
Abstract
Parkinson’s disease is characterized by the selective degeneration of neuromelanin-containing dopaminergic neurons in the substantia nigra and locus coeruleus. Although the cause of this disease remains unknown, several transition metals, including manganese and copper, have been associated with the development of the atypical form of Parkinsonism, and iron accumulation has been associated with the development of Parkinson’s disease. Manganese3+ is a strong oxidizing agent, which oxidizes dopamine to aminochrome (dopaminochrome), the precursor of neuromelanin. Aminochrome formation in cell culture medium induces acute cell death in cells that uptake aminochrome, explaining the role of manganese in the development of atypical Parkinsonism. Copper accumulation in Wilson’s disease also induces Parkinsonism as one of the main symptoms, and an atypical Parkinsonism has also been observed in young copper miners. Interestingly, copper is able to complex with dopamine, which can be taken up by cells expressing the dopamine transporter, inducing caspase-independent cell death with formation of autophagic vacuoles. Iron is also able to form a complex with dopamine, the neurotoxic action of which also depends on the cellular expression of the dopamine transporter. The neurotoxicities of these transition metals to cells expressing the dopamine transporter all involve dopamine oxidation to quinones and require the inhibition of DT-diaphorase.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
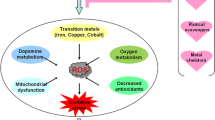


Keywords
Introduction
The possible role of the ions of the metals manganese, copper, and iron in the neurodegenerative process has been associated with the formation of reactive oxygen species. Manganese2+ is able to react with superoxide radicals generating hydrogen peroxide, the precursor of hydroxyl radicals, and both Fe2+ and Cu+ are able to catalyze the Fenton reaction with the formation of hydroxyl radicals in the presence of hydrogen peroxide. This review discusses other mechanisms of dopamine neuron degeneration induced by the ions of the metals copper, manganese, and iron, where the formation of reactive oxygen species is only a part of the mechanism that induces cell death. The degeneration of dopamine neurons is associated with the motoric symptoms observed in Parkinsonism induced by manganese and copper. The association of iron with Parkinson’s disease is based on the accumulation of iron in the affected regions of the brain.
Manganese
Exposure to excessive levels of manganese (Mn) can induce severe psychiatric and extrapyramidal motor dysfunction closely resembling Parkinson’s disease. The clinical manifestations of manganese toxicity arise from focal injury to the basal ganglia. This region, characterized by intense oxygen consumption and significant dopamine content, can undergo mitochondrial dysfunction, depletion of peroxidase and catalase, and imbalances in catecholamine levels following manganese exposure [1]. Welding fumes have been associated with the development of Parkinsonism, and the evaluation of rats exposed to gas metal arc-mild steel with low manganese, or to manual metal arc-hard surfacing with high manganese, revealed a loss of tyrosine hydroxylase in the striatum and midbrain [2].
A brief exposure to Mn2+ increases reactive oxygen species levels and glutathione production, decreases oxygen consumption affecting mitochondrial membrane potential, and induces dopaminergic neuronal death [3]. Manganese chloride (Mn2+)- and LPS-induced reactive oxygen species and nitric oxide, cytokine release, and dopaminergic neurotoxicity in microglia were significantly attenuated by pre-treatment with the potential anti-inflammatory agents minocycline and naloxone [4, 5]. A sub-chronic exposure to Mn during fetal development leads to temporally distinct patterns of glial activation, which result in elevated nitric oxide stress in distinct populations of basal ganglia neurons [6]. Treatment with minocycline, an inhibitor of microglial activation, prior to manganese exposure attenuated microglial activation and mitigated IL-1beta, TNF-alpha, and iNOS production as well as dopaminergic neurotoxicity [7].
Tyrosine hydroxylase is the rate-limiting enzyme in dopamine synthesis and is regulated acutely by phosphorylation at Ser40 and chronically by protein synthesis. Sustained phosphorylation of tyrosine hydroxylase at Ser40 and the consequent stimulation of enzyme activity both occurred at low concentrations of Mn2+, and this effect represents a potential mechanism for Mn2+-induced neuronal toxicity that does not involve H2O2-mediated cell death [8]. Acute Mn2+ exposure induces cytoskeleton dysfunction prior to degeneration, and chronic Mn2+ exposure results in neurochemical dysfunction with features overlapping those of Parkinson’s disease [9]. Induction of NF-kappaB and activation of nitric oxide synthase through reactive oxygen species was suggested to be involved in Mn2+-induced apoptosis in primary mesencephalic cells exposed to dopamine prior to Mn2+ [10].
