
Abstract
The relationship of sphingolipids with human disease first arose from the study of sphingolipid storage diseases over 50 years ago. Most of these disorders are due to inherited deficiencies of specific sphingolipid hydrolases, although a small number also result from defects in sphingolipid transport or activator proteins. Due to the primary protein deficiencies sphingolipids and other macromolecules accumulate in cells and tissues of affected patients, leading to a diverse presentation of clinical abnormalities. Over 25 sphingolipid storage diseases have been described to date. Most of the genes have been isolated, disease-causing mutations have been identified, the recombinant proteins have been produced and characterized, and animal models exist for most of the human diseases. Since most sphingolipid hydrolases are enriched within the endosomal/lysosomal system, macromolecules first accumulate within these compartments. However, these abnormalities rapidly spread to other compartments and cause a wide range of cellular dysfunction. This review focuses on the genetics of sphingolipid storage diseases and related hydrolytic enzymes with an emphasis on the relationship between genetic mutations and human disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction and Overview
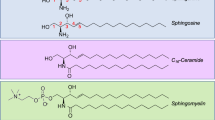
Sphingolipids are a diverse group of over 100 bioactive lipids involved in many aspects of cellular function (see Chen et al. 2012; Furuya et al. 2011; Petrache et al. 2011; Podbielska et al. 2012 for recent reviews). They all share a common sphingosine (2-amino-4-octadecene-1,3-diol) backbone, which may be linked via an amide bond to a fatty acid (forming ceramide), or phosphorlyated to form sphingosine-1-phosphate. Ceramide itself also may be modified to form glycosphingolipids, sphingomyelin, or ceramide-1-phosphate.
The metabolism of sphingolipids is complex and carefully regulated (e.g., Mullen et al. 2012; Wennekes et al. 2009; Worgall 2011). Abnormal metabolism can have profound effects on cellular function, leading to enhanced cell death, proliferation, and/or abnormal cellular differentiation. In general, the synthesis and breakdown of sphingolipids occur in distinct cellular compartments. For example, numerous enzymes contribute to the de novo synthesis of sphingolipids, mostly found in the endoplasmic reticulum and Golgi apparatus. In contrast, the breakdown of sphingolipids occurs primarily within endosomes and lysosomes by a series of hydrolytic enzymes that function optimally at acidic pH. Hydrolysis of sphingolipids also may occur in non-endosomal/lysosomal compartments, for example by enzymes located at the plasma membrane or within mitochondria. Overall, over 100 enzymes participate in the synthesis or breakdown of sphingolipids.
Historically, most information concerning the importance of sphingolipids in human disease stems from studies of the sphingolipid storage diseases (Schulze and Sandhoff 2011). These disorders are each due to deficient function of one or more of the hydrolytic enzymes found within the endosomal/lysosomal system, resulting in sphingolipid accumulation (Cox and Cachón-González 2012). Each of these diseases is inherited as a Mendelian trait (recessive or X-linked), the cDNAs/genes encoding the respective enzymes have been isolated, and many mutations causing the human diseases have been identified. In addition, genetic abnormalities in several of these enzymes/genes have now been linked to more common diseases (e.g., Parkinson’s disease and beta-glucocerebrosidase) (Young et al. 2012). Table 1 lists the sphingolipid storage diseases and the respective enzymes involved.
This review focuses on the genetics of sphingolipid storage diseases and related hydrolytic enzymes. Each of the diseases/enzymes is briefly reviewed, with an emphasis on the importance of genetic mutations in disease.
2 Acid Ceramidase Deficiency: Farber Disease
Patients with Farber disease (also known as Farber lipogranulomatosis) usually present within the first few months of life with a triad of clinical manifestations, including painful and progressively deformed joints, subcutaneous nodules, particularly near joints, and progressive hoarseness due to laryngeal involvement (Farber et al. 1957). About half of the patients have neurological deficits (Ahmad et al. 2009). As with all of the sphingolipid storage disorders, there is considerable clinical heterogeneity among Farber disease patients, and later onset, attenuated cases also have been described. Farber disease is the least common of the sphingolipid storage diseases, likely because severe mutations in the acid ceramidase (AC) gene (ASAH1) lead to embryonic lethality (see below).
Acid ceramidase (E.C. 3.5.1.23) was first identified and purified in the 1960s. It was recognized as the enzyme deficient in Farber disease in the 1970s (Sugita et al. 1972). The full-length cDNAs and genes encoding AC have been isolated from humans, mice, and other species. The human cDNA encodes a 395 amino acid precursor polypeptide, and the gene is located on chromosomal region 8p22. Of note, the newly synthesized AC precursor is inactive, and only assumes an active form after N-glycosylation and the internal cleavage of a peptide bond, resulting in alpha and beta subunits. Interestingly, this cleavage event is carried out by AC itself, and it is therefore considered a “self-regulating” enzyme. In addition, AC associates with other lipid hydrolases within lysosomes and other cell compartments, including acid sphingomyelinase (see below). The importance of this multienzyme complex on AC activity, as well as on sphingolipid metabolism in general, is not clearly understood. Finally, full AC activity, at least in lysosomes, depends on the expression of a sphingolipid activator protein (SAP-D), which is expressed by a distinct gene (see Sect. 10, below).
Acid ceramidase is an “amidase” that hydrolyzes the amide bond between sphingosine and the fatty acid in ceramide. Ceramides are a heterogenous group of lipids defined by the length of their fatty acid chains. While the substrate specificity of AC is not entirely clear, ceramides with fatty acid chains shorter than 12 carbons cannot be efficiently hydrolyzed by the enzyme in vitro. In vivo, it is likely that AC can recognize and cleave ceramides of varying chain lengths. The ceramides that accumulate in Farber disease patients are primarily found within lysosomes, leading to the original designation of AC as a lysosomal enzyme. AC also has maximal activity at lysosomal (acidic) pH, although it has been localized to sites other than the lysosomes (e.g., the spindle in oocytes) as well. It is therefore not entirely clear to what extent the biological activity of AC depends on the acidic environment, and in fact a “reverse” ceramidase reaction has been attributed to the enzyme at more neutral pH (Okino et al. 2003). Numerous in vitro assays have been developed for AC, and some can be used to diagnose Farber disease in cultured cells from suspected patients, or prenatally using aminiocytes or cultured chorionic villi. However, due to the very hydrophobic nature of ceramides, it remains one of the more difficult diagnostic enzyme assays to carry out.
To date, only about 100 Farber disease patients have been reported in the literature (Park and Schuchman 2006). Several cases of hydrops fetalis also have been reported. Complete inactivation of the AC gene in mice leads to very early embryonic lethality (four-cell stage), suggesting that severe mutations in the human ASAH1 gene will lead to embryonic lethality as well (Li et al. 2002). Of note, mouse embryos lacking AC can be rescued by the addition of recombinant AC (obtained from overexpressing Chinese hamster ovary [CHO] cells) to the culture media. Moreover, addition of recombinant AC to the culture media during in vitro fertilization (mouse, bovine) enhances embryo production and quality, resulting in more live births (Eliyahu et al. 2010).
Thus, AC is an essential enzyme required for very early mammalian development. One explanation for the lethal phenotype in mice is that in the absence of this enzymatic activity, early-stage embryos (four cells in mice) undergo apoptosis due to ceramide accumulation. It should be recognized, however, that an additional consequence of such AC “loss of function” during embryogenesis is reduced production of sphingosine, which in turn is converted to S1P, an important mitogenic/proliferative lipid. Thus, AC mutations also may reduce S1P production, contributing to the lethality. Indeed, embryos lacking AC can be partially rescued by S1P as well.
As noted above, the first relationship between AC and human disease arose from studies on Farber disease. To date over 20 mutations have been described in the ASAH1 gene leading to Farber disease (Park and Schuchman 2006). Most are point mutations, although several splice variants leading to small deletions have been described as well. To date, no patients have been found homozygous for a complete loss-of-function mutation, likely because such individuals do not survive embryonic development. Farber disease is inherited as an autosomal recessive disorder, and to develop the disorder mutations must be inherited on both ASAH1 alleles. No reports of clinical findings in the parents of affected patients (obligate carriers) have been published; however such individuals are not usually followed by clinicians and follow-up analysis is lacking.
Recently, a “knock-in” mouse model of Farber disease has been reported where a specific human mutation was expressed (add ref). Preliminary analysis of these animals indicates that they develop a Farber disease-like phenotype. In addition, a conditional AC knockout mouse has been developed where AC can be inactivated at various stages of development or in specific tissues.
Other than Farber disease, which is due to the deficiency of AC, constitutive overexpression of the AC gene also occurs in many types of cancer (Park et al. 2005; Beckham et al. 2012). This may promote tumorigenesis and/or metastasis due to overproduction of S1P, or may lead to chemoresistance due to an enhanced ability to hydrolyze pro-apoptotic ceramide (Flowers et al. 2012; Furuya et al. 2011). The molecular basis of AC overexpression in cancer has not been studied extensively, but at least in some cases it appears to be due to elevated transcription. Gain-of-function mutations also could explain this finding, although to date these have not been described. In addition, a recent report showed that the ASAH1 gene was down-regulated in a Chinese population of schizophrenic patients, and was associated with two single nucleotide polymorphisms in this gene (Zhang et al. 2012).
Finally, it must be noted that at least six other ceramidase activities have been described in man, and at least four other genes have been cloned (Mao and Obeid 2008). These ceramidases are defined by their unique pH, subcellular location, and other biological properties. These enzymes also play important roles in sphingolipid metabolism and/or signaling, and are likely to contribute to human disease pathogenesis. The biology of the other ceramidases has been reviewed extensively elsewhere (e.g., Mao and Obeid 2008).
3 Acid Sphingomyelinase Deficiency: Types A and B Niemann–Pick Disease
Types A and B Niemann–Pick Disease (NPD) result from mutations in the gene (SMPD1) encoding acid sphingomyelinase (E.C. 3.1.4.12) (McGovern and Schuchman 2009; Schuchman 2009). Patients with Type A NPD present in early infancy with hepatosplenomegaly and failure to thrive, and develop a rapidly degenerative neurological phenotype that leads to death by 3 years of age. In contrast, patients with Type B NPD have a later onset disease, and usually lack neurological findings. Such individuals also frequently present with hepatosplenomegaly, and may have pulmonary involvement as well. Dyslipidemia (high LDL-C, low HDL-C, high triglycerides) also is common. Patients with Type B NPD may survive into adulthood, although their life span is generally shortened by the disease. The cause of death is unknown, but may occur from complications related to liver disease, trauma-induced bleeding episodes due to thrombocytopenia, early-onset cardiovascular disease, or pulmonary disease.
Acid sphingomyelinase (ASM) was found to be deficient in Type A and B NPD patients in the late 1960s. Although the enzyme had been discovered several years earlier in rat tissues, it was not substantially purified until the late 1980s. The cDNAs and genes encoding human and mouse ASM were cloned in the early 1990s. The human gene is located on chromosome 11p15.1. Of interest, among genes encoding lysosomal enzymes, it is the only one for which genomic imprinting has been described. This form of genetic regulation is generally reserved for genes with important developmental roles.
