
Abstract
Owing to their properties, dendritic cells (DCs) are often called “nature’s adjuvants,” and thus have become the natural targets for antigen delivery. DCs provide an essential link between the innate and the adaptive arms of the immune system. DCs control both tolerance and immunity. Therefore, DCs are key targets for both preventive and therapeutic vaccination. Herein, we will discuss the physiology of DCs as it applies to immunotherapy of cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Introduction
The immune system has the potential to eliminate neoplastic cells. Perhaps the most compelling evidence of tumor immunosurveillance in humans is provided by paraneoplastic diseases which link neurological disorders to an anti-tumor response [1]. Onconeural antigens, normally expressed on neurons, can also be expressed in breast cancer cells [2]. Some patients develop a strong antigen-specific CD8+ T cell response which controls cancer but concomitantly results in autoimmune cerebellar degeneration, causing a severe neurologic disease [3, 4].
The adoptive transfer of cancer antigen-specific effector T cells in patients can result in rejection of established tumors, thereby illustrating the potential of tumor immunotherapy [5]. Ideally, one would like to directly induce efficient tumor-specific T cells through vaccination, including effector T cells able to reject tumors, and memory T cells able to prevent tumor relapse. DCs are essential in generation of immune responses, and as such represent targets and vectors for vaccination. Furthermore, murine models demonstrate that the generation of protective anti-tumor immunity depends on the presentation of tumor antigens (Ags) by DCs [6, 7]. There, DCs can capture tumor antigens released from tumor cells, either alive or dying, and cross-present these antigens to T cells in tumor,draining lymph nodes. This antigen presentation results in the generation of tumor-specific effector T cells that contribute to tumor rejection [6, 7]. Thus, DCs represent important targets for therapeutic interventions in cancer.
Preventive vaccines are designed to initiate protective humoral immune responses. Today, more than 70 preventive vaccines have been licensed for use against approximately 30 microbes, sparing countless lives [8]. However, effective vaccines remain elusive for diseases such as human immunodeficiency virus (HIV)-induced acquired immune deficiency syndrome, plasmodium-induced malaria, virus-induced hepatitis C, and Mycobacterium-induced tuberculosis, to cite a few [8]. These require therapeutic vaccines able to elicit strong cellular immunity, in particular in cytotoxic T cells, which is necessary to eliminate existing disease, i.e., the cells that are infected with the causative agent and/or malignant cells (Fig. 4.1). Here, we will discuss the biology of human DC subsets in the context of vaccination.

Therapeutic vaccines. Therapeutic vaccines are designed to elicit cellular immunity, that is to say, prime new T cells, as well as induce a transition from chronically activated non-protective CD8+ T cells to healthy CD8+ T cells. The features of healthy CD8+ T cells include their ability to generate cytotoxic T lymphocytes (CTLs) that reject cancer and to provide long-lived memory CD8+ T cells able to rapidly generate new effector T cells secreting cytotoxic molecules thereby preventing relapse. Numerous approaches to therapeutic vaccines are being pursued. Their common denominator is the action via DCs
4.2 The Biology of DC Subsets
4.2.1 Basics of DC Biology
DCs are a rare cell type that was discovered by Ralph Steinman in 1973. After four decades of research, it is now clear that DCs are at the center of the immune system through their ability to control both tolerance and immunity [9, 10]. DCs are bone-marrow-derived cells that seed all tissues (reviewed in [10]). They are poised to sample the environment and transmit the gathered information to cells of the adaptive immune system, i.e., T cells and B cells [9, 10]. In peripheral tissues, DCs capture Ags through several complementary mechanisms. DCs launch the immune response by presenting the captured Ag in the form of peptide-MHC complexes to naïve, i.e., antigen-inexperienced T cells in lymphoid tissues. Upon interaction with DCs, naïve CD4+ and CD8+ T cells differentiate into antigen-specific memory T cells with different functions. CD4+ T cells can become Th1, Th2, Th17, and T follicular helper (Tfh) cells that help B cells differentiate into antibody-secreting cells, as well as regulatory T cells (Tregs) that downregulate the functions of other lymphocytes. Naïve CD8+ T cells can give rise to cytotoxic effector lymphocytes (CTLs). The plasticity of DCs in response to extrinsic signals, and the existence of distinct DC subsets with distinct functions, contribute to the mounting of highly diverse immune responses. DCs are essential in pathogen resistance including different viruses, bacteria, and parasites, as demonstrated using DC-depleted mice [11].
