
Abstract
There are several well-established mechanisms involved in radiation-induced cell death in mammalian cell systems. The p53-mediated apoptotic pathway is the most widely recognized mechanism (Lowe et al. Nature 362:847–849, 1993), although apoptosis has long been considered a less relevant mechanism of radiation-induced cell death (Steel, Acta Oncol 40:968–975, 2001; Brown and Wouters, Cancer Res 59:1391–1399, 1999; Olive and Durand, Int J Radiat Biol 71:695–707, 1997). We and others have recently focused instead on the emerging links between radiation, apoptosis, and ceramide and showed that ceramide is a sphingolipid-derived second messenger capable of initiating apoptotic cascades in response to various stress stimuli, including radiation.
Ceramide, the backbone of all sphingolipids, is synthesized by a family of ceramide synthases (CerS), each using acyl-CoAs of defined chain length for N-acylation of the sphingoid long-chain base. Six mammalian CerS homologs have been cloned that demonstrated high selectivity towards acyl-CoAs (Lahiri et al. FEBS Lett 581:5289–5294, 2007), and more recently, it was shown that their activity can be modulated by dimer formation (Mesicek et al. Cell Signal 22:1300–1307, 2010; Laviad et al. J Biol Chem 283:5677–5684, 2008).
This de novo ceramide synthesis has been observed in irradiated cells through a pathway normally suppressed by ataxia telangiectasia-mutated (ATM) protein, a key component of the cellular response to DNA double-strand breaks (Liao et al. J Biol Chem 274:17908–17917, 1999). ATM is not the sole factor known to affect apoptotic potential by modulating CerS activity. Recent work has also implicated protein kinase Cα (PKCα) as a potential CerS activator (Truman et al. Cancer Biol Ther 8:54–63, 2009).
In this review, we summarize involvement of CerS in sphingolipid-mediated apoptosis in irradiated human prostate cancer cells and discuss future directions in this field.
The authors have no conflict of interest or financial ties to disclose.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Radioresistance in Prostate Cancer: From Bedside to Bench
1.1 Local Relapse After Radiation Therapy
Prostate cancer is the most commonly diagnosed non-skin cancer in men in the United States. It is estimated that 217,730 men were diagnosed with prostate cancer during 2010. About 32,050 died of the disease the same year (Jemal et al. 2010). Radiotherapy (RT) is a widely used modality for men with prostate cancer, but although radiation is capable of permanently eradicating localized prostate tumors, nearly 30 % of patients treated with potentially curative doses relapse at the sites of the irradiated tumors (Scardino and Wheeler 1988; Crook et al. 1995; Zelefsky et al. 1998). More recently, continued progress in RT delivery techniques has improved outcome through dose escalation. Nonetheless, although higher radiation dose levels were consistently associated with improved biochemical control outcomes and reduction in distant metastases, the biochemical relapse rate was still greater than 60 % at 5 years in high-risk patients (Zelefsky et al. 2008). These data indicate that prostate tumors vary in sensitivity to ionizing radiation. Furthermore, clinical data show that in patients who relapse locally after RT, initial treatment eliminates the great majority of the tumor cells, whereas a small fraction of tumor clonogens survive the lethal effects of radiation and eventually repopulate the irradiated site. This observation indicates that there are variations in clonal sensitivity to the lethal effects of radiation even within a given tumor. Thus far, there have been neither a criteria for predicting the presence or prevalence of radiation-resistant tumor clones nor an effective approach to modulate the radiation response of human prostate tumor cells. Improved understanding of pathways of radiation-induced cell death and signaling systems that regulate these pathways may yield opportunities for pharmacological modulation of radiation resistance in prostate cancer.
1.2 Human Prostate Cancer Cell Lines’ Response to Radiation
Cell lines derived from human prostate cancer are regarded as relatively resistant to both radiation-induced clonogenic death and apoptotic death (Royai et al. 1996). The best-characterized human prostate cell lines include the PC-3, DU-145, CWR22-Rv1, and LNCaP cells that were established from metastatic human tumor lesions (Stone et al. 1978; Kaighn et al. 1979; Horoszewicz et al. 1983). In general, these cell lines are among the most radioresistant human tumor cells, as assessed by the clonogenic assay (Wollin et al. 1989; Leith et al. 1993; Leith 1994; DeWeese et al. 1998). However, the dose-survival data do indicate distinct differences between these cell lines, as expressed by the Dq, D0, SF-2, and the linear quadratic α and β exponents. There are also differences in the apoptotic response to radiation. Several studies reported lack of apoptosis in PC-3 cells up to 72 h after exposure to doses of 10–30 Gy (Algan et al. 1996; Li and Franklin 1998) and an incidence of 10–15 % apoptosis in DU-145 cells at 72 h after 10–12 Gy (Algan et al. 1996; Bowen et al. 1998). However, one study reported 40 % apoptosis in PC-3 cells at 72 h after 20 Gy (Kyprianou et al. 1997). This study is also the only study that has thus far reported apoptosis in LNCaP cells, occurring at a rate of 35 % at 72 h after exposure to 20 Gy (Kyprianou et al. 1997). Altogether, these observations indicate clone-specific sensitivities of human prostate tumor cells to radiation. The pleiotropic nature of death pathways induced by radiation suggests that radiation resistance is likely to be regulated by a variety of mechanisms, each of which is associated with a specific death pathway. Whether radiation resistance of human prostate tumor clones is associated with a single mechanism or a spectrum of mechanisms is unknown. These data also suggest that an approach to reduce radiation resistance clinically might require the use of combinations of chemical and biological modifiers to cover a spectrum of resistance mechanisms that may operate concomitantly in prostate cancer.