Over 100 mutations have been found in the SMPD1 gene leading to Types A or B NPD (http://www.hgmd.org). Of note, several of these mutations are common within specific regions or ethnic populations, allowing genetic screening programs to be established. Homozygosity for these mutations is required to develop NPD, and it is inherited as an autosomal recessive trait. However, heterozygous carriers of SMPD1 mutations may develop dyslipidemia and late-onset features of the disorder. In part, the development of a phenotype in the carrier individuals may be related to their imprinting status (i.e., whether the mutations are inherited from the mother or the father) (Simonaro et al. 2006). Unlike acid ceramidase and several other sphingolipid hydrolases, ASM does not require an exogenous activator protein to achieve full function. Thus, mutations in the sphingolipid activator protein gene (see below) do not affect ASM activity or cause Types A and B NPD. Of interest, the lysosomal lipid, bis(monacyl) phosphate, may activate the enzyme in vitro and in vivo.
In addition to ASM, several other sphingomyelinases (nonacidic) exist in mammalian cells (Clarke et al. 2011). These are defined by their unique subcellular location or other biochemical properties. At least four human genes have been isolated encoding these sphingomyelinases. Definitive proof that mutations in the SMPD1 gene were solely responsible for Types A and B NPD came from studies in the knockout mouse model (ASMKO), where neutral and other sphingomyelinase activities were normal, despite the fact that these animals completely lacked ASM and developed NPD-like pathology and clinical features (Horinouchi et al. 1995). In addition, these early studies in the ASMKO mice showed that the previously identified zinc-activated, serum (secreted) form of ASM was encoded by the same SMPD1 gene as lysosomal ASM (Schissel et al. 1998). It is now known that ASM is, in fact, a zinc-requiring enzyme. However, the lysosomal form of the enzyme has high levels of zinc bound, and thus does not require exogenous zinc when measured in vitro. In contrast, the serum form of the enzyme requires exogenous zinc in the assay systems routinely used.
Another early finding in the ASMKO mouse was that ceramide, the product of sphingomyelin hydrolysis by ASM, also was elevated in tissues of these animals. While initially a surprising result, it is likely that the elevated ceramide derives from breakdown of the accumulating sphingomyelin in NPD tissues by other sphingomyelinases. Sphingomyelin levels may be up to 30-fold above normal in the ASMKO mice, and ceramide two- to fivefold above normal. Most recently, sphingosine levels also were found to be high in tissues from ASMKO mice, probably due to breakdown of the accumulating ceramide by ceramidases (Schuchman et al., unpublished observations). Of interest, the degree of sphingosine accumulation in these animals (>20-fold above normal) cannot be explained by ceramide alone, perhaps suggesting some defect in converting the sphingosine to S1P.
The ASMKO mice develop a phenotype intermediate between Types A and B NPD. They die prematurely at ~6–8 months, secondary to neurological disease. They have been extensively used to study the pathobiology of this disease, and to develop/evaluate new therapies. In particular, enzyme replacement therapy (ERT) using recombinant ASM produced in CHO cells has been developed in the mouse model (Miranda et al. 2000), and a phase 1 clinical trial in adult patients with Type B NPD was recently completed (http://clinicaltrials.gov). A phase 2 trial is planned for 2012. Of interest, in the ASMKO mice dose-related toxicity was observed (which was not seen in healthy animals) using recombinant ASM, presumably due to rapid breakdown of the accumulating sphingomyelin to ceramide. This hypothesis was confirmed by pretreating animals with low dose of enzyme (“debulking” the accumulating sphingomyelin), which prevented any toxicity. In the patients treated by ERT, transient changes in several inflammatory biomarkers were observed, corresponding to dose and serum levels of ceramide.
Beginning in the late 1990s, the ASMKO mice also were extensively used to investigate the role of ASM in cell signaling. This is the subject of several reviews, and is not focused on in this chapter. However, it is now clear that ASM plays an important role in stress-induced ceramide generation and apoptosis, and that this occurs through hydrolysis of sphingomyelin at the cell surface within lipid rafts (Zeidan and Hannun 2010). How ASM, which has an acidic pH optimum in vitro and functions within lysosomes, participates in these processes has been the subject of much debate. It is now known that after the induction of stress or developmental signals, ASM may be rapidly translocated to lipid rafts at the cell surface. The mechanism underlying this rapid translocation is not entirely clear, but phosphorylation within the ER/Golgi may be responsible in part (Zeidan et al. 2008). It is also known that although ASM requires an acidic pH for optimal activity in vitro, in vivo it is capable of efficiently hydrolyzing sphingomyelin at physiological pH. This highlights the complex biology of this and other sphingolipid hydrolases, particularly in terms of pH, subcellular location, and other properties.
These observations on the ASMKO mice have also highlighted the important role of ASM in human disease, beyond NPD (e.g., Becker et al. 2010; Bikman and Summers 2011; Truman et al. 2011). Elevated ASM activity has now been detected in numerous common diseases, including diabetes, Alzheimer disease, cardiovascular diseases, pulmonary diseases, depression, and others. These will also be reviewed elsewhere and not discussed further here. At the present time it is not known if the elevated ASM found in these diseases is due to an activating genetic mutation, enhanced transcription, and/or posttranslational modification of the enzyme.
4 Beta-Glucocerebrosidase Deficiency: Gaucher Disease
Gaucher disease, due to the deficient activity of acid beta-glucocerebrosidase (also known as beta-glucosidase [E.C. 3.2.1.45]), is the most common sphingolipid storage disorder (Campbell and Choy 2012; Lee et al. 2012). There are three clinical subtypes of Gaucher disease, including an early-onset, neurological form (Type 2), an intermediate form (Type 3), and a late-onset, non-neurological form (Type 1) (Lo et al. 2012; Pastores 2010). All Gaucher patients present with hepatosplenomegaly, but the progression of the disease and survival are markedly different. For example, Type 3 children may only survive until 2–3 years of age, while Type 1 patients may survive into adulthood. If left untreated (see below), the hematological and bone lesions in Type 1 patients progress with age, seriously limiting quality of life. A massively enlarged spleen and frequent bone fractures may become common, requiring splenectomy, joint replacement, or other surgical interventions (Mikosch and Hughes 2010; Goker-Alpan 2011). Bleeding also is a common presenting feature of Gaucher disease due to thrombocytopenia. Pulmonary involvement may similarly occur. Of note, neoplastic disorders, particularly of the blood-forming organs, are more common in Gaucher disease patients than in the general population. These include chronic lymphocytic leukemia, multiple myeloma, and Hodgkin and non-Hodgkin lymphoma. The molecular basis of this increased cancer risk remains unknown (Choy and Campbell 2011).
All Gaucher patients are characterized by the presence of “Gaucher” cells in various organs, lipid-filled cells derived from the monocyte/macrophage system. Gaucher cells have a distinctive appearance by light microscopy, and can be readily distinguished from cells of other sphingolipid storage disorders. Gaucher cells are distributed throughout the body, wherever macrophages reside. The largest numbers are found in the spleen, sinusoids of the liver, bone marrow, and lymph nodes.
The major accumulating lipid in Gaucher disease is N-acyl-sphingosyl-1-0-β-d-glucoside (glucosylceramide, also known as glucosylcerebroside, GC). GC is widely distributed in mammalian tissues in small quantities as a metabolic intermediate in the synthesis and degradation of complex glycosphingolipids, such as gangliosides or globoside. The deacylated form of GC, glucosylsphingosine, is not normally found in tissues, but also accumulates in Gaucher disease cells. GC levels in plasma may be 2- to 20-fold above normal.
The major site of GC turnover in cells is the lysosome. Beta-glucocerebrosidase is normally found in this organelle and functions optimally at an acidic pH. As with the other lipid hydrolases, in vitro detection of this enzymatic activity can be difficult, and there have been various factors (lipids, protein, sugars) shown to influence the assay systems. In vivo, sphingolipid activator-derived peptides, particularly saposin (SAP) C, appear to be required for full enzymatic activity (see below). Nonacidic glucocerebrosidases also have been described and are encoded by distinct genes. Whether these activities can be modifying factors for the Gaucher phenotype has been the subject of much discussion, but remains unclear.
The gene encoding beta-glucocerebrosidase (GBA1) has been mapped to chromosome 1q21. It is ~7 kb and contains 11 exons. Of note, a 5 kb “pseudogene” is located immediately downstream from the GBA1 gene. This pseudogene has maintained a high degree of homology with the functional gene and is transcribed. However, the nucleotide changes within the pseudogene sequence prevent the occurrence of a long open reading frame, precluding the possibility of expressing an active beta-glucocerebrosidase (Brown et al. 2006). Full-length cDNAs from many species also have been obtained that can be used to express the active enzyme.
Numerous mutations have been found in the GBA1 gene causing Gaucher disease (Hruska et al. 2008). As described for Niemann–Pick disease above, several common mutations are found in specific populations, and DNA-based screening is now routinely carried out in the Ashkenazi Jewish population (Scott et al. 2010). Estimates of the Gaucher disease carrier frequency in this population range from ~1:10 to 1:25. Some of the mutations also can be used to predict the occurrence of severe or mild Gaucher phenotypes, assisting in family planning and genetic counseling.
Recently, mutations in the GBA1 gene also have been associated with the occurrence of Parkinson’s disease, although the precise relationship between the GBA1 gene, glucocerebrosidase activity, and Parkinson’s disease remains uncertain (Kinghorn 2011). One of the first clues suggesting this linkage came from the small number of Gaucher patients who exhibited Parkinson-like features, while stronger associations later came from Gaucher relatives who were carriers for GBA1 mutations. Currently, heterozygous GBA1 mutations are considered to be one of the most common genetic risk factor for Parkinson’s disease (Anheim et al. 2012).
A complete knockout mouse model for Gaucher was constructed and had an early neonatal lethal phenotype, dying within 34 days after birth. These animals had massive GC storage, and while the cause of death was not entirely clear, skin abnormalities were prevalent. Several knock-in models also have been constructed, and some exhibited longer survival and later onset GC storage in macrophages, more closely resembling Gaucher disease patients (Farfel-Becker et al. 2011; Sardi et al. 2012).
Gaucher disease was the first sphingolipid storage disease for which ERT was developed in the early 1990s, first using beta glucocerebrosidase purified from placentae (Ceredase), and subsequently using recombinant enzyme produced in CHO cells (Cerezyme) (Hughes and Pastores 2010; Lachmann 2011). For non-neurological patients, ERT was life changing, resulting in reduction in spleen size and correction of the hematological abnormalities. Bone disease has been much less responsive to ERT, and remains a challenge in the treatment of Gaucher disease, as does treatment of the brain disease (Hughes and Pastores 2010). Another treatment strategy currently available for Gaucher patients uses a small molecule inhibitor of GC synthesis (Miglustat) to slow the accumulation of this lipid (Abian et al. 2011; Benito et al. 2011). The target of this inhibitor is the enzyme glucosylceramide synthase.
Of note, treatment of Gaucher patients either by ERT or with Miglustat should elevate ceramide, and could potentially result in ceramide-mediated toxicity. To date, however, such toxicity has not been observed. There are several factors that might explain this observation. First, compared to sphingomyelin, GC is generally found in small quantities in cells, and is not a major structural component of cell membranes. Thus, accumulation of GC in Gaucher cells is more exclusively found in the lysosomes than sphingomyelin in Niemann–Pick cells, where substantial amounts of the accumulating lipid also are found in the plasma membrane. Thus, ceramide-mediated raft reorganization and signaling are less likely to become activated in Gaucher disease compared to NPD. In addition, ceramide produced by ASM following ERT in NPD is likely to remain within the cell membrane, while the ceramide produced following ERT with Cerezyme or by inhibiting GC synthesis with Miglustat may be found in different cellular compartments, and therefore more readily accessible for biosynthetic pathways. Lastly, the ERT dose used to treat Gaucher patients (1.6 mg/kg, every 2 weeks) may be below the threshold needed to induce ceramide-mediated toxicity.