Weanling rats chronically exposed to Mn exhibited significant Mn accumulation in several brain regions. However, rats receiving the selective dopamine transporter (DAT) inhibitor GBR12909 had significantly lower Mn levels only in the globus pallidus compared with saline-treated rats. These data show that inhibition of the DAT exclusively inhibits Mn accumulation in the globus pallidus during chronic exposure to the metal [11]. Postnatal Mn exposure caused persistent declines in DAT protein expression and [3H]dopamine uptake in the striatum and nucleus accumbens, as well as long-term reductions in striatal dopamine efflux [12]. However, Mn accumulated to similar levels in PC12 cells overexpressing the DAT and control PC12 cells following incubation with manganese chloride, suggesting that the DAT is not sufficient for manganese cytotoxicity [13]. In general, Mn2+ has been used to study the mechanism of manganese neurotoxicity, and the possible involvement of the DAT in Mn uptake was found to be restricted to the globus pallidus region [11]. However, it is worth noting that the divalent metal transporter 1 (DMT1) is able to take up Mn2+ into various tissues given its wide expression. DMT1 has been reported to be expressed in the striatum, ventral mesencephalon, substantia nigra, hippocampus, caudate nucleus, putamen, corpus callosum, and cerebellum; in neurons in the cortex, striatum, cerebellum, and thalamus; in ependymal cells lining the third ventricle; and in vascular cells throughout the brain. DMT1 is also expressed in several cell lines, such as MES23.5, SH-SY5Y, PC12, RCSN-3 (derived from rat substantia nigra), rat C6 astrocytoma, human U87 glioblastoma, and hippocampal cells [14–24]. Therefore, manganese does not accumulate selectively in dopaminergic neurons, and its neurotoxic effect will differentially affect various neuronal systems. The wide expression of DMT1 may explain why manganese induces an atypical Parkinsonism. Manganese induces a consistent pattern characterized by damage to the globus pallidus, sparing of the substantia nigra pars compacta, and absence of Lewy bodies. This finding contrasts what is seen in Parkinson’s disease, in which there is preferential degeneration of dopaminergic neurons in the substantia nigra pars compacta coupled with Lewy bodies and preservation of the pallidum [25]. The uptake of Mn2+ mediated by DMT1 is probably not the most neurotoxic event because manganese can be oxidized to Mn3+ in the presence of superoxide (Fig. 1). Auto-oxidation of dopamine to aminochrome was found to be considerably potentiated by Mn2+, suggesting that Mn2+ is contributing dopamine oxidation as a consequence of its oxidation to Mn3+ [26].

Manganese uptake and intracellular manganese oxidation. Mediated by DMT1 transporter manganese2+ is taken up into the cytosol of the cell where it can be oxidized to manganese3+ catalyzed by superoxide radicals with concomitant formation of hydrogen peroxide
Manganese3+ is a potent oxidizing agent that is able to oxidize dopamine, and dopamine oxidation catalyzed by Mn3+ does not require oxygen (Fig. 2). In addition, Mn3+ was far more efficient than Mn2+, Mn4+ (MnO2), O2 −, or H2O2 in oxidizing the catecholamine [27].

Dopamine oxidation to aminochrome catalyzed by manganese3+. Manganese3+ oxidizes dopamine to dopamine o-quinone that at physiological pH spontaneously cyclizes to leukoaminochrome that doesn’t require oxygen to form aminochrome since manganese3+ is able to catalyze this reaction both in aerobic and anaerobic conditions [28]
Dopamine is oxidized by Mn3+ to yield dopamine o-quinone, which is the precursor of aminochrome. This is a transient compound at physiological pH because its amino chain undergoes cyclization to leukoaminochrome, which is oxidized by Mn3+ to aminochrome in the absence of oxygen. Stable dopamine o-quinone can only be found when the pH is lower than 2.0 [28]. However, Mn3+ can also oxidize thiol groups of proteins or hydroxyl groups of amino acids, thereby inactivating enzymes not only in dopaminergic neurons, but also in other neurons that express DMT1. Therefore, manganese is not a selective neurotoxic agent for dopaminergic neurons, explaining the atypical Parkinsonism resulting from the neurotoxic effects of manganese in different neuronal systems. This non-specific action of manganese explains why depletion of dopamine does not prevent manganese neurotoxicity and why, in the presence of 5 mM GSH and 10 mM N-acetylcysteine, cells otherwise sensitive to manganese are protected [29]. Intracellular dopamine oxidation and the oxidation of the thiol and hydroxyl groups of cysteine and serine, respectively, can participate in a dopamine-independent mechanism of manganese neurotoxicity, although both mechanisms result in the concomitant formation of reactive oxygen species, generating a redox cycling between Mn2+ and Mn3+ that potentiates the neurotoxic effects of manganese (Fig. 3).