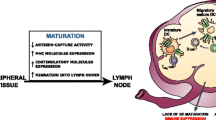
In the steady state, non-activated (immature) DCs present self-antigens to T cells, thereby leading to tolerance through either T-cell deletion or differentiation of regulatory/suppressor T cells. These immature DCs have special characteristics, including: (1) the ability to efficiently capture antigens, (2) accumulation of MHC class II molecules in the late endosome–lysosomal compartment, (3) low levels of costimulatory molecule expression, (4) a unique set of chemokine receptors that allow their migration to lymphoid tissues (e.g., CCR7), and (5) a limited capacity to secrete cytokines [12]. In contrast, mature antigen-loaded DCs can launch the differentiation of antigen-specific T cells into effector cells with unique functions and cytokine profiles (Fig. 4.2). Indeed, immature DCs promptly respond to environmental signals, and differentiate into mature DCs. DC maturation is associated with (1) the down-regulation of antigen-capture activity, (2) the increased expression of surface MHC class II molecules and costimulatory molecules, (3) the ability to secrete cytokines [12], and (4) the acquisition of CCR7, which allows migration of the DC into the draining lymph node [12]. However, DC maturation does not result in a unique phenotype. Rather, in response to different signals that are provided by different microbes either directly or through the surrounding cells, DCs acquire distinct phenotypes that eventually contribute to diverse immune responses. In addition to cytokines or direct microbial signals, the ligation of CD40 represents an essential signal for the differentiation into fully mature DCs able to launch adaptive T cell immunity [13].

Launching of the immune response. Antigen(s) can reach lymph nodes via two pathways: one pathway is via lymphatics where the antigen is captured by lymph-node resident DCs. The other pathway is mediated by tissue resident DCs. There, immature DCs capture antigens; tissue- and/or pathogen-derived signals trigger DC activation, their migration towards secondary lymphoid organs and simultaneous maturation. DCs display antigens in the context of classical MHC class I and class II or non-classical CD1 molecules, which allow selection of rare antigen-specific T lymphocytes. Activated T cells drive DCs towards their terminal maturation, which induces further expansion and differentiation of lymphocytes into effector cells. If DCs do not receive maturation signals, they will remain immature, and antigen presentation will lead to immune regulation and/or suppression
4.2.2 Basics of DC Subsets
Human blood DC subsets can be distinguished by the differential expression of three surface molecules: CD303 (BDCA-2), CD1c (BDCA-1), and CD141 (BDCA3). CD303+ pDCs represent a front line of anti-viral immunity through their ability to secrete large amounts of type I interferon (IFN) in response to virus encounter [14]. Their pre-synthesized stores of MHC class I may permit a rapid initial CD8+ T cell response to viral infections [15]. pDCs-derived type I IFN may promote the immunogenic maturation of other DC populations [16], thereby helping in the activation of novel T-cell clones. In their resting state, pDCs are considered as playing an important role in tolerance, including oral tolerance [16].
Human CD141+CD1c– DCs uniquely express Toll-like receptor (TLR) 3, produce interleukin (IL) 12, and efficiently cross-prime CD8+ T cells when activated with poly I:C [17–23]. However, other human DCs such as epidermal Langerhans cells (LCs) [24, 25] and CD1c+ DCs also cross-present antigens to CD8+ T cells [19, 21, 22].
The human skin hosts epidermal LCs and dermal interstitial DCs (dermal DCs). The dermal DCs can be further subdivided into CD1a+ DCs and CD14+ DCs. Earlier studies of human cutaneous DCs demonstrated their phenotypic and functional heterogeneity with regard to cellular immunity and priming of highly efficient CTLs [26]. Our studies concluded that human CD14+ DCs can directly help activated B cells as well as induce naïve T cells to differentiate into cells with properties of T follicular helper cells (Tfh) [24]. They may thus be specialized for the development of humoral responses [24]. On the contrary, LCs are more efficient in cross-presenting peptides from protein antigens to CD8+ T cells and prime CD8+ T cells into potent CTLs.
The DC subsets that sit in tissues under the steady state are dependent upon fms-related tyrosine kinase 3 (FLT3) and macrophage colony-stimulating factor receptor (MCSF-R). However, inflammatory processes such as those initiated by microbial invasion, or vaccine adjuvant, substantially alter the DC compartments. The origin of DCs that are recruited to inflammation sites is still under investigation, though it is clear that monocytes give rise to inflammatory DCs in vivo [27]. DCs express numerous non-clonal recognition receptors, including lectins, TLRs, NOD-like receptors (NLRs), and helicases through which they can sense microbes and microbial products such as, for example, nucleic acids, thereby allowing the launching of protective type I interferon production [28, 29]. Indeed the experimental adjuvants CpG and imiquimod bind to TLR9 and TLR7/8 respectively [29]. Most recently biochemical approaches revealed novel sensors of nucleic acid function from the DExD/H-box helicase family molecules in DCs [30, 31].
