
Abstract
Members of the transforming growth factor-β (TGF-β) family exert their effect virtually on all cell types in the body, producing diverse and complex cellular responses. TGF-β signaling is deregulated and hyperactive in many malignant conditions, making it an appealing target in the combat of cancer disease. The predominantly endothelial TGF-β receptors, ALK1 and endoglin, which are activated during neoangiogenesis both during development and pathological conditions, pose attractive modulating opportunities to impair tumor vessel formation and cancer progression. However, the precise function of TGF-β family signaling in ECs is difficult to predict, as it appears highly context dependent due to the many ligands and receptors influencing the final outcome. Furthermore, TGF-β is involved in autocrine and intricate dynamic paracrine signaling events in the context of the tumor microenvironment. Pharmacological inhibitors for ALK1, endoglin, and TGF-β or its receptors have been developed and will facilitate more comprehensive studies on the exact function of the TGF-β family in the endothelium, and more specifically in tumor angiogenesis. Here, we will summarize the current knowledge on TGF-β signaling in the regulation of the formation and function of the vascular network.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
1.1 The TGF-β-Rich Tumor Microenvironment
The tumor microenvironment, or stroma, influences the growth of the tumor and its ability to progress and metastasize. The stroma thus constitutes an important aspect to consider when developing therapeutic approaches, as it can alter interstitial fluid pressure, limit the access of therapeutics to the tumor, change drug metabolism, or even contribute to the development of drug resistance (Egeblad et al. 2010).
Despite the importance of tumor–host stroma interactions, there is limited understanding of the stromal milieu composition, and the complexity and dynamics in the relationship between the tumor malignant cells and the surrounding host cells. It is, however, acknowledged that tumor cells and their stroma coevolve during tumorigenesis and progression (Pietras and Östman 2010). Nonetheless, the precise nature of the cells that comprise the normal stroma, how these cells or newly recruited cells are altered during tumor progression, and how they reciprocally influence tumor initiation and progression, is poorly understood.
The large family of TGF-β extracellular pleiotropic cytokines exerts influence essentially on all cellular strata in the body, namely in epithelial cells, fibroblasts, immune, endothelial, lymphatic and perivascular cells (Fig. 14.1) (Elliott and Blobe 2005). TGF-β is the prototypical element of an extensive ligand group that also includes bone morphogenetic proteins (BMPs), activins, inhibins, nodal, and growth and differentiation factors (GDF)s that elicit signaling activity through a collection of five type II receptors and seven type I receptors (Padua and Massague 2009). Different ligand-receptor II–receptor I combinations can be assembled, hence delineating one of the TGF-β family hallmarks of complexity.

(a) TGF-β secreted from various sources is acting on a wide range of cell types in the tumor microenvironment. TGF-β promotes tumor epithelial cell invasiveness and metastasis in an autocrine fashion. TGF-β also affects tumor matrix remodeling by cancer-associated fibroblasts and regulates angiogenesis by acting on ECs and pericytes. Tumor infiltration by leukocytes, macrophages, and bone marrow-derived endothelial, mesenchymal, and myeloid precursor cells is also mediated by TGF-β. TGF-β suppresses proliferation and differentiation of lymphocytes including T cells, natural killer cells, and macrophages, preventing immune surveillance control over the developing tumor. (b) Immunostaining of an experimental pancreatic neuroendocrine tumor from RIP1-Tag2 mice for TGF-β (red) and the endothelial cell marker CD31 (green) illustrates the abundance of TGF-β throughout the tumor microenvironment
In normal, unstressed tissue, sustained basal release of TGF-β by local sources regulates homeostasis. In pathological conditions, TGF-β is abundantly released in the tumor microenvironment (Fig. 14.1), initially as a signal to avoid premalignant progression, but eventually as a cue that cancer cells utilize to their own advantage in later stages of malignancy (Massague 2008; Wakefield and Roberts 2002). Most human tumors overproduce TGF-β whose autocrine and paracrine actions promote tumor epithelial cell invasiveness and metastasis, functioning as a differentiation switch required for transient but reversible invasiveness of carcinoma cells through epithelial-to-mesenchymal mechanisms (Pardali and Moustakas 2007). In addition to eliciting mitogenic signals toward the carcinoma cells, TGF-β also affects cancer-associated fibroblast induction of tumor matrix remodeling and regulates angiogenesis by acting on endothelial cells (ECs) and pericytes. Finally, TGF-β suppresses proliferation and differentiation of lymphocytes including T cells, natural killer cells, and macrophages, thus preventing immune surveillance control over the developing tumor.
In a nutshell, TGF-β signaling is intimately implicated in tumor development and contributes to most hallmarks of cancer described by Hanahan and Weinberg (2000, 2011). It is, thus, of vital importance to carefully analyze the role of the TGF-β family members in the tumor microenvironment and how signaling circuits arising from tumor and stromal interactions can be efficiently modulated in cancer therapy.
1.2 Involvement of TGF-β in Vascular Syndromes
In vascular biology, TGF-β has traditionally been seen as a differentiation regulator for vascular smooth muscle cells (VSMCs), ultimately contributing to vessel stabilization and maturation, by inducing ECM deposition and inhibiting EC migration and proliferation.
The critical relevance of TGF-β signaling in vascular development was, however, recognized by identification of mutations in TGF-β receptor genes in familial vascular syndromes, in the type I endothelial specific receptor, activin receptor-like kinase (ALK)1, and the type III or co-receptor, endoglin (ten Dijke and Arthur 2007). Germline loss-of-function mutations in ALK1 or endoglin are causal in the development of the human syndrome of hereditary hemorrhagic telangiectasia (HHT). HHT is characterized by cutaneous telangiectases and gastrointestinal hemorrhage (Berg et al. 1997; McAllister et al. 1994). In addition, major arteriovenous malformations occur in lung, liver, or brain, which may ultimately cause severe morbidity and mortality. While Alk1 and endoglin null mice die at midgestation as a result of severe arteriovenous malformations (AVMs) (Arthur et al. 2000; Oh et al. 2000), mice lacking one copy of the gene for either Alk1 (Alk1+/−; HHT2) or endoglin (Eng+/−; HHT1) recapitulate with age the HHT phenotype (Srinivasan et al. 2003; Torsney et al. 2003).
There is additional strong evidence gathered from in vivo studies on genetically manipulated mouse models, for a prominent role of the TGF-β pathway in vasculo- and angiogenesis mechanisms (ten Dijke and Arthur 2007).
Nearly half of TGF-β1−/− and 25 % of TGF-β1+/− mouse embryos die in utero due to defective hematopoiesis and vasculogenesis (Dickson et al. 1995). TGF-β1-deficient mice that do survive until birth die at about 3 weeks of age of a multifocal inflammatory disorder, primarily affecting heart and lungs, although mild liver inflammation has been also observed (Shull et al. 1992). Supporting these reports, the classical TGF-βRII and TGF-βRI/ALK5 receptors also exhibit defective vasculogenesis and lead to mid gestation lethality (Larsson et al. 2001; Oshima et al. 1996).
In fact, deletion of TGF-β signaling mediators results (Table 14.1), for most cases, in serious implications on the developing embryonic vasculature leading ultimately to lethality in mice (Bertolino et al. 2005; ten Dijke and Arthur 2007).
A lot of attention has been focused on the endothelial cell-specific TGF-β receptors ALK1 and endoglin not only because they already have documented involvement in the human syndrome HHT, but more importantly they hold therapeutic promise in pathological conditions, such as cancer, where their upregulated expression in the vascular component plays a critical role in cancer development.
1.3 TGF-β Signaling in ECs: ALK1/ALK5 Interconnecting Crosstalk
Following synthesis, secretion, and activation, the mature TGF-β dimeric ligand is released from the ECM to trigger specific serine/threonine type I and type II kinase receptor heterotetrameric complexes (Wrana et al. 1994). In ECs, TGF-β signaling has been described to signal via the globally expressed type I receptor, ALK5 or alternatively through the predominantly endothelial type I receptor, ALK1. The prevailing recruited type I receptor dictates the activation of a particular Smad transducing cascade. ALK1 activation generates phosphorylation of Smad1, 5, and 8, whereas ALK5 leads to Smad 2 and 3 signaling activation (Goumans et al. 2002). The selected activated Smad subset independently forms a heteromeric complex with a related molecule, Smad4, which translocates the complexes into the nucleus to launch transcription of specific target genes (Massague 2000; ten Dijke et al. 2000), involved in distinct angiogenic responses.
