
Abstract
The tumour microenvironment is a heterogeneous and complex environment, characterized by the presence of malignant cells and non-neoplastic cellular elements, including immune and stromal cells, as well as blood vessels. Tumour progression is associated with profound alteration of myelopoiesis, which gives origin to myeloid-derived suppressor cells (MDSCs) from immature myeloid progenitors. MDSCs accumulate in the blood, secondary lymphoid organs, bone marrow, and at tumour sites, as has been observed in different cancer patients and experimental tumour models in response to pro-inflammatory cytokines and growth factors released by tumour. Upon recruitment, MDSCs exert various immunosuppressive effects to block innate and adaptive anti-tumour responses. This chapter reviews the origin and features of MDSCs, as well as the immunosuppressive mechanisms used by these cells in order to promote tumour progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Nitric Oxide
- Vascular Endothelial Growth Factor
- Stem Cell Factor
- Myeloid Progenitor
- Immature Myeloid Cell
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 Introduction
Established tumours are heterogeneous and complex masses characterized by the presence of malignant proliferating cells and non-transformed cellular elements including stromal cells, blood vessels, and inflammatory cells [1–3]. There is now considerable evidence that non-neoplastic cells present in the tumour microenvironment functionally interact with tumour cells, and in this way promote tumour progression and metastasis [4]. Various cells belonging to the adaptive immune system such as T and B lymphocytes, and to the innate immune system including macrophages, dendritic cells (DCs), polymorphonuclear neutrophils (PMNs), natural killer (NK) cells, eosinophils, mast cells, and myeloid-derived suppressor cells (MDSCs), have been identified [5].
Unlike cells found in secondary lymphoid organs, T lymphocytes present in tumours are often disregulated and unable to mount specific responses against neoplastic cells [3, 6]. This latter effect is due to immunosuppressive molecules released, such as transforming growth factor-β (TGF-β) and interleukin-10 (IL-10), and proteins expressed (galectin-1 and indoleamine-2,3-dioxigenase: IDO) by the tumour itself [7–9]. In addition, malignant cells may inhibit the development of fully differentiated immune cells, and participate in the generation of immature non-functional immune cells such as immature dendritic cells (iDCs). With regard to different factors released by tumour cells such as vascular endothelial growth factor (VEGF), interleukin-6 (IL-6), IL-10, and macrophage-colony stem factor (M-CSF), they activate the transcriptional factor signal transducer and activator of transcription (STAT) 3 and consequently inhibit maturation of DCs [10, 11]. Additional immunosuppressive immune cells found in tumours include regulatory T cells (Tregs) expressing CD4, CD25, and Foxp3 markers, T natural killer (NKT), and MDSCs.
Treg have been demonstrated to inhibit antitumor immune responses through different mechanisms, including: (1) secretion of TGF-β and IL-10, which inhibit antitumour effector responses promoted by CD4+, CD8+ and NK cells, (2) metabolic disruption through deprivation of cytokines such as IL-2, or generation of immunosuppressive adenosine by the ectoenzymes CD39 and CD73, (3) inhibition of DC maturation and functions, and (4) induction of cytolysis of CD8+ lymphocytes by granzymes A or B and perforin [12]. Several studies of mice and human models revealed higher number of Treg both in the periphery and within tumours of different histology and demonstrated that depletion of Treg significantly improved antitumoural immunity [13–15].
The NKT represent a immune cell population involved in different diseases such as autoimmune disorders, infections, and cancer. NKT express an invariant α/β TCR α24β11 which recognizes glycolipids associated to CD1d molecule. Upon direct or indirect activation by DCs, the NKT secrete Th1 and Th2 cytokines including interferon-γ (IFN-γ), IL-3, and IL-14, and are involved in immunosuppressive cell recruitment [16]. An additional immunosuppressive effect mediated by tumour cells is represented by alterations of myeloid cell differentiation. As a result, normal pathways involved in the generation of DCs, granulocytes, and macrophages are blocked, and/or the development of monocyte MDSCs (M-MDSCs), granulocyte MDSCs (G-MDSCs), suppressive DCs and tumour-associated macrophages (TAMs) is promoted [10, 17].
The MDSCs represent a heterogeneous population of cells of myeloid origin that are expanded and activated in response to growth factors and cytokines released by tumours. Once MDSCs are activated, they accumulate in lymphoid organs and tumours, where they exert T-cell immunosuppression. In the following section we will discuss the origin, the functions, and the mechanisms of action of MDSCs, as well as the strategies to target these cells for the therapeutic benefit of cancer patients.