1.3 Radiation-Induced DNA Damage
It is well accepted that radiation-induced cell death stems from lethal DNA damage. When cells are irradiated, X-rays induce numerous DNA breaks. All break types do not have the same biologic consequence as far as cell killing is concerned. Many DNA single-strand breaks are readily repaired using the opposite DNA strand as a template. Breaks in both strands, if well separated, are also readily repaired because they are handled independently. Breaks in both strands that are opposite or separated by only a few base pairs may lead to double-strand breaks (DSBs). There is increasing amount of evidence that DSBs rather than single-strand breaks lead to important biologic endpoints, including “mitotic catastrophe” and loss of replicative potential (Hall and Giaccia 2006a). Such lesions are produced in the DNA by direct interaction with X-rays or with reactive oxygen intermediates generated within the cell by the radiation. DNA DSBs, the most lethal form of ionizing radiation-induced damage, are repaired by nonhomologous end-joining (NHEJ) repair in the G1 phase of the cell cycle and homologous recombination (HR) repair pathway in the S/G2 phase of the cell cycle. Whereas most radiation-induced DNA DSBs are rapidly repaired by constitutively expressed DNA repair mechanisms, residual unrepaired or misrepaired breaks lead to genetic instability and to increased frequency of mutations and chromosomal aberrations. Lethal mutations or dysfunctional chromosomal aberrations eventually lead to either progeny cell death, usually after several mitotic cycles (also termed reproductive or postmitotic), or to p53-mediated apoptosis (Hall and Giaccia 2006b).
1.4 Radiation-Induced Apoptosis
Radiation-induced apoptosis has long been considered a less relevant mechanism in cell loss from normal tissues and tumors based on published data comparing apoptosis response and cell survival responses in tumor cells that have generally failed to find a causal relationship. Moreover, modulating apoptotic potential usually had little impact on cellular radiosensitivity (Steel 2001; Brown and Wouters 1999; Olive and Durand 1997; Kyprianou et al. 1997; Aldridge et al. 1995; Lock and Ross 1990). Apoptosis or programmed cell death is an intermitotic (interphase) inducible death pathway of sequential biochemical events that are constitutively expressed in an inactive form in most, if not all, mammalian cells. Also defined as a mechanism of cellular suicide, apoptosis occurs in response to a variety of physiological or environmental stresses impacting distinct cellular targets to initiate cell type-specific apoptotic signaling pathways. The various upstream signaling cascades converge downstream to activate a common final caspase-dependent effector mechanism eventually leading to activation of a calcium–magnesium-dependent endonuclease that cleaves the nuclear chromatin at selective inter-nucleosomal linker sites, thus dismantling the dying cell. Chromosomal fragmentation, cytoplasmic blebbing, and apoptotic bodies are consequently seen during apoptosis, which ultimately results in the condensation of the nucleus and shrinking of the cell. Apoptosis is characteristically different from cell necrosis in morphology and biochemistry, and its end result is cell death without inflammation of the surrounding tissue. After a few decades and after quite a few debates, the case against apoptosis is no longer successfully defended. Today, we acknowledge apoptosis, or programmed cell death, not only as the process leading to disorders of normal tissues (Orrenius 1995; Fadeel et al. 1999; Reed 2002; Mullauer et al. 2001) but also as a form of death in response to both chemotherapy and radiation therapy for cancer, in particular for hormone-dependent cancers such as prostate cancer (Wu 1996; Olson and Kornbluth 2001; Ameisen 2002; Meyn et al. 2009). As a matter of fact, facilitation of apoptosis in vivo has been shown to effectively increase the number of apoptotic cells in tumors (Dubray et al. 1998; Jansen et al. 2000), and the detected early apoptotic response correlates well with subsequent outcome (Symmans et al. 2000; Meyn et al. 1995; Ellis et al. 1997). Proapoptotic antibodies, anti-CD-95 antibodies, are considered to be used as new therapeutic agents for tumor treatment. Moreover, the emerging links between radiation, apoptosis, and ceramide suggest that ceramide-mediated apoptosis following ionizing radiation might explain part of normal tissue and tumor cell death.
2 Activation of the Ceramide Synthase Pathway in Response to Radiation
We have previously shown that DNA DSBs induce ceramide generation via the de novo ceramide synthesis pathway involving activation of CerS enzyme in bovine aortic endothelial cells (BAEC) (Liao et al. 1999). Actually, a variety of ceramide species syntheses are catalyzed by six CerS enzyme isotypes (Table 1). CerS are integral membrane proteins of the endoplasmic reticulum. Once activated, these enzymes catalyze the condensation of sphinganine and fatty acyl-CoA to form dihydroceramide, which is rapidly oxidized to ceramide. Originally, we showed that daunorubicin stimulates CerS and generates ceramide resulting in apoptosis in P388 murine leukemia cells and U937 human monoblastic leukemia cells (Bose et al. 1995). An obligatory role for CerS was thus defined, since its natural specific inhibitor, Fumonisin B1 (FB1), blocked daunorubicin-induced ceramide elevation and apoptosis. This study was the first to demonstrate that CerS activity is regulated in eukaryotes and constituted definitive evidence for a requirement for ceramide elevation in the induction of apoptosis.