TGF-β ligands also interact with co-receptors or type III receptors, represented by betaglycan (TGF-βRIII) and the typically endothelial endoglin. However, because these type III receptors lack the kinase domain, they essentially hold an accessory role in ligand binding and signaling activation, adding yet another level of regulation to the TGF-β complex signaling web.
Far from consensual, the TGF-β contribution to vascular biology knowledge has been constantly under debate due to numerous paradoxical reports (Goumans et al. 2002; Lamouille et al. 2002; Valdimarsdottir et al. 2002).
A crucial role in angiogenesis for ALK1 was first described in a study reporting ALK1 as pivotal for SMC recruitment, implying a vital role for TGF-β/ALK1 signaling axis in the maturation phase of angiogenesis (Oh et al. 2000). On the other hand, signaling derived from ALK5 was more pronounced during the activation phase of angiogenesis, when ECs degrade perivascular basement membrane, invade and migrate into the newly available space, through active proliferation and lumen formation. The balance theory was then hypothesized for the first time, speculating that different levels of TGF-β ligand availability would determine the sequential angiogenic fate and control the properties of the endothelium during angiogenesis. Also, in parallel studies another laboratory presented evidence for development of shunts between arteries and veins and severe arteriovenous malformations due to fusion of major arteries and veins in mice lacking ALK1 (Urness et al. 2000).
The balance working model was quickly challenged when another laboratory proposed that TGF-β engages in the activation of ALK1 signaling via Smad1/5, which concomitantly inhibits ALK5 signaling through Smad2/3 (Goumans et al. 2002). ALK5, while critical for ALK1 signaling, demonstrated by studies on ALK5-deficient mouse embryonic ECs, commits to an anti-angiogenic cascade of events, while ALK1 mediates pro-angiogenic activation (Goumans et al. 2003). These studies indicated that TGF-β stimulatory effects on either ALK5 or ALK1 are mutually exclusive inducing differential transcriptional activation of PAI-1 and Id1, respectively, which ultimately elicit a different set of physiological responses.
Motivated by the fact that both ALK1 and ALK5 null mice render an embryonic lethal phenotype due to extensive vascular abnormalities (Larsson et al. 2001; Oh et al. 2000; Urness et al. 2000), ALK1 dependency on ALK5, by means of signaling or by mere anchoring, has been questioned and addressed with reservation by multiple laboratories.
Transcriptional profiling of human umbilical vein ECs expressing constitutively active adenoviral constructs of ALK1 or ALK5 demonstrated substantial differences in the transcriptional output from either signaling pathway (Ota et al. 2002), validating previously described downstream gene regulation. Interestingly, the non-overlapping expression patterns of ALK1 and ALK5 in vivo (Seki et al. 2006) by thorough analysis of a knockin mouse line carrying a lacZ reporter in the Alk5 gene locus (Alk5lacZ), also lends support to divergent roles in vascular development for each of the two type I receptors expressed by ECs.
ALK5 suppression, by genetic silencing or small molecule inhibition, was shown not to interfere with BMP9/ALK1-induced phosphorylation of Smad1/5/8 in bovine aortic ECs (BAECs) (Shao et al. 2009). Instead, silencing of Alk1 or any of its downstream molecular effectors, by means of siRNA transfection, rather induce a potent ALK5 signaling upregulation. In agreement with the ALK5-independent action of ALK1 is the notion that ALK5 is present in ECs in vivo either at low levels, or only expressed by the neighboring VSMCs, suggesting that ALK5 may only participate in ALK1-dependent angiogenesis in a paracrine fashion (Park et al. 2008; Shao et al. 2009). Congruent with these results, EC-specific ablation of Alk5 does not inflict vascular abnormalities in mice or zebrafish (Park et al. 2008). However, embryos from knockin mice carrying a mutation on L45 loop in Alk5 rescued to some extent the earliest vascular defects observed in ALK5 mouse knockouts (Itoh et al. 2009), probably because this mutation, yet interfering with ALK5 kinase ability to phosphorylate Smad2, inherently preserves ALK5 competence to mediate non-Smad signaling and lateral signaling to ALK1. In agreement with these findings, ALK1 signal inhibition proved to interfere, in pathological conditions, not only with its own target genes but ALK5 signal transduction also exhibited a suppressive modulation in a model of pancreatic neuroendocrine cancer (Cunha et al. 2010).
More recently, it has been demonstrated that selective deletion of ALK5 in ECs using an Alk5GFPCre mouse line resulted in embryonic lethality due to brain vessel pathological morphology and intracerebral hemorrhage (Nguyen et al. 2011). Independent observations of EC-specific deletion of Smad2/3 using Tie2-Cre transgenic mice revealed critical hemorrhaging and embryonic lethality around E12.5. In this study, vascular maturation was incomplete owing to inadequate assembly of mural cells in the vasculature, most likely because of impaired expression of PDGF-B by the Smad2/3 ablated endothelium (Itoh et al. 2012). These observations substantiate the vital relevance of ALK5 in the endothelium either as a signaling anchor or by actively participating in the vasculogenic process.
Collectively, these reports demonstrate that ALK5 signaling is indeed relevant for endothelial homeostasis. Further studies aiming at dichotomizing ALK1 versus ALK5 signaling in endothelial and in perivascular cells during development and in the tumor microenvironment are thus required to clarify and reconciliate previous paradoxical observations and infer about the benefit or risk of clinically targeting such pathways without proper amendments. It is plausible that the relative stoichiometry of ALK1 and ALK5 signaling may be crucial for proper regulation of gene expression (Cunha and Pietras 2011).
1.4 Bone Morphogenetic Proteins
The BMPs, a subcategory of the TGF-β superfamily, were first identified in extracts from bone matrix and characterized by their ability to induce ectopic bone formation when implanted subcutaneously in rats (Wozney et al. 1988). It soon became clear that BMPs play a key role in vertebrate organogenesis, as well as in embryonic vascularization (Kishigami and Mishina 2005; Zhao 2003).
The BMP family, including the GDFs, comprises a group of 20 ligands that activate a classical BMP pathway in vertebrates (Lowery and de Caestecker 2010; Wagner et al. 2010). In the canonical BMP signaling pathway, three type II receptors (BMPRII, ActRIIa, and ActRII2b) and four type I receptors (ALK1, ALK2, ALK3, and ALK6) can be activated (Miyazono et al. 2010; Moustakas and Heldin 2009). In addition to primarily triggering Smad1, 5, and 8, BMP cues may also activate Smad2 (Upton et al. 2009) and Smad-independent signaling (Sieber et al. 2009).
BMP9 has been implicated in hematopoiesis, hepato-, osteo-, chondro-, and adipogenesis (Canalis et al. 2003; Li et al. 2003; Lord et al. 2010; Ploemacher et al. 1999; Sieber et al. 2009). It has also been described as a regulator of glucose metabolism (Chen et al. 2003) and as a differentiation factor for cholinergic neurons in the central nervous system (Lopez-Coviella et al. 2000). More recently, BMP9 was pinpointed as the physiologically functional high affinity ligand for the predominantly endothelial receptor, ALK1. This fact highlighted BMP9 as a critical modulator of angiogenesis (David et al. 2007, 2008; Scharpfenecker et al. 2007).
BMP9 was originally cloned from a rodent cDNA library obtained from mouse liver, where it was shown to be highly expressed (Miller et al. 2000). Accordingly, the liver was characterized as the major source of human and mouse BMP9, expressed by hepatocytes and intrahepatic biliary epithelial cells, while brain and lung only express it at much lower levels (Bidart et al. 2012). In line with these observations, the Human Protein Atlas profile for BMP9 in normal tissues indicates that it is highly expressed in liver, pancreas, placenta, lung, epididimus, gastrointestinal tract, gall bladder, and thyroid, but also in hematopoietic cells (www.proteinatlas.org).