5 Galactocerebrosidase Deficiency: Krabbe Disease/Globoid Cell Leukodystrophy
Infantile globoid cell leukodystrophy (GLD), also known as Krabbe disease, is a rapidly progressive, fatal neurological disease of infants (Kohlschütter and Eichler 2011). The disease usually begins between 3 and 6 months of life with symptoms that may include irritability or hypersensitivity to stimuli. Patients rapidly exhibit severe mental and motor deterioration, and rarely survive beyond the second year (Duffner et al. 2011). Clinical manifestations in classical metachromatic leukodystrophy (MLD) patients are limited to the nervous system. Blindness and deafness are common, and peripheral neuropathy is almost always detectable. Patients with later onset forms of the disease, including adult-onset forms, may present with blindness, spastic paraparesis, and dementia (Malandrini et al. 2013; Perlman and Mars 2012).
The presence of numerous multinucleated globoid cells, almost total loss of myelin and oligodendrocytes, and astrocytic gliosis in the white matter are the morphologic basis of the disease. “Globoid cells” in Krabbe disease are enlarged macrophages that contain undigested galacotsylceramide and other lipids. Demyelination, axonal degeneration, fibrosis, and histiocytic infiltration are also common in the peripheral nervous system. Consistent with the myelin loss, the white matter is severely depleted of all lipids, particularly glycolipids. Under normal circumstances, galactosylceramide is present almost exclusively in the myelin sheath. The ratio of galactosylceramide to sulfatide (3-0 sulfogalactosylceramide) is abnormally high in MLD.
The genetic cause of Krabbe disease is mutations in the gene encoding galactocerebrosidase beta-galactosidase (GALC; E.C. 3.2.1.46) (see below). This lysosomal enzyme normally degrades galactosylceramide to ceramide and galactose. It is postulated that accumulation of a toxic metabolite, psychosine (galactosylsphingosine), which is also a substrate for the missing enzyme, leads to early destruction of the oligodendrocytes (see below). The total brain content of galactosylceramide is not increased in patients with Krabbe disease, supporting this hypothesis. It should be noted that mammalian tissues contain two genetically distinct lysosomal beta-galactosidases, GALC and GM1-ganglioside beta-galactosidase (see Sect. 8, below). These two enzymes have overlapping substrate specificities in vitro, which can present complications when attempting to make the enzymatic diagnosis of Krabbe disease (Sakai 2009).
The human GALC gene has been mapped to chromosome 14q24.3-32.1, and the genomic and/or full-length cDNA sequences have been cloned from multiple species. The human cDNA encodes a 669 amino acid polypeptide with six potential N-glycosylation sites. The fully glycosylated precursor polypeptide is ~85 kDa, and is processed into ~50 and 30 kDa subunits. To date, over 70 mutations in the GALC gene have been identified in infantile and late-onset GALC-deficient patients (http://www.hgmd.org). Most of these mutations are “private,” occurring in individual families, but some may be commonly found in specific regions or ethnicities. Among these, the most common mutation occurs in patients of European ancestry, and deletes a large segment of the GALC gene. Several polymorphisms also have been found in the GALC gene, and some of these may alter amino acids and thus potentially affect the function of the GALC polypeptide, although not enough to cause GLD.
In general, carriers of GALC mutations are considered clinically normal, although one interesting, recent report did show an association between the risk for primary open-angle glaucoma and a heterozygous deletion of the GALC gene (Liu et al. 2011). Another interesting recent paper also showed that GALC contributed to the maintenance of a functional hematopoietic stem cell niche, suggesting that genetic alterations in the GALC gene may be associated with hematological conditions as well. Indeed, hematopoietic stem cells retrieved from Twitcher mice (GALC deficient, see below) had defects in their frequency, proliferation, clonogenic potential, and engraftment.
As noted above, in 1972 Miyatake and Suzuki formulated a hypothesis to explain the unusually rapid and complete destruction of oligodendrocytes in GLD. It was already known at that time that psychosine (galactosyl sphingosine), due to its free amino group, was highly cytotoxic, and that this lipid served as a substrate for GALC, but not GM1 beta-galactosidase. In the central nervous system, psychosine is primarily formed within oligodendrocytes, the site of myelin synthesis, and this may explain the selective destruction of these cells in GLD brains. Indeed, in the white matter of brains from Krabbe patients, psychosine levels may be elevated up to 100-fold, while galactosylceramide levels are either normal or only elevated slightly.
GALC deficiency occurs in a number of naturally occurring animal models, including mice, sheep, several breeds of dogs, and the rhesus monkey. The mouse model in particular (also known as the “Twitcher” mouse) has been exploited for numerous therapeutic studies (Luzi et al. 2001). Affected mice develop clinical signs at ~20 days, with stunted growth, twitching, and hind leg weakness. By 40 days they reach a near-terminal stage. Twitcher mice have been treated by ERT, gene therapies, BMT, and other modalities (Neri et al. 2011; Reddy et al. 2011; Lin et al. 2011). BMT in particular may have positive therapeutic effects; however all of the treated mice eventually die of characteristic pathological findings. Thus, this and the other available therapies may slow the disease progress to some degree, but not prevent it.
Since 2006, New York State in the USA has screened all newborns for the occurrence of Krabbe disease. Pilot screening programs are now under way in other states and parts of the world. This screening is accomplished by GALC activity assays on dried blood spots, followed by DNA analyses. The rationale for this screening program is that the early identification of infants with Krabbe disease will permit early BMT and other therapeutic interventions, improving the quality of life (Duffner et al. 2012). However, since BMT has not been curative in the animal models, screening for severe neurological disorders such as this remains controversial.
6 Arylsulfatase A Deficiency: Metachromatic Leukodystrophy
MLD is an autosomal recessively inherited disorder in which the desulfation of 3-0-sulfogalactosylsphingosine or 3-0-sulfogalactosyl glycolipids is deficient. Among the accumulating glycolipids in MLD are sulfatide (galactosyceramide-3-sulfate), lysosulfatide (i.e., similar to sulfatide but lacking the fatty acid in the ceramide moiety), lactosylceramide-3-sulfate, and others. These sulfated glycolipids normally occur in the myelin sheaths in the central and peripheral nervous systems, and to a lesser extent in visceral organs.
The clinical onset and severity of MLD are highly variable (Renaud 2012). The late infantile form is usually recognized in the second year of life and fatal in early childhood. The juvenile form presents between age 4 and puberty; the adult form may occur at any age after puberty. In both of the later onset variants, gait disturbance and mental regression are the earliest signs. In the childhood variants, other common signs are blindness, loss of speech, quadriparesis, peripheral neuropathy, and seizures (Kehrer et al. 2011).
In 1963, Austin et al. reported the deficiency of arylsulfatase A (ASA, E.C. 3.1.6.8) activity in MLD, also known as sulfatide sulfatase, and mutations in this gene (ARSA) account for the vast majority of MLD patients. There also are two other minor, genetic causes of MLD. One is due to mutations in the gene (PSAP, see Sect. 10, below) encoding the sphingolipid activator protein, saposin B. Mutations in the saposin B encoding region of the PSAP gene lead to deficient ASA activity, and a clinical picture indistinguishable from MLD. In fact, these observations in MLD patients provided the first in vivo proof that such sphingolipid activator proteins were required for in vivo activity of this and other sphingolipid hydrolases. In addition to ASA and SAP B mutations, patients with multiple sulfatase deficiency, due to mutations in the gene encoding sulfatase-modifying factor 1 (SUMF-1) (see below), also have a reduction in ASA and other sulfatase activities, and develop features of MLD.
As with the other sphingolipid hydrolases, full-length genomic and cDNA sequences encoding ASA have been isolated from several species. The human ARSA gene has been mapped to chromosomal region 22q13. It is a remarkably simple gene, and encompasses only 3.2 kb divided into eight exons. As noted above, almost all cases of MLD are due to mutations in this gene, except for the rare cases of PSAP or SUMF-1 mutations. The human ARSA gene encodes a 507 amino acid polypeptide precursor that undergoes posttranslational modifications much like other lysosomal enzymes (e.g., N-glycosylation). Also, as with all known eukaryotic sulfatases, a formylglycine residue is substituted for cysteine 69 in human ASA. The formylglycine residue is generated by oxidation of the thiol group to an aldehyde. Mutations in the gene encoding the cysteine-modifying enzyme, SUMF-1, lead to the synthesis of inactive sulfatases, and are the cause for multiple sulfatase deficiency (Gieselmann et al. 1994).
Over 100 MLD-causing mutations have been described in the ARSA gene. The most common mutation in patients with late-infantile MLD is a G-to-A transition that eliminates the donor splice site at the start of intron 2 (Ługowska et al. 2011). This causes a loss of all ASA enzymatic activity, producing a severe, early-onset phenotype. In contrast, in adults with MLD the most frequent mutation is a C-to-T transition that results in substitution of a leucine for proline in amino acid residue 426. Patients with this mutation have a small amount of residual enzyme activity and therefore exhibit late-onset MLD. Common mutations in specific populations also have been described.
Notably, there also are two mutations that cause “pseudodeficiency” of ASA activity. The term “pseudodeficiency” reflects the fact that these mutations cause severe reduction of ASA activity in vitro, but do not result in MLD (Tinsa et al. 2010). One of these mutations alters an N-glycosylation site, but this has no effect on enzyme function. The other affects a polyadenylation site in the mRNA. The pseudodeficiency alleles may be found in ~3–8 % of the normal population, and therefore pose a diagnostic challenge since the artificial in vitro assay systems may reveal very low ASA activity, even though the individual is highly unlikely to develop MLD.
Alterations in the ARSA gene also have been linked with the occurrence of several common diseases. For example, as early as 1991, Kappler et al. found that among a small set of patients with multiple sclerosis (MS), there was a higher incidence of the ASA pseudodeficiency alleles than in the general population (Baronica et al. 2011). In addition, variants of the ASA protein and/or gene have been associated with depression and alcoholism. MLD itself also has been associated with several psychiatric disorders, and there is at least one report where polymorphisms in the genes encoding the serotonin and dopamine pathways affected the MLD phenotype (Kumperscak et al. 2008).
Other than humans, naturally occurring mutations causing MLD do not occur in other species. ASA knockout mice have been constructed that exhibit a complete loss of ASA activity and sulfatide storage multiple organs (Geiselmann et al. 1998). While these animals exhibit some neurological deficits, they do not develop the predicted severe phenotype of MLD and live a near-normal life span. They also do not develop demyelinating disease. Thus, these observations in the mouse suggest the existence of compensating pathways in this species that do not exist in humans, perhaps providing a link to novel therapies. Several therapies have been evaluated in the MLD knockout mouse model, including BMT, ERT, and gene therapies (Matthes et al. 2012; Stroobants et al. 2011). BMT also has been extensively used in MLD patients. While it is not effective in the infantile cases, BMT is often recommended for the later onset cases (Solders et al. 1998; Gassas et al. 2011). However, the data supporting an improved clinical outcome in the late-onset MLD cases are fragmented and incomplete, and it is unclear whether the benefit outweighs the potential risks.