5.2 Origin and Features of MDSCs
The bone marrow (BM) represents the site in which myelopoiesis takes place under the control of different soluble factors such as cytokines, IL-3, and growth factors: granulocyte/macrophage colony-stimulating factor (GM-CSF), macrophage CSF (M-CSF), and stem cell factor (SCF). In particular, hematopoietic stem cells give origin to common myeloid progenitors from which immature myeloid cells (IMC) are generated [10]. In healthy individuals, IMCs migrate to peripheral organs and differentiate into mature granulocytes, DCs, and macrophages. In contrast, under pathological conditions such as cancer, infections, trauma, or sepsis, specific factors inhibit IMC differentiation into mature myeloid cells, and stimulate MDSC expansion and activation [10]. Tumour-bearing mice represent suitable models where it’s possible to identify and isolate MDSCs that preferentially accumulate in the BM, spleen, and peripheral blood, and to a lesser extent in lymph nodes [18, 19]. In contrast, in cancer patients MDSCs have been preferentially found in peripheral blood (PB) and tumours [10].
The soluble factors involved in expansion and activation of MDSCs can be divided into two main groups. The first group includes molecules primarily produced by tumour cells that mediate MDSC expansion through stimulation of myelopoiesis. These factors include VEGF, SCF, GM-CSF, granulocyte CSF (G-CSF), M-CSF, gangliosides, prostaglandins, IL-6, IL-10, IL-12, metalloproteinase 9 (MMP9), and CCL2 [10, 20, 21]. Most of these factors converge on the activation of the STAT3 that has a crucial role in the following processes: (1) MDSC expansion, (2) contribution of MDSCs to angiogenesis, (3) MDSC accumulation in cancer patients, and (4) MDSC immune suppressive activity [22]. The second group of soluble factors, implicated in MDSC activation, is produced by tumour stromal cells and activated T cells. These factors, including IFN-γ, ligands for Toll-like receptors (TLRs), IL-4, IL-13, and TGF-β, are responsible for activation of different transcription factors such as STAT6, STAT1, and nuclear factor-κB (NFκB) [10]. It is noteworthy that MDSCs acquire immunosuppressive activity only after their activation.
The MDSCs have been identified in the spleen of tumour-bearing mice on the basis of their expression of two specific markers, i.e., CD11b and Gr1, and also their ability to inhibit CD8+ T lymphocyte activation through different mechanisms [23]. Murine MDSCs are also F4/80int CD11clowMHC-IIlow. In addition, some markers, including IL-4Rα, the receptor for M-CSF, and the co-stimulatory molecule CD80, have been used in order to identify an immunosuppressive MDSC fraction [24–26]. However, these latter molecules are strictly related to the tumour model used and cannot be used as general markers for MDSC identification.
More recently, different groups have shown that the antibody against Gr1 made it possible to distinguish two different MDSC fractions based on their intensity of Gr1 expression, i.e., Gr1high which express Gr1 at high intensity and prevalently constituted by granulocytes, and Gr1low with a low intensity mainly characterized by monocytes and other myeloid immature cells [24, 27]. In addition, the antibody against Gr1 molecule binds two different molecules belonging to the Ly6 super-family, Ly6G and Ly6C, which reside on the surface on granulocytes and monocytes respectively [18, 28]. On these bases, two major classes of MDSCs, i.e., G-MDSCs, consisting of CD11b+ Ly6Ghigh Ly6Clow, and M-MDSCs, which are CD11b+ Ly6Glow Ly6Chigh, have been identified in the spleen of tumour-bearing mice. In most tumour models, the G-MDSC subset is the predominant, representing almost 70–80 % of tumour-derived MDSCs [18, 28]. While G-MDSCs produce high levels of reactive oxygen species (ROS) and low levels of nitric oxide (NO), due to the increased activity of STAT3 and NADPH oxidase, M-MDSC subset has up-regulated expression of STAT1 and inducible nitric oxide synthase (iNOS) with consequent high levels of NO and low concentrations of ROS [18, 28].
More recent studies have described a novel immunomagnetic method which made it possible to separate MDSCs into three fractions, i.e., Gr1low, Gr1int, and Gr1high [29]. The fraction constituted by Gr1high characterized by granulocytes Ly6Ghigh was shown to possess a weak immunosuppressive effect on the response mediated by allogeneic or antigen-specific T lymphocytes [29]. In contrast, the fractions Gr1low and Gr1int, composed of monocytes and immature myeloid cells with a ring shape nucleus respectively, were highly immunosuppressive [29]. Similarly to the splenic M-MDSCs and G-MDSCs, analogous subsets have been also identified in tumour infiltrates. In two different tumour models, more than 90 % were M-MDSCs CD11b+Gr1lowF4/80+IL4-Rα+CCR2+CX3CR1+ , and the remainder were G-MDSCs Gr1highF4/80low [30].
5.3 Mechanisms of MDSC Immunosuppressive Activity
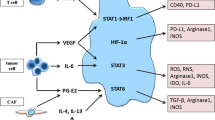
MDSCs suppress multiple effectors of adaptive and innate immunity (Fig. 5.1) [17]. In particular, MDSCs have been shown to:

Immunosuppressive mechanisms mediated by myeloid-derived suppressor cells. CD4+ and CD8+ T cell activation is inhibited by arginase 1, inducible nitric oxide synthase (iNOS), generation of reactive oxygen species (ROS) and cysteine deprivation, and induction of T regulatory cells (Tregs) is mediated by IL-10 and transforming growth factor-β (TGF-β). Innate immunity is suppressed by down-regulation of dendritic cell (DC), production of IL-12 by macrophages, and by inhibition of natural killer (NK) cell cytotoxicity. Myeloid-derived suppressor cells (MDSCs) produce a high amount of IL-10 that induces Treg and Th2 cells and inhibits IL-12 production
-
1.