Interestingly, it is now clearly described that pulmonary and cerebral arterial malformations occur more often in HHT1, while hepatic arterial malformations are more frequent in HHT2 (Letteboer et al. 2006). In fact, in HHT2, the frequency of hepatic arterial malformations is between 38 % and 41 %, while in HHT1, it ranges between 2.5 % and 8 % (Gallione et al. 2006). The specific expression of the ALK1 ligand, BMP9, predominantly in the liver reflects a seemingly tissue-specific manifestation in HHT2.
The modus operandus of BMP ligand interaction with their receptors differs from that of TGF-β: while TGF-β exhibits higher affinity for type II receptors and do not stably interact with type I receptors alone, BMPs bind independently to both type I and type II receptors (Groppe et al. 2008; Lin et al. 2006). The BMP ligands can also display affinity to the co-receptors endoglin and betaglycan (Lowery and de Caestecker 2010). In fact, BMP9 can directly bind endoglin (Scharpfenecker et al. 2007).
BMP9 is synthesized as a precursor protein, which is then cleaved by furin, a serine-endoprotease, forming a short dimeric mature form to which the prodomain can remain non-covalently associated (Bidart et al. 2012). Until recently, neither BMP9 nor its closely related family member BMP10 was found to be negatively regulated by common BMP pathway antagonists (David et al. 2008; Seemann et al. 2009). However, recent studies show that ALK1 activation by BMP9 induces expression of matrix Gla protein and crossveinless 2 (CV2), both known as antagonists of BMP4-induced angiogenesis (Yao et al. 2006, 2011, 2012). CV2, a member of the Chordin family, preferentially binds and inhibits BMP9 thereby providing strong feedback inhibition on ALK1 (Yao et al. 2012), suggesting a critical mutual regulation by BMP9 and CV2 in vasculature regulation.
Analogous to TGF-β signaling mediated by ALK1, BMP9 has also been reported to have incongruent effects on ECs. For example, BMP9 exhibits anti-angiogenic effects counteracting fibroblast growth factor (FGF)-induced angiogenesis in ex vivo metatarsal models (David et al. 2007; Scharpfenecker et al. 2007) and acts as a circulating vascular quiescent factor (David et al. 2008). Nevertheless, multiple types of ECs activate their proliferative status in vitro in response to BMP9, which pro-angiogenic properties also activate matrigel plug vascularization and tumor angiogenesis in a pancreatic cancer xenograft model (Suzuki et al. 2010).
In order to unmistakably clarify the effects on the endothelium by BMP9 stimulation and its specific downstream mediators, an extensive analysis of BMP9 downstream activation in comparison to other ligands on ECs is mandatory in the field. According to the present knowledge, BMP-induced responses have as common denominator the Smad1/5/8 pathway and Id1, 2, and 3 as target genes, suggesting other differentially responsive genes may exist, more specifically induced by each BMP. In a recent study such efforts were initiated where EC-specific Smad1/5 target genes were characterized and upregulation of Notch signaling-related genes were identified upon BMP9 stimulation (Morikawa et al. 2011).
Despite the paucity of detailed studies, the Human Protein Atlas profile for BMP9 in cancer disease indicates that BMP9 expression is increased in colorectal cancer, head and neck squamous carcinoma, and pancreatic and liver cancers (www.proteinatalas.com). Interestingly, BMP9 is primarily expressed in the islets of Langerhans by the tumor cellular compartment in mouse neuroendocrine pancreatic tumors (Cunha et al. 2010).
Similar to BMP9, BMP10 has also been identified as a functional activator of ALK1 in ECs, inducing comparable cellular effects. In agreement with a 65 % amino acid sequence homology between both ligands, BMP10, much like BMP9, exhibits angiostatic properties on dermal HMVEC (David et al. 2007). Nevertheless, BMP10 binds to ALK1 with lower affinity than BMP9 (David et al. 2007) and is mainly expressed in the murine developing and postnatal heart. The impaired lethal cardiac growth and physiology in the BMP10 knockout mouse coupled to normal vascular development of embryo and yolk sac propose a critical role for BMP10 in cardiogenesis (Chen et al. 2004). Interestingly, it has also been demonstrated that BMP10 can additionally bind to ALK3 (Mazerbourg et al. 2005). Of note, ALK3 targeted deletion in neural crest cells generates embryonic heart failure (Stottmann et al. 2004). All in all, these observations suggest the cardiac-specific nature of BMP10 signaling most likely through ALK3, rather than ALK1. However, the direct effect of BMP10 on the vasculature should not be overlooked, as it has potential to cooperate or even compensate for BMP9 signaling through ALK1. In fact, very recently Ricard et al. unveiled that Bmp9-KO mice do not exhibit defective vascularization in the retina (Ricard et al. 2012). However, injection of the extracellular domain of ALK1 or a neutralizing anti-BMP10 antibody impaired retinal vascularization in Bmp9-KO neonates, reducing retinal vascular expansion and exacerbating vascular density (Ricard et al. 2012). These data thus sustain a cooperative or compensatory role for BMP9 and BMP10 in postnatal vascular remodeling of the retina. Whether this cooperative role occurs in the context of cancer remains to be determined.
1.5 Interplay Between Type I Receptors
As mentioned earlier, ALK1 shares similar properties in terms of BMP-dependent activation of Smad1/5/8 signaling with the related BMP type I receptors ALK2, ALK3, and ALK6 (Fig. 14.2). Ligand specificity has not been carefully elucidated, and many ligands, including BMP2, BMP4, BMP6, BMP7, BMP9, and BMP10, exhibit a multitude of effects on ECs, ranging from metabolism, endothelial-to-mesenchymal transition (EndMT), and tumor angiogenesis (Fig. 14.2) (Bostrom et al. 2011; Heinke et al. 2008; Langenfeld and Langenfeld 2004; Medici et al. 2010; Ramoshebi and Ripamonti 2000; Yao et al. 2008; Zeisberg et al. 2007). While described as the physiological ligand for ALK1, BMP9 has also documented binding ability toward ALK2 in non-EC, such as myoblasts and breast tumor cells (Scharpfenecker et al. 2007), with the BMP9/ALK2 signaling axis being also linked to promotion of proliferation of ovarian cancer cells (Herrera et al. 2009). Lending support to the need of substantial analysis of the signaling arising from these receptors in the endothelium, vascular ECs have been shown to transform into multipotent stem-like cells in an ALK2-dependent fashion, in lesions from individuals with fibrodysplasia ossificans progressiva (FOP) (Medici et al. 2010). This disabling disorder occurs as a result of gain-of-function mutations in ALK2 in humans or mirrored in mice by constitutive activation of ALK2 signaling on chondrocytes and osteoblasts. Lineage tracing of heterotopic ossification in mice using a Tie2-Cre construct disclosed the endothelial origin of these cell types (Medici et al. 2010). In agreement with this finding, ECs conditionally deficient for ALK2 do not succeed to undergo EndMT during endocardial cushion formation in embryogenesis (Wang et al. 2005). Of note, ALK2 has been demonstrated to upregulate ALK1 in ECs in response to high-density lipoproteins, after which ALK1 in turn promotes survival by inducing expression of vascular endothelial growth factor (VEGF)-A (Yao et al. 2008). Glucose level augmentation co-regulates ALK1 and ALK2 expression in human aortic ECs (Bostrom et al. 2011). Also, BMP/TGF-β receptors appear to be activated and function sequentially: ALK3, ALK2, ALK1, and ALK5, where each receptor can possibly entail a distinct function and correlate to a specific stage in vascular growth and development (Bostrom et al. 2011; Shao et al. 2009; Yao et al. 2008).

Multiple TGF-β family ligands and receptors are acting on endothelial cells to shape the angiogenic response
Thus, the interplay and/or compensatory crosstalk primarily between ALK1 and ALK2, but also with ALK3 and ALK6, which is of critical importance in a therapeutic context, promptly begs for more detailed tumor studies.
1.6 TGF-β and BMP Signaling Pathways: Competitive or Synergistic?
Closely connected to receptor interplay is the role specifically played by the ligands. Classically, BMPs and TGF-βs have long been described to exert parallel antagonistic effect on the other pathway in a variety of biological contexts (Gronroos et al. 2012).