7 Alpha Galactosidase A Deficiency: Fabry Disease
Fabry disease in inherited as an X-linked recessive trait, the only such X-linked disease among the sphingolipid storage disorders (Tarabuso 2011; Toyooka 2011). Therefore, all males (“hemizygotes”) carrying mutations in the Fabry gene (GLA, see below) develop symptoms of the disease, although the severity may vary depending on the nature of the individual mutation. Female heterozygotes who inherit one copy of the mutant gene on one of their X chromosomes also may exhibit features of the disease depending on the pattern of X-inactivation (“lyonization”) that occurs in their individual tissues.
The gene responsible for Fabry disease encodes an enzyme, alpha-galactosidase A (E.C. 3.2.1.22), that is required to hydrolyze glycosphingolipids with terminal alpha galactosyl moieties, predominately globotriosylceramide, and to a lesser extent, galabiosylceramide and blood group B substances. Affected males have extensive deposition of these lipids in body fluids and in the lysosomes of endothelial, perithelial, and smooth muscle cells of blood vessels. Deposition also occurs in ganglion cells, and in many cell types in the heart, kidney, eyes, and most other tissues (Aerts et al. 2011).
Clinical manifestations in classically affected hemizygotes with no detectable alpha galactosidase A activity include the onset during childhood or adolescence of pain and paresthesias in the extremities, angiokeratomas in the skin and mucous membranes, and hypohidrosis. Corneal and lenticular opacities also are early findings. With increasing age, proteinuria, hyposthenuria, and lymphedema appear. Severe renal impairment leads to hypertension and uremia. Death usually occurs from renal failure or from cardiac or cerebrovascular disease. Atypical variant males with residual alpha galactosidase A activity may be asymptomatic or have late-onset disease. Heterozygous females may have an attenuated form of the disease depending on the pattern of X-inactivation. They usually are asymptomatic, although rarely present with clinical disease as severe as hemizygous males.
As noted above, in Fabry disease glycosphingolipids with terminal alpha-d-galactosyl residues are not properly broken down due to the alpha galactosidase A enzyme defect, and these lipids therefore accumulate in cells. The predominant accumulating lipid is globotriaosylceramide (Gal(α1-4)Gal(α1-4)Glc(β1-1′)Cer). Since alpha galactosidase A is predominately found in lysosomes, the major site of lipid accumulation is this organelle. Elevated globotriaosylceramide is found in most tissues, but predominantly in kidney.
Other minor accumulating lipids include galabiosylceramide (Galα1-4)(Gal(β1-1′)Cer) and the blood group B and P glycosphingolipids (Linthorst et al. 2004). In human erythrocytes there are two neutral glycosphingolipids with terminal alpha galactosyl residues that inhibit blood group B-specific hemagglutination. The structure of these blood group B glycosphingolipids has been determined, and they are found at high levels in Fabry patients. This raises an interesting and unique point about Fabry disease. Fabry hemizygotes and heterozygotes who have blood group B and AB accumulate four glycosphingolipids, while those who are blood group A or O only accumulate two (globotriaosylceramide and galabiosylceramide). A fifth neutral glycosphingolipid that can accumulate in Fabry is the P1 blood group antigen, which also has terminal alpha galactosyl residues.
In man, there are actually two alpha galactosidases (alpha galactosidases A and B). Fabry disease is caused by mutations in the gene (GLA) encoding the “A” type (see below) (Tomasic et al. 2010). However, when measuring alpha galactosidase activity in blood from Fabry patients in vitro, classic hemizygotes may exhibit up to 25 % of normal activity due to the presence of the “B” form. Alpha galactosidase B is encoded by a distinct gene (NAGA), and is now known to be an alpha-N-acetylgalactosaminidase that recognizes these moieties in glycoproteins, complex carbohydrates, and other molecules. Because the artificial substrates routinely used to measure alpha galactosidase activity do not readily distinguish these enzymes, substantial misdiagnosis can occur by simple enzyme testing.
The human gene encoding alpha galactosidase A resides on chromosomal region Xq22. It encompasses ~12 kb and contains seven exons. The full-length human cDNA contains a 1,290 bp open reading frame that encodes an unglycosylated precursor polypeptide of ~48 kDa. As with most lysosomal hydrolases, there are several N-glycosylation sites in the predicted polypeptide sequence and the oligosaccharide chains undergo mannose-6-phosphate modifications, which facilitates targeting of the enzyme to lysosomes. The mature, fully glycosylated alpha galactosidase A in lysosomes is ~51 kDa. Genomic and cDNA sequences encoding alpha galactosidases have now been isolated from many species. An unusual feature of most human alpha galactosidase A cDNAs is the lack of a 3′ untranslated region. The polyadenylation signal sequence is often actually found in the coding region, 12 bp from the termination codon, followed by a poly(A) tract.
The full-length cDNA encoding human alpha galactosidase A had been used to express and purify the recombinant enzyme from CHO cells. This enzyme was extensively characterized and used in the Fabry disease mouse model to evaluate ERT (see below). Based on these results and after extensive clinical testing, a recombinant enzyme drug (Fabrazyme) was approved for use in human Fabry disease patients in 2003. This represents the second sphingolipid storage disorder for which ERT became available (Gaucher disease was the first). ERT in Fabry patients reduces pain, proteinuria, and endothelial cell storage of glycolipids, and improves the quality of life for Fabry patients (Rozenfeld and Neumann 2011). Since no naturally occurring animal model of Fabry disease has been found, the preclinical evaluation of ERT was performed in a knockout mouse model. These animals exhibit age-dependent glycosphingolipid storage, but there are only a few clinical findings of Fabry disease.
Numerous mutations have been found in the GLA gene causing Fabry disease, spread throughout the enzyme-coding region (Schaefer et al. 2005). Several regulatory (promoter) and splice site mutations also have been found. As noted above, the presence of one GLA mutation in hemizygous males will cause disease, but the severity will depend on the effect of this mutation on the residual alpha galactosidase A activity. In contrast, in female heterozygotes, because of X-inactivation, only some may develop disease (Bouwman et al. 2012). It should be noted that such “Fabry” females carrying single mutations will be essentially “mosaics” for alpha galactosidase A, with some cells expressing the mutant allele and others the normal allele (due to random inactivation of one X chromosome). Because the enzyme can be released from cells at low levels (as with all lysosomal hydrolases), and then taken up by neighboring cells, “cross-correction” of the lipid storage is possible. Aside from Fabry disease, there has been some evidence that mutations in the GLA gene also may be a previously unrecognized cause of stroke. Indeed, newborn screening programs have revealed that in some populations the incidence of GLA mutations is unexpectedly high, suggesting that this gene/disease association should be studied further as a potential risk factor for this and other common disorders of the vasculature (Spáčil et al. 2011).
8 Beta Galactosidase Deficiency: GM1 Gangliosidosis
An inherited deficiency of lysosomal acid beta galactosidase (E.C. 3.2.1.23) is expressed as two clinically distinct diseases, GM1 gangliosidosis and Morquio B disease (Brunetti-Pierri and Scaglia 2008; Morita et al. 2009; Caciotti et al. 2011). GM1 gangliosidosis affects both neurological and somatic tissues, occurring mainly in early infancy (type 1). Developmental arrest is usually observed a few months after birth, followed by progressive neurologic deterioration and generalized rigospasticity with sensorimotor and psycho/intellectual dysfunction. As with many of the other neurodegenerative lysosomal storage diseases, cherry-red maculae are common, as is facial dysmorphism, hepatosplenomegaly, and generalized skeletal dysplasia. Later onset, juvenile and adult forms of beta galactosidase deficiency also have been described (types 2 and 3, respectively). They are observed as progressive neurologic diseases in childhood or in young adults. Dysmorphic skeletal changes are less prominent or absent in these clinical forms. A protracted course, mainly presenting as dystonia, is the major neurological manifestation in adults with GM1 gangliosidosis.
Morquio B disease is clinically expressed as a generalized skeletal dysplasia with corneal clouding, resulting in short stature, pectus carinatum, platyspondylia, odontoid hypoplasia, kyphoscoliosis, and genu valgum. There is no central nervous system involvement, although spinal cord compression may occur at late stages of the disease. Intelligence is normal and hepatosplenomegaly is not present.
In man, two lysosomal enzymes are known to be responsible for the hydrolysis of terminal beta-linked galactose residues at acidic pH in various glycoconjugates. One is the enzyme being discussed here, beta galactosidase (E.C. 3.2.1.23), and the other is galactocerebrosidase (E.C. 3.2.1.46), whose primary substrates include galactosylceramide, galactosylsphingosine, and other lipids. This is the enzyme deficient in globoid cell leukodystrophy (Krabbe disease), and was discussed in the section above. Beta galactosidase activity is severely deficient in cells and tissues from patients with both GM1 gangliosidosis and Morquio B disease, and both diseases are now known to be due to genetic mutations in the same gene (GLB1) (Hofer et al. 2009). The primary substrates for beta galactosidase include galactose-containing oligosaccharides (e.g., keratan sulfate), and GM1 ganglioside. The asialo derivative of GM1, GA1 ganglioside, also may be a substrate for this enzyme.
As expected, tissues from patients with beta galactosidase deficiency exhibit a broad spectrum of accumulating galactose-containing macromolecules, including GM1, GA1, and keratan sulfate. Gangliosides are normal components of plasma membranes, concentrated in neuronal membranes, especially in the regions of nerve endings and dendrites. GM1 is the major ganglioside in the brains of vertebrates. Gangliosides display a broad spectrum of interactions, and may act as binding molecules for toxins and hormones, and also are involved in cell differentiation, cell–cell interactions, and cell signaling (see below).
In the brains of patients with GM1 gangliosidosis, storage of GM1 is the most prominent observation (Nada et al. 2011). The accumulating GM1 has the same fatty acid composition, sugar composition and sequence, and glycosidic linkages as normal GM1. Visceral organs also show storage of GM1 ganglioside, but to a lesser degree. Also to a lesser degree, GA1 ganglioside may accumulate in the brain of GM1 gangliosidosis patients, as do glycosylceramide, lactosylceramide, and GM2 ganglioside. White matter shows chemical manifestations of myelin breakdown, including low proteolipid protein, low total lipid, and the presence of esterified cholesterol (Steenweg et al. 2010).
Other than gangliosides, the other major storage molecule in beta galactosidase-deficient patients is keratan sulfate (Ohto et al. 2012). This glycosaminoglycan normally exists in a proteoglycan linked with chondrotin sulfate. It is found primarily within connective tissues, particularly cartilage, explaining the skeletal dysplasia typical of many beta galactosidase-deficient patients. After proteolysis and release from the proteoglycan, free keratan sulfate chains are hydrolyzed by a series of exoenzymes, including beta galactosidase.
It should be noted that another protein, referred to as protective protein/cathepsin A (PPCA), has been associated with both beta galactosidase and another lysosomal hydrolase, neuraminidase, and stabilizes and in some cases activates the enzymes (Tatano et al. 2006). A genetic deficiency of this protective protein results in combined deficiency of beta galactosidase and neuraminidase (galactosialidosis). The gene encoding PPCA is distinct from both of these enzymes, and is not discussed further here.
The gene encoding beta galactosidase (GLB1) is located in chromosomal region 3p21.33. Genes encoding this enzyme also have been isolated from mice and several other species. The full-length human GLB1 cDNA encodes a 677 amino acid polypeptide with beta galactosidase activity. Mutations in the GLB1 gene are responsible for both GM1 gangliosidosis and Morquio B disease. As with other lysosomal diseases, most are point mutations altering single amino acids, although small deletions, splice site mutations, and other alterations also have been identified. To date, no correlation has been made between the location or type of GLB1 mutations and the occurrence of GM1 gangliosidosis or Morquio B disease.