Intracellular effect of manganese2+. Manganese2+ uptake induces intracellular dopamine oxidation and protein oxidation by oxidizing the thiol and hydroxyl groups of cysteine and serine
One mechanism of manganese neurotoxicity involves Mn3+-dependent oxidation of thiol groups in cysteine-containing enzymes, which leads to a loss of activity leading to loss of function. The total thiol content was lowered 40 % in cultures of the neuroblastoma clone N1E 115 treated with manganese. This decreased thiol content was also reflected by the reduced activity of the thiolenzyme glyceraldehyde-3-phosphate dehydrogenase in manganese-exposed cells [30]. Mn3+, a powerful oxidizing agent, is able to oxidize hydroxyl groups of amino acids such as serine, threonine, and tyrosine present in the polypeptide chain of enzymes, which may result in a loss of enzymatic activity. Manganese treatment of PC12 cells induces inhibition of proteasome activity with significant accumulation of protein carbonyls arising from damage to proteins [31].
The dopamine-dependent mechanism of manganese neurotoxicity involves Mn3+-catalyzed dopamine oxidation to aminochrome (Fig. 4). Studies in RCSN-3 cells of dopamine oxidation catalyzed by Mn3+ in the cell culture medium prior to addition to the cells showed that aminochrome neurotoxicity was dependent on DT-diaphorase inhibition and resulted in the formation of a leukoaminochrome-o-semiquinone radical. The Mn3+ neurotoxicity induced the formation of intracellular hydroperoxides and hydroxyl radicals measured by electron spin resonance with an absence of apoptosis, a strong decrease of mitochondrial membrane potential, and a swelling and disruption of the outer and inner mitochondrial membranes (assessed by transmission electron microscopy) [32]. These results support the proposed role of leukoaminochrome-o-semiquinone radical as a neurotoxin in dopaminergic neurons and of DT-diaphorase as a neuroprotective enzyme [33–44].

The dopamine-dependent mechanism of manganese neurotoxicity
Copper
Several important enzymes within the brain, such as dopamine β-mono-oxygenase, peptidyl-α-amidating mono-oxygenase, Cu/Zn superoxide dismutase, cytochrome c oxidase, and lysine oxidase, require a copper cofactor for their enzymatic activity. On the other hand, accumulation or deficiency of copper is associated with neurodegenerative disorders, such as Wilson’s and Menke’s diseases [45].
Copper induces an increase in oxidative stress, resulting in DNA damage and activation of p53-dependent cell death [46]. Neuronal cell death induced by GSH depletion is dependent on trace levels of extracellular copper in the culture medium [47]. Cultured astrocytes and neurons treated with copper reveal that mitochondrial membrane permeability transition and oxidative and nitrosative stress represent major factors in copper-induced toxicity, whereas oxidative and nitrosative stress appear to play a major role in neuronal injury [48]. In vivo, copper treatment alone caused no significant learning and memory impairments in behavioral tasks [49]. However, trace amounts of copper in cholesterol-fed mice induced significant learning and memory impairments accompanied by the upregulation of amyloid precursor protein, which contributes to amyloid beta-peptide deposition, increased protein expression of tumor necrosis factor-alpha, and degradation of IkappaB protein. Furthermore, an increased production of inducible nitric oxide synthase and cyclo-oxygenase-2 was observed in the hippocampus and cerebral cortex [50]. Copper concentrations are elevated in amyloid plaques, and copper binds with high affinity to the Abeta peptide, promoting its oligomerization and neurotoxicity [51].