4.2.3 Human DC Subsets and Humoral Immune Responses
T helper (Th) subsets, specialized for promoting particular types of immune responses and eventually inflammations, function through their secretion of cytokines, enabling unique immune responses (reviewed in [32]). Among those, T follicular helper (Tfh) cells help B cells to differentiate into antibody-secreting cells and govern the germinal center reaction, the main site of immunoglobulin somatic mutation and isotype switching [33, 34]. Human blood CXCR5+CD4+ T cells represent circulating memory Tfh cells. Blood CXCR5+CD4+ T cells comprise three subsets: Tfh1, Tfh2, and Tfh17 cells. Tfh2 and Tfh17 cells efficiently induced naive B cells to produce immunoglobulins via IL-21 [35]. In contrast, Tfh1 cells have been found to lack the capacity to help naïve B cells [35]. In-vitro studies demonstrated that Tfh development is regulated by a specific DC subset, interstitial CD14+ DCs [24]. This requires IL-12 both in vitro [36] and in vivo, as IL-12Rβ1 deficient humans have displayed substantially less circulating memory Tfh and memory B cells than control subjects [37]. Importantly in the context of vaccination, expansion of Tfh1 cells at day 7 correlates with protective antibody titers at day 28 after influenza vaccination in healthy adults and children [38]. Whether the induction of Tfh differentiation depends on the same mechanisms in mice remains to be established.
4.2.4 Human DC Subsets and Cellular Immune Responses
CD8+ T cells recognize peptide-MHC (pMHC) class I molecules expressed by DC, and develop into CTLs able to kill cells presenting a specific pMHC complex [10]. The ideal properties of vaccine-elicited CD8+ T cells include: (1) high avidity for pMHC on tumor cells, (2) high levels of granzyme and perforin, molecules essential for cytotoxic activity against cancer/infected cells, (3) expression of surface molecules allowing trafficking into the tumor; and (4) resistance to regulatory mechanisms present in the tumor [24, 39]. At least four components of the immune response are necessary for that ideal response to happen: (1) the presence of antigen presenting DCs, (2) the quality of induced CD4+ helper T cells [40], (3) the elimination of Tregs which can inhibit CTLs via the secretion of various cytokines including transforming growth factor β (TGF-β), and can compete with CD8+ T cells for IL-2 via constitutive expression of CD25 [41], and (4) the breakdown of the immunosuppressive tumor microenvironment. As discussed earlier, our studies with human Langerhans cells and interstitial DCs showed their specialization in priming CD8+ T cell immunity and humoral immunity respectively. Skin LC efficiency in priming naïve CD8+ T can be at least partially explained by their surface expression of IL-15 [42, 43] and/or upregulation of CD70 upon viral exposure [44]. Furthermore, interstitial DCs play a major role in generation of suppressor CD8+ T cells [45]. Recent studies have further analyzed lymph-node-resident and skin-migratory DC subsets in the human [23, 46]. Both CD1c+ and CLEC9A-expressing CD141+ DCs isolated from human lymph nodes were able to cross-present long peptides (requiring processing) of melanoma-tissue-derived antigen (MART-1) to T cell lines [46], whereas blood DCs can cross-present when activated via Toll-like receptor ligands [18, 19].
Antigen-specific CTLs must traffic into the tumor bed, an area that is not clearly understood [39]. A dysregulation of chemokine homeostasis might prevent the CD8+ T cells to enter the tumor bed. Tumors might actively repulse CD8+ T cell [47]. Finally, the tumor-infiltrating myeloid-derived suppressor cells [48, 49] might inhibit effector CD8+ T cell functions. The negative cues of the tumor environment can be counteracted by a series of therapeutic modalities. These include antibodies neutralizing cytokines such as IL-10, IL-13, and TGF-β [50]. Antibodies such as anti-CTLA-4 and anti-programmed cell death protein-1 ligand 1 (PD-L1) which block the immune-inhibitory signals in lymphocytes can also be used in combinations with cancer vaccines to off-set regulatory mechanisms [40]. The roles of DCs in the regulation of tumor microenvironment are discussed later.