In collaboration with Anthony Futerman’s group [Weizmann Institute of Science, Israel (Lahiri et al. 2007)], we demonstrated that despite the high selectivity towards acyl-CoAs, mammalian CerS have a very similar K m value towards sphinganine, strengthening the notion that the main biochemical difference between CerS is in their specificity towards acyl-CoAs. In addition, in these studies, conditions for assaying CerS activity were optimized, and a K m value of all six mammalian CerS towards sphinganine in the low μM range (2–5 μM) was demonstrated, which is significantly lower than that suggested using a variety of other assays and tissue sources.
Subsequently, we reported that ionizing radiation also induces de novo synthesis of ceramide in HeLa cells resulting in apoptosis by specifically activating CerS5 and CerS6 (Mesicek et al. 2010). Overexpression of CerS2 resulted in partial protection from RT-induced apoptosis, whereas overexpression of CerS5 generated the apoptogenic ceramide species C16-ceramide and increased apoptosis in these cells. Knockdown studies determined that CerS2 is responsible for all observable RT-induced C(24:0) CerS activity, and while CerS5 and CerS6 each confer approximately 50 % of the C(16:0) CerS baseline synthetic activity, both are required for RT-induced activity. Additionally, co-immunoprecipitation studies suggest that CerS2, 5, and 6 might exist as heterocomplexes in HeLa cells, providing further insight into the regulation of CerS enzymes. Moreover, CerS were shown to have additional subcellular localizations, such as perinuclear membranes and the mitochondria-associated membrane (MAM). The interplay between long-chain C16-ceramide and very long-chain C24:1- and C24-ceramides has come into recent spotlight regarding their roles in maintaining cellular homeostasis. These data add to the growing body of evidence demonstrating interplay among the CerS isozymes in a stress stimulus-, cell type-, and subcellular compartment-specific manner.
CerS can be phosphorylated (Sridevi et al. 2009) and glycosylated (Mizutani et al. 2006), and it was recently demonstrated that CerS can form both homo- and heterodimers (Laviad et al. 2008) and that the activity of one member of a heterodimer depends upon, and can be modulated by, the activity of the other member. These studies suggest a rapid and reversible mechanism of regulating CerS activity by dimer formation.
CerS engagement in mammalian apoptosis in response to ionizing radiation, drugs, and cytokines (Truman et al. 2009; Garcia-Bermejo et al. 2002; Canman and Kastan 1995; Kastan et al. 1992) is a cell type-specific phenomena. The biological significance of CerS as a transducer of apoptosis has also been confirmed in vivo, as CerS transactivates disease pathogenesis in gastric ulcer, ischemia/reperfusion, and emphysema rodent models of human disease (Lowe et al. 1993; Spiegel et al. 1996; Haimovitz-Friedman et al. 1997; Lin et al. 2000; Mathias et al. 1998; Pena et al. 1997). In these reports, epithelial apoptosis occurred 18–24 h after radiation exposure or about 6–10 h after CerS activation and was inhibited by FB1.
3 Modulation of Ceramide Synthase Activity in Prostate Cancer Cells
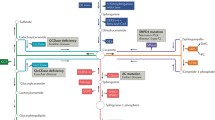
There are several well-established mechanisms involved in radiation-induced apoptosis in mammalian cell systems. The p53-mediated pathway is the most widely recognized mechanism (Lowe et al. 1993). LNCaP and CWR22-Rv1 human prostate cell lines express wild-type p53 and exhibit extreme resistance to radiation-induced apoptosis. Yet, we found that these cells display high sensitivity to radiation-induced apoptosis both in vitro and in vivo when pretreated with TPA (12-O-tetradecanoylphorbol-13-acetate) or other PKC activators (Truman et al. 2009). Studies in our laboratory focus on ceramide-mediated apoptosis, a response to radiation that is distinct from the classical p53-mediated response. Our work indicates that the apoptotic pathway involved in LNCaP and CWR22-Rv1 radiosensitization is mediated by activation of CerS (Fig. 1), and not by other known pathways of ceramide-mediated apoptosis involving the activation of either acid sphingomyelinase (ASMase) or neutral sphingomyelinase (NSMase) (Garzotto et al. 1998, 1999).

Integration of radiation- and PKCα-induced and apoptosis (Asterisk denotes activated).TPA and DAG-Lactone simultaneously down-regulated ATM levels and enhance CerS activity in human prostate cancer cells via activation of PKCα. Radiation acts synergistically on CerS activation by inducing DNA DSBs, once the ATM inhibition is removed. This results in higher accumulation of ceramide levels in these cells and radiation-induced apoptosis both in vitro and in vivo [modified from Truman et al. (2009)]
3.1 PKCα Activation of Ceramide Synthase in Prostate Cancer Cells
TPA treatment of LNCaP cells activates PKCα and PKCδ to induce CerS activation (Garzotto et al. 1998) with rapid but progressive ceramide generation. This was followed by a delayed form of apoptosis that reached maximal levels at 48 h. Investigations into the mechanism of TPA-induced ceramide generation revealed that ASMase and NSMase activities were not enhanced. In contrast, TPA induced an increase in CerS activity that persisted for at least 16 h. Treatment with FB1 abrogated both TPA-induced ceramide production and apoptosis. Thus, CerS activation appears to be required for TPA-induced apoptosis in LNCaP cells (Garzotto et al. 1998).