Wilson’s disease is a copper deposition disorder caused by a genetic mutation in a copper transporter ATPase (ATP7B), which is essential for the maintenance of intracellular copper concentration. This mutation prevents the excretion of excess copper from the liver to the bile, resulting in the hepatic accumulation of copper. Interestingly, the major impact of this copper overload is not in the liver but in the brain, leading to neurologic Wilson’s disease. A study on the clinical presentations of Wilson’s disease in 282 patients showed that 69 % of the patients were classified as having neurologic Wilson’s disease, the predominant neurologic feature of which was Parkinsonism (62.3 %) [52]. The fact that Parkinsonism is one of the major neurologic symptoms of Wilson’s disease opens the question as to why copper overload in the brain affects dopaminergic neurons so extensively. Young workers in the copper refining industry exposed to high concentrations of copper developed an irreversible atypical Parkinsonism [53]. The ability of reduced copper to catalyze the Fenton reaction is a non-specific mechanism and cannot explain the selective action of copper on dopaminergic neurons. One possible explanation for the selectivity of copper resulting in extensive neurodegeneration of dopaminergic neurons is the ability of Cu2+ to form a complex with dopamine (reaction 1) based on the formation of ionic binding between the positive charge of Cu2+ and the negative charges of dissociated hydroxyl groups [34].
The DAT is able to take up the Cu–dopamine complex into cells (reaction 2) that express the protein, conferring specificity to dopaminergic neurons or cells expressing DAT. However, uptake of the Cu–dopamine complex is not restricted to dopaminergic neurons, which may explain why Parkinsonism is the main symptom of Wilson’s disease. Dopamine is oxidized to aminochrome (reaction 3) inside cells when Cu+ is released from the Cu–dopamine complex and chelated by proteins (reaction 4). Aminochrome can be reduced with two electrons to yield leukoaminochrome via a reaction catalyzed by DT-diaphorase (reaction 5), thus preventing aminochrome from participating in such neurotoxic reactions as one-electron reduction to leukoaminochrome o-semiquinone radical (reaction 6), which is extremely reactive with oxygen [54]. This reaction is catalyzed by flavoenzymes that reduce quinones with one electron using NADH or NADPH. Leukoaminochrome o-semiquinone radical reduces oxygen, generating superoxide radicals (reaction 7) that spontaneously or enzymatically (via superoxide dismutase) generate hydrogen peroxide (reaction 8). When NADH is used in this redox cycling, both oxygen and NADH are depleted, generating an energy collapse because of ATP depletion, which induces cell death. However, when the flavoenzymes use NADPH in this redox cycling, there results both a NADPH depletion affecting the reduction of oxidized glutathione and an oxygen depletion, which also affects the electron transport chain in the mitochondria and leads to ATP depletion (Fig. 5). Under these conditions, the Cu–dopamine complex induces caspase-independent cell death with formation of autophagic vacuoles [39]. Animals subjected to a unilateral intranigral injection of CuSO4 and dicoumarol, an inhibitor of DT-diaphorase, presented a significant and characteristic contralateral rotational behavior and a loss of tyrosine hydroxylase-positive staining [55]. Interestingly, the formation of Cu–dopamine complex requires DT-diaphorase inhibition both in cell culture and in vivo, supporting the proposed neuroprotective role of DT-diaphorase [39, 55]. Copper inhibits the vesicular H(+)-ATPase required for DAT into monoaminergic vesicles mediated by VMAT-2, resulting in an increase of free cytosolic dopamine and its subsequent oxidation to aminochrome [56], supporting the role of dopamine in copper neurotoxicity.

The dopamine-dependent mechanism of copper neurotoxicity
Iron
Iron is essential for life because it is an essential component of the heme group of hemoglobin, cytochromes, and enzymes, but it is also involved in neurodegenerative mechanisms, such as protein aggregation, free radical generation, and oxidative stress. The role of iron in the pathophysiology of Parkinson’s disease has been extensively studied and supported by the finding that iron accumulates in the brain regions affected by this disease [57]. The iron concentration in the substantia nigra correlates with UPDRS motor score, indicating that iron concentration can function as an in vivo biomarker to objectively evaluate the status of Parkinson’s disease [58]. Iron accumulation in dopaminergic neurons and glial cells in the substantia nigra of Parkinson’s disease patients may contribute to the neurodegenerative process in this disease, but the mechanisms involved in iron accumulation remain unclear. The major transport protein responsible for uptake of iron is the DMT1, and it is expressed in rat substantia nigra both with and without the iron regulatory element (IRE) in neurons, astrocytes, and microglia, but not in oligodendrocytes [59]. Recent studies demonstrate that the 1B isoform is regulated post-translationally by degradation via the proteasome pathway, and overexpression of parkin results in a decrease in the 1B isoform of DMT1 [60].