4.3 Cancer Immunotherapy via DCs
4.3.1 Vaccination via DCs
DCs can be engaged in the action of therapeutic vaccines in a number of ways, including indirect involvement as, for example, with GVAX [51] or listeria-based vaccines [52] to name a few (Fig. 4.3). DC can also be used directly following their generation ex vivo and injection to patients (reviewed in [53]). These studies concluded that DC-based vaccines are safe and can induce the expansion of circulating CD4+ and CD8+ T cells that are specific for tumor Ags [53–56]. Objective clinical responses have been observed in some patients. A recent study focused on optimizing vaccine immunogenicity, and demonstrated in phase I/II clinical trials that provision of MHC class II epitopes from defined melanoma tumor antigens results in improved immunogenicity [57]. Furthermore, novel approaches are being developed, including the pre-operative vaccination of patients with her2+ breast cancer [58] as well as combination therapies in ovarian cancer utilizing autologous DC vaccines and adoptive T-cell transfer to enhance vaccine efficacy [59]. More recent studies have utilized pDCs as cell-based vaccines [60]. Patients with metastatic melanoma received intranodal injections of activated and tumor antigen-associated peptide loaded-pDCs. Several patients mounted vaccine antigen-specific CD4+ and CD8+ T-cell responses. Despite the limited number of administered pDCs, an IFN signature was observed after each vaccination [60]. Whereas the clinical efficacy of elicited immunity will need to be determined in larger cohorts and long-term follow-up, type IFN response is highly desirable in melanoma [7, 61].

DCs and cancer immunotherapy. DCs can be exploited for cancer immunotherapy in numerous ways including: (1) their random targeting in “endogenous” vaccination resulting from in-vivo antigen release in a process of immunogenic cell death in response to chemotherapy, radiotherapy as well as immune modulation approaches targeted at T cells, (2) vaccines based on ex-vivo generated tumor antigen-loaded DCs that are injected back into patients, (3) vaccines based on specific in-vivo DC targeting with anti-DC antibodies fused with antigens and with DC activators, and (4) targeting DCs in the tumor environment to reprogram pro-tumor inflammation towards tumor rejection
4.3.2 Vaccination via DCs: In Vivo DC Targeting
Following the pioneering studies from Ralph Steinman and Michel Nussenzweig labs with anti-DEC 205 antibodies [62–64], numerous studies performed in mouse models and in human in vitro systems have demonstrated the efficacy of targeting DCs [10]. Most particularly, targeting antigens through the DC surface lectins DCIR [25, 65], DC-SIGN [66], Dectin [67], Clec9A [68], and Langerin [69] results in humoral and/or cellular CD4+ and/or CD8+ T-cell responses. In the absence of adjuvants, targeting DEC205+ DCs in vivo can induce tolerance [62]. Provision of adjuvants such as TLR3 or TLR7/8 agonists or DC activation signal via CD40 enables the concomitant maturation of vaccine-engulfing DCs [70]. Furthermore, targeting different DC receptors generates quantitatively and qualitatively different immune responses [64, 71]. Injection of antigens coupled to antibodies against DC surface molecule Clec9A results in production of strong antibody responses, even without co-administration of adjuvants [72]. That happens via antigen presentation by DC on MHC class II and consequent Tfh expansion [73]. These results in the mouse are in line with prior studies showing the essential role of DCs in the generation of antibody responses, and show that these can be amplified by targeting antigen to DC surface receptors in vivo. Importantly, CLEC9a is also a receptor for necrotic cells, and has been shown to facilitate cross-presentation [74]. As opposed to antibody response, CLEC9A-dependent generation of CD8+ T-cell responses requires adjuvant. Generation of different responses by targeting distinct DC receptors is further exemplified by recent studies targeting DC-asialoglycoprotein receptor (DC-ASGPR), a lectin-like receptor, which is a known scavenger receptor. Targeting antigens to human DCs via DC-ASGPR in vitro but not lectin-like oxidized-low density lipoprotein (LDL) receptor, Dectin-1, or DC-specific ICAM-3-grabbing non-integrin favored the generation of antigen-specific suppressive CD4+ T cells that produce IL-10 [75]. Furthermore, comparing the cross-presentation of identical antigens conjugated with antibodies against different DC receptors that are targeted to early or late endosomes at distinct efficiencies revealed remarkable differences [76]. Thus, in human BDCA1+ and monocyte-derived DCs, CD40 and mannose receptor targeted antibody conjugates to early endosomes, whereas DEC205 targeted antigen primarily to late compartments. Surprisingly, the receptor least efficient at internalization, CD40, was the most efficient at cross-presentation. This did not reflect DC activation by CD40, but rather its relatively poor uptake or intra-endosomal degradation compared with mannose receptor or DEC205 [76]. DC targeting-based vaccination studies in non-human primates demonstrated robust T-cell immunity in prime-boost design with HIV gag-DEC205 targeting vaccine [77]. Early clinical trials in the human analyzed the delivery of gonadotropin [hCG-b]) to antigen-presenting cells (APCs) by antibody-mediated targeting of a mannose receptor [78]. Delivery of this product with granulocyte–macrophage colony-stimulating factor (GM-CSF) and TLR3/TLR7/8 agonists induced consistent humoral and cellular immune responses to hCG-b [78]. Several studies are ongoing testing the immune efficacy of HIV antigens or NY-ESO1 cancer antigen targeted via DEC-205 in healthy individuals and in cancer patients (clinicaltrials.gov).