Moreover, in physiological conditions, cells in the body are exposed to multiple ligands simultaneously, which may render alternative responses than what is customarily studied when analyzing the effects of each ligand in isolation. In an attempt to clarify the role of ALK1 signaling in EC, we recently described an unanticipated synergistic effect of TGF-β with BMP9 on tumor angiogenesis. We demonstrated in vitro and in vivo in various systems, that while either cytokine on its own exerted suppressive action on the endothelium, both ligands in combination boosted the EC response toward other pro-angiogenic stimuli (Cunha et al. 2010). On a molecular level, simultaneous ECs induction with TGF-β and BMP9 induces a synergistic response on ALK5 target gene expression (e.g., PAI-1 and PDGF-B).
Another publication demonstrated that BMP2 synergistically enhances TGF-β3-induced initial phenotypic changes associated with EndMT, taking place during endocardial cushion formation (Yamagishi et al. 1999). Of note, BMPs and their receptors are expressed at many sites in which epithelial or endothelial-to-mesenchymal transition occurs during developmental organogenesis (Dewulf et al. 1995; Jones et al. 1991; Lyons et al. 1989).
TGF-β and BMP7 also coadjuvantly stimulate angiogenesis in the chick chorioallantoic membrane assay (Ramoshebi and Ripamonti 2000), and collaborately activate prostate cancer cells (Buijs et al. 2007) and osteoblast differentiation. In contrast, BMP7 counteracts TGF-β-induced EndMT in a model of cardiac fibrosis, rendering the EC capable of preserving their endothelial identity (Zeisberg et al. 2007), suggesting a context dependency of the synergy between TGF-βs and BMPs.
The numerous BMP ligands and type I receptors exert a variety of effects on ECs, yet the fact that different ligands utilize common pathway components raises important questions, which may have been neglected until recently: how cells respond specifically to individual ligands, and how cells integrate and interpret signals received from multiple ligands? Concerning this context, worthy of note are studies suggesting that preformed BMP receptor complexes or BMP-induced oligomerization of type I and type II receptors predominantly activate Smad-dependent and -independent signaling, respectively (Nohe et al. 2002; 2004). Also, the choice of type II receptor can persuade the signaling outcome of BMP stimulation as downstream-specific binding of Limk1 to the BMP type II receptor, but not to TGF-β or activin type II receptors (Foletta et al. 2003; Lee-Hoeflich et al. 2004). More recently, different R-Smad complex formation, Smad1/5–Smad2 versus Smad1/5–Smad3, was described (Gronroos et al. 2012), opening the possibility that the novel complexes may be the source of antagonistic versus synergistic responses in different studies.
Evidently, signaling through non-Smad effectors, the recruitment of distinct type II receptors and perhaps more importantly the variability created by alternative Smad complex formation should be further examined as the explanations for the diverse effects.
2 Physiological Role of the TGF-β Family in the Vasculature
2.1 Physiological Role of ALK1 in the Vasculature
The importance of this receptor became obvious, when Alk1 loss of function studies revealed that its complete loss causes embryonic lethality at midgestation, due to severe vascular abnormalities, which included vessels hyperdilation, AVMs resulting from fusion of major arteries and veins, and impaired recruitment of VSMC (Oh et al. 2000; Urness et al. 2000). Mutations in the Alk1 gene have been identified as an underlying cause for development of HHT, a rare, human autosomal dominant disease characterized by the presence of recurrent epistaxis and small characteristic malformations of the peripheral blood vessels near the surface of the skin or mucosal linings (Geirdal et al. 2012). AVMs of the lung, liver, and central nervous system are also known clinical findings. Interestingly, EC-specific deletion of the Alk1 gene in the mouse results in neonatal lethality at P5, with the pups exhibiting hemorrhaging brain, lung, and gastrointestinal tract (Park et al. 2008). In attempts to evaluate the contribution of ALK1 to vascular homeostasis in adult mice, Park et al. (2009) deleted the Alk1 gene by tamoxifen administration in 2 months old R26+/CreER Acvrl12loxP/loxP mice. Tamoxifen-induced Alk1 deletion resulted in severe internal hemorrhage in lung, small intestine, and uterine vessels, and ultimately fatality.
Strong expression of ALK1 has been reported during developmental and neonatal stages, while suppressed during adulthood, except in certain organs, e.g., the lungs (Park et al. 2008). Supportive of that is the observation that ALK1 is fundamental for umbilical and placental blood vessel formation (Hong et al. 2007). However, ALK1 expression is induced in feeding arteries and newly formed blood vessels during wound healing, in adult subdermal blood vessels (Park et al. 2009). AVMs appearing only in subdermal blood vessels where a wound was inflicted provides in vivo experimental evidence that genetic predisposition by endoglin or ALK1 mutations is not enough for development of de novo AVMs in HHT (Park et al. 2008). Interestingly, only selected vascular beds in HHT patients develop telangiectasias or AVM lesions, while other areas (>99.9 %) remain normal (Sadick et al. 2006).
Inhibition of ALK1 by systemic injection of an ALK1 soluble extracellular domain efficiently impaired retinal neonatal angiogenesis, described and validated in three independent studies (Larrivee et al. 2012; Niessen et al. 2010; Ricard et al. 2012). These data implied that ALK1 signaling blockade induced retinal hypervascularization and appearance of AVMs in neonatal mice. Incidentally, the most recent studies also report a cooperative effect of ALK1 and Notch signaling pathways (Larrivee et al. 2012; Morikawa et al. 2011; Ricard et al. 2012). The reported synergy between ALK1 and Notch pathways generated exacerbation of the hypervascularization phenotype, inducing potentiated expression of Notch target genes in the stalk cells, which concomitantly suppress VEGF signaling to the endothelial tip cell (Larrivee et al. 2012). In parallel studies, endothelial-specific inactivation of Smad1/5 in mouse embryonic development yields impaired Dll4/Notch signaling and augmented tip cell in detriment to stalk cell number (Moya et al. 2012). These studies put forward a regulatory crosstalk loop among BMP9/ALK1/Smad1/5 and Notch signaling coordinating tip versus stalk cell specification.
Additionally to being expressed by blood ECs, ALK1 is also expressed by lymphatic ECs (Niessen et al. 2010). In vitro stimulation of lymphatic ECs by BMP9 generates downstream target gene transactivation. Furthermore, inhibition of ALK1 signaling by means of an ALK1-Fc soluble fusion protein diminishes neonatal retinal lymphangiogenesis, while the use of ALK1 targeting monoclonal antibody also impairs Lyve-1 positive lymphangiogenesis in mammary fat pad–implanted MDA-MB-231 breast carcinoma xenografts (Hu-Lowe et al. 2011). Of note, lymph vessel development comprises coordinate and synergistic ALK1 and VEGFR3 signaling regulation, evocative of the crosstalk observed between ALK1/ALK5, ALK1/ALK2, and VEGF-receptor signaling in blood vessel angiogenesis (Cunha et al. 2010; Mitchell et al. 2010).
2.2 Physiological Role of Endoglin in the Vasculature
Endoglin, an auxiliary receptor for TGF-β, is required for angiogenesis during development (Li et al. 1999). It is expressed primarily in ECs and its expression is substantially incremented during EC activation, inflammation, ischemia, and tumor angiogenesis (Docherty et al. 2006; Jonker and Arthur 2002; Torsney et al. 2002). The mechanisms involved in endoglin upregulation are presumably multifactorial, but hypoxia is a probable inducer as it prevails in most pathophysiological environments where endoglin is enhanced (Bernabeu et al. 2009).
Endoglin associates with type II receptors of the TGF-β family in the presence of ligand and with the type I signaling receptors, ALK1 and ALK5, even in the absence of exogenous ligand (Barbara et al. 1999). Despite possessing no enzymatic activity, endoglin has been reported to be necessary to modulate ligand-receptor interaction in ALK1, but not in ALK5 signaling (Cheifetz et al. 1992; Lebrin et al. 2004; Pece-Barbara et al. 2005; Scharpfenecker et al. 2007). More recently, ALK5 was shown to phosphorylate the cytoplasmic domain of endoglin in ECs (Ray et al. 2010). Depending on the serine phosphorylation status, only on 646 or on both 646 and 649 serine residues, results in loss of endoglin-mediated inhibition or activation of Smad1/5/8 signaling, respectively, in response to TGF-β/BMP9 signaling (Ray et al. 2010). Taken together, these results indicate that endoglin phosphorylation by ALK5 is an important mechanism for regulating TGF-β and BMP signaling in ECs.