Whereas LNCaP cells failed to respond to radiation with ceramide generation and apoptosis, pretreatment with TPA or diacylglycerol (DAG)-lactone, a potent and specific PKCα activator in LNCaP cells, converted this pattern, enabling radiation to signal CerS activation and apoptosis (Fig. 1) (Truman et al. 2005). The catalytic domain of activated PKCα is required for this event, since LNCaP cells transfected with a kinase-dead dominant-negative mutant of PKCα showed no increase in ceramide generation or in apoptosis levels, indicating that PKCα plays a role in CerS activation. Furthermore, treatment of nude mice with intravenous TPA or DAG-lactone elicited specific PKCα activation leading to downregulation of ataxia telangiectasia-mutated (ATM) protein in prostate tumors in vivo and consequently enhanced radiation-induced tumor response in orthotopically implanted LNCaP cells (Fig. 2). This represents the first description of a signaling-based therapy designed to overcome one form of radiation resistance expressed preferentially in human prostate cancer cells (Truman et al. 2009).

Effect of PKCα stimulation on orthotopic prostate cancer model. (a) DAG-lactone was injected twice i.p. into orthotopically-implanted nude mice at 24 h intervals at a concentration of 12 mg/kg per day. Prostate tumors were removed 48 h after the first injection, homogenized and prepared for Western blot as described in the “Materials and Methods.” The density of the bands was measured and is shown in comparison to the control. The results shown are representative of three separate experiments. (b) Western blots for prostate tumor samples were probed with either phospho-serine specific Abs to PKCα or PKCδ, and compared with total levels of PKCα or PKCδ as a control. The results are representative of two separate experiments. (c) PSA levels in TPA-treated mice. Mice were injected i.p. with 40 μg/kg TPA or DMSO solvent control for 5 days. Radiation-treated mice were locally irradiated with 4 Gy 16 h after each TPA injection for a total of 20 Gy. Control non-irradiated mice n = 20, radiated-treated control mice n = 12, TPA non-irradiated mice n = 17, and TPA-treated irradiated mice n = 10. (d) PSA levels in DAG-lactone treated mice. Mice were injected i.p. with 12 mg/kg per day of DAG-lactone or with DMSO solvent control for 10 days. Radiation-treated mice were locally irradiated at 1 Gy 16 h after each injection to a total of 10 Gy. Control non-irradiated mice n = 14, radiation-treated control mice n = 14, DAG-lactone treated mice n = 13, radiation-treated DAG-lactone mice n = 12. The values represent the mean ± standard errors (Truman et al. 2009)
3.2 ATM Negatively Regulates Ceramide Synthase Activity
By use of (Liao et al. 1999) metabolic incorporation of 125I-labeled 5-iodo-2′deoxyuridine ([125I] dURd), which produces DNA DSBs and external beam irradiation, de novo ceramide synthesis was initiated by posttranslational activation of CerS in BAEC. In the same study, it was shown that ATM negatively regulates CerS activity (Liao et al. 1999). Subsequently, it was shown that TPA decreases ATM protein levels in LNCaP and CWR22Rv1 cells, an event crucial for the induction of apoptosis and mediating radiosensitization of these cells. The alteration in ATM protein levels appeared to be due to inhibition of ATM transcription via decreased binding of transcription factor Sp1 to the ATM promoter. The additional TPA-mediated effector in downregulating ATM transcription remains unknown, nor is there information on the precise mechanism by which TPA affects Sp1 binding. The catalytic domain of activated PKCα is required for this event, since LNCaP cells transfected with a kinase-dead dominant-negative mutant of PKCα showed no decrease in ATM protein. The enabling of CerS-mediated apoptosis in both androgen-sensitive (LNCaP, CWR22-Rv1) and androgen-insensitive (PC-3, DU-145) prostate cancer cells via reduction of ATM protein levels indicates that the interaction between ATM kinase and CerS may represent a generic mode of regulation of radiation-induced death in these cells (Fig. 1). Whereas ATM kinase normally represses CerS activity, a reduction in ATM protein by TPA or antisense ATM oligonucleotide AS-ATM-ODN treatment attenuates CerS repression, enabling ceramide synthesis and a proapoptotic state. However, the experiments with AS-ATM-ODN indicated that ATM reduction by itself is not sufficient to induce CerS activation and that a second signal, such as that provided by radiation, is required. Consistent with this observation, TPA mimicked radiation in inducing apoptosis in AS-ATM-ODN-treated LNCaP cells.
ATM inactivation by AS-ATM-ODN conferred extreme radiation hypersensitivity, since the level of apoptosis attained in AS-ATM-ODN-treated LNCaP exposed to 2 Gy was already at 72 % of the maximum effect observed with 20 Gy (32 ± 0.73 %), both significantly higher than the 3.4 ± 0.2 % observed after 20 Gy exposure of ODN-untreated cells (Truman et al. 2005). The mechanism by which ATM kinase regulates CerS remains unknown. Whereas ATM inhibition has been shown to affect progression through the G2/M checkpoint in some cell types (Shiloh 2001), our studies indicate that deregulation G2/M is not the mechanism of prostate cancer cell radiosensitization. The significant apoptotic response observed using clinically relevant doses of 1 Gy and 2 Gy in AS-ATM-ODN-treated cells suggests ATM as a potential molecular target for clinical application. Further development of AS reagents, siRNA, or small molecules aimed at ATM inactivation would appear warranted in the treatment of prostate cancer.