The role of transferrin and its receptor in iron accumulation was discarded, and the overexpression of lactoferrin receptors and DMT1 has been reported. A mutation in DMT1, which impairs iron transport, protected rodents against the Parkinsonism-inducing neurotoxins 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine hydrochloride (MPTP) and 6-hydroxydopamine [61, 62]. Ferroportin1 and hephaestin, two iron export proteins, regulate iron export in the gut and are expressed by astrocytes, microglia, oligodendrocytes, and neurons in the substantia nigra [63]. It was reported that 6-hydroxydopamine induced the downregulation of the iron transporters ferroportin 1 and hephaestin, resulting in the decreased iron efflux and iron accumulation in primary ventral mesencephalic neurons and in MES23.5 dopaminergic cells. However, under iron-overload conditions, ferroportin 1 showed dose-dependent upregulation, whereas hephaestin showed no response, indicating that 6-hydroxydopamine-induced downregulation was not caused by increased intracellular iron content [64]. Overexpression of the human ferritin heavy chain (H-ferritin) in dopamine neurons exerts significant neuroprotection, possibly by modulating iron homeostasis and restoring ubiquitin proteasome activity [65]. Reduction in the levels of glutathione in immortalized midbrain-derived dopaminergic neurons leads to increases in the cellular pool of labile iron that are independent of the induction of either iron regulatory protein/iron regulatory element (IRP/IRE) or hypoxia inducible factor, but are dependent on hydrogen peroxide and protein synthesis. A link between glutathione depletion in dopaminergic neurons with an increase in iron levels based on translational activation of transferrin receptor-1 has been suggested [66]. l-Ferritin was found to be localized in neuromelanin granules [67].
In MPTP- and 6-hydroxydopamine-induced dopaminergic neurotoxicity and in Parkinson’s disease, iron accumulates in the substantia nigra pars compacta. Pretreatment with iron chelators such as desferal, clioquinol, VK-28, and M30 is neuroprotective in both neurotoxin models [68]. The naturally occurring iron chelator phytic acid decreased 1-methyl-4-phenylpyridinium [MPP(+)]-induced caspase-3 activation and DNA fragmentation, and increased cell survival in immortalized rat mesencephalic/dopaminergic cells exposed to excess iron [69]. 6-Hydroxymelatonin reduces Fe2+-induced neurotoxicity in the rat hippocampus [70]. Dopamine and Fe2+ in combination induce apoptosis with significantly increased concentrations of hydroxyl radicals and malondialdehyde in SH-SY5Y cells [71].
Neuromelanin blocks hydroxyl radical production by Fenton’s reaction and also inhibits the iron-mediated oxidation of ascorbic acid. The sequestration of iron into a stable iron–neuromelanin complex prevents dopamine oxidation, inhibiting the formation of neurotoxic dopamine quinones [72]. l-Ferritin was found to be localized in neuromelanin granules [67]. Lisuride, a dopamine agonist, protects against iron-induced lesions [73].
The formation of a Fe–dopamine complex was reported to be a mechanism for specific neurotoxicity in cells expressing DAT such as dopamine, norepinephrine(NET), and serotonin(SERT) transporters [35, 74]. It was reported that iron forms a complex with dopamine [75] and that the Fe–dopamine complex protected isolated hepatocytes against hypoxia-re-oxygenation-induced injury [76] because these cells do not express DAT. It was also reported that iron accumulates into dopamine neurovesicles, and the inhibition of dopamine synthesis results in a decreased vesicular storage of iron [77]. These results suggest that VMAT-2 transports the Fe–dopamine complex into monoaminergic vesicles, supporting the idea that the Fe–dopamine complex can be formed in cells that synthesize dopamine. The precursor of dopamine, l-dopa, also forms a complex with Fe3+ [78].