Even though endoglin has an undeniably well-documented connection to ALK1 and its signaling enhancement (Blanco et al. 2005; Lebrin et al. 2004), it is imperative to mention that it interacts not only with other ligands than TGF-β and BMP9 (TGF-β3, activin A, BMP2, and BMP7) but also with several different type I and type II receptors involved in BMP and TGF-β signaling (Barbara et al. 1999). Alternatively, endoglin has also been implicated in interactions with cytoplasmic proteins such as Zyxin, ZRP-1, β-Arrestin, and Tctex2β, which may further generate additional cellular outcomes (Bernabeu et al. 2009). The signaling unrestrictiveness of endoglin consequently adds an extra degree of intricacy to the elaborated signaling networks deriving from the TGF-β family in the angiogenesis field. Moreover, the fact that endoglin is positively associated with ECs proliferation, while weakly expressed in quiescent endothelium, has focused the interest on endoglin as a potential target for cancer in vivo.
2.3 Physiological Role of TGF-β in the Vasculature
TGF-β effect in vascular biology is traditionally regarded as an accessory pathway that primarily participates in VMCs differentiation and maturation. However, apart from the genetic evidence for TGF-β relevant direct role in the ECs, there is additional in vitro evidence revealing that several key angiogenic mediators such as VEGF and connective tissue growth factor are direct targets of the TGF-β signaling pathway (Padua and Massague 2009; Sanchez-Elsner et al. 2001). Interestingly, hypoxic conditions present at the core of a tumor juxtaposed to TGF-β signaling can induce robust levels of VEGF mRNA through the activation of hypoxia-inducible factor (HIF)-1 and Smad proteins (Sanchez-Elsner et al. 2001). TGF-β can also regulate the expression, secretion, and activity of matrix metalloproteinase (MMP)-2 and MMP-9, and downregulate the expression of the protease inhibitor TIMP in the tumor and ECs (Derynck et al. 2001; Hagedorn et al. 2001). This TGF-β-mediated metalloprotease activity can subsequently enhance the migratory and invasive properties of ECs required for angiogenesis.
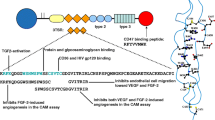
Multiple genetic mouse models suggestive of a direct role for TGF-β signaling pathway in angiogenesis further corroborate the need to more thoroughly analyze this pathway in the tumor context. Increased expression of TGF-β in either prostate carcinoma cells or Chinese hamster ovary cells resulted in robust angiogenic responses, which could be blocked by TGF-β neutralizing antibodies (Tuxhorn et al. 2002; Ueki et al. 1992). In sharp contrast, LNCaP human prostate xenografts treated with TGF-β neutralizing antibodies exhibited a 2- to 3.5-fold reduction in microvessel density vessels relative to control animals, whereas blood lakes were found in some areas of the tumors (Tuxhorn et al. 2002). Additionally, a TGF-β small molecule inhibitor LY364947 impaired tumor angiogenesis and growth in a transgenic multistep pancreatic islet tumor model (Cunha, SI, unpublished observation; Fig. 14.3). Another study using the orthotopic xenograft model of breast cancer MDA-MB-231 reported that ALK5-mediated TGF-β signaling is critical for metastasis dissemination and tumor angiogenesis in MMP9-regulated fashion (Safina et al. 2007). In parallel, Smad2 and Smad3 were reported to have opposing roles in breast cancer bone metastasis by differentially affecting VEGF-mediated tumor angiogenesis in an experimental mouse model of bone metastasis, where osteotropic MDA-MB-231-luc cells were intracardially inoculated (Petersen et al. 2010).

Inhibition of ALK5 signaling by the small molecule LY364947 inhibits tumor angiogenesis in the RIP1-Tag2 mouse model of pancreatic neuroendocrine cancer (immunostaining for the endothelial cell marker podocalyxin, green and the pericyte marker NG2, red)
To sum up, both direct and indirect effects of TGF-β on the tumor microenvironment stimulate tumor angiogenesis.
TGF-β affects tumor angiogenesis per se but it also regulates vascular permeability. Neutralizing antibodies to TGF-β1 or an ALK5 inhibitor significantly augment vessel permeability. In two distinct transgenic mouse tumor models, MMTV-PyMT and K14-HPV16, breast and epidermal squamous cell cancers, respectively, inhibition of ALK5 further enhanced vascular leakage into the interstitium and facilitated increased delivery of high molecular weight compounds into premalignant and malignant tissue (Lammerts et al. 2002; Sounni et al. 2010). These data strongly suggest that ALK5 antagonists can be therapeutically exploited to improve the delivery of drugs or molecular contrast agents into tissues where chronic damage or neoplastic disease limits their efficient delivery. Along with these observations, another study compared the effects of two different tyrosine kinase inhibitors, imatinib and sorafenib, with an ALK5 inhibitor (LY364947) on extravasation of a modeled nanoparticle, 2 MDa dextran in two tumor models: the CT26 colon cancer model and the BxPC3 pancreatic cancer model (Kano et al. 2009). In fact, sorafenib most potently enhanced the accumulation of nanoparticles in the CT26 colon cancer model, whereas TGF-β inhibitor exhibited a stronger effect on the BxPC3 pancreatic cancer model, suggesting that while ALK5 inhibitors are an appropriate strategy to enhance delivery of nanoparticle-delivered drugs in pancreatic tumors, this may not be longitudinally optimal for all tumor types.
A small molecule ALK5 inhibitor at a low dose was used for treating several experimental intractable solid tumors, including pancreatic adenocarcinoma and diffuse-type gastric cancer, characterized by hypovascularity and thick fibrotic tumor stroma. Low doses of ALK5 inhibitor altered TGF-β signaling neither in the malignant cells nor in the cancer-associated fibroblast components. However, the ALK5 inhibitor decreased pericyte coverage on the endothelium without reducing endothelial area specifically in the tumor vasculature. As a result, ALK5 inhibition promoted accumulation of anticancer nanocarriers in the tumors. In the absence of ALK5 inhibitor, the anticancer nanocarriers exhibited poor growth-inhibitory effects (Kano et al. 2007).
The vasculature of solid tumors is abnormal, both in terms of vessel architecture and blood flow dynamics. Permeable heterogeneous vessel walls permit the leakage of proteins and fluid that, coupled with inefficient lymphatic drainage, hinder drug delivery. As acute blockade of TGF-β signaling transiently alters vessel stability, permeabilization of tumor vascular beds improves intravenous delivery of high molecular weight compounds to the tumor such as antibodies or nanoparticles that are therapeutically selective, collectively rendering an improved clinical outcome. This effect has particular relevance when treating stroma-rich intractable solid tumors. Furthermore, by promotion of a more pervious tumor vessel phenotype, not only the delivery of standard therapeutic agents can be improved but also diagnostic molecular imaging agents can be more easily monitored in tumor tissue where TGF-β signaling has been transiently impaired.
Most studies on the role of TGF-β in cancer primarily rely on the effect of this cytokine on the cancerous epithelial cells and on how it affects cellular plasticity, more specifically epithelial-to-mesenchymal transition (EMT) and metastatic dissemination. Here, we focus essentially on the effects of TGF-β signaling on the tumor vasculature; however some of the studies on metastasis may have affected TGF-β-mediated vasculature leniency to tumor cells intra- and extravasation. In fact, some laboratories have shown that the metastatic spread of the 4 T1 breast cancer transplantable model of metastatic breast cancer can be efficiently suppressed by administering an antibody that targets all three isoforms of the TGF-β ligand. This work went on to show that TGF-β neutralizing antibodies have multiple cooperative effects on angiogenesis, immune cell function, and tumor cell viability, eventually leading to effective tumor growth control and reduction of metastatic foci (Nam et al. 2008).
3 Perspective
The formation of new blood vessels in pathological processes, such as cancer, leads to unstable, proliferative suboptimally functional vessels in a dynamic state of remodeling. Tumor vascular networks support tumor growth, in spite of a range of abnormal features that compromise their physiology. Tumor vessels are commonly tortuous and leaky, causing hemorrhage and increased interstitial fluid pressure (Carmeliet and Jain 2000). Inefficient blood flow caused by poor hierarchical anatomy and organization of the tumor vasculature leads to ischemia and necrosis, which are common characteristics of rapidly growing tumors.