4.3.3 Modulating DCs in Tumor Environment
Another approach to immunotherapy via DCs is focused on exploiting DCs in the tumor microenvironment (Fig. 4.3). Indeed, DCs are found in most tumors in humans and mice. DCs can sample tumor antigens through the capture of dying tumor cells and through the nibbling of live tumor cells (reviewed in [79]). Tumors can prevent antigen presentation and the establishment of tumor-specific immunity through a variety of mechanisms. By converting immature DCs into macrophages, i.e., through IL-6 and M-CSF, tumors can prevent the priming of tumor-specific T cells [80, 81]. Alternatively, the tumor glycoproteins carcinoembryonic antigen (CEA) and MUC-1 that are endocytosed by DCs stay confined in the early endosomes, therefore preventing efficient processing and presentation to T cells [82].
Tumors can inhibit DC maturation, for example through the secretion of IL-10, leading to Ag-specific anergy [83]. Tumor-derived factors can alter DCs maturation so as to yield cells that indirectly help tumor growth (“pro-tumor” inflammation) [84]. As an example, we have shown that thymic stromal limphopoietin (TSLP) that is produced by tumor cells, induces DCs to express OX40L that directs the generation of Th2 cells [85, 86]. These skewed CD4+ T cells accelerate breast tumor development through the secretion of IL-4/IL-13 [85, 86]. These cytokines prevent tumor cell apoptosis and promote the proliferation of cancer cells indirectly by stimulating tumor-associated macrophages to secrete EGF [87]. A similar pathway operates in pancreatic cancer [88].
pDCs that are infiltrating breast carcinomas produce little type I interferon upon TLR ligation [89]. These pDCs induce naïve CD4+T cells to differentiate into IL-10-producing T cells with suppressive functions. Such inhibition of type I interferon secretion might also impact the generation of effector T cells, as DCs require type I interferon signals to cross-present tumor antigens [6, 7]. Whether this mechanism explains why pDC are associated to poor prognosis [90] remains to be determined. Finally, DCs can have direct pro-tumor effects. In multiple myeloma mDCs directly promote the survival and clonogenicity of tumor cells [91]. In ovarian cancer, pDCs contribute to tumor angiogenesis by secreting pro-angiogenic cytokines [92]. Thus, understanding the functions of DCs in the tumor bed might represent a rich field of investigation. Ultimately, rewiring the “pro-tumor” DCs into “anti-tumor” DCs might represent a novel approach for cancer immunotherapy.
4.4 Final Remarks
Immunotherapy is moving to the forefront of cancer therapy, owing to recent progresses in the field. For example, an antibody that blocks the function of CTLA-4, a molecule providing negative regulatory feedback in T-cell activation, has been approved in 2011 by the Food and Drug Administration (FDA) for the treatment of melanoma [93]. The classical cancer therapies that are based on chemotherapy might in fact be effective through the engagement of the immune system. For example, chemotherapeutic agents such as anthracyclines and oxaliplatin induce cancer cells to undergo apoptosis, which is associated with cell surface exposure of calreticulin. Surface calreticulin might contribute to the capture of apoptotic bodies by DCs and the elicitation of tumor-specific CD8+ T cell immunity. These T cells might contribute to the elimination of the tumor cells [94] that have not responded to the chemotherapy. There is now strong evidence that antibody therapy with agents such as anti-CD20 and anti-HER 2 involve the adaptive immune system beyond the elicitation of antibody-dependent cytotoxicity (ADCC). Indeed, antibodies against Her-2 can enhance cross-presentation of tumor antigens, most likely by DCs, leading to the break of tolerance against this antigen [95]. Accordingly, patients responding to trastuzumab (Herceptin) show enhanced CD8+ T cell immunity to Her-2 [95].