Inhibit CD4+ and CD8+ activation and proliferation in a major histocompatibility complex (MHC) restricted or unrestricted and antigen-specific manner [31, 32]
-
2.
Indirectly affect T-cell activation by inducing Treg expansion thanks to the production of IL-10 and TGF-β or arginase 1 (ARG1) [33]
-
3.
Stimulate the conversion of macrophages into M2 phenotype through the secretion of IL-10 and downregulation of M1 macrophage production of IL-12 [34]
-
4.
Inhibit cytotoxicity of NK cells and their IFN-γ production [35]. However, the role of MDSCs on NK cell activity is controversial, since it has been reported that MDSCs expressing Rae-I, the ligand for NKG2D, can activate NK cells as well [36]
-
5.
Interact with type II iNKT that facilitate tumour progression by producing IL-13, which induces the accumulation of MDSCs [27, 32, 37]
Multiple mechanisms by which MDSCs mediate immune evasion have been elucidated in the mouse models and will be discussed below.
5.3.1 T-Cell Deprivation of Essential Amino-acids
The MDSCs cause the depletion of two amino-acids required by lymphocytes for their growth and differentiation, i.e., l-arginine and l-cysteine. Extracellular l-arginine is depleted through its consumption by ARG1, which is induced in the cytosol of MDSCs by Th2 cytokines such as IL-4 and IL-13 and TGF-β in a STAT6-dependent and -independent manner [38]. MDSC activation of ARG1 reduces the extracellular levels of l-arginine, which leads to down-regulation of the CD3ζ chain of the T-cell receptor (TCR) and its signal transduction [39, 40]. In addition, the depletion of l-arginine can cause a G0-G1 cell cycle arrest through inhibition of the phosphoinositide 3-kinase (PI3K)/mammalian target of the rapamycin (mTOR) pathway [10, 41].
Cysteine is an essential amino-acid that serves as a fundamental substrate for generation of glutathione, a major intracellular molecule that protects cells from oxidative stress [42, 43]. Cysteine can be synthesized from intracellular cystine through the action of cystathionase, or alternatively can be imported as the oxidized form of cystine through ASC neutral amino-acid plasma membrane transporter. T cells lack cystathionase, and have a defective cystine transporter [42]. As a result, T cells must obtain cysteine from extracellular sources. DCs and macrophages normally have large amounts of cysteine that in part derives from the import of cystine, which is sequentially reduced, and in part is intracellularly synthesized through the enzyme cystathionase. During antigen presentation, DCs, which are in close proximity to lymphocytes, release the surplus cysteine that is readily taken up by T cells. In contrast to DCs, MDSCs, which do not express cystathionase and ASC transporter, generate cysteine from imported cystine. As a result, MDSCs deplete the environment of cystine, do not export cysteine and consequently prevent T-cell proliferation and activation [38].
5.3.2 Generation and Release of Oxidizing Molecules
The MDSCs express different enzymes involved in the production of ROS and NO, including NADPH oxidase (also known as NOX2) and iNOS respectively [10]. MDSCs also express the calcium-binding proteins S100A8 and S100A9, which together with gp91phox are part of the NOX complex, responsible for the increased production of ROS. ROS include superoxide anion (O2 −) that is converted to hydrogen peroxide (H2O2) [23]. The latter molecule is then involved in the downregulation of the CD3ζ chain of TCR, thereby inhibiting T-cell activation through TCR [44]. iNOS is responsible for NO production, which interacts with O2 − to form the highly reactive peroxynitrite anion (ONOO−). NO is able to block the phosphorylation and subsequent activation of proteins associated to IL-2 receptor such as Janus kinase-1 (JAK-1), JAK-3, STAT5, extracellular-signal-regulated kinases (ERK), and AKT [45–47]. Moreover, NO can also decrease the stability of mRNA of IL-2 and the release of IL-2 [48]. It is worthy of note that in the presence of low cytosolic levels of l-arginine, iNOS activity can be modified and converted to induce the production of O2 − together with NO [49, 50]. Peroxynitrite anion and NO cause the nitration or nitrosylation of TCR, CD8, and CD3 chains, thereby blocking T-cell activation. In particular, the first studies performed by Kusmartsev et al. [51] demonstrated that peroxynitrite may cause apoptosis of activated T lymphocytes through the inhibition of phosphorylation events in T cell signal transduction. Recently, it has been shown that peroxynitrite can also impair the TCR recognition of MHC-I-peptides due to nitrosylation of TCR tyrosines [31]. Furthermore, CCL2, a chemokine involved in inducing T-cell migration upon interaction with CCR2 expressed by T cells, can be modified by nitrosylation and inhibited in its function of T lymphocyte recruitment [52].