3.3 PKCα Downregulation of ATM in Prostate Cancer In Vivo
PKCα activation was shown to radiosensitize prostate cancer in vivo (Fig. 2). While fractionated radiation alone generated only a delayed tumor growth response in xenografts of human prostate tumors growing in nude mice, pretreatment with DAG-lactone followed by radiation resulted in permanent local tumor control. The detailed mechanistic pathway involved in this response remains unknown. We are currently trying to determine whether the radiation component of the apoptotic response in LNCaP cells is synergistic with the PKCα component or whether it promotes an autonomous hypersensitized response, from CerS, perhaps via direct activation of radiation-sensitive CerS homologs, whose activation is also enabled by PKCα-mediated ATM downregulation. Therefore, activation of PKCα regulates two main events necessary for the apoptotic response in these human prostate cells (1) ATM downregulation and (2) CerS activation. Since ATM is overexpressed in these cells, radiation is incapable of activating CerS unless a significant amount of ATM protein is downregulated (Fig. 1). In contrast to radiation-induced CerS activation, PKCα-mediated CerS activation does not depend on generation of DNA DSBs in these cells (Galvin and Haimovitz-Friedman, unpublished observations).
3.4 TNF-α and Fas Enhance Radiation-Induced Apoptosis in Prostate Cells
TNF-α was also shown to sensitize LNCaP prostate cancer cells to γ-radiation-induced apoptosis when added 24 h prior to radiation (Kimura et al. 1999). Simultaneous exposure of LNCaP cells to TNF-α and 8 Gy resulted in increased ceramide generation that correlated with a threefold increase in apoptotic cells within 72 h compared to TNF-α treatment alone. LNCaP cells could also be sensitized, although to a lesser degree, by the agonistic FAS antibody, CH-11 (Kimura and Gelmann 2000). In this study, TNF-α increased production of ceramide in LNCaP cells 48 h after exposure. Moreover, nontoxic levels of exogenous C2-ceramide sensitized LNCaP cells to radiation similarly to TNF-α, suggesting that increased intracellular ceramide could explain the mechanism by which LNCaP cells were sensitized to radiation and even to chemotherapy. Further studies proved that TNF-α induced enhanced activation of the intrinsic apoptotic pathway and enhanced cell death, with or without γ-irradiation, yet CerS activity was not reported. Both TNF-α and γ-irradiation elevated levels of endogenous ceramide and activated the intrinsic cell death pathway in a synergistic fashion (Kimura et al. 1999, 2003).
Moreover, it was shown in LNCaP cells that androgen inhibits apoptosis induced by both TNF-α and by CD95 activation with or without concomitant irradiation. This was thought to be mediated by androgen blockade of caspase activation in both intrinsic and extrinsic cell death pathways, but whether SMase is involved in LNCaP cell apoptosis remains to be proven (Kimura et al. 2003). In addition, TNF-α and radiation induced a significant increase in sphingosine levels and markedly reduced sphingosine-1-phosphate (S1P).The increase in sphingosine levels either by exogenous sphingosine or by treatment with the sphingosine kinase (SphK) inhibitor induced apoptosis and also radiosensitized LNCaP cells in this study.
Increasing amount of data now suggest that the relative levels of proapoptotic sphingolipid metabolites, such as certain ceramide species and sphingosine, and the levels of the antiapoptotic sphingolipid metabolites, such as S1P, might play a role in determining the radiosensitivity of prostate cancer cells (Nava et al. 2000). In fact, SphK1 and S1P receptors are highly expressed in chemotherapy-resistant prostate cancer PC3 cells and are upregulated by anticancer drug camptothecin (Akao et al. 2006). Pchejetsky et al. (2008) showed that selective pharmacologic inhibition of SphK1 triggers apoptosis in LNCaP and PC3 cells, an effect reversed by SphK1 enforced expression. Nevertheless, further investigations are necessary to clarify the role of SphK1 and S1P in apoptosis in prostate cells.
3.5 Modulation of Prostate Cancer Cell Response via Acid Ceramidase Activity
Acid ceramidase (AC) converts ceramide into sphingosine and was found to be overexpressed in 60 % of primary prostate cancer tissues (Liu et al. 2009; Norris et al. 2006; Seelan et al. 2000). Conversely, upregulation of AC in prostate cancer cells conferred resistance to both chemo- and radiotherapy (Saad et al. 2007; Mahdy et al. 2009). Along the same line, autophagy was increased in prostate cancer cells overexpressing AC, thereby enhancing resistance to C6-ceramide. This resistance was overcome via modulation of radiation effect by using AC inhibitors (Liu et al. 2009). Interestingly, in another hormone-regulated cancer, of the breast, C6−ceramide and targeted inhibition of AC were also shown to induce synergistic decreases in the cancer cell growth (Flowers et al. 2012).
Resveratrol (3,5,4′-trans-trihydroxystilbene), a natural product from grapes and present in red wine, was shown to be synergistic with radiation in androgen-independent and otherwise radioresistant DU145 human prostate cancer cell line by promoting de novo ceramide biosynthesis in these cells, but no mechanism was suggested (Scarlatti et al. 2007).
4 Summary and Future Directions
In conclusion, both DNA DSB-induced damage and non-DNA DSB-induced damage contribute to cell killing of human prostate cancer cells after exposure to ionizing irradiation. The relative contribution from each mode of cell death may differ with dose and from one cell type to another, relative to their inherent and inducible capacities to overcome each of these types of lethal radiation damage and according to their microenvironment. Utilization of the SMase pathway for induction of apoptosis in response to cell death receptor signals and ionizing radiation has now been demonstrated in a large number of mammalian cells. An alternative mechanism to SMase-mediated generation of ceramide in response to stress is a pathway that involves de novo synthesis of ceramide, via activation of CerS. CerS activation was shown to be of particular importance in a variety of human prostate cancer cell lines (LNCaP, CWR22-Rv1, PC3, and DU145) that under normal conditions exhibit extreme resistance to radiation-induced apoptosis. Yet we found that these cells display high sensitivity to radiation-induced apoptosis both in vitro and in vivo when pretreated with specific PKCα activators. We have also demonstrated that PKCα activation suppresses ATM, derepressing CerS activity, thus enabling generation of apoptogenic ceramide.