Nearly 40 years after their discovery, the importance that DCs have achieved in physiology and medicine has been recognized by the award to Ralph Steinman of the Nobel Prize for Medicine and Physiology. The new knowledge represents a fertile ground to work on to design better strategies for intervening in numerous clinical situations. The capacity of LCs and CD14+ DCs to preferentially prime cellular immunity and humoral immunity respectively has significant implications, most particularly in the context of novel human vaccines. Thus, DCs are moving to the forefront of cancer immunotherapy.
References
Roberts WK, Darnell RB (2004) Neuroimmunology of the paraneoplastic neurological degenerations. Curr Opin Immunol 16(5):616–622
Anderson NE, Rosenblum MK et al (1988) Paraneoplastic cerebellar degeneration: clinical–immunological correlations. Ann Neurol 24(4):559–567
Albert ML, Darnell JC et al (1998) Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat Med 4(11):1321–1324
Darnell JC, Albert ML et al (2000) Cdr2, a target antigen of naturally occurring human tumor immunity, is widely expressed in gynecological tumors. Cancer Res 60(8):2136–2139
June CH (2007) Principles of adoptive T cell cancer therapy. J Clin Invest 117(5):1204–1212
Diamond MS, Kinder M et al (2011) Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208(10):1989–2003
Fuertes MB, Kacha AK et al (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 208(10):2005–2016
Nabel GJ (2013) Designing tomorrow’s vaccines. N Engl J Med 368(6):551–560
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392(6673):245–252
Steinman RM (2011) Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 30:1–22
Bar-On L, Jung S (2010) Defining dendritic cells by conditional and constitutive cell ablation. Immunol Rev 234(1):76–89
Trombetta ES, Mellman I (2005) Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol 23:975–1028
Caux C, Massacrier C et al (1994) Activation of human dendritic cells through CD40 cross-linking. J Exp Med 180(4):1263–1272
Siegal FP, Kadowaki N et al (1999) The nature of the principal type 1 interferon-producing cells in human blood. Science 284(5421):1835–1837
Di Pucchio T, Chatterjee B et al (2008) Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat Immunol 9(5):551–557
Liu YJ (2005) IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol 23:275–306
Bachem A, Guttler S et al (2010) Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 207(6):1273–1281
Crozat K, Guiton R et al (2010) The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med 207(6):1283–1292
Jongbloed SL, Kassianos AJ et al (2010) Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med 207(6):1247–1260
Lauterbach H, Bathke B et al (2010) Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J Exp Med 207(12):2703–2717
Poulin LF, Salio M et al (2010) Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med 207(6):1261–1271
Mittag D, Proietto AI et al (2011) Human dendritic cell subsets from spleen and blood are similar in phenotype and function but modified by donor health status. J Immunol 186(11):6207–6217
Haniffa M, Shin A et al (2012) Human tissues contain CD141(hi) cross-presenting dendritic cells with functional homology to mouse CD103(+) nonlymphoid dendritic cells. Immunity 37(1):60–73
Klechevsky E, Morita R et al (2008) Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity 29(3):497–510
Klechevsky E, Flamar AL et al (2010) Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood 116(10):1685–1697
Joffre OP, Segura E et al (2012) Cross-presentation by dendritic cells. Nat Rev Immunol 12(8):557–569
Segura E, Touzot M et al (2013) Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 38(2):336–348
Barber GN (2011) Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol 23(1):10–20
Desmet CJ, Ishii KJ (2012) Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat Rev Immunol 12(7):479–491
Zhang Z, Kim T et al (2011) DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34(6):866–878
Zhang Z, Bao M et al (2013) The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat Immunol 14(2):172–178
Bluestone JA, Mackay CR et al (2009) The functional plasticity of T cell subsets. Nat Rev Immunol 9(11):811–816
Vinuesa CG, Cyster JG (2011) How T cells earn the follicular rite of passage. Immunity 35(5):671–680
Ma CS, Deenick EK et al (2012) The origins, function, and regulation of T follicular helper cells. J Exp Med 209(7):1241–1253
Morita R, Schmitt N et al (2011) Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 34(1):108–121
Schmitt N, Morita R et al (2009) Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity 31(1):158–169