Fe–dopamine complex formation occurs in the extracellular space (Fig. 6, reaction 1), and the uptake of the Fe–dopamine complex into cells is mediated by DAT, NAT and SERT. Once inside the cell, the Fe–dopamine complex undergoes dopamine oxidation to dopamine o-quinone (Fig. 6, reaction 2), which at physiological pH spontaneously cyclizes to aminochrome in a two-step reaction (reaction 3). Aminochrome can be reduced to leukoaminochrome in a reaction catalyzed by DT-diaphorase, and the product prevents aminochrome from participating in neurotoxic reactions, such as the one-electron reduction catalyzed by flavoenzymes that use NADH or NADPH as an electron donor (reaction 4). The one-electron reduction of aminochrome results in the formation of leukoaminochrome o-semiquinone radical (reaction 5), which is extremely reactive with oxygen [54] and neurotoxic [32, 34–40, 43, 79]. The auto-oxidation of leukoaminochrome o-semiquinone radical in the presence of oxygen generates a redox cycling (reaction 6) with concomitant formation of a superoxide radical, which spontaneously or enzymatically dismutates to H2O2. This redox cycling will proceed until oxygen, NADH, or NADPH is depleted. The depletion of NADH and oxygen will affect the mitochondrial electron transport chain, thereby inhibiting ATP formation, which induces an energy collapse and cell death. The depletion of NADPH prevents the reduction of oxidized glutathione and a subsequent depletion of GSH, which is an important antioxidant. Upon oxidation of the Fe–dopamine complex, Fe3+ is reduced to Fe2+ (reaction 2), which is able to catalyze the formation of a hydroxyl radical (reaction 8) from the hydrogen peroxide generated by the dismutation of superoxide radicals (reaction 7). The Fenton reaction oxidizes Fe2+ to Fe3+, which is able to form a new complex with intracellular dopamine, potentiating the neurotoxic effect of the Fe–dopamine complex. The inhibition of ATP formation resulting from NADH and oxygen depletion generating an energy collapse and the oxidative stress caused by hydroxyl radicals and depletion of reduced glutathione eventually lead to cell death [35, 74]. It is important to note that Fe–dopamine complex neurotoxicity requires the expression of DAT and the inhibition of DT-diaphorase. Interestingly, norepinephrine is able to compete with dopamine to form a complex with Fe3+ and prevents Fe–dopamine complex neurotoxicity because the Fe–norepinephrine complex is not neurotoxic, despite the fact that cells are able to take up the Fe–norepinephrine complex.

The dopamine-dependent mechanism of iron neurotoxicity
Conclusions
The transition metals manganese and copper have been involved in neurodegenerative processes in dopaminergic neurons related to atypical Parkinson’s disease and iron accumulation has been associated with Parkinson’s disease. Interestingly, these metals are able to induce neurotoxicity by reacting with the neurotransmitter dopamine. Mn3+ is able to oxidize dopamine to aminochrome and potentiate aminochrome neurotoxicity. Cu2+ and Fe3+ are able to form complexes with dopamine and are able to induce neurotoxicity in cells expressing DAT, resulting in the selective action of copper on dopaminergic neurons. However, the neurotoxicity induced by these metals was dependent on the one-electron reduction of aminochrome in conditions where DT-diaphorase was inhibited, supporting the proposed neuroprotective role of DT-diaphorase in dopaminergic neurons.
References
HaMai D, Bondy SC (2004) Ann N Y Acad Sci 1012:129
Sriram K, Lin GX, Jefferson AM, Roberts JR, Chapman RS, Chen BT, Soukup JM, Ghio AJ, Antonini JM (2010) Arch Toxicol 84:521
Settivari R, Levora J, Nass R (2009) J Biol Chem 284:35758