Our in vivo data using the prostate orthotopic model suggest that development of radiation response modulators for human prostate cancer affecting the ATM/CerS pathway is essential to overcome radiation resistance in this cancer. Recently, a selective tissue and subcellular distribution of the six mammalian CerS isoforms, combined with distinct fatty acyl chain length substrate preferences, has been described (Table 1) (Mesicek et al. 2010). This implicates differential functions of specific ceramide species in cellular signaling in a stress stimulus-, cell type-, and subcellular compartment-specific manner. Understanding the contribution of the different CerSs homologs and their mode of activation within the different prostate cancer cell types may provide new therapeutic molecular targets to manipulate the radiation response of these cells. These studies may pave the way for future studies looking at DNA-damage-inducing chemotherapeutic drugs and radiation to be used in combination with specific PKCα activators and molecular targets within the sphingolipids metabolic pathways to combat metastatic prostate cancer. Using DAG-lactone as a radiosensitizer via activation of PKCα is currently being considered as a therapeutic approach for prostate cancer at our center.
References
Akao Y, Banno Y, Nakagawa Y, Hasegawa N, Kim TJ, Murate T, Igarashi Y, Nozawa Y (2006) High expression of sphingosine kinase 1 and S1P receptors in chemotherapy-resistant prostate cancer PC3 cells and their camptothecin-induced up-regulation. Biochem Biophys Res Commun 342:1284–1290
Aldridge DR, Arends MJ, Radford IR (1995) Increasing the susceptibility of the rat 208F fibroblast cell line to radiation-induced apoptosis does not alter its clonogenic survival dose–response. Br J Cancer 71:571–577
Algan O, Stobbe CC, Helt AM, Hanks GE, Chapman JD (1996) Radiation inactivation of human prostate cancer cells: the role of apoptosis. Radiat Res 146:267–275
Ameisen JC (2002) On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ 9:367–393
Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R (1995) Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell 82:405–414
Bowen C, Spiegel S, Gelmann EP (1998) Radiation-induced apoptosis mediated by retinoblastoma protein. Cancer Res 58:3275–3281
Brown JM, Wouters BG (1999) Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res 59:1391–1399
Canman CE, Kastan MB (1995) Induction of apoptosis by tumor suppressor genes and oncogenes [Review]. Semin Cancer Biol 6:17–25
Crook JM, Perry GA, Robertson S, Esche BA (1995) Routine prostate biopsies following radiotherapy for prostate cancer: results for 226 patients. Urology 45:624–631
DeWeese TL, Shipman JM, Dillehay LE, Nelson WG (1998) Sensitivity of human prostatic carcinoma cell lines to low dose rate radiation exposure. J Urol 159:591–598
Dubray B, Breton C, Delic J, Klijanienko J, Maciorowski Z, Vielh P, Fourquet A, Dumont J, Magdelenat H, Cosset JM (1998) In vitro radiation-induced apoptosis and early response to low-dose radiotherapy in non-Hodgkin’s lymphomas. Radiother Oncol 46:185–191
Ellis PA, Smith IE, McCarthy K, Detre S, Salter J, Dowsett M (1997) Preoperative chemotherapy induces apoptosis in early breast cancer. Lancet 349:849
Fadeel B, Orrenius S, Zhivotovsky B (1999) Apoptosis in human disease: a new skin for the old ceremony. Biochem Biophys Res Commun 266:699–717
Flowers M, Fabriás G, Delgado A, Casas J, Abad JL, Cabot MC (2012) C6-Ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res Treat 133:447–458
Garcia-Bermejo ML, Leekow FC, Fujii T, Wang Q, Blumberg PM, Ohba M, Kuroki J, Han K-C, Lee J, Marquez VE et al (2002) Diacylglycerol (DAG)-lactones, a new class of protein kinase C (PKC) agonists, induce apoptosis in LNCaP prostate cancer cells by selective activation of PKCα. J Biol Chem 277:645–655
Garzotto M, White-Jones M, Jiang Y, Ehleiter D, Liao WC, Haimovitz-Friedman A, Fuks Z, Kolesnick R (1998) 12-O-tetradecanoylphorbol-13-acetate-induced apoptosis in LNCaP cells is mediated through ceramide synthase. Cancer Res 58:2260–2264
Garzotto M, Haimovitz-Friedman A, Liao WC, White-Jones M, Huryk R, Heston DWW, Cardon-Cardo C, Kolesnick R, Fuks Z (1999) Reversal of radiation resistance in LNCaP cells by targeting apoptosis through ceramide synthase. Cancer Res 59:5194–5201
Haimovitz-Friedman A, Kolesnick RN, Fuks Z (1997) Ceramide signaling in apoptosis. Br Med Bull 53:539–553
Hall EJ, Giaccia AJ (2006a) DNA strand breaks and chromosomal aberrations. In: McAllister L, Bierig L, Barrett K (eds) Radiobiology for the radiologist. Lippincott, Williams & Wilkins, Philadelphia, PA, pp 16–29
Hall EJ, Giaccia AJ (2006b) Repair of radiation damage and the dose-rate effect. In: McAllister L, Bierig L, Barrett K (eds) Radiobiology for the radiologist. Lippincott, Williams & Wilkins, Philadelphia, PA, pp 85–105
Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA, Murphy GP (1983) LNCaP model of human prostatic carcinoma. Cancer Res 43:1809–1818
Jansen B, Wacheck V, Heere-Ress E, Schlagbauer-Wadl H, Hoeller C, Lucas T, Hoermann M, Hollenstein U, Wolff K, Pehamberger H (2000) Chemosensitization of malignant melanoma by BCL2 antisense therapy. Lancet 356:1728–1733
Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics. CA Cancer J Clin 60:277–300
Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW (1979) Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest Urol 17:16–23
Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ Jr (1992) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71:587–597
Kimura K, Gelmann EP (2000) Tumor necrosis factor-alpha and Fas activate complementary Fas-associated death domain-dependent pathways that enhance apoptosis induced by gamma-irradiation. J Biol Chem 275:8610–8617
Kimura K, Bowen C, Spiegel S, Gelmann EP (1999) Tumor necrosis factor-alpha sensitizes prostate cancer cells to gamma-irradiation-induced apoptosis. Cancer Res 59:1606–1614
Kimura K, Markowski M, Edsall LC, Spiegel S, Gelmann EP (2003) Role of ceramide in mediating apoptosis of irradiated LNCaP prostate cancer cells. Cell Death Differ 10:240–248
Kyprianou N, King ED, Bradbury D, Rhee JG (1997) Bcl-2 over-expression delays radiation-induced apoptosis without affecting the clonogenic survival of human prostate cancer cells. Int J Cancer 70:341–348
Lahiri S, Lee H, Mesicek J, Fuks Z, Haimovitz-Friedman A, Kolesnick RN, Futerman AH (2007) Kinetic characterization of mammalian ceramide synthases: determination of K(m) values toward sphinganine. FEBS Lett 581:5289–5294
Laviad EL, Albee L, Pankova-Kholmyansky I, Epstein S, Park H, Merrill AH Jr, Futerman AH (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem 283:5677–5684
Leith JT (1994) In vitro radiation sensitivity of the LNCaP prostatic tumor cell line. Prostate 24:119–124
Leith JT, Quaranto L, Padfield G, Michelson S, Hercbergs A (1993) Radiobiological studies of PC-3 and DU-145 human prostate cancer cells: X-ray sensitivity in vitro and hypoxic fractions of xenografted tumors in vivo. Int J Radiat Oncol Biol Phys 25:283–287
Li WX, Franklin WA (1998) Radiation- and heat-induced apoptosis in PC-3 prostate cancer cells. Radiat Res 150:190–194
Liao W-C, Haimovitz-Friedman A, Persaud R, McLoughlin M, Ehleiter D, Zhang N, Gatei M, Lavin M, Kolesnick R, Fuks Z (1999) Ataxia Telangiectasia-mutated gene product inhibits DNA damage-induced apoptosis via ceramide synthase. J Biol Chem 274:17908–17917
Lin T, Genestier L, Pinkoski MJ, Castro A, Nicholas S, Mogil R, Paris F, Fuks Z, Schuchman EH, Kolesnick RN et al (2000) Role of acidic sphingomyelinase in Fas/CD95-mediated cell death. J Biol Chem 275:8657–8663
Liu X, Cheng JC, Turner LS, Elojeimy S, Beckham TH, Bielawska A, Keane TE, Hannun YA, Norris JS (2009) Acid ceramidase upregulation in prostate cancer: role in tumor development and implications for therapy. Expert Opin Ther Targets 13:1449–1458
Lock RB, Ross WE (1990) Inhibition of p34cdc2 kinase activity by etoposide or irradiation as a mechanism of G2 arrest in Chinese hamster ovary cells. Cancer Res 50:3761–3766
Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T (1993) p53 is required for radiation-induced apoptosis in mouse thymocytes [see comments]. Nature 362:847–849
Mahdy AE, Cheng JC, Li J, Elojeimy S, Meacham WD, Turner LS, Bai A, Gault CR, McPherson AS, Garcia N et al (2009) Acid ceramidase upregulation in prostate cancer cells confers resistance to radiation: AC inhibition, a potential radiosensitizer. Mol Ther 17:430–438
Mathias S, Pena LA, Kolesnick RN (1998) Signal transduction of stress via ceramide. Biochem J 335:465–480
Mesicek J, Lee H, Feldman T, Jiang X, Skobeleva A, Berdyshev EV, Haimovitz-Friedman A, Fuks Z, Kolesnick R (2010) Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell Signal 22:1300–1307
Meyn RE, Stephens LC, Hunter NR, Milas L (1995) Apoptosis in murine tumors treated with chemotherapy agents. Anticancer Drugs 6:443–450
Meyn RE, Milas L, Ang KK (2009) The role of apoptosis in radiation oncology. Int J Radiat Biol 85:107–115
Mizutani Y, Kihara A, Igarashi Y (2006) LASS3 (longevity assurance homologue 3) is a mainly testis-specific (dihydro)ceramide synthase with relatively broad substrate specificity. Biochem J 398:531–538
Mullauer L, Gruber P, Sebinger D, Buch J, Wohlfart S, Chott A (2001) Mutations in apoptosis genes: a pathogenetic factor for human disease. Mutat Res 488:211–231
Nava VE, Cuvillier O, Edsall LC, Kimura K, Milstien S, Gelmann EP, Spiegel S (2000) Sphingosine enhances apoptosis of radiation-resistant prostate cancer cells. Cancer Res 60:4468–4474
Norris JS, Bielawska A, Day T, El-Zawahri A, ElOjeimy S, Hannun Y, Holman D, Hyer M, Landon C, Lowe S et al (2006) Combined therapeutic use of AdGFPFasL and small molecule inhibitors of ceramide metabolism in prostate and head and neck cancers: a status report. Cancer Gene Ther 13:1045–1051