Its inherent instability should make tumor vascular beds distinct from normal vessels and therefore more prone to be exquisitely affected by selective targeting anti-angiogenic drugs (Baluk et al. 2005). During the past few years we have witnessed a suboptimal outcome from anti-angiogenic treatment of human cancer implying the need for improvement and suggesting that alternative therapeutic avenues should be explored. Better knowledge of molecular and cellular mechanisms taking place in the tumor microenvironment and in the tumor vasculature is essential to achieve such goals.
The complexity of the TGF-β and BMP pathways by means of redundancy, cooperation, or by simply having different levels of regulation reflects the uniqueness and tight tissue specificity of this intriguing pathway. However, despite being complex, the TGF-β signaling pathway may hold central roles in tumor angiogenesis, as suggested by studies targeting its endothelial-specific receptors ALK1 and endoglin. Inhibitors for these receptors have already been generated and they hold great promise in impairment of de novo formed tumor vessels, while leaving normal idle vasculature undisturbed. The preclinical development and clinical use of inhibitors of pro-angiogenic TGF-β signaling will be discussed in detail in Chap. 19.
Abbreviations
- ALK:
-
Activin receptor-like kinase
- AVM:
-
Arteriovenous malformations
- BMP:
-
Bone morphogenetic proteins
- EC:
-
Endothelial cell
- EMT:
-
Epithelial-to-mesenchymal transition
- EndMT:
-
Endothelial-to-mesenchymal transition
- FGF:
-
Fibroblast growth factor
- GDF:
-
Growth and differentiation factor
- HHT:
-
Hereditary hemorrhagic telangiectasia
- MMP:
-
Matrix metalloproteinase
- TGF-β:
-
Transforming growth factor-β
- VEGF:
-
Vascular endothelial growth factor
- VSMC:
-
Vascular smooth muscle cell
References
Anderson L, Lowery JW, Frank DB et al (2010) Bmp2 and Bmp4 exert opposing effects in hypoxic pulmonary hypertension. Am J Physiol Regul Integr Comp Physiol 298:R833–R842
Arthur HM, Ure J, Smith AJ et al (2000) Endoglin, an ancillary TGFβ receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol 217:42–53
Baluk P, Hashizume H, McDonald DM (2005) Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15:102–111
Barbara NP, Wrana JL, Letarte M (1999) Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-β superfamily. J Biol Chem 274:584–594
Beppu H, Ichinose F, Kawai N et al (2004) BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol 287:L1241–L1247
Berg JN, Gallione CJ, Stenzel TT et al (1997) The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 61:60–67
Bernabeu C, Lopez-Novoa JM, Quintanilla M (2009) The emerging role of TGF-β superfamily coreceptors in cancer. Biochim Biophys Acta 1792:954–973
Bertolino P, Deckers M, Lebrin F, ten Dijke P (2005) Transforming growth factor-β signal transduction in angiogenesis and vascular disorders. Chest 128:585S–590S
Bidart M, Ricard N, Levet S et al (2012) BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell Mol Life Sci 69:313–324
Blanco FJ, Santibanez JF, Guerrero-Esteo M et al (2005) Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-β receptor complex. J Cell Physiol 204:574–584
Bostrom KI, Jumabay M, Matveyenko A et al (2011) Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res 108:446–457
Bourdeau A, Faughnan ME, Letarte M (2000) Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia. Trends Cardiovasc Med 10:279–285
Buijs JT, Rentsch CA, van der Horst G et al (2007) BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am J Pathol 171:1047–1057
Canalis E, Economides AN, Gazzerro E (2003) Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr Rev 24:218–235
Carmeliet P, Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407:249–257
Carta L, Pereira L, Arteaga-Solis E et al (2006) Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem 281:8016–8023
Chang H, Huylebroeck D, Verschueren K et al (1999) Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 126:1631–1642
Cheifetz S, Bellon T, Cales C et al (1992) Endoglin is a component of the transforming growth factor-β receptor system in human endothelial cells. J Biol Chem 267:19027–19030
Chen C, Grzegorzewski KJ, Barash S et al (2003) An integrated functional genomics screening program reveals a role for BMP-9 in glucose homeostasis. Nat Biotechnol 21:294–301
Chen H, Shi S, Acosta L et al (2004) BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 131:2219–2231
Compton LA, Potash DA, Brown CB, Barnett JV (2007) Coronary vessel development is dependent on the type III transforming growth factor β receptor. Circ Res 101:784–791
Crawford SE, Stellmach V, Murphy-Ullrich JE et al (1998) Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell 93:1159–1170
Cunha SI, Pietras K (2011) ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood 117:6999–7006
Cunha SI, Pardali E, Thorikay M et al (2010) Genetic and pharmacological targeting of activin receptor-like kinase 1 impairs tumor growth and angiogenesis. J Exp Med 207:85–100
David L, Mallet C, Mazerbourg S et al (2007) Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109:1953–1961
David L, Mallet C, Keramidas M et al (2008) Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ Res 102:914–922
Derynck R, Akhurst RJ, Balmain A (2001) TGF-β signaling in tumor suppression and cancer progression. Nat Genet 29:117–129
Dewulf N, Verschueren K, Lonnoy O et al (1995) Distinct spatial and temporal expression patterns of two type I receptors for bone morphogenetic proteins during mouse embryogenesis. Endocrinology 136:2652–2663
Dickson MC, Martin JS, Cousins FM et al (1995) Defective haematopoiesis and vasculogenesis in transforming growth factor-β 1 knock out mice. Development 121:1845–1854
Docherty NG, Lopez-Novoa JM, Arevalo M et al (2006) Endoglin regulates renal ischaemia-reperfusion injury. Nephrol Dial Transplant 21:2106–2119
Egeblad M, Nakasone ES, Werb Z (2010) Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 18:884–901
El-Bizri N, Guignabert C, Wang L et al (2008a) SM22α-targeted deletion of bone morphogenetic protein receptor 1A in mice impairs cardiac and vascular development, and influences organogenesis. Development 135:2981–2991
El-Bizri N, Wang L, Merklinger SL et al (2008b) Smooth muscle protein 22α-mediated patchy deletion of Bmpr1a impairs cardiac contractility but protects against pulmonary vascular remodeling. Circ Res 102:380–388
Elliott RL, Blobe GC (2005) Role of transforming growth factor β in human cancer. J Clin Oncol 23:2078–2093
Foletta VC, Lim MA, Soosairajah J et al (2003) Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol 162:1089–1098
Gallione CJ, Repetto GM, Legius E et al (2004) A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 363:852–859
Gallione CJ, Richards JA, Letteboer TG et al (2006) SMAD4 mutations found in unselected HHT patients. J Med Genet 43:793–797
Galvin KM, Donovan MJ, Lynch CA et al (2000) A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet 24:171–174