Zhang P, Lokuta KM, Turner DE, Liu B (2010) J Neurochem 112:434
Zhang P, Wong TA, Lokuta KM, Turner DE, Vujisic K, Liu B (2009) Exp Neurol 217:219
Moreno JA, Streifel KM, Sullivan KA, Legare ME, Tjalkens RB (2009) Toxicol Sci 112:405
Zhao F, Cai T, Liu M, Zheng G, Luo W, Chen J (2009) Toxicol Sci 107:156
Posser T, Franco JL, Bobrovskaya L, Leal RB, Dickson PW, Dunkley PR (2009) J Neurochem 110:848
Stanwood GD, Leitch DB, Savchenko V, Wu J, Fitsanakis VA, Anderson DJ, Stankowski JN, Aschner M, McLaughlin B (2009) J Neurochem 110:378
Prabhakaran K, Ghosh D, Chapman GD, Gunasekar PG (2008) Brain Res Bull 76:361
Anderson JG, Cooney PT, Erikson KM (2007) Environ Toxicol Pharmacol 3:179
McDougall SA, Reichel CM, Farley CM, Flesher MM, Der-Ghazarian T, Cortez AM, Wacan JJ, Martinez CE, Varela FA, Butt AE, Crawford CA (2008) Neuroscience 154:848
Hirata Y, Suzuno H, Tsuruta T, Oh-hashi K, Kiuchi K (2008) Toxicology 244:249
Williams BB, Kwakye GF, Wegrzynowicz M, Li D, Aschner M, Erikson KM, Bowman AB (2010) Toxicol Sci 117:169
Hirsch EC (2009) Parkinsonism Relat Disord 15:S209
Du T, Ciccotosto GD, Cranston GA, Kocak G, Masters CL, Crouch PJ, Cappai R, White AR (2008) Free Radic Biol Med 44:44
Zhang S, Wang J, Song N, Xie J, Jiang H (2009) Neurobiol Aging 30:1466
Huang E, Ong WY, Go ML, Connor JR (2006) Exp Brain Res 170:376
Huang E, Ong WY, Connor JR (2004) Neuroscience 128:487
Aguirre P, Mena N, Tapia V, Arredondo M, Núñez MT (2005) BMC Neurosci 6:3
Lis A, Barone TA, Paradkar PN, Plunkett RJ, Roth JA (2004) Brain Res Mol Brain Res 122:62
Knutson M, Menzies S, Connor J (2004) J Neurosci Res 76:633
Wang XS, Ong WY, Connor JR (2003) 120:21
Burdo JR, Menzies SL, Simpson IA, Garrick LM, Garrick MD, Dolan KG, Haile DJ (2001) J Neurosci Res 66:1198
Perl DP, Olanow CW (2007) J Neuropathol Exp Neurol 66:675
Donaldson J, LaBella FS, Gesser D (1981) Neurotoxicology 2:53
Archibald FS, Tyree C (1987) Arch Biochem Biophys 256:638
Segura-Aguilar J, Lind C (1989) Chem Biol Interact 72:309
Stredrick DL, Stokes AH, Worst TJ, Freeman WM, Johnson EA, Lash LH
Eriksson H, Heilbronn E (1983) Arch Toxicol 54:53
Cai T, Yao T, Li Y, Chen Y, Du K, Chen J, Luo W (2007) Brain Res 1185:359
Arriagada C, Paris I, Sanchez de las Matas MJ, Martinez-Alvarado P, Cardenas S, Castañeda P, Graumann R, Perez-Pastene C, Olea-Azar C, Couve E, Herrero MT, Caviedes P, Segura-Aguilar J (2004) Neurobiol Dis 16:468
Segura-Aguilar J, Metodiewa D, Baez S (2001) Neurotox Res 3:157
Paris I, Dagnino-Subiabre A, Marcelain K, Bennett LB, Caviedes P, Caviedes R, Azar CO, Segura-Aguilar J (2001) J Neurochem 77:519
Paris I, Martinez-Alvarado P, Perez-Pastene C, Vieira MN, Olea-Azar C, Raisman-Vozari R, Cardenas S, Graumann R, Caviedes P, Segura-Aguilar J (2005) J Neurochem 92:1021
Paris I, Cardenas S, Lozano J, Perez-Pastene C, Graumann R, Riveros A, Caviedes P, Segura-Aguilar J (2007) Neurotox Res 12:125
Paris I, Lozano J, Perez-Pastene C, Muñoz P, Segura-Aguilar J (2009) Neurotox Res 16:271
Paris I, Perez-Pastene C, Cardenas S, Iturriaga-Vasquez P, Muñoz P, Couve E, Caviedes P, Segura-Aguilar J (2010) Neurotox Res 18:82
Paris I, Perez-Pastene C, Couve E, Caviedes P, Ledoux S, Segura-Aguilar J (2009) J Biol Chem 284:13306
Fuentes P, Paris I, Nassif M, Caviedes P, Segura-Aguilar J (2007) Chem Res Toxicol 20:776
Segura-Aguilar J, Diaz-Veliz G, Mora S, Herrera-Marschitz M (2002) Neurotox Res 4:127
Paris I, Lozano J, Cardenas S, Perez-Pastene C, Saud K, Fuentes P, Caviedes P, Dagnino-Subiabre A, Raisman-Vozari R, Shimahara T, Kostrzewa JP, Chi D, Kostrzewa RM, Caviedes R, Segura-Aguilar J (2008) Neurotox Res 13:221
Díaz-Véliz G, Mora S, Gómez P, Dossi MT, Montiel J, Arriagada C, Aboitiz F, Segura-Aguilar J (2004) Pharmacol Biochem Behav 77:245
