach JR, Jiang A, Bundman DS, Krieg T, Derynck R, Roop DR (1997) Expression of a dominant-negative type II transforming growth factor β (TGF-β) receptor in the epidermis of transgenic mice blocks TGF-β-mediated growth inhibition. Proc Natl Acad Sci USA 94:2386–2391
Wang XJ, Han G, Owens P, Siddiqui Y, Li AG (2006) Role of TGF β-mediated inflammation in cutaneous wound healing. J Investig Dermatol Symp Proc 11:112–117
Weeks BH, He W, Olson KL, Wang XJ (2001) Inducible expression of transforming growth factor β1 in papillomas causes rapid metastasis. Cancer Res 61:7435–7443
Wei J, Bhattacharyya S, Tourtellotte WG, Varga J (2011) Fibrosis in systemic sclerosis: emerging concepts and implications for targeted therapy. Autoimmun Rev 10:267–275
Weinstein M, Yang X, Li C, Xu X, Gotay J, Deng CX (1998) Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc Natl Acad Sci USA 95:9378–9383
Whitby DJ, Ferguson MW (1991) Immunohistochemical localization of growth factors in fetal wound healing. Dev Biol 147:207–215
Wu CS, Wu PH, Fang AH, Lan CC (2012) FK506 inhibits the enhancing effects of transforming growth factor-β1 on collagen expression and TGF-β/Smad signaling in keloid fibroblasts: implication for new therapeutic approach. Br J Dermatol 167(3):532–541
Wynn TA (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 117:524–529
Yamamoto T (2010) Animal model of systemic sclerosis. J Dermatol 37:26–41
Yang X, Li C, Xu X, Deng C (1998) The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci USA 95:3667–3672
Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C (1999) Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J 18:1280–1291
Yang L, Chan T, Demare J, Iwashina T, Ghahary A, Scott PG, Tredget EE (2001) Healing of burn wounds in transgenic mice overexpressing transforming growth factor-β 1 in the epidermis. Am J Pathol 159:2147–2157
Yang L, Mao C, Teng Y, Li W, Zhang J, Cheng X, Li X, Han X, Xia Z, Deng H, Yang X (2005) Targeted disruption of Smad4 in mouse epidermis results in failure of hair follicle cycling and formation of skin tumors. Cancer Res 65:8671–8678
Yu H, Bock O, Bayat A, Ferguson MW, Mrowietz U (2006) Decreased expression of inhibitory SMAD6 and SMAD7 in keloid scarring. J Plastic Reconstr Aesthet Surg 59:221–229
Yuspa SH (1986) Cutaneous chemical carcinogenesis. J Am Acad Dermatol 15:1031–1044
Zhang K, Garner W, Cohen L, Rodriguez J, Phan S (1995) Increased types I and III collagen and transforming growth factor-β 1 mRNA and protein in hypertrophic burn scar. J Invest Dermatol 104:750–754
Zhang W, Ou J, Inagaki Y, Greenwel P, Ramirez F (2000) Synergistic cooperation between Sp1 and Smad3/Smad4 mediates transforming growth factor β1 stimulation of α 2(I)-collagen (COL1A2) transcription. J Biol Chem 275:39237–39245
Zhu Y, Richardson JA, Parada LF, Graff JM (1998) Smad3 mutant mice develop metastatic colorectal cancer. Cell 94:703–714
Zhu H, Li Y, Qu S, Luo H, Zhou Y, Wang Y, Zhao H, You Y, Xiao X, Zuo X (2012) MicroRNA expression abnormalities in limited cutaneous scleroderma and diffuse cutaneous scleroderma. J Clin Immunol 32:514–522
Acknowledgments
The original work from the Wang laboratory is supported by NIH grants CA79998, CA87849 and AR061792. The authors thank Pamela Garl for carefully proofreading this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer
About this chapter
Cite this chapter
Han, G., Han, Z., Wang, XJ. (2013). TGF-β in Skin Cancer and Fibrosis. In: Moustakas, A., Miyazawa, K. (eds) TGF-β in Human Disease. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54409-8_9
Download citation
DOI: https://doi.org/10.1007/978-4-431-54409-8_9
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54408-1
Online ISBN: 978-4-431-54409-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)