The pathogenesis of beta galactosidase deficiency is complex. GM1 ganglioside stimulates neurite outgrowth and affects neuronal differentiation, and enhances the action of nerve growth factor (van der Voorn et al. 2004). Golgi and electron microscopic studies of cortical neurons from this and several other neurological lysosomal diseases exhibit large neural processes (meganeurites), which may be explained by GM1 accumulation. The extent of meganeurite development is related to the onset, severity, and clinical course of the disease. In other studies, significant changes in neuronal connectivity were found in the cerebral cortex of animals with beta galactosidase deficiency, and cholinergic function was altered as well. Phosphoinositol-specific phospholipase C and adenyl cyclase activities were altered in the membranes of cerebral cortex from GM1 gangliosidosis cats. In addition, the beta subunit of cholera toxin, which binds specifically to GM1 in the outer leaflet of the cell membrane, was found to induce an increase of calcium and manganese influx in N18 cells, and this has been associated with the activation of an L-type voltage-dependent calcium channel. This channel has important implications for neural development, and its reliance on GM1 ganglioside is also likely related to the pathogenesis of GM1 storage. These and other data suggest that various morphologic and metabolic aberrations occur in the brains of GM1 gangliosidosis patients and animals, and can be linked to the biology of GM1.
Naturally occurring animal models of GM1 gangliosidosis have been reported in cats, dogs, sheep, and calves. The cat model in particular has been studied in some detail, and is a neurologic disorder with clinical, morphological, biochemical, and genetic similarities to human GM1 gangliosidosis (Claro et al. 1991). Affected kittens appear normal at birth, but tremors of the head and hind limbs are first noted at 2–3 months of age, followed by generalized dysmetria and spastic quadriplegia. GM1 ganglioside and other galactose-containing oligosaccharides accumulate in the tissues of affected animals. A genetic knockout of beta galactosidase activity also has been generated in mice by gene targeting (Itoh et al. 2001). Progressive and severe neurodegeneration occurs in these animals, and GM1 and GA1 gangliosides accumulate in the brain. Unlike patients, GA1 ganglioside may accumulate in beta galactosidase deficiency mice to a greater extent than GM1, which may be explained by the fact that there is a unique neuraminidase in mice that converts GM1 to GA1. Skeletal dysplasia, which is characteristic of patients with beta galactosidase deficiency, does not occur in the mice. This also is due to the unique metabolism of keratan sulfate in mice compared to humans.
9 Hexosaminidase A and B Deficiency: GM2 Gangliosidoses
The GM2 gangliosidoses are a group of genetic disorders caused by excessive accumulation of GM2 ganglioside and related glycolipids in lysosomes, mainly of neuronal cells (Maegawa 2012). The general cause of GM2 gangliosidosis is deficient activity of the enzyme beta hexosaminidase (E.C. 3.2.1.30). The enzymatic hydrolysis of GM2 ganglioside by this enzyme requires that it be complexed with a substrate-specific cofactor, the GM2 activator. There are two isozymes of beta hexosaminidase, Hex A, which is composed of alpha and beta subunits, and Hex B, which is composed of two beta subunits. Hex A and B can only act on the GM2/GM2 activator complex.
Defects in any of the three genes can lead to GM2 gangliosidosis, HEXA, which encodes the alpha subunit of Hex A, HEXB, which encodes the beta subunit of Hex A and Hex B, or GM2A, which encodes the GM2 activator protein (Cordeiro et al. 2000). Three clinical forms of GM2 gangliosidosis occur from mutations in these genes: Tay–Sachs disease and variants, resulting from mutations of the HEXA gene, and resulting in deficient activity of Hex A; Sandhoff disease and variants, resulting from mutations of the HEXB gene, and resulting in deficient activity of both Hex A and Hex B; and GM2 activator deficiency, caused by mutations in the GM2A gene and characterized by normal Hex A and Hex B, but the inability to form a functional ganglioside GM2/GM2 activator complex.
GM2 gangliosidosis can present with a wide spectrum of severity. These range from infantile-onset, rapidly progressive neurodegenerative disease culminating in death before the age of 4 years (classical Tay–Sachs and Sandhoff diseases and GM2 activator deficiency) to adult-onset, slowly progressive neurological conditions compatible with long survival with little or no effect on intellect (Bley et al. 2011; Smith et al. 2012; Jain et al. 2010; Osher et al. 2011). The clinical phenotypes of the acute, infantile form of any of the three genetic GM2 gangliosidosis (HEXA, HEXB, or GM2A mutations) are essentially indistinguishable. Affected infants generally appear normal at birth. The earliest signs of disease are often mild motor weakness, beginning at 3–5 months of age. An exaggerated startle response is also commonly observed at this early stage as well. Soon after, regression and loss of already acquired mental and motor skills become obvious. The disease is rapidly progressive and leads to death in early childhood.
As noted above, later onset forms of GM2 gangliosidosis also may occur. The clinical phenotype varies widely among these patients, and onset can occur at any time from the late infantile period to adults. In general, in the later onset cases involvement of the deeper brain structures is more prominent compared to the overwhelming generalized gray matter involvement in the infantile form. Manifestations include dystonia, other extrapyramidal signs such as ataxia, choreoathetoid movements, signs of spinocerebellar degeneration, and ALS-like motor neuron involvement. Psychotic manifestations are not uncommon as well. It should also be noted that in a small number of later onset cases, mental capacity is well preserved, although severe dysarthria often masks the preserved intelligence.
Like most lysosomal glycosidases, beta hexosaminidase hydrolyzes a broad spectrum of substrates. It is specific for only the terminal nonreducing sugar (GlcNac or GalNac) in beta linkage. The primary substrates for Hex A and Hex B are glycoproteins, oligosaccharides, glycosaminoglycans, and glycolipids, including the ganglioside GM2 when complexed with the GM2 activator protein. It should be noted that a minor isoform composed of two alpha subunits (Hex S) also has been identified, but the biochemical and clinical significance of this isoform remains unclear. Both the alpha and beta subunits of beta hexosaminidase possess an active site, although dimerization is required for activity. The substrate specificity of the two subunits differs however; the beta subunit prefers neutral, water-soluble substrates, while the alpha subunit also hydrolyzes negatively charged substrates such as GM2 gangliosides or glycosaminoglycans. The fact that mutations in the HEXA gene, in which the Hex B isozyme (composed of two beta subunits) is normal, lead to accumulated GM2 ganglioside demonstrates that the preferred substrate for the alpha subunit is GM2 (see below). However, as noted above, the Hex A isozyme can only hydrolyze GM2 if complexed with the GM2 activator protein.
Mutations in the alpha subunit encoding the HEXA gene are therefore primarily characterized by accumulation of GM2 ganglioside. Indeed, GM2 ganglioside is the primary neuronal storage material in all three genetic causes, hence their grouping as GM2 gangliosidoses. HEXA mutations are responsible for classical Tay–Sachs disease and the later onset Tay–Sachs variants. The pathology of classical Tay–Sachs disease has been described extensively (add ref). The brain is atrophic during the early stage, but usually increases over time until death. Histological analysis shows classical neuronal storage disease in essentially all neurons of the CNS and the peripheral nervous system as well, including swollen retinal ganglion cells. The membranous storage bodies characteristic of Tay–Sachs disease upon electron microscopic examination are composed of cholesterol, phospholipid, and GM2 ganglioside. Increased apoptotic neurons also have been observed. The neuronal pathology in the acute or subacute forms of Tay–Sachs disease is generally less severe than that in the infantile form, and tends to be more prominent in the hypothalamus, cerebellum, brain stem, and spinal cord.
In the Sandhoff disease variants, due to mutations in the HEXB gene, the pathological and clinical findings are very similar to those of Tay–Sachs (Maegawa 2012). However, yellowing of the cerebral cortex and deeper structures has been documented in Sandhoff disease, possibly due to accumulated asialoganglioside. Also, additional accumulation of sphingoglycolipids with a terminal hexosamine residue and fragments of undigested glycoprotein in systemic organs has been found as well. Patients with GM2A mutations are indistinguishable from those with HEXA or HEXB mutations.
The HEXA gene, encoding the alpha subunit, is located within the chromosomal region 15q23–q24. It is ~35 kb in length and contains 14 exons. The HEXB gene, encoding the beta subunit, is located at chromosomal region 5q13 and is ~45 kb in length that is divided into 14 exons. Analysis of these genes strongly suggests that they arose from a common ancestor. They share a very common exon structure, and in both genes the promoter activity resides within ~150 bp of the initiating ATG codon. Overall, the alpha and beta subunits share ~57 % amino acid identity. The GM2A gene maps to chromosomal region 5q31.1–31.3. In addition, an evolutionarily related pseudogene, GM2AP, maps to chromosome 3. The GM2A gene is ~16 kb and contains four exons.
Numerous mutations have been described in the HEXA gene responsible for Tay–Sachs disease and its variants. Infantile Tay–Sachs disease occurs most commonly among individuals of Ashkenazi Jewish ancestry (carrier frequency ~1:25), and there are two mutations that are the most frequent; one is a 4 bp insertion in exon 11, and the second is a donor splice site mutation in intron 11 (Frisch et al. 2004). Both of these mutations result in severely deficient or absent HEXA mRNA expression, leading to absent Hex A and Hex S activities. Another common HEXA mutation causing infantile Tay–Sachs disease occurs in French Canadian patients, and is due to a deletion that extends from ~2 kb upstream of the 5′ end of the gene into intron 1 (De Braekeleer et al. 1992; Martin et al. 2007). This also results in the absence of Hex A mRNA and protein. Many other mutations in the HEXA gene have been described causing classical Tay–Sachs disease, effecting protein processing, catalytic activity, and/or mRNA processing. In addition, mutations causing the later onset forms are known, and generally result in expression of HEXA mRNA and residual enzyme activity. An important HEXA mutation also is known as the B1 variant, in which there is normal activity towards the artificial substrates generally used to measure beta hexosaminidase activity, but no activity of the mutant enzyme towards GM2 ganglioside. Individuals with this mutation pose a unique diagnostic challenge since they will appear enzymatically normal, but develop severe disease. Many mutations in the HEXB gene causing Sandhoff disease and its variants also have been described. A much small number of mutations in the GM2A gene are known. As noted above, the fact that mutations in this gene caused a severe clinical disease provided the first proof of the physiological significance of the GM2/GM2 activator complex (Peleg et al. 1995; Tanaka et al. 2003).
Naturally occurring mutations causing GM2 gangliosidosis have been described in dogs, cats, and pigs (Yamato et al. 2002; Kosanke et al. 1979; Bradbury et al. 2009). In all species, abundant neuronal storage is the major finding. Visceral storage is only found in the feline model however. Meganurite formation, which has been observed in human GM2 gangliosidosis patients, occurs in the dog and cat models. A number of groups also have independently generated murine models of Tay–Sachs, Sandhoff, and GM2 activator deficiency in mice using gene targeting strategies. In general, knockout of the HEXA gene in mice (Tay–Sachs disease) results in a much milder neurological disease than predicted from the human cases (Gulinello et al. 1994). Mutations in the HEXB gene in mice result in a more severe phenotype, with the onset of clinical disease at ~3 months associated with excessive neuronal GM2 storage (Yamanaka et al. 2008). Mutations in the GM2A gene of mice also resulted in a much milder disease than predicted (Phaneuf et al. 1996).