These observation prove that ARG1 and iNOS represent an important immunosuppressive mechanism mediated by MDSCs, and that the simultaneous inhibition of these enzymes may in part reconstitute T lymphocyte responsiveness [40].
5.3.3 Induction of Development and Expansion of Treg
In an experimental model of murine colon carcinoma, MDSCs have been shown to induce the expansion of Treg by IL-10, TGF-β, decrease of l-arginine and upregulation of CD40–CD40L interactions (essential for Treg activation) in an IFN-γ-dependent manner [25, 53]. In the model of ovarian carcinoma, MDSCs expressing high levels of CD80 cooperate with CD152+ Treg in order to inhibit T-cell activation [26]. Additionally, MDSCs mediate Treg induction with a mechanism that requires ARG1 but is transforming growth factor-beta independent [33]. Interestingly, human CD14+HLA-DRlow/− MDSCs promote the transdifferentiation of TH17 cells into FOXP3−-induced Treg by producing TGF-β and retinoic acid [33].
5.3.4 Interference with T-Cell Migration and Viability
The expression of metalloproteinase ADAM17 by MDSCs induces the cleavage of CD62L, which is necessary for T-cell migration to draining lymph nodes [54]. Furthermore, MDSCs express galectin 9 which binds to T immunoglobulin and mucin domain-containing protein 3 (TIM3) on lymphocytes and induces T cell apoptosis [55]. As discussed above, CCL2 may be modified by MDSC-derived peroxynitrite, thereby impairing CD8+ migration to the tumour core [52].
5.4 Molecular Mechanisms of MDSC Activation in Cancer
Expansion and activation of MDSCs are promoted by different soluble factors which can be classified into two groups. The first group includes tumour-derived soluble factors (TDSF) that induce MDSC expansion through stimulation of myelopoiesis and inhibition of differentiation of mature myeloid cells [11]. In contrast, the second group is characterized by factors released by activated T lymphocytes and tumour-derived stromal cells implicated in MDSC activation [11].
Typical TDSF are represented by cytokines (IL-3, IL-1β, IL-6, IL-10), growth factors (GM-CSF, VEGF, SCF, M-CSF, TGF-β), chemokines (CCL2), prostaglandins (PGE2), and proinflammatory proteins such as S100A8/S100A9. In general, none of these factors is sufficient to induce and activate MDSCs by itself [11].
IL-3 represents one of the first cytokines able to induce myelopoiesis in tumour-bearing mice and in particular to promote the expansion of immunosuppressive MDSCs [56]. GM-CSF, commonly released by different human tumours and murine tumour cell lines, has been shown to induce CD11b+Gr1+ without the participation of other cytokines. Accordingly, the administration of recombinant GM-CSF in mice affected by cancer promotes recruitment of MDSCs and their immunosuppressive function [56–59]. The presence of GM-CSF has been proposed as a negative prognostic factor in patients with head and neck squamous carcinoma. Thus patients with tumours releasing relevant amounts of GM-CSF showed higher incidence of relapse and metastases than those characterized by low GM-CSF production [60]. However, it is worth mentioning that GM-CSF is also used to transfect tumour cells administered as potent vaccines, since this cytokine is also able to recruit and expand professional antigen-presenting cells (APC) [61]. The contradictory effect mediated by GM-CSF seems to be dependent on the dosage used; only at high doses could GM-CSF mobilize myeloid precursors from the bone marrow, and induce immunosuppressive MDSC accumulation in the PB and lymphoid organs [62]. VEGF, which is secreted by various tumours and is often associated with poor prognosis, has been shown to inhibit in-vivo DC differentiation and promote MDSC Gr1+ expansion [63–66]. The use of amino-biphosphonates, inhibitors of MMP-9 which regulates VEGF bioavailability, made it possible to reduce the accumulation of MDSCs [67].
Another factor able to induce MDSC recruitment is SCF, whose silencing or neutralization by specific antibodies is shown to reduce CD11b+Gr-1+CD115+ in the BM of tumour-bearing mice, to decrease the tumour progression and angiogenesis, and to restore the proliferation of T lymphocytes [68].
PGE2, produced by cyclooxygenase 2 (COX2), represents a crucial factor in inducing MDSC expansion [69, 70]. PGE2 interacts with different PGE2 receptors (EP1, EP2, and EP4), among which EP4 seems to be involved in the MDSC induction of ARG1 [70]. Similarly, knock-out mice for EP2 showed decreased tumour growth and less accumulation of intra-tumour MDSCs [17].