Geirdal AO, Dheyauldeen S, Bachmann-Harildstad G, Heimdal K (2012) Quality of life in patients with hereditary hemorrhagic telangiectasia in Norway: A population based study. Am J Med Genet A 158A:1269–1278
George EL, Georges-Labouesse EN, Patel-King RS et al (1993) Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 119:1079–1091
Goumans MJ, Mummery C (2000) Functional analysis of the TGFβ receptor/Smad pathway through gene ablation in mice. Int J Dev Biol 44:253–265
Goumans MJ, Valdimarsdottir G, Itoh S et al (2002) Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J 21:1743–1753
Goumans MJ, Valdimarsdottir G, Itoh S et al (2003) Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol Cell 12:817–828
Gronroos E, Kingston IJ, Ramachandran A et al (2012) TGF-β inhibits BMP-induced transcription through novel phosphorylated Smad1/5-Smad3 complexes. Mol Cell Biol 32:2904–2916
Groppe J, Hinck CS, Samavarchi-Tehrani P et al (2008) Cooperative assembly of TGF-β superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol Cell 29:157–168
Hagedorn HG, Bachmeier BE, Nerlich AG (2001) Synthesis and degradation of basement membranes and extracellular matrix and their regulation by TGF-β in invasive carcinomas (Review). Int J Oncol 18:669–681
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Heinke J, Wehofsits L, Zhou Q et al (2008) BMPER is an endothelial cell regulator and controls bone morphogenetic protein-4-dependent angiogenesis. Circ Res 103:804–812
Herrera B, van Dinther M, Ten Dijke P, Inman GJ (2009) Autocrine bone morphogenetic protein-9 signals through activin receptor-like kinase-2/Smad1/Smad4 to promote ovarian cancer cell proliferation. Cancer Res 69:9254–9262
Hong KH, Seki T, Oh SP (2007) Activin receptor-like kinase 1 is essential for placental vascular development in mice. Lab Invest 87:670–679
Huang Z, Wang D, Ihida-Stansbury K et al (2009) Defective pulmonary vascular remodeling in Smad8 mutant mice. Hum Mol Genet 18:2791–2801
Hu-Lowe DD, Chen E, Zhang L et al (2011) Targeting activin receptor-like kinase 1 inhibits angiogenesis and tumorigenesis through a mechanism of action complementary to anti-VEGF therapies. Cancer Res 71:1362–1373
Itoh F, Itoh S, Carvalho RL et al (2009) Poor vessel formation in embryos from knock-in mice expressing ALK5 with L45 loop mutation defective in Smad activation. Lab Invest 89:800–810
Itoh F, Itoh S, Adachi T et al (2012) Smad2/Smad3 in endothelium is indispensable for vascular stability via S1PR1 and N-cadherin expressions. Blood 119:5320–5328
Jones CM, Lyons KM, Hogan BL (1991) Involvement of Bone Morphogenetic Protein-4 (BMP-4) and Vgr-1 in morphogenesis and neurogenesis in the mouse. Development 111:531–542
Jonker L, Arthur HM (2002) Endoglin expression in early development is associated with vasculogenesis and angiogenesis. Mech Dev 110:193–196
Judge DP, Biery NJ, Keene DR et al (2004) Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest 114:172–181
Kano MR, Bae Y, Iwata C et al (2007) Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-β signaling. Proc Natl Acad Sci USA 104:3460–3465
Kano MR, Komuta Y, Iwata C et al (2009) Comparison of the effects of the kinase inhibitors imatinib, sorafenib, and transforming growth factor-β receptor inhibitor on extravasation of nanoparticles from neovasculature. Cancer Sci 100:173–180
Kishigami S, Mishina Y (2005) BMP signaling and early embryonic patterning. Cytokine Growth Factor Rev 16:265–278
Lammerts E, Roswall P, Sundberg C et al (2002) Interference with TGF-β1 and -β3 in tumor stroma lowers tumor interstitial fluid pressure independently of growth in experimental carcinoma. Int J Cancer 102:453–462
Lamouille S, Mallet C, Feige JJ, Bailly S (2002) Activin receptor-like kinase 1 is implicated in the maturation phase of angiogenesis. Blood 100:4495–4501
Lan Y, Liu B, Yao H et al (2007) Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol Cell Biol 27:7683–7692
Langenfeld EM, Langenfeld J (2004) Bone morphogenetic protein-2 stimulates angiogenesis in developing tumors. Mol Cancer Res 2:141–149
Larrivee B, Prahst C, Gordon E et al (2012) ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell 22:489–500
Larsson J, Goumans MJ, Sjostrand LJ et al (2001) Abnormal angiogenesis but intact hematopoietic potential in TGF-β type I receptor-deficient mice. EMBO J 20:1663–1673
Lawler J, Sunday M, Thibert V et al (1998) Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest 101:982–992
Lebrin F, Goumans MJ, Jonker L et al (2004) Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J 23:4018–4028
Lechleider RJ, Ryan JL, Garrett L et al (2001) Targeted mutagenesis of Smad1 reveals an essential role in chorioallantoic fusion. Dev Biol 240:157–167
Lee-Hoeflich ST, Causing CG, Podkowa M et al (2004) Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J 23:4792–4801
Letteboer TG, Mager JJ, Snijder RJ et al (2006) Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J Med Genet 43:371–377
Li DY, Sorensen LK, Brooke BS et al (1999) Defective angiogenesis in mice lacking endoglin. Science 284:1534–1537
Li JZ, Hankins GR, Kao C et al (2003) Osteogenesis in rats induced by a novel recombinant helper-dependent bone morphogenetic protein-9 (BMP-9) adenovirus. J Gene Med 5:748–756
Lin SJ, Lerch TF, Cook RW et al (2006) The structural basis of TGF-β, bone morphogenetic protein, and activin ligand binding. Reproduction 132:179–190
Lopez-Coviella I, Berse B, Krauss R et al (2000) Induction and maintenance of the neuronal cholinergic phenotype in the central nervous system by BMP-9. Science 289:313–316
Lord E, Bergeron E, Senta H et al (2010) Effect of BMP-9 and its derived peptide on the differentiation of human white preadipocytes. Growth Factors 28:149–156
Lowery JW, de Caestecker MP (2010) BMP signaling in vascular development and disease. Cytokine Growth Factor Rev 21:287–298
Lyons KM, Pelton RW, Hogan BL (1989) Patterns of expression of murine Vgr-1 and BMP-2a RNA suggest that transforming growth factor-β-like genes coordinately regulate aspects of embryonic development. Genes Dev 3:1657–1668
Mahmoud M, Allinson KR, Zhai Z et al (2010) Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res 106:1425–1433
Massague J (2000) How cells read TGF-β signals. Nat Rev Mol Cell Biol 1:169–178
Massague J (2008) TGFβ in Cancer. Cell 134:215–230
Mazerbourg S, Sangkuhl K, Luo CW et al (2005) Identification of receptors and signaling pathways for orphan bone morphogenetic protein/growth differentiation factor ligands based on genomic analyses. J Biol Chem 280:32122–32132
McAllister KA, Grogg KM, Johnson DW et al (1994) Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 8:345–351
Medici D, Shore EM, Lounev VY et al (2010) Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16:1400–1406
Miller AF, Harvey SA, Thies RS, Olson MS (2000) Bone morphogenetic protein-9. An autocrine/paracrine cytokine in the liver. J Biol Chem 275(24):17937–17945.