In part, the distinction between the mouse models and the human disorder is due to the distinct degradation of GM2 ganglioside by these species (Baek et al. 2009). In humans, GM2 is degraded nearly exclusively by Hex A in collaboration with the activator protein to yield the ganglioside GM3. In contrast, in mice GM2 can be degraded by two different pathways. One is identical to the human pathway, and the second is essentially unique to the mouse and is initiated by sialidase acting on GM2 to yield GA2 ganglioside. The GA2 is then degraded by either Hex A with activator protein or to a lesser extent Hex B. This explains the mild phenotype due to alpha subunit mutations (HEXA), since the Hex B can act on the GA2 produced by sialidase. In contrast, HEXB mutations result in deficiency of both enzymes, and a complete block of degradation.
10 Sphingolipid Activator Proteins
As discussed in the sections above, the lysosomal degradation of sphingolipids requires small nonenzymatic glycoproteins, referred to as “sphingolipid activator proteins” (SAPs) (Sandhoff and Kolter 1998; Kolter and Sandhoff 2010). These include the saposin proteins (SAP A–D, and GM2 activator protein). There are two genes responsible for these proteins. One is GMA2, which encodes the GM2 activator protein and is located on chromosome 5 (Wendeler et al. 2003), and the other is PSAP, located on chromosome 10 and encoding the saposin precursor protein and the individual SAP A–D, which are derived from proteolytic cleavage of the precursor (Misasi et al. 2009). The GM2 activator protein and resulting mutations (responsible for the “A/B” Tay–Sachs variant) were discussed in the section above and will not be discussed further here.
The PSAP gene encodes the SAP precursor protein (prosaposin), with a total of 524 amino acids and five N-glycosylation sites (Misasi et al. 2009). There are four homologous domains located within the precursor protein, each of ~80 amino acids. A major portion of the newly synthesized precursor is exported to the cell surface and then imported into the lysosomal compartment, where it is processed to the mature SAP A–D proteins. Notably, unlike most lysosomal proteins the prosaposin protein is transported to the lysosome via its interaction with the alternative receptor, sortilin (Wähe et al. 2010; Yuan and Morales 2011). The occurrence of the non-processed SAP precursor in body fluids and its neurotrophic properties indicates that its function may not be restricted to being the precursor of the individual SAPs. For example, prosaposin has been found in milk, serum, and seminal fluid, and treatment of cell lines with PSAP increased cell survival and was anti-apoptotic. Indeed, serum prosapsin levels are increased in patients with advanced prostate cancer. Of interest is whether these effects of PSAP on cell survival are direct effects of the protein itself, or rather the effects of the individual processed SAPs on the activation of the sphingolipid hydrolases.
In vitro, SAP A stimulates beta glucocerebrosidase (the enzyme deficient in Gaucher disease) and beta galactocerebrosidase (the enzyme deficient in Krabbe disease) activities in the presence of detergents. SAP B is a nonspecific glycolipid-binding protein that stimulates the hydrolysis of ~20 or more glycolipids by different enzymes in vitro, including the hydrolysis of sulfatide by ASA (the enzyme deficient in MLD). SAP C stimulates the in vitro activities of glucosyl- and galactosylcerebrosidases, as well as sphingomyelin by ASM (the enzyme deficient in Types A and B Niemann–Pick disease) (Kölzer et al. 2004). SAP D is required for the degradation of ceramide by AC (the enzyme deficient in Farber disease) (Linke et al. 2001).
Analysis of mutations in the genes encoding the PSAP protein has provided important insights regarding the role of the SAP proteins for the in vivo hydrolysis of sphingolipids, and their relationship to human disease. Of note, a SAP precursor deficiency was found in a child who died at 16 weeks and who was homozygous for a mutation in the initiation codon of the PSAP gene. The complete absence of the precursor protein and four resultant SAPs led to a generalized accumulation of ceramide, glucosylceramide, galactosylceramide, sulfatide, lactosylceramide, digalactosylceramide, and other sphingolipids. The symptoms of the disease resembled those of type 2 Gaucher disease. Other point mutations within the region of the PSAP gene encoding SAP B caused accumulation of sulfatide and some other sphingolipids, and a clinical course resembling that of juvenile MLD. Mutations in the SAP C region only led to accumulation of glucosylceramide, similar to juvenile Gaucher disease (Vaccaro et al. 2010). No sphingomyelin storage was observed in individuals with SAP C mutations, and this has been explained by the fact that there is a SAP-like domain within ASM that compensates for the loss of SAP C in these patients. Patients with mutations in the SAP A region of the PSAP gene resemble those with Krabbe disease (GLD), and accumulate galactosylcerebroside within the CNS and peripheral nervous system (Grossi et al. 2008). There are no human patients with SAP D mutations, but the requirement by AC for SAP D has been confirmed in vivo in mutant mice, which accumulate ceramides with alpha hydroxylated fatty acids mainly in the brain and kidney, and develop Purkinje cell loss and defects in the urinary system (Oya et al. 1998; Tohyama et al. 1999).
A mouse model of SAP precursor deficiency also has been constructed by gene targeting, and the clinical and biochemical features resembled those of the human disease (Matsuda et al. 2007). Mice developed massive sphingolipid storage and neurological disease by ~20 days, and died by ~35–38 days. Several knock-in models expressing specific human mutations also have been constructed, confirming the in vivo importance of these proteins (Sun et al. 2008, 2010).
References
Abian O, Alfonso P, Velazquez-Campoy A, Giraldo P, Pocovi M, Sancho J (2011) Therapeutic strategies for Gaucher disease: miglustat (NB-DNJ) as a pharmacological chaperone for glucocerebrosidase and the different thermostability of velaglucerase alfa and imiglucerase. Mol Pharm 8(6):2390–2397
Aerts JM, Boot RG, van Eijk M, Groener J, Bijl N, Lombardo E, Bietrix FM, Dekker N, Groen AK, Ottenhoff R, van Roomen C, Aten J, Serlie M, Langeveld M, Wennekes T, Overkleeft HS (2011) Glycosphingolipids and insulin resistance. Adv Exp Med Biol 721:99–119
Ahmad A, Mazhar AU, Anwar M (2009) Farber disease: a rare neurodegenerative disorder. J Coll Physicians Surg Pak 19(1):67–68
Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, Pollak P, Bonaïti B, Bonaïti-Pellié C, Brice A (2012) Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78(6):417–420
Baek RC, Martin DR, Cox NR, Seyfried TN (2009) Comparative analysis of brain lipids in mice, cats, and humans with Sandhoff disease. Lipids 44(3):197–205
Baronica KB, Mlinac K, Ozretić D, Vladić A, Bognar SK (2011) Arylsulfatase a gene polymorphisms in relapsing remitting multiple sclerosis: genotype-phenotype correlation and estimation of disease progression. Coll Antropol 35(Suppl 1):11–16
Becker KA, Grassmé H, Zhang Y, Gulbins E (2010) Ceramide in Pseudomonas aeruginosa infections and cystic fibrosis. Cell Physiol Biochem 26(1):57–66
Beckham TH, Lu P, Cheng JC, Zhao D, Turner LS, Zhang X, Hoffman S, Armeson KE, Liu A, Marrison T, Hannun YA, Liu X (2012) Acid ceramidase-mediated production of sphingosine 1-phosphate promotes prostate cancer invasion through upregulation of cathepsin B. Int J Cancer 131:2034–2043. doi:10.1002/ijc.27480
Benito JM, García Fernández JM, Ortiz Mellet C (2011) Pharmacological chaperone therapy for Gaucher disease: a patent review. Expert Opin Ther Pat 21(6):885–903
Bikman BT, Summers SA (2011) Sphingolipids and hepatic steatosis. Adv Exp Med Biol 721: 87–97
Bley AE, Giannikopoulos OA, Hayden D, Kubilus K, Tifft CJ, Eichler FS (2011) Natural history of infantile G(M2) gangliosidosis. Pediatrics 128(5):e1233–e1241
Bouwman MG, Rombach SM, Schenk E, Sweeb A, Wijburg FA, Hollak CE, Linthorst GE (2012) Prevalence of symptoms in female Fabry disease patients: a case-control survey. J Inherit Metab Dis 35:891–898
Bradbury AM, Morrison NE, Hwang M, Cox NR, Baker HJ, Martin DR (2009) Neurodegenerative lysosomal storage disease in European Burmese cats with hexosaminidase beta-subunit deficiency. Mol Genet Metab 97(1):53–59
Brown JT, Lahey C, Laosinchai-Wolf W, Hadd AG (2006) Polymorphisms in the glucocerebrosidase gene and pseudogene urge caution in clinical analysis of Gaucher disease allele c.1448T > C (L444P). BMC Med Genet 7:69
Brunetti-Pierri N, Scaglia F (2008) GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol Genet Metab 94(4):391–396
Caciotti A, Garman SC, Rivera-Colón Y, Procopio E, Catarzi S, Ferri L, Guido C, Martelli P, Parini R, Antuzzi D, Battini R, Sibilio M, Simonati A, Fontana E, Salviati A, Akinci G, Cereda C, Dionisi-Vici C, Deodato F, d’Amico A, d’Azzo A, Bertini E, Filocamo M, Scarpa M, di Rocco M, Tifft CJ, Ciani F, Gasperini S, Pasquini E, Guerrini R, Donati MA, Morrone A (2011) GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim Biophys Acta 1812(7):782–790
Campbell TN, Choy FY (2012) Gaucher disease and the synucleinopathies: refining the relationship. Orphanet J Rare Dis 7:12
Chen H, Tran JT, Brush RS, Saadi A, Rahman AK, Yu M, Yasumura D, Matthes MT, Ahern K, Yang H, LaVail MM, Mandal MN (2012) Ceramide signaling in retinal degeneration. Adv Exp Med Biol 723:553–558
Choy FY, Campbell TN (2011) Gaucher disease and cancer: concept and controversy. Int J Cell Biol 2011:150450
Clarke CJ, Wu BX, Hannun YA (2011) The neutral sphingomyelinase family: identifying biochemical connections. Adv Enzyme Regul 51(1):51–58
Claro E, Wallace MA, Fain JN, Nair BG, Patel TB, Shanker G, Baker HJ (1991) Altered phosphoinositide-specific phospholipase C and adenylyl cyclase in brain cortical membranes of cats with GM1 and GM2 gangliosidosis. Brain Res Mol Brain Res 11(3–4):265–271
Cordeiro P, Hechtman P, Kaplan F (2000) The GM2 gangliosidoses databases: allelic variation at the HEXA, HEXB, and GM2A gene loci. Genet Med 2(6):319–327
Cox TM, Cachón-González MB (2012) The cellular pathology of lysosomal diseases. J Pathol 226(2):241–254