Pro-inflammatory cytokines such as IL-1β and IL-6, which are present in the microenvironment of many tumours, have been shown to dramatically increase the rate of immunosuppressive MDSC accumulation [71–73]. IL-1β also increases MDSC suppression of innate immunity by facilitating cross-talk between MDSCs and macrophages. Similarly, the pro-inflammatory S100A8/A9 proteins, highly expressed by tumour-infiltrating leucocytes, participate in an autocrine loop produced by MDSCs and involved in the recruitment of these latter cells [74–76]. In this manner, an immunosuppressive tumour microenvironment is maintained. Most of the factors listed above converge toward the activation of JAK proteins and STAT3 transcription factor, which represent the main regulatory factors of MDSC expansion [77–79]. In this connection, STAT3 has been demonstrated to upregulate different target proteins including:
-
S100A8, S100A9, two calcium-binding proinflammatory proteins involved in MDSC accumulation [75, 76, 80]
-
iNOS and NADPH oxidase (NOX2), related to NO and ROS production by MDSCs [81]
-
Cyclin-D, MYC, BCL-XL, which promote MDSC survival [11]
-
CCAAT/enhancer-binding protein-β (C/EBPβ), an important regulator of differentiation of myeloid progenitors to functional MDSCs [82]
Reduced expansion of MDSCs in STAT3−/− conditional knockout mice, as well as normal mice, treated with specific inhibitors of STAT3, supports the hypothesis that STAT3 has a crucial role in MDSC expansion and activation [78, 83]. Moreover, persistent activation of STAT3 in myeloid progenitors prevents differentiation into mature myeloid elements and promotes expansion of MDSCs [80].
The transcription factors STAT1 (mainly activated by IFN-γ) and STAT6 (activated by IL-4 and IL-13) are additional important regulators of MDSC activation through its effects on iNOS and ARG1 expression, whereas STAT5 (activated by GM-CSF) is involved in promoting MDSC expansion through the induction of cyclins, survivin, B cell lymphoma XL (BCL-XL), and MYC [11, 84–86]. Finally, NF-κB, upon activation by TLR ligands, has a relevant role in inducing the mobilization of MDSCs to sites of tumour growth, probaly through targeting ARG1 and iNOS and the pro-inflammatory mediators PGE2 and COX2, which enhance MDSC accumulation and suppressive activity [11, 70, 87]. Figure 5.2 summarizes the molecular mechanisms involved in tumour-mediated induction of MDSCs.

Molecular mechanisms involved in tumour-mediated induction of myeloid-derived suppressor cells. Tumour cells release different pro-inflammatory cytokines and growth factors that are responsible for the induction of multiple transcription factors including signal transducer and activator of transcription (STAT)1, STAT3, STAT5, STAT6, nuclear factor-κB (NF-κB). Each of these factors regulate the expression of proteins involved in myeloid-derived suppressor cell (MDSC)-mediated functions such as inducible nitric oxide synthase (iNOS), NADPH oxidase (NOX2), arginase 1 (ARG1), S100A8, S100B, B-cell lymphoma XL (BCL-XL), cyclin D1, survivin, MYC, CCAAT/enhancer-binding protein-β (C/EBPβ), prostaglandin E2 (PGE2). G-CSF granulocyte colony-stimulating factor, GM-CSF granulocyte–macrophage colony-stimulating factor, IFNγ interferon-γ, IL interleukin, ROS reactive oxygen species, TLR Toll-like receptor, VEGF vascular endothelial growth factor
5.5 MDSCs in Cancer Patients
MDSCs have been identified in the peripheral blood and in tumour infiltrates of patients affected by different tumour types; however, the MDSC phenotype in humans is less definite in comparison to that found in mice. Similarly to MDSCs found in mice, in humans there have been two different MDSC subtypes identified so far, for the monocyte and the granulocyte. The prevalence of M-MDSCs compared to G-MDSCs depends on the tumour type; for example the PB of patients affected by head and neck carcinoma, non small lung cancer, prostate and breast carcinoma, and multiple myeloma is characterized by the presence of M-MDSCs that are able to inhibit allogeneic and antigen-specific T responses, and may differentiate into mature DCs in presence of GM-CSF e IL-4 [59, 88, 89]. More recently, an highly immunosuppressive MDSC population has been identified in patients affected by metastatic melanoma. These cells are CD14+HLA-DR−/low, and induce immunosuppression through a mechanism mediated by TGF-β independently from ARG1 and NOS [59]. What is more, CD14+HLA-DR−/low have been also isolated from the PB of hepatocarcinoma patients, where they inhibit lymphocyte proliferation in an ARG1-dependent manner and induce Treg [90].
The first evidence of the presence of G-MDSCs in cancer patients was found in the research that characterized cells CD11b+CD15+CD14− expressing ARG1 in patients affected by renal carcinoma [91]. Subsequently, the same group defined more precisely that G-MDSCs in renal carcinoma patients belonged to a subset of activated granulocytes CD66b+VEGFR1+CD62LlowCD16low able to secrete ARG1 after degranulation [92]. Additional studies revealed the presence of IL4Rα marker (typically expressed in MDSCs isolated from different experimental tumour models developed in mice) also in G- and M-MDSCs defined in melanoma and colon carcinoma patients [93]. However, the immunosuppressive activity was identified only in the granulocytic CD14+ fraction [93].
Other important issues addressed in cancer patients are whether MDSC presence correlates with clinical cancer stage, and whether MDSC presence in PB can be modulated by chemotherapy. In this regard, an interesting paper by Diaz-Montero et al. demonstrated that MDSCs, defined as Linlow/−, HLADR−, CD33+, CD11b+, were present at higher percentages in the peripheral blood of cancer patients than in that of healthy donors [94]. Moreover, among stage IV patients, those with extensive metastatic burden had the highest number of MDSCs. Two chemotherapy regimens were evaluated, and in both cases MDSCs were increased after therapy [94].