Schmitt N, Bustamante J et al (2013) IL-12 receptor beta1 deficiency alters in vivo T follicular helper cell response in humans. Blood 121(17):3375–3385
Bentebibel SE, Lopez S et al (2013) Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med 5(176):176ra32
Appay V, Douek DC et al (2008) CD8+ T cell efficacy in vaccination and disease. Nat Med 14(6):623–628
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12(4):252–264
Kastenmuller W, Gasteiger G et al (2011) Regulatory T cells selectively control CD8+ T cell effector pool size via IL-2 restriction. J Immunol 187(6):3186–3197
Banchereau J, Thompson-Snipes L et al (2012) The differential production of cytokines by human Langerhans cells and dermal CD14(+) DCs controls CTL priming. Blood 119(24):5742–5749
Romano E, Cotari JW et al (2012) Human Langerhans cells use an IL-15R-alpha/IL-15/pSTAT5-dependent mechanism to break T-cell tolerance against the self-differentiation tumor antigen WT1. Blood 119(22):5182–5190
van der Aar AM, de Groot R et al (2011) Cutting edge: virus selectively primes human Langerhans cells for CD70 expression promoting CD8+ T cell responses. J Immunol 187(7):3488–3492
Banchereau J, Zurawski S et al (2012) Immunoglobulin-like transcript receptors on human dermal CD14+ dendritic cells act as a CD8-antagonist to control cytotoxic T cell priming. Proc Natl Acad Sci USA 109(46):18885–18890
Segura E, Valladeau-Guilemond J et al (2012) Characterization of resident and migratory dendritic cells in human lymph nodes. J Exp Med 209(4):653–660
Le Floc’h A, Jalil A et al (2007) Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med 204(3):559–570
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9(3):162–174
Menetrier-Caux C, Gobert M et al (2009) Differences in tumor regulatory T-cell localization and activation status impact patient outcome. Cancer Res 69(20):7895–7898
Terabe M, Ambrosino E et al (2009) Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin Cancer Res 15(21):6560–6569
Le DT, Pardoll DM et al (2010) Cellular vaccine approaches. Cancer J 16(4):304–310
Le DT, Dubenksy TW Jr et al (2012) Clinical development of Listeria monocytogenes-based immunotherapies. Semin Oncol 39(3):311–322
Palucka K, Banchereau J (2012) Cancer immunotherapy via dendritic cells. Nat Rev Cancer 12(4):265–277
Schuler G (2010) Dendritic cells in cancer immunotherapy. Eur J Immunol 40(8):2123–2130
Kalinski P, Edington H et al (2011) Dendritic cells in cancer immunotherapy: vaccines or autologous transplants? Immunol Res 50(2–3):235–247
Galluzzi L, Senovilla L et al (2012) Trial watch: Dendritic cell-based interventions for cancer therapy. Oncoimmunology 1(7):1111–1134
Aarntzen EH, De Vries IJ et al (2013) Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res 73(1):19–29
Sharma A, Koldovsky U et al (2012) HER-2 pulsed dendritic cell vaccine can eliminate HER-2 expression and impact ductal carcinoma in situ. Cancer 118(17):4354–4362
Kandalaft LE, Powell DJ Jr et al (2013) Autologous lysate-pulsed dendritic cell vaccination followed by adoptive transfer of vaccine-primed ex vivo co-stimulated T cells in recurrent ovarian cancer. Oncoimmunology 2(1):e22664
Tel J, Aarntzen EH et al (2013) Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res 73(3):1063–1075
Gajewski TF (2007) Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res 13(18 Pt 1):5256–5261
Hawiger D, Inaba K et al (2001) Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med 194:769–780
Bonifaz L, Bonnyay D et al (2002) Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med 196(12):1627–1638
Soares H, Waechter H et al (2007) A subset of dendritic cells induces CD4+ T cells to produce IFN-{gamma} by an IL-12-independent but CD70-dependent mechanism in vivo. J Exp Med 204(5):1095–1106
Meyer-Wentrup F, Cambi A et al (2009) DCIR is endocytosed into human dendritic cells and inhibits TLR8-mediated cytokine production. J Leukoc Biol 85(3):518–525
Dakappagari N, Maruyama T et al (2006) Internalizing antibodies to the C-type lectins, L-SIGN and DC-SIGN, inhibit viral glycoprotein binding and deliver antigen to human dendritic cells for the induction of T cell responses. J Immunol 176(1):426–440
Ni L, Gayet I et al (2010) Concomitant activation and antigen uptake via human dectin-1 results in potent antigen-specific CD8+ T cell responses. J Immunol 185(6):3504–3513
Sancho D, Mourao-Sa D et al (2008) Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest 118(6):2098–2110
Flacher V, Sparber F et al (2009) Targeting of epidermal Langerhans cells with antigenic proteins: attempts to harness their properties for immunotherapy. Cancer Immunol Immunother 58(7):1137–1147