De Braekeleer M, Hechtman P, Andermann E, Kaplan F (1992) The French Canadian Tay-Sachs disease deletion mutation: identification of probable founders. Hum Genet 89(1):83–87
Duffner PK, Barczykowski A, Jalal K, Yan L, Kay DM, Carter RL (2011) Early infantile Krabbe disease: results of the World-Wide Krabbe Registry. Pediatr Neurol 45(3):141–148
Duffner PK, Granger C, Lyon N, Niewczyk P, Barczykowski A, Bauer S (2012) Msall ME (2012) developmental and functional outcomes in children with a positive newborn screen for Krabbe disease: a pilot study of a phone-based interview surveillance technique. J Pediatr 161:258–263
Eliyahu E, Shtraizent N, Martinuzzi K, Barritt J, He X, Wei H, Chaubal S, Copperman AB, Schuchman EH (2010) Acid ceramidase improves the quality of oocytes and embryos and the outcome of in vitro fertilization. FASEB J 24(4):1229–1238
Farber S, Cohen J, Uzman LL (1957) Lipogranulomatosis; a new lipo-glycoprotein storage disease. J Mt Sinai Hosp NY 24(6):816–837
Farfel-Becker T, Vitner EB, Futerman AH (2011) Animal models for Gaucher disease research. Dis Model Mech 4(6):746–752
Flowers M, Fabriás G, Delgado A, Casas J, Abad JL, Cabot MC (2012) C6-Ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res Treat 133:447–458
Frisch A, Colombo R, Michaelovsky E, Karpati M, Goldman B, Peleg L (2004) Origin and spread of the 1278insTATC mutation causing Tay-Sachs disease in Ashkenazi Jews: genetic drift as a robust and parsimonious hypothesis. Hum Genet 114(4):366–376
Furuya H, Shimizu Y, Kawamori T (2011) Sphingolipids in cancer. Cancer Metastasis Rev 30(3–4):567–576
Gassas A, Raiman J, White L, Schechter T, Clarke J, Doyle J (2011) Long-term adaptive functioning outcomes of children with inherited metabolic and genetic diseases treated with hematopoietic stem cell transplantation in a single large pediatric center: parents’ perspective. J Pediatr Hematol Oncol 33(3):216–220
Geiselmann V, Matzner, U, Hess B, Lullmann-Rauch R, Coenen R, Hartmann D, D’Hooge R, DeDeyn P, Nagels G (1998) Metachromatic leukodystrophy: molecular genetics and an animal model. J. Inherit Metab Dis:564–574
Gieselmann V, Zlotogora J, Harris A, Wenger DA, Morris CP (1994) Molecular genetics of metachromatic leukodystrophy. Hum Mutat 4(4):233–242
Goker-Alpan O (2011) Therapeutic approaches to bone pathology in Gaucher disease: past, present and future. Mol Genet Metab 104(4):438–447
Grossi S, Regis S, Rosano C, Corsolini F, Uziel G, Sessa M, Di Rocco M, Parenti G, Deodato F, Leuzzi V, Biancheri R, Filocamo M (2008) Molecular analysis of ARSA and PSAP genes in twenty-one Italian patients with metachromatic leukodystrophy: identification and functional characterization of 11 novel ARSA alleles. Hum Mutat 29(11):E220–E230
Gulinello M, Chen F, Dobrenis K (1994) Targeted disruption of the Hexa gene results in mice with biochemical and pathologic features of Tay-Sachs disease. Proc Natl Acad Sci U S A 91(21):9975–9979
Hofer D, Paul K, Fantur K, Beck M, Bürger F, Caillaud C, Fumic K, Ledvinova J, Lugowska A, Michelakakis H, Radeva B, Ramaswami U, Plecko B, Paschke E (2009) GM1 gangliosidosis and Morquio B disease: expression analysis of missense mutations affecting the catalytic site of acid beta-galactosidase. Hum Mutat 30(8):1214–1221
Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH (1995) Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet 10(3):288–293
Hruska KS, LaMarca ME, Scott CR, Sidransky E (2008) Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 29(5):567–583
Hughes DA, Pastores GM (2010) The pathophysiology of GD - current understanding and rationale for existing and emerging therapeutic approaches. Wien Med Wochenschr 160(23–24):594–599
Itoh M, Matsuda J, Suzuki O, Ogura A, Oshima A, Tai T, Suzuki Y, Takashima S (2001) Development of lysosomal storage in mice with targeted disruption of the beta-galactosidase gene: a model of human G(M1)-gangliosidosis. Brain Dev 23(6):379–384
Jain A, Kohli A, Sachan D (2010) Infantile Sandhoff’s disease with peripheral neuropathy. Pediatr Neurol 42(6):459–461
Kehrer C, Blumenstock G, Gieselmann V, Krägeloh-Mann I, Leukonet G (2011) The natural course of gross motor deterioration in metachromatic leukodystrophy. Dev Med Child Neurol 53(9):850–855
Kinghorn KJ (2011) Pathological looping in the synucleinopathies: investigating the link between Parkinson’s disease and Gaucher disease. Dis Model Mech 4(6):713–715
Kohlschütter A, Eichler F (2011) Childhood leukodystrophies: a clinical perspective. Expert Rev Neurother 11(10):1485–1496
Kolter T, Sandhoff K (2010) Lysosomal degradation of membrane lipids. FEBS Lett 584(9): 1700–1712
Kölzer M, Ferlinz K, Bartelsen O, Hoops SL, Lang F, Sandhoff K (2004) Functional characterization of the postulated intramolecular sphingolipid activator protein domain of human acid sphingomyelinase. Biol Chem 385(12):1193–1195
Kosanke SD, Pierce KR, Read WK (1979) Morphogenesis of light and electron microscopic lesions in porcine GM2-gangliosidosis. Vet Pathol 16(1):6–17
Kumperscak HG, Dolzan V, Videtic A, Plesnicar BK (2008) Polymorphisms in genes encoding the serotonin and dopamine pathways in two sisters with metachromatic leukodystrophy. J Int Med Res 36(5):1123–1128
Lachmann RH (2011) Enzyme replacement therapy for lysosomal storage diseases. Curr Opin Pediatr 23(6):588–593
Lee JY, Lee BH, Kim GH, Jung CW, Lee J, Choi JH, Yoo HW (2012) Clinical and genetic characteristics of Gaucher disease according to phenotypic subgroups. Korean J Pediatr 55(2): 48–53
Li CM, Park JH, Simonaro CM, He X, Gordon RE, Friedman AH, Ehleiter D, Paris F, Manova K, Hepbildikler S, Fuks Z, Sandhoff K, Kolesnick R, Schuchman EH (2002) Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics 79(2):218–224
Lin DS, Hsiao CD, Liau I, Lin SP, Chiang MF, Chuang CK, Wang TJ, Wu TY, Jian YR, Huang SF, Liu HL (2011) CNS-targeted AAV5 gene transfer results in global dispersal of vector and prevention of morphological and function deterioration in CNS of globoid cell leukodystrophy mouse model. Mol Genet Metab 103(4):367–377
Linke T, Wilkening G, Sadeghlar F, Mozcall H, Bernardo K, Schuchman E, Sandhoff K (2001) Interfacial regulation of acid ceramidase activity. Stimulation of ceramide degradation by lysosomal lipids and sphingolipid activator proteins. J Biol Chem 276(8):5760–5768
Linthorst GE, Hollak CE, Donker-Koopman WE, Strigland A, Aerts JM (2004) Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase slpha and beta. Kidney Int 66: 1589–95
Liu Y, Gibson J, Wheeler J, Kwee LC, Santiago-Turla CM, Akafo SK, Lichter PR, Gaasterland DE, Moroi SE, Challa P, Herndon LW, Girkin CA, Budenz DL, Richards JE, Allingham RR, Hauser MA (2011) GALC deletions increase the risk of primary open-angle glaucoma: the role of Mendelian variants in complex disease. PLoS One 6(11):e27134
Lo SM, Choi M, Liu J, Jain D, Boot RG, Kallemeijn WW, Aerts JM, Pashankar F, Kupfer GM, Mane S, Lifton RP, Mistry PK (2012) Phenotypic diversity in type 1 Gaucher disease: discovering the genetic basis of Gaucher disease/hematological malignancy phenotype by individual genome analysis. Blood 119:4731–4740
Ługowska A, Ponińska J, Krajewski P, Broda G, Płoski R (2011) Population carrier rates of pathogenic ARSA gene mutations: is metachromatic leukodystrophy underdiagnosed? PLoS One 6(6):e20218
Luzi P, Rafi MA, Zaka M, Curtis M, Vanier MT, Wenger DA (2001) Generation of a mouse with low galactocerebrosidase activity by gene targeting: a new model of globoid cell leukodystrophy (Krabbe disease). Mol Genet Metab 73(3):211–223
Maegawa G (2012) GM2 gangliosidosis: the prototype of lysosomal storage disorders. Dev Med Child Neurol 54(2):104–105
Malandrini A, D’Eramo C, Palmeri S, Gaudiano C, Gambelli S, Sicurelli F, Berti G, Formichi P, Kuqo A, Dotti MT, Federico A (2013) Peripheral neuropathy in late-onset Krabbe disease: report of three cases.Neurol Sci 34:79–83
Mao C, Obeid L (2008) Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine and sphingosine-1-phosphate. Biochim Biophys Acta 1781(9):424–434
Martin DC, Mark BL, Triggs-Raine BL, Natowicz MR (2007) Evaluation of the risk for Tay-Sachs disease in individuals of French Canadian ancestry living in new England. Clin Chem 53(3): 392–398
Matsuda J, Yoneshige A, Suzuki K (2007) The function of sphingolipids in the nervous system: lessons learnt from mouse models of specific sphingolipid activator protein deficiencies. J Neurochem 103(Suppl 1):32–38
Matthes F, Stroobants S, Gerlach D, Wohlenberg C, Wessig C, Fogh J, Gieselmann V, Eckhardt M, D’Hooge R, Matzner U (2012) Efficacy of enzyme replacement therapy in an aggravated mouse model of metachromatic leukodystrophy declines with age. Hum Mol Genet 21:2599–2609
McGovern MM, Schuchman EH (2009) Acid sphingomyelinase defieciency. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds) Gene Reviews™ [Internet]. University of Washington, Seattle, WA
Mikosch P, Hughes D (2010) An overview on bone manifestations in Gaucher disease. Wien Med Wochenschr 160(23–24):609–624
Miranda SR, He X, Simonaro CM, Gatt S, Dagan A, Desnick RJ, Schuchman EH (2000) Infusion of recombinant human acid sphingomyelinase into niemann-pick disease mice leads to visceral, but not neurological, correction of the pathophysiology. FASEB J 14(13):1988–1995
Misasi R, Hozumi I, Inuzuka T, Capozzi A, Mattei V, Kuramoto Y, Shimeno H, Soeda S, Azuma N, Yamauchi T, Hiraiwa M (2009) Biochemistry and neurobiology of prosaposin: a potential therapeutic neuro-effector. Cent Nerv Syst Agents Med Chem 9(2):119–131
Miyatake T, Suzuki K (1972) Globoid cell leukodystrophy: additional deficiency of psychosine galactosidase. Biochem Biophys Res Commun 48(3):539–543
Morita M, Saito S, Ikeda K, Ohno K, Sugawara K, Suzuki T, Togawa T, Sakuraba H (2009) Structural bases of GM1 gangliosidosis and Morquio B disease. J Hum Genet 54(9):510–515
Mullen TD, Hannun YA, Obeid LM (2012) Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem J 441(3):789–802
Nada R, Gupta K, Lal SB, Vasishta RK (2011) An autopsy case of infantile GM1 gangliosidosis with adrenal calcification. Metab Brain Dis 26(4):307–310
Neri M, Ricca A, di Girolamo I, Alcala’-Franco B, Cavazzin C, Orlacchio A, Martino S, Naldini L, Gritti A (2011) Neural stem cell gene therapy ameliorates pathology and function in a mouse model of globoid cell leukodystrophy. Stem Cells 29(10):1559–1571