Olive PL, Durand RE (1997) Apoptosis: an indicator of radiosensitivity in vitro? Int J Radiat Biol 71:695–707
Olson M, Kornbluth S (2001) Mitochondria in apoptosis and human disease. Curr Mol Med 1:91–122
Orrenius S (1995) Apoptosis: molecular mechanisms and implications for human disease. J Intern Med 237:529–536
Pchejetski D, Doumerc N, Golzio M, Naymark M, Teissié J, Kohama T, Waxman J, Malavaud B, Cuvillier O (2008) Chemosensitizing effects of sphingosine kinase-1 inhibition in prostate cancer cell and animal models. Mol Cancer Ther 7:1836–1845
Pena LA, Fuks Z, Kolesnick R (1997) Stress-induced apoptosis and the sphingomyelin pathway. Biochem Pharmacol 53:615–621
Reed JC (2002) Apoptosis-based therapies. Nat Rev Drug Discov 1:111–121
Royai R, Lange PH, Vessella R (1996) Preclinical models of prostate cancer. Semin Oncol 23:35–40
Saad AF, Meacham WD, Bai A, Anelli V, Elojeimy S, Mahdy AE, Turner LS, Cheng J, Bielawska A, Bielawski J et al (2007) The functional effects of acid ceramidase overexpression in prostate cancer progression and resistance to chemotherapy. Cancer Biol Ther 6:1455–1460
Scardino PT, Wheeler TM (1988) Local control of prostate cancer with radiotherapy: frequency and prognostic significance of positive results of postirradiation prostate biopsy. NCI Monogr 7:95–103
Scarlatti F, Sala G, Ricci C, Maioli C, Milani F, Minella M, Botturi M, Ghidoni R (2007) Resveratrol sensitization of DU145 prostate cancer cells to ionizing radiation is associated to ceramide increase. Cancer Lett 253:124–130
Schenck M, Carpinteiro A, Grassmé H, Lang F, Gulbins E (2007a) Ceramide: physiological and pathophysiological aspects. Arch Biochem Biophys 462:171–175
Seelan RS, Qian C, Yokomizo A, Bostwick DG, Smith DI, Liu W (2000) Human acid ceramidase is overexpressed but not mutated in prostate cancer. Genes Chromosomes Cancer 29:137–146
Shiloh Y (2001) ATM (ataxia telangiectasia mutated): expanding roles in the DNA damage response and cellular homeostasis. Biochem Soc Trans 29:661–666
Spiegel S, Foster D, Kolesnick R (1996) Signal transduction through lipid second messengers. Curr Opin Cell Biol 8:159–167
Sridevi P, Alexander H, Laviad EL, Pewzner-Jung Y, Hannink M, Futerman AH, Alexander S (2009) Ceramide synthase 1 is regulated by proteasomal mediated turnover. Biochim Biophys Acta 1793:1218–1227
Steel GG (2001) The case against apoptosis. Acta Oncol 40:968–975
Stone KR, Mickey DD, Wunderli H, Mickey GH, Paulson DF (1978) Isolation of a human prostate carcinoma cell line (DU 145). Int J Cancer 21:274–281
Symmans WF, Volm MD, Shapiro RL, Perkins AB, Kim AY, Demaria S, Yee HT, McMullen H, Oratz R, Klein P et al (2000) Paclitaxel-induced apoptosis and mitotic arrest assessed by serial fine-needle aspiration: implications for early prediction of breast cancer response to neoadjuvant treatment. Clin Cancer Res 6:4610–4617
Truman JP, Gueven N, Lavin M, Leibel S, Kolesnick R, Fuks Z, Haimovitz-Friedman A (2005) Down-regulation of ATM protein sensitizes human prostate cancer cells to radiation-induced apoptosis. J Biol Chem 280:23262–23272
Truman JP, Rotenberg SA, Kang JH, Lerman G, Fuks Z, Kolesnick R, Marquez VE, Haimovitz-Friedman A (2009) PKCalpha activation downregulates ATM and radio-sensitizes androgen-sensitive human prostate cancer cells in vitro and in vivo. Cancer Biol Ther 8:54–63
Wollin M, FitzGerald TJ, Santucci MA, Menon M, Longcope C, Reale F, Carlson J, Sakakeeny MA, Greenberger JS (1989) Radiosensitivity of human prostate cancer and malignant melanoma cell lines. Radiother Oncol 15:285–293
Wu J (1996) Apoptosis and angiogenesis: two promising tumor markers in breast cancer. Anticancer Res 16:2233–2239
Zelefsky MJ, Leibel SA, Gaudin PB, Kutcher GJ, Fleshner NE, Venkatramen ES, Reuter VE, Fair WR, Ling CC, Fuks Z (1998) Dose escalation with three-dimensional conformal radiation therapy affects the outcome in prostate cancer. Int J Radiat Oncol Biol Phys 41:491–500
Zelefsky MJ, Reuter VE, Fuks Z, Scardino P, Shippy A (2008) Influence of local tumor control on distant metastases and cancer related mortality after external beam radiotherapy for prostate cancer. J Urol 179:1368–1373 (discussion 1373)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this chapter
Cite this chapter
Hajj, C., Haimovitz-Friedman, A. (2013). Sphingolipids’ Role in Radiotherapy for Prostate Cancer. In: Gulbins, E., Petrache, I. (eds) Sphingolipids in Disease. Handbook of Experimental Pharmacology, vol 216. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1511-4_6
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1511-4_6
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1510-7
Online ISBN: 978-3-7091-1511-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)