Tacken PJ, Figdor CG (2011) Targeted antigen delivery and activation of dendritic cells in vivo: steps towards cost effective vaccines. Semin Immunol 23(1):12–20
Dudziak D, Kamphorst AO et al (2007) Differential antigen processing by dendritic cell subsets in vivo. Science 315(5808):107–111
Caminschi I, Vremec D et al (2012) Antibody responses initiated by Clec9A-bearing dendritic cells in normal and Batf3(−/−) mice. Mol Immunol 50(1–2):9–17
Lahoud MH, Ahmet F et al (2011) Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J Immunol 187(2):842–850
Sancho D, Joffre OP et al (2009) Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458(7240):899–903
Li D, Romain G et al (2012) Targeting self- and foreign antigens to dendritic cells via DC-ASGPR generates IL-10-producing suppressive CD4+ T cells. J Exp Med 209(1):109–121
Chatterjee B, Smed-Sorensen A et al (2012) Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 120(10):2011–2020
Flynn BJ, Kastenmuller K et al (2011) Immunization with HIV Gag targeted to dendritic cells followed by recombinant New York vaccinia virus induces robust T-cell immunity in nonhuman primates. Proc Natl Acad Sci USA 108(17):7131–7136
Morse MA, Chapman R et al (2011) Phase I study utilizing a novel antigen-presenting cell-targeted vaccine with Toll-like receptor stimulation to induce immunity to self-antigens in cancer patients. Clin Cancer Res 17(14):4844–4853
Dhodapkar MV, Dhodapkar KM et al (2008) Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differ 15(1):39–50
Chomarat P, Banchereau J et al (2000) IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol 1(6):510–514
Chomarat P, Dantin C et al (2003) TNF skews monocyte differentiation from macrophages to dendritic cells. J Immunol 171(5):2262–2269
Hiltbold EM, Vlad AM et al (2000) The mechanism of unresponsiveness to circulating tumor antigen MUC1 is a block in intracellular sorting and processing by dendritic cells. J Immunol 165(7):3730–3741
Steinbrink K, Jonuleit H et al (1999) Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood 93:1634–1642
Coussens LM, Zitvogel L et al (2013) Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 339(6117):286–291
Aspord C, Pedroza-Gonzalez A et al (2007) Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J Exp Med 204(5):1037–1047
Pedroza-Gonzalez A, Xu K et al (2011) Thymic stromal lymphopoietin fosters human breast tumor growth by promoting type 2 inflammation. J Exp Med 208(3):479–490
DeNardo DG, Barreto JB et al (2009) CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16(2):91–102
De Monte L, Reni M et al (2011) Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med 208(3):469–478
Cao W, Bover L et al (2009) Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J Exp Med 206(7):1603–1614
Treilleux I, Blay JY et al (2004) Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res 10(22):7466–7474
Kukreja A, Hutchinson A et al (2006) Enhancement of clonogenicity of human multiple myeloma by dendritic cells. J Exp Med 203(8):1859–1865
Curiel TJ, Cheng P et al (2004) Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res 64(16):5535–5538
Hodi FS, O’Day SJ et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723
Zitvogel L, Kepp O et al (2010) Immunogenic tumor cell death for optimal anticancer therapy: the calreticulin exposure pathway. Clin Cancer Res 16(12):3100–3104
Taylor C, Hershman D et al (2007) Augmented HER-2 specific immunity during treatment with trastuzumab and chemotherapy. Clin Cancer Res 13(17):5133–5143
Acknowledgements
We thank all the patients and volunteers who participated in our studies and clinical trials. We thank former and current members of the Institute for their contributions to our progress. Our studies have been supported by the NIH (P01 CA084514, U19 AIO57234, R01 CA089440, CA078846 and CA140602), the Dana Foundation, the Susan Komen Foundation, the Baylor Health Care System; the Baylor Health Care System Foundation, the ANRS and the INSERM. KP holds the Michael A. Ramsay Chair for Cancer Immunology Research. Due to space limitations, we have only been able to cite a fraction of the vast number of publications.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Wien
About this chapter
Cite this chapter
Palucka, K., Banchereau, J. (2014). Cancer Immunotherapy via Dendritic Cells. In: Klink, M. (eds) Interaction of Immune and Cancer Cells. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1300-4_4
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1300-4_4
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1299-1
Online ISBN: 978-3-7091-1300-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)