Ohto U, Usui K, Ochi T, Yuki K, Satow Y, Shimizu T (2012) Crystal structure of human β-galactosidase: structural basis of Gm1 gangliosidosis and morquio B diseases. J Biol Chem 287(3):1801–1812
Okino N, He X, Gatt S, Sandhoff K, Schuchman EH (2003) The reverse activity of human acid ceramidase. J Biol Chem 278(32):29948–29953
Osher E, Fattal-Valevski A, Sagie L, Urshanski N, Amir-Levi Y, Katzburg S, Peleg L, Lerman-Sagie T, Zimran A, Elstein D, Navon R, Stern N, Valevski A (2011) Pyrimethamine increases β-hexosaminidase A activity in patients with Late Onset Tay Sachs. Mol Genet Metab 102(3):356–633
Oya Y, Nakayasu H, Fujita N, Suzuki K, Suzuki K (1998) Pathological study of mice with total deficiency of sphingolipid activator proteins (SAP knockout mice). Acta Neuropathol 96(1): 29–40
Park JH, Eliyahu E, Narla G, DiFeo A, Martignetti JA, Schuchman EH (2005) KLF6 is one transcription factor involved in regulating acid ceramidase gene expression. Biochim Biophys Acta 1732(1–3):82–87
Park JH, Schuchman EH (2006) Acid ceramidase and human disease. Biochim Biophys Acta 1758(12):2133–2138
Pastores GM (2010) Neuropathic Gaucher disease. Wien Med Wochenschr 160(23–24):605–608
Perlman SJ, Mar S (2012) Leukodystrophies. Adv Exp Med Biol 724:154–171
Peleg L, Meltzer F, Karpati M, Goldman B (1995) GM2 gangliosidosis B1 variant: biochemical and molecular characterization of hexosaminidase A. Biochem Mol Med 54(2):126–132
Petrache I, Petrusca DN, Bowler RP, Kamocki K (2011) Involvement of ceramide in cell death responses in the pulmonary circulation. Proc Am Thorac Soc 8(6):492–496
Phaneuf D, Wakamatsu N, Huang JQ, Borowski A, Peterson AC, Fortunato SR, Ritter G, Igdoura SA, Morales CR, Benoit G, Akerman BR, Leclerc D, Hanai N, Marth JD, Trasler JM, Gravel RA (1996) Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Hum Mol Genet 5(1):1–14
Podbielska M, Krotkiewski H, Hogan EL (2012) Signaling and regulatory functions of bioactive sphingolipids as therapeutic targets in multiple sclerosis. Neurochem Res 37:1154–1169
Reddy AS, Kim JH, Hawkins-Salsbury JA, Macauley SL, Tracy ET, Vogler CA, Han X, Song SK, Wozniak DF, Fowler SC, Klein RS, Sands MS (2011) Bone marrow transplantation augments the effect of brain- and spinal cord-directed adeno-associated virus 2/5 gene therapy by altering inflammation in the murine model of globoid-cell leukodystrophy. J Neurosci 31(27):9945–9957
Renaud DL (2012) Lysosomal disorders associated with leukoencephalopathy. Semin Neurol 32(1):51–54
Rozenfeld P, Neumann PM (2011) Treatment of Fabry disease: current and emerging strategies. Curr Pharm Biotechnol 12:916–22
Sakai N (2009) Pathogenesis of leukodystrophy for Krabbe disease: molecular mechanism and clinical treatment. Brain Dev 31(7):485–487
Sandhoff K, Kolter T (1998) Processing of sphingolipid activator proteins and the topology of lysosomal digestion. Acta Biochim Pol 45(2):373–384
Sardi SP, Singh P, Cheng SH, Shihabuddin LS, Schlossmacher MG (2012) Mutant GBA1 expression and synucleinopathy risk: first insights from cellular and mouse models. Neurodegener Dis 10:195–202
Schaefer E, Mehta A, Gal A (2005) Genotype and phenotype in Fabry disease; analysis of the Fabry outcome survey. Acta Paediatr Suppl 94:87–92
Schissel SL, Keesler GA, Schuchman EH, Williams KJ, Tabas I (1998) The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J Biol Chem 273(29):18250–18259
Schuchman EH (2009) The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. Int J Clin Pharmacol Ther 47(Suppl 1):S48–S57
Schulze H, Sandhoff K (2011) Lysosomal lipid storage diseases. Cold Spring Harb Perspect Biol 3(6):pii: a0084804. doi:10.1101/cschperspect.a004804
Scott SA, Edelmann L, Liu L, Luo M, Desnick RJ, Kornreich R (2010) Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum Mutat 31(11):1240–1250
Simonaro CM, Park JH, Eliyahu E, Shtraizent N, McGovern MM, Schuchman EH (2006) Imprinting at the SMPD1 locus: implications for acid sphingomyelinase-deficient Niemann-Pick disease. Am J Hum Genet 78(5):865–870
Smith NJ, Winstone AM, Stellitano L, Cox TM, Verity CM (2012) GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol 54(2):176–182
Solders G, Celsing G, Hagenfeldt L, Ljungman P, Isberg B, Ringdén O (1998) Improved peripheral nerve conduction, EEG and verbal IQ after bone marrow transplantation for adult metachromatic leukodystrophy. Bone Marrow Transplant 22(11):1119–1122
Spáčil Z, Elliott S, Reeber SL, Gelb MH, Scott CR, Tureček F (2011) Comparative triplex tandem mass spectrometry assays of lysosomal enzyme activities in dried blood spots using fast liquid chromatography: application to newborn screening of Pompe, Fabry, and Hurler diseases. Anal Chem 83(12):4822–4828
Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM, van Wieringen WN, Barkhof F, Wolf NI, van der Knaap MS (2010) Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 133(10):2971–2982
Stroobants S, Gerlach D, Matthes F, Hartmann D, Fogh J, Gieselmann V, D’Hooge R, Matzner U (2011) Intracerebroventricular enzyme infusion corrects central nervous system pathology and dysfunction in a mouse model of metachromatic leukodystrophy. Hum Mol Genet 20(14): 2760–2769
Sugita M, Dulaney JT, Moser HW (1972) Ceramidase deficiency in Farber’s disease (lipogranulomatosis). Science 178(4065):1100–1102
Sun Y, Ran H, Zamzow M, Kitatani K, Skelton MR, Williams MT, Vorhees CV, Witte DP, Hannun YA, Grabowski GA (2010) Specific saposin C deficiency: CNS impairment and acid beta-glucosidase effects in the mouse. Hum Mol Genet 19(4):634–647
Sun Y, Witte DP, Ran H, Zamzow M, Barnes S, Cheng H, Han X, Williams MT, Skelton MR, Vorhees CV, Grabowski GA (2008) Neurological deficits and glycosphingolipid accumulation in saposin B deficient mice. Hum Mol Genet 17(15):2345–2356
Tanaka A, Hoang LT, Nishi Y, Maniwa S, Oka M, Yamano T (2003) Different attenuated phenotypes of GM2 gangliosidosis variant B in Japanese patients with HEXA mutations at codon 499, and five novel mutations responsible for infantile acute form. J Hum Genet 48(11):571–574
Tarabuso AL (2011) Fabry disease. Skinmed 9(3):173–177
Tatano Y, Takeuchi N, Kuwahara J, Sakuraba H, Takahashi T, Takada G, Itoh K (2006) Elastogenesis in cultured dermal fibroblasts from patients with lysosomal beta-galactosidase, protective protein/cathepsin A and neuraminidase-1 deficiencies. J Med Invest 53(1–2):103–112
Tinsa F, Caillaud C, Vanier MT, Bousnina D, Boussetta K, Bousnina S (2010) An unusual homozygous arylsulfatase: a pseudodeficiency in a metachromatic leukodystrophy Tunisian patient. J Child Neurol 25(1):82–86
Tohyama J, Oya Y, Ezoe T, Vanier MT, Nakayasu H, Fujita N, Suzuki K (1999) Ceramide accumulation is associated with increased apoptotic cell death in cultured fibroblasts of sphingolipid activator protein-deficient mouse but not in fibroblasts of patients with Farber disease. J Inherit Metab Dis 22(5):649–662
Tomasic IB, Metcalf MC, Guce AI, Clark NE, Garman SC (2010) Interconversion of the specificities of human lysosomal enzymes associated with Fabry and Schindler diseases. J Biol Chem 285(28):21560–21566
Toyooka K (2011) Fabry disease. Curr Opin Neurol 24(5):463–468
Truman JP, Al Gadban MM, Smith KJ, Hammad SM (2011) Acid sphingomyelinase in macrophage biology. Cell Mol Life Sci 68(20):3293–3305
Vaccaro AM, Motta M, Tatti M, Scarpa S, Masuelli L, Bhat M, Vanier MT, Tylki-Szymanska A, Salvioli R (2010) Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum Mol Genet 19(15):2987–2997
van der Voorn JP, Kamphorst W, van der Knaap MS, Powers JM (2004) The leukoencephalopathy of infantile GM1 gangliosidosis: oligodendrocytic loss and axonal dysfunction. Acta Neuropathol 107(6):539–545
Wähe A, Kasmapour B, Schmaderer C, Liebl D, Sandhoff K, Nykjaer A, Griffiths G, Gutierrez MG (2010) Golgi-to-phagosome transport of acid sphingomyelinase and prosaposin is mediated by sortilin. J Cell Sci 23(Pt 14):2502–2511
Wendeler M, Lemm T, Weisgerber J, Hoernschemeyer J, Bartelsen O, Schepers U, Sandhoff K (2003) Expression of recombinant human GM2-activator protein in insect cells: purification and characterization by mass spectrometry. Protein Expr Purif 27(2):259–266
Wennekes T, van den Berg RJ, Boot RG, van der Marel GA, Overkleeft HS, Aerts JM (2009) Glycosphingolipids–nature, function, and pharmacological modulation. Angew Chem Int Ed Engl 48(47):8848–8869
Worgall TS (2011) Sphingolipid synthetic pathways are major regulators of lipid homeostasis. Adv Exp Med Biol 721:139–148
Yamanaka S, Johnson MD, Grinberg A, Westphal H, Crawley JN, Taniike M, Suzuki K, Proia RL (2008) Early deficits in motor coordination and cognitive dysfunction in a mouse model of the neurodegenerative lysosomal storage disorder, Sandhoff disease. Behav Brain Res 193(2): 315–319
Yamato O, Matsuki N, Satoh H, Inaba M, Ono K, Yamasaki M, Maede Y (2002) Sandhoff disease in a golden retriever dog. J Inherit Metab Dis 25(4):319–320
Young SA, Mina JG, Denny PW, Smith TK (2012) Sphingolipid and ceramide homeostasis: potential therapeutic targets. Biochem Res Int 2012:248135
Yuan L, Morales CR (2011) Prosaposin sorting is mediated by oligomerization. Exp Cell Res 317(17):2456–2467
Zeidan YH, Hannun YA (2010) The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr Mol Med 10(5):454–466
Zeidan YH, Wu BX, Jenkins RW, Obeid LM, Hannun YA (2008) A novel role for protein kinase C delta-mediated phosphorylation of acid sphingomyelinase in UV light-induced mitochondrial injury. FASEB J 22(1):183–193
Zhang H, Li D, Su Y, Jiang S, Xu Y, Jiang K, Cui D (2012) Identification of the N-acylsphingosine amidohydrolase 1 gene (ASAH1) for susceptibility to schizophrenia in a Han Chinese population. World J Biol Psychiatry 13(2):106–113
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Schuchman, E.H., Simonaro, C.M. (2013). The Genetics of Sphingolipid Hydrolases and Sphingolipid Storage Diseases. In: Gulbins, E., Petrache, I. (eds) Sphingolipids: Basic Science and Drug Development. Handbook of Experimental Pharmacology, vol 215. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1368-4_1
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1368-4_1
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1367-7
Online ISBN: 978-3-7091-1368-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)