Díaz-Véliz G, Mora S, Dossi MT, Gómez P, Arriagada C, Montiel J, Aboitiz F, Segura-Aguilar J (2002) Pharmacol Biochem Behav 73:843
Strausak D, Mercer JF, Dieter HH, Stremmel W, Multhaup G (2001) Brain Res Bull 55:175
Du F, Qian ZM, Zhu L, Wu XM, Yung WH, Tsim TY, Ke Y (2009) PLoS One 4:e4593
White AR, Cappai R (2003) J Neurosci Res 71:889
Reddy PV, Rao KV, Norenberg MD (2008) Lab Invest 88:816
Lu J, Zheng YL, Wu DM, Sun DX, Shan Q, Fan SH (2006) FEBS Lett 580:6730
Lu J, Wu DM, Zheng YL, Sun DX, Hu B, Shan Q, Zhang ZF, Fan SH (2009) Brain Behav Immun 23:193
Zheng Z, White C, Lee J, Peterson TS, Bush AI, Sun GY, Weisman GA, Petris MJ (2010) J Neurochem 114:1630
Taly AB, Meenakshi-Sundaram S, Sinha S, Swamy HS, Arunodaya GR (2007) Medicine (Baltimore) 86:112
Caviedes P, Segura-Aguilar J (2001) Neuroreport 12:A25
Segura-Aguilar J, Metodiewa D, Welch CJ (1998) Biochim Biophys Acta 1381:1
Díaz-Véliz G, Paris I, Mora S, Raisman-Vozari R, Segura-Aguilar J (2008) Chem Res Toxicol 21:1180
Wimalasena DS, Wiese TJ, Wimalasena K (2009) J Neurochem 101:313
Kume A, Shiratori M, Takahashi A, Kato T, Ito K, Tadokoro M, Sakuma S (1992) J Neurol Sci 110:37
Zhang J, Zhang Y, Wang J, Cai P, Luo C, Qian Z, Dai Y, Feng H (2010) Brain Res 1330:124
Song N, Jiang H, Wang J, Xie JX (2007) J Neurosci Res 85:3118
Roth JA, Singleton S, Feng J, Garrick M, Paradkar PN (2010) J Neurochem 113:454
Hirsch EC (2009) Parkinsonism Relat Disord 15:S209
Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nuñez MT, Garrick MD, Raisman-Vozari R, Hirsch EC (2008) Proc Natl Acad Sci USA 105:18578
Wang J, Jiang H, Xie JX (2007) Eur J Neurosci 25:2766
Song N, Wang J, Jiang H, Xie J (2010) Free Radic Biol Med 48:332
Zhu W, Li X, Xie W, Luo F, Kaur D, Andersen JK, Jankovic J, Le W (2010) Neurobiol Dis 37:307
Kaur D, Lee D, Ragapolan S, Andersen JK (2009) Free Radic Biol Med 46:593
Tribl F, Asan E, Arzberger T, Tatschner T, Langenfeld E, Meyer HE, Bringmann G, Riederer P, Gerlach M, Marcus K (2009) Mol Cell Proteomics 8:1832
Youdim MB, Grünblatt E, Mandel S (2007) J Neural Transm 114:205
Xu Q, Kanthasamy AG, Reddy MB (2008) Toxicology 245:101
Maharaj DS, Maharaj H, Daya S, Glass BD (2006) J Neurochem 96:78
Wang R, Qing H, Liu XQ, Zheng XL, Deng YL (2008) Neurosci Bull 24:125
Zecca L, Casella L, Albertini A, Bellei C, Zucca FA, Engelen M, Zadlo A, Szewczyk G, Zareba M, Sarna T (2008) J Neurochem 106:1866
Double KL, Halliday GM, Henderson J, Griffiths FM, Heinemann T, Riederer P, Gerlach M (2003) Exp Neurol 184:530
Paris I, Martinez-Alvarado P, Cárdenas S, Perez-Pastene C, Graumann R, Fuentes P, Olea-Azar C, Caviedes P, Segura-Aguilar J (2005) Chem Res Toxicol 18:415
Arreguin S, Nelson P, Padway S, Shirazi M, Pierpont C (2009) J Inorg Biochem 103:87
Siraki AG, Smythies J, O’Brien PJ (2000) Neurosci Lett 296:37
Ortega R, Cloetens P, Devès G, Carmona A, Bohic S (2007) PLoS One 2:e925
Mohamed GG, Zayed MA, El-Dien FA, El-Nahas RG (2004) Spectrochim Acta A 60:1775
Arriagada C, Dagnino-Subiabre A, Caviedes P, Armero JM, Caviedes R, Segura-Aguilar J (2000) Amino Acids 18:363
Acknowledgments
Supported by FONDECYT 1100165.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Wien
About this chapter
Cite this chapter
Paris, I., Segura-Aguilar, J. (2012). The role of metal ions in dopaminergic neuron degeneration in Parkinsonism and Parkinson’s disease. In: Linert, W., Kozlowski, H. (eds) Metal Ions in Neurological Systems. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1001-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1001-0_3
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1000-3
Online ISBN: 978-3-7091-1001-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)