Mitchell D, Pobre EG, Mulivor AW et al (2010) ALK1-Fc inhibits multiple mediators of angiogenesis and suppresses tumor growth. Mol Cancer Ther 9:379–388
Miyazono K, Kamiya Y, Morikawa M (2010) Bone morphogenetic protein receptors and signal transduction. J Biochem 147:35–51
Morikawa M, Koinuma D, Tsutsumi S et al (2011) ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res 39:8712–8727
Moustakas A, Heldin CH (2009) The regulation of TGFβ signal transduction. Development 136:3699–3714
Moya IM, Umans L, Maas E et al (2012) Stalk cell phenotype depends on integration of Notch and Smad1/5 signaling cascades. Dev Cell 22:501–514
Nam JS, Terabe M, Mamura M et al (2008) An anti-transforming growth factor β antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res 68:3835–3843
Nguyen HL, Lee YJ, Shin J et al (2011) TGF-β signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab Invest 91:1554–1563
Niessen K, Zhang G, Ridgway JB et al (2010) ALK1 signaling regulates early postnatal lymphatic vessel development. Blood 115:1654–1661
Nohe A, Hassel S, Ehrlich M et al (2002) The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem 277:5330–5338
Nohe A, Keating E, Knaus P, Petersen NO (2004) Signal transduction of bone morphogenetic protein receptors. Cell Signal 16:291–299
Oh SP, Seki T, Goss KA et al (2000) Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA 97:2626–2631
Oshima M, Oshima H, Taketo MM (1996) TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev Biol 179:297–302
Ota T, Fujii M, Sugizaki T et al (2002) Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-β in human umbilical vein endothelial cells. J Cell Physiol 193:299–318
Padua D, Massague J (2009) Roles of TGFβ in metastasis. Cell Res 19:89–102
Pardali K, Moustakas A (2007) Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta 1775:21–62
Park C, Lavine K, Mishina Y et al (2006) Bone morphogenetic protein receptor 1A signaling is dispensable for hematopoietic development but essential for vessel and atrioventricular endocardial cushion formation. Development 133:3473–3484
Park SO, Lee YJ, Seki T et al (2008) ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood 111:633–642
Park SO, Wankhede M, Lee YJ et al (2009) Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest 119:3487–3496
Pece-Barbara N, Vera S, Kathirkamathamby K et al (2005) Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor β1 with higher affinity receptors and an activated Alk1 pathway. J Biol Chem 280:27800–27808
Petersen M, Pardali E, van der Horst G et al (2010) Smad2 and Smad3 have opposing roles in breast cancer bone metastasis by differentially affecting tumor angiogenesis. Oncogene 29:1351–1361
Pietras K, Östman A (2010) Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res 316:1324–1331
Ploemacher RE, Engels LJ, Mayer AE et al (1999) Bone morphogenetic protein 9 is a potent synergistic factor for murine hemopoietic progenitor cell generation and colony formation in serum-free cultures. Leukemia 13:428–437
Ramoshebi LN, Ripamonti U (2000) Osteogenic protein-1, a bone morphogenetic protein, induces angiogenesis in the chick chorioallantoic membrane and synergizes with basic fibroblast growth factor and transforming growth factor-β1. Anat Rec 259:97–107
Ray BN, Lee NY, How T, Blobe GC (2010) ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis 31:435–441
Ricard N, Ciais D, Levet S et al (2012) BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 119(25):6162–6171
Rudarakanchana N, Flanagan JA, Chen H et al (2002) Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet 11:1517–1525
Sadick H, Sadick M, Gotte K et al (2006) Hereditary hemorrhagic telangiectasia: an update on clinical manifestations and diagnostic measures. Wien Klin Wochenschr 118:72–80
Safina A, Vandette E, Bakin AV (2007) ALK5 promotes tumor angiogenesis by upregulating matrix metalloproteinase-9 in tumor cells. Oncogene 26:2407–2422
Sanchez-Elsner T, Botella LM, Velasco B et al (2001) Synergistic cooperation between hypoxia and transforming growth factor-β pathways on human vascular endothelial growth factor gene expression. J Biol Chem 276:38527–38535
Sanford LP, Ormsby I, Gittenberger-de Groot AC et al (1997) TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 124:2659–2670
Scharpfenecker M, van Dinther M, Liu Z et al (2007) BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J Cell Sci 120:964–972
Seemann P, Brehm A, Konig J et al (2009) Mutations in GDF5 reveal a key residue mediating BMP inhibition by NOGGIN. PLoS Genet 5:e1000747
Seki T, Hong KH, Oh SP (2006) Nonoverlapping expression patterns of ALK1 and ALK5 reveal distinct roles of each receptor in vascular development. Lab Invest 86:116–129
Shao ES, Lin L, Yao Y, Bostrom KI (2009) Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood 114:2197–2206
Shore EM, Xu M, Feldman GJ et al (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38:525–527
Shull MM, Ormsby I, Kier AB et al (1992) Targeted disruption of the mouse transforming growth factor-β 1 gene results in multifocal inflammatory disease. Nature 359:693–699
Sieber C, Kopf J, Hiepen C, Knaus P (2009) Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev 20:343–355
Song Y, Jones JE, Beppu H et al (2005) Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation 112:553–562
Sounni NE, Dehne K, van Kempen L et al (2010) Stromal regulation of vessel stability by MMP14 and TGFβ. Dis Model Mech 3:317–332
Srinivasan S, Hanes MA, Dickens T et al (2003) A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet 12:473–482
Stenvers KL, Tursky ML, Harder KW et al (2003) Heart and liver defects and reduced transforming growth factor β2 sensitivity in transforming growth factor β type III receptor-deficient embryos. Mol Cell Biol 23:4371–4385
Stottmann RW, Choi M, Mishina Y et al (2004) BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development 131:2205–2218
Suzuki Y, Ohga N, Morishita Y et al (2010) BMP-9 induces proliferation of multiple types of endothelial cells in vitro and in vivo. J Cell Sci 123:1684–1692
ten Dijke P, Arthur HM (2007) Extracellular control of TGFβ signalling in vascular development and disease. Nat Rev Mol Cell Biol 8:857–869
ten Dijke P, Miyazono K, Heldin CH (2000) Signaling inputs converge on nuclear effectors in TGF-β signaling. Trends Biochem Sci 25:64–70
Thomas PS, Sridurongrit S, Ruiz-Lozano P, Kaartinen V (2012) Deficient signaling via Alk2 (Acvr1) leads to bicuspid aortic valve development. PLoS One 7:e35539
Todorovic V, Frendewey D, Gutstein DE et al (2007) Long form of latent TGF-β binding protein 1 (Ltbp1L) is essential for cardiac outflow tract septation and remodeling. Development 134:3723–3732
Torsney E, Charlton R, Parums D et al (2002) Inducible expression of human endoglin during inflammation and wound healing in vivo. Inflamm Res 51:464–470
Torsney E, Charlton R, Diamond AG et al (2003) Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation 107:1653–1657
Tuxhorn JA, McAlhany SJ, Yang F et al (2002) Inhibition of transforming growth factor-β activity decreases angiogenesis in a human prostate cancer-reactive stroma xenograft model. Cancer Res 62:6021–6025
Ueki N, Nakazato M, Ohkawa T et al (1992) Excessive production of transforming growth-factor β 1 can play an important role in the development of tumorigenesis by its action for angiogenesis: validity of neutralizing antibodies to block tumor growth. Biochim Biophys Acta 1137:189–196
Upton PD, Davies RJ, Trembath RC, Morrell NW (2009) Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J Biol Chem 284:15794–15804
Urness LD, Sorensen LK, Li DY (2000) Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet 26:328–331
Valdimarsdottir G, Goumans MJ, Rosendahl A et al (2002) Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation 106:2263–2270
Wagner DO, Sieber C, Bhushan R et al (2010) BMPs: from bone to body morphogenetic proteins. Sci Signal 3:mr1
Wakefield LM, Roberts AB (2002) TGF-β signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev 12:22–29
Wang J, Sridurongrit S, Dudas M et al (2005) Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Dev Biol 286:299–310
Wozney JM, Rosen V, Celeste AJ et al (1988) Novel regulators of bone formation: molecular clones and activities. Science 242:1528–1534
Wrana JL, Attisano L, Wieser R et al (1994) Mechanism of activation of the TGF-β receptor. Nature 370:341–347
Yamagishi T, Nakajima Y, Miyazono K, Nakamura H (1999) Bone morphogenetic protein-2 acts synergistically with transforming growth factor-β3 during endothelial-mesenchymal transformation in the developing chick heart. J Cell Physiol 180:35–45
Yang X, Castilla LH, Xu X et al (1999) Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development 126:1571–1580
Yao Y, Zebboudj AF, Shao E et al (2006) Regulation of bone morphogenetic protein-4 by matrix GLA protein in vascular endothelial cells involves activin-like kinase receptor 1. J Biol Chem 281:33921–33930
Yao Y, Shao ES, Jumabay M et al (2008) High-density lipoproteins affect endothelial BMP-signaling by modulating expression of the activin-like kinase receptor 1 and 2. Arterioscler Thromb Vasc Biol 28:2266–2274
Yao Y, Jumabay M, Wang A, Bostrom KI (2011) Matrix Gla protein deficiency causes arteriovenous malformations in mice. J Clin Invest 121:2993–3004
Yao Y, Jumabay M, Ly A et al (2012) Crossveinless 2 regulates bone morphogenetic protein 9 in human and mouse vascular endothelium. Blood 119:5037–5047
Zacchigna L, Vecchione C, Notte A et al (2006) Emilin1 links TGF-β maturation to blood pressure homeostasis. Cell 124:929–942
Zeisberg EM, Potenta S, Xie L et al (2007) Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 67:10123–10128
Zhang H, Bradley A (1996) Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development 122:2977–2986
Zhao GQ (2003) Consequences of knocking out BMP signaling in the mouse. Genesis 35:43–56
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer
About this chapter
Cite this chapter
Cunha, S.I., Pietras, K. (2013). TGF-β Signaling in Physiological and Pathological Angiogenesis. In: Moustakas, A., Miyazawa, K. (eds) TGF-β in Human Disease. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54409-8_14
Download citation
DOI: https://doi.org/10.1007/978-4-431-54409-8_14
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54408-1
Online ISBN: 978-4-431-54409-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)