5.6 Therapeutic Targeting of MDSCs
It has become increasingly clear that successful cancer immunotherapy relies on the elimination of the immunosuppressive barrier induced by different elements including MDSCs [10]. For this purpose, different therapeutic approaches are currently being explored in order to:
-
Induce myeloid differentiation
-
Inhibit MDSC expansion
-
Eliminate MDSCs
-
Inhibit MDSC functions
One of the most promising approaches is based on the promotion of myeloid differentiation. Vitamin A has been identified as a compound that mediates this effect. Specifically, vitamin A metabolites such as retinoic acid have been shown to promote the differentiation of myeloid progenitors into DCs and macrophages [95]. Furthermore, administration of therapeutic concentrations of all-trans retinoic acid (ATRA) in renal-cell cancer patients induced reduction of MDSC numbers and promoted MDSC differentiation into DCs and macrophages [95–97]. Similar effects have been obtained in tumour-bearing mice in which ATRA eliminated immature myeloid cells and improved the effect of vaccination [97]. The mechanism of ATRA effect on MDSCs occurred through upregulation of glutathione synthesis and reduction of ROS [98]. Vitamin D3 is another compound that has the potential to decrease MDSC numbers and promote myeloid differentiation in cancer patients [99].
Since MDSC expansion is regulated by soluble factors released by tumours, a promising approach is the use of agents able to neutralize the effect of these factors. In this regard, it has been shown that inhibitors of SCF signaling decreased MDSC expansion, an antibody against VEGF (Avastin) reduced the size of CD11b+VEGFR1+ population in PB of cancer patients, and an inhibitor of MMP9 diminished the number of MDSCs in the spleen and tumours of cancer-bearing mice [65, 67]. An additional approach is the use of agents that inhibit MDSC function, such as inhibitors of COX2 and ROS. COX2 is a well-known enzyme involved in the production of PGE2, which upregulates ARG1 expression by MDSCs [69, 70]. Accordingly, COX2 inhibitors were found to downregulate the expression of ARG1 by MDSCs and to improve the therapeutic efficacy of immunotherapy [91, 100]. Nitroaspirin, a non steroidal anti-inflammatory drug, and phosphodiesterase 5 (PDE5) inhibitors such as sildenafil have been found to limit the activity of ARG1 and iNOS in splenic MDSCs and to improve the function of tumour-antigen-specific T cells [89, 101].
Finally, MDSCs can be eliminated using some chemotherapeutic drugs. For example, gemcitabine reduced the number of MDSCs and improved T-cell responses in cancer-bearing mice [102]. MDSC recruitment can also be reduced by the use of STAT3 inhibitors such as sunitinib that decreased the number of MDSCs in patients with renal cancer [103].
The identification of different MDSC subpopulations with different immunosuppressive functions will permit the design of more specific therapeutic strategies able to inhibit only the most immunosuppressive population, thereby avoiding the induction of strong myeloablation.
Abbreviations
- APC:
-
Antigen-presenting cells
- ARG1:
-
Arginase 1
- ATRA:
-
All-trans retinoic acid
- BCL-XL:
-
B cell lymphoma XL
- BM:
-
Bone marrow
- C/EBPβ:
-
CCAAT/enhancer-binding protein-β
- COX2:
-
Cyclooxygenase 2
- DCs:
-
Dendritic cells
- ERK:
-
Extracellular-signal-regulated kinases
- G-CSF:
-
Granulocyte CSF
- GM-CSF:
-
Granulocyte/macrophage colony-stimulating factor
- G-MDSCs:
-
Granulocyte MDSCs
- iDCs:
-
Immature dendritic cells
- IFN-γ:
-
Interferon-γ
- IL:
-
Interleukin
- IMC:
-
Immature myeloid cells
- iNOS:
-
Inducible nitric oxide synthase
- JAK-1:
-
Janus kinase-1
- M-CSF:
-
Macrophage CSF
- MDSCs:
-
Myeloid-derived suppressor cells
- MHC:
-
Major histocompatibility complex
- M-MDSCs:
-
Monocyte-MDSCs
- MMP9:
-
Metalloproteinase 9
- mTOR:
-
Mammalian target of rapamycin
- NFκB:
-
Nuclear factor-κB
- NK:
-
Natural killer
- NKT:
-
T natural killer
- NO:
-
Nitric oxide
- PB:
-
Peripheral blood
- PDE5:
-
Phosphodiesterase 5
- PGE2:
-
Prostaglandin E2
- PI3K:
-
Phosphoinositide 3-kinase
- PMNs:
-
Polymorphonuclear neutrophils
- ROS:
-
Reactive oxygen species
- SCF:
-
Stem cell factor
- STAT 3:
-
Transcriptional factor signal transducer and activator of transcription 3
- TAMs:
-
Tumour-associated macrophages
- TDSF:
-
Tumour-derived soluble factors
- TGF-β:
-
Transforming growth factor-β
- TLRs:
-
Toll-like receptors
- Treg:
-
Regulatory T cells
- VEGF:
-
Vascular endothelial growth factor
References
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454:436–444
Mantovani A, Sica A (2010) Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol 22:231–237
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Joyce JA, Pollard JW (2009) Microenvironmental regulation of metastasis. Nat Rev Cancer 9:239–252
Kerkar SP, Restifo NP (2012) Cellular constituents of immune escape within the tumor microenvironment. Cancer Res 72:3125–3130
Yu P, Rowley DA, Fu YX, Schreiber H (2006) The role of stroma in immune recognition and destruction of well-established solid tumors. Curr Opin Immunol 18:226–231
Khong HT, Restifo NP (2002) Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol 3:999–1005
Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, Mordoh J, Fainboim L, Podhajcer OL, Rabinovich GA (2004) Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell 5:241–251
Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ (2003) Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 9:1269–1274
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268
Vignali D (2008) How many mechanisms do regulatory T cells need? Eur J Immunol 38:908–911
Zou W (2006) Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 6:295–307
Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B (2003) Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res 9:606–612
Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E (1999) Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res 59:3128–3133
Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, Carbone DP, Paul WE, Berzofsky JA (2000) NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol 1:515–520
Ostrand-Rosenberg S, Sinha P (2009) Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 182:4499–4506
Youn JI, Nagaraj S, Collazo M, Gabrilovich DI (2008) Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 181:5791–5802
Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V (2010) Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol 22:238–244
Talmadge JE (2007) Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res 13:5243–5248
Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V (2008) Tumor-induced tolerance and immune suppression by myeloid-derived suppressor cells. Immunol Rev 222:162–179
Condamine T, Gabrilovich DI (2010) Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 32:19–25
Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI (2004) Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol 172:989–999
Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P, Bicciato S, Bronte V (2006) Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 116:2777–2790
Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH (2006) Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 66:1123–1131
Yang R, Cai Z, Zhang Y, Yutzy WH, Roby KF, Roden RB (2006) CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res 66:6807–6815
Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, Letterio JJ, Paul WE, Berzofsky JA (2003) Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med 198:1741–1752
Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA (2008) Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 111:4233–4244
Dolcetti L, Peranzoni E, Ugel S, Marigo I, Fernandez Gomez A, Mesa C, Geilich M, Winkels G, Traggiai E, Casati A, Grassi F, Bronte V (2010) Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol 40:22–35
Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R, Shibata T, Takami T (2008) Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol 83:1136–1144
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI (2007) Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 13:828–835
Sinha P, Clements VK, Ostrand-Rosenberg S (2005) Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res 65:11743–11751
Serafini P, Mgebroff S, Noonan K, Borrello I (2008) Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res 68:5439–5449
Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S (2007) Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 179:977–983
Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, Zhang HG (2007) Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 109:4336–4342
Nausch N, Galani IE, Schlecker E, Cerwenka A (2008) Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood 112:4080–4089
Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, Godfrey DI, Ostrand-Rosenberg S, Smyth MJ, Berzofsky JA (2005) A nonclassical non-Valpha14Jalpha18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med 202:1627–1633
Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res 70:68–77
Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC (2002) Regulation of T cell receptor CD3zeta chain expression by L-arginine. J Biol Chem 277:21123–21129
Bronte V, Zanovello P (2005) Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol 5:641–654
Rodriguez PC, Quiceno DG, Ochoa AC (2007) L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 109:1568–1573
Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother 59:1593–1600
Sakakura Y, Sato H, Shiiya A, Tamba M, Sagara J, Matsuda M, Okamura N, Makino N, Bannai S (2007) Expression and function of cystine/glutamate transporter in neutrophils. J Leukoc Biol 81:974–982
Schmielau J, Finn OJ (2001) Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res 61:4756–4760
Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM (2002) Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol 168:689–695
Bingisser RM, Tilbrook PA, Holt PG, Kees UR (1998) Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol 160:5729–5734
Pericle F, Kirken RA, Bronte V, Sconocchia G, DaSilva L, Segal DM (1997) Immunocompromised tumor-bearing mice show a selective loss of STAT5a/b expression in T and B lymphocytes. J Immunol 159:2580–2585
Macphail SE, Gibney CA, Brooks BM, Booth CG, Flanagan BF, Coleman JW (2003) Nitric oxide regulation of human peripheral blood mononuclear cells: critical time dependence and selectivity for cytokine versus chemokine expression. J Immunol 171:4809–4815
Xia Y, Roman LJ, Masters BS, Zweier JL (1998) Inducible nitric-oxide synthase generates superoxide from the reductase domain. J Biol Chem 273:22635–22639
Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P (2003) L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol 24:302–306
Kusmartsev SA, Li Y, Chen SH (2000) Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J Immunol 165:779–785
Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M, Savino B, Colombo P, Jonjic N, Pecanic S, Lazzarato L, Fruttero R, Gasco A, Bronte V, Viola A (2011) A chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med 208:1949–1962
Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen SH (2010) Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res 70:99–108
Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S (2009) Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol 183:937–944
Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK (2011) Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol 32:345–349
Young MR, Wright MA, Young ME (1991) Antibodies to colony-stimulating factors block Lewis lung carcinoma cell stimulation of immune-suppressive bone marrow cells. Cancer Immunol Immunother 33:146–152
Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, Restifo NP (1999) Unopposed production of granulocyte–macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol 162:5728–5737
Fu YX, Watson G, Jimenez JJ, Wang Y, Lopez DM (1990) Expansion of immunoregulatory macrophages by granulocyte–macrophage colony-stimulating factor derived from a murine mammary tumor. Cancer Res 50:227–234
Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L (2007) Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte–macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol 25:2546–2553
Young MR, Wright MA, Pandit R (1997) Myeloid differentiation treatment to diminish the presence of immune-suppressive CD34+ cells within human head and neck squamous cell carcinomas. J Immunol 159:990–996
Dranoff G (2002) GM-CSF-based cancer vaccines. Immunol Rev 188:147–154
Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I (2004) High-dose granulocyte–macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res 64:6337–6343
Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP (1996) Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med 2:1096–1103
Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP (1998) Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 92:4150–4166
Kusmartsev S, Eruslanov E, Kubler H, Tseng T, Sakai Y, Su Z, Kaliberov S, Heiser A, Rosser C, Dahm P, Siemann D, Vieweg J (2008) Oxidative stress regulates expression of VEGFR1 in myeloid cells: link to tumor-induced immune suppression in renal cell carcinoma. J Immunol 181:346–353
Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N (2007) Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol 25:911–920
Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP (2007) Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor–bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res 67:11438–11446
Pan PY, Wang GX, Yin B, Ozao J, Ku T, Divino CM, Chen SH (2008) Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood 111:219–228
Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S (2007) Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 67:4507–4513
Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC (2005) Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med 202:931–939
Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S (2007) Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 67:10019–10026
Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S (2006) Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol 176:284–290
Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, Voronov E, Apte RN (2005) CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol 175:8200–8208
Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA (2003) Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol 170:3233–3242
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI (2008) Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 205:2235–2249
Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G (2008) Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol 181:4666–4675
Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D (2004) Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol 172:464–474
Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI (2005) Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res 65:9525–9535
Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R (2010) Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res 70:4335–4345
Foell D, Wittkowski H, Vogl T, Roth J (2007) S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol 81:28–37
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI (2009) Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol 182:5693–5701
Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P, Cozzi E, Mandruzzato S, Bronte V (2010) Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 32:790–802
Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, Jove R, Pardoll D, Yu H (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 11:1314–1321
Kusmartsev S, Gabrilovich DI (2005) STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol 174:4880–4891
Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, Colombo MP, Zanovello P (2003) IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol 170:270–278
Munera V, Popovic PJ, Bryk J, Pribis J, Caba D, Matta BM, Zenati M, Ochoa JB (2010) Stat 6-dependent induction of myeloid derived suppressor cells after physical injury regulates nitric oxide response to endotoxin. Ann Surg 251:120–126
Bunt SK, Clements VK, Hanson EM, Sinha P, Ostrand-Rosenberg S (2009) Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J Leukoc Biol 85:996–1004
Vuk-Pavlovic S, Bulur PA, Lin Y, Qin R, Szumlanski CL, Zhao X, Dietz AB (2010) Immunosuppressive CD14+HLA-DRlow/− monocytes in prostate cancer. Prostate 70:443–455
Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I (2006) Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med 203:2691–2702
Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, Greten TF, Korangy F (2008) A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 135:234–243
Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O’Neill A, Mier J, Ochoa AC (2005) Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res 65:3044–3048
Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, Ochoa AC (2009) Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res 69:1553–1560
Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, Zanon A, Rossi CR, Nitti D, Bronte V, Zanovello P (2009) IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol 182:6562–6568
Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ (2009) Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother 58:49–59
Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM (2001) Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol 166:5398–5406
Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI (2001) Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol 166:678–689
Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich D (2003) All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res 63:4441–4449
Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI (2007) Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res 67:11021–11028
Lathers DM, Clark JI, Achille NJ, Young MR (2004) Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3. Cancer Immunol Immunother 53:422–430
Talmadge JE, Hood KC, Zobel LC, Shafer LR, Coles M, Toth B (2007) Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int Immunopharmacol 7:140–151
De Santo C, Serafini P, Marigo I, Dolcetti L, Bolla M, Del Soldato P, Melani C, Guiducci C, Colombo MP, Iezzi M, Musiani P, Zanovello P, Bronte V (2005) Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci USA 102:4185–4190
Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM (2005) Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res 11:6713–6721
Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, Dreicer R, Bukowski R, Finke JH (2009) Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res 15:2148–2157
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Wien
About this chapter
Cite this chapter
Raffaghello, L., Bianchi, G. (2014). Myeloid-Derived Suppressor Cells and Tumor Growth. In: Klink, M. (eds) Interaction of Immune and Cancer Cells. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1300-4_5
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1300-4_5
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1299-1
Online ISBN: 978-3-7091-1300-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)