
Abstract
Since the first report in 1964 (Guroff 1964) of a cytosolic Ca2+-dependent neutral proteolytic activity, now known as calpain-1 (also called μ-calpain), other members of the calpain family of proteinases have been discovered (Croall and DeMartino 1991; Molinari and Carafoli 1997; Goll et al. 2003; Bertipaglia and Carafoli 2007; Croall and Ersfeld 2007; Sorimachi et al. 2010, 2011a). The calpains are defined as having amino acid (aa) sequences significantly similar to that of the protease domain of human calpain-1. Calpains also contain a variety of protein structural motifs including C2, calpain-type β-sandwich (CBSW), Zn-finger and penta-EF-hand (PEF) domains. This important class of intracellular cysteine proteases is found in almost all eukaryotes and some bacteria. To date, 15 calpain genes have been identified in humans (see Fig. 12.1). However, mammalian calpain-1 and -2, sometimes referred to as the “ubiquitous,” “conventional,” or “classical” calpains, are the best-characterized members of the calpain superfamily (Clan CA, family C02, EC 3.4.22.17). The conventional calpains have a very specific proteinaceous inhibitor in vivo called calpastatin. The ubiquitous calpain-1 and -2 and their natural inhibitor calpastatin are the three members of the calpain system that have been studied most extensively at the structure-functional level. Under normal physiological conditions, calpains exist at very low activity in cells and are proposed to participate in important cellular activities, including signal transduction, cell motility and apoptosis. However, excessive calpain activation following ischemic and traumatic cell injury or aberrant expression of components of the calpain system results in cell death or impaired cellular function. This review summarizes current knowledge of the biological significance of regulated calpain activity and the pathological consequences of uncontrolled or excessive calpain activation, with special emphasis on the pathophysiological roles of the calpains in ischemic and traumatic brain injury and some tissue-specific diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Traumatic Brain Injury
- Central Nervous System Injury
- Calpain Inhibitor
- Autolytic Activity
- Calpain System
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
12.1 Introduction
Since the first report in 1964 (Guroff 1964) of a cytosolic Ca2+-dependent neutral proteolytic activity, now known as calpain-1 (also called μ-calpain ), other members of the calpain family of proteinases have been discovered (Croall and DeMartino 1991; Molinari and Carafoli 1997; Goll et al. 2003; Bertipaglia and Carafoli 2007; Croall and Ersfeld 2007; Sorimachi et al. 2010, 2011a, b). The calpains are defined as having amino acid (aa) sequences significantly similar to that of the protease domain of human calpain-1. Calpains also contain a variety of protein structural motifs including C2, calpain-type β-sandwich (CBSW; once called C2-like), Zn-finger and penta-EF-hand (PEF ) domains. This important class of intracellular cysteine proteases is found in almost all eukaryotes and some bacteria. To date, 15 calpain genes have been identified in humans (see Fig. 12.1). However, mammalian calpain-1 and calpain-2 (also called m-calpain, calpain-II or mCANP), sometimes referred to as the “ubiquitous,” “conventional,” or “classical” calpains , are the best-characterized members of the calpain superfamily (Clan CA, family C02, EC 3.4.22.17). The conventional calpains have a very specific proteinaceous inhibitor in vivo called calpastatin . The ubiquitous calpain-1 and -2 and their natural inhibitor calpastatin are the three members of the calpain system that have been studied most extensively at the structure-functional level. Under normal physiological conditions, calpains exist at very low activity in cells and are proposed to participate in important cellular activities, including signal transduction, cell motility and apoptosis. However, excessive calpain activation following ischemic and traumatic cell injury or aberrant expression of components of the calpain system results in cell death or impaired cellular function. This review summarizes current knowledge of the biological significance of regulated calpain activity and the pathological consequences of uncontrolled or excessive calpain activation, with special emphasis on the pathophysiological roles of the calpains in ischemic and traumatic brain injury and some tissue-specific diseases.

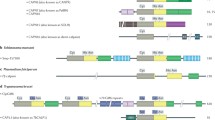
Schematic structures of human calpains and the related molecules. (a) Schematic structures of 15 human calpains. Names for calpains are by the general nomenclature of gene product (e.g., CAPN1 → CAPN1) used in this review, and their previous names, if any, are shown after the general names. Black and reversed letters indicate ubiquitous and tissue/organ-specific calpains, respectively. Symbols: PC1 and PC2, protease core domains 1 and 2 in the calpain protease (CysPc) domain; CBSW calpain-type β-sandwich domain; PEF(L/S), penta-EF-hand domains in the large(L)/small(S) subunit; GR, glycine-rich hydrophobic domain; MIT, microtubule interacting and transport motif; C2, C2 domain; Zn, Zn-finger motif; SOH, SOL-homology domain; IQ, a motif interactive with calmodulin; NS/IS1/IS2, CAPN3/p94-characteristic sequences. (b) Domain architecture of human calpain-1 and calpain-2. Calpain-1 and -2 are heterodimers composed of a common regulatory small subunit (CAPNS1/30K) and each catalytic large subunit (CAPN1/μCL and CAPN2/mCL, respectively). See (a) for abbreviations of domain names. (c) Domain architecture of calpastatin
12.2 Conventional Calpains
12.2.1 Calpain-1/μ-Calpain and Calpain-2/m-Calpain
12.2.1.1 Discovery and Nomenclature
Ca2+-activated neutral proteinase (CANP ) activity was first discovered in the soluble fraction of rat brain in 1964 (Guroff 1964). Similar observations were subsequently made in rabbit, human and chicken skeletal muscle as well as in rat liver (Meyer et al. 1964; Huston and Krebs 1968; Busch et al. 1972; Reddy et al. 1975; Kar and Pearson 1976; Takai et al. 1977; Ishuira et al. 1978). Further characterization of the biochemical properties of the Ca2+-dependent proteolytic activity led to the isolation of distinct molecules of the calpain system. The cDNA for calpain was finally cloned by Ohno et al. (1984). Hundreds of calpain-related genes, including tissue-specific and non-classical forms, have been cloned and sequenced since the original cloning of the ubiquitously expressed calpain, but the gene products of a majority of them remain to be characterized. The conventional calpains are represented by two isoforms of calpains, calpain-1 and calpain-2, that are activated at neutral pH by micro- and milli-molar concentrations of Ca2+ in vitro, respectively, and are ubiquitously expressed in all eukaryotic cells examined so far (Croall and DeMartino 1991; Goll et al. 2003; Croall and Ersfeld 2007; Sorimachi et al. 2010). They are also called μ-calpain and m-calpain, calpain-I and calpain II, or μCANP and mCANP, respectively. An endogenous specific inhibitor of calpain-1 and -2, called calpastatin, was later discovered in the late 1970s (Dayton et al. 1976; Suzuki and Murachi 1977; Nishiura et al. 1978). To date, 15 human calpain genes have been discovered and numbered CAPN 1–3 and 5–16, as shown in Fig. 12.1a. μCL and mCL correspond to the gene products of CAPN1 and CAPN2, and the systematic names, calpain-1 and calpain-2, have gained popularity. However, since “calpain” originally was a term for an enzyme consisting of two distinct subunits, this nomenclature is confusing and misleading. Therefore, here, the formal gene product nomenclature, i.e., CAPN1 and CAPN2 , is used. Accordingly, calpain-1 (= μ-calpain) is a heterodimer of CAPNS1 and CAPN1. In this review, to include the older name, CAPN1/μCL and CAPN2/mCL, is used for clarity.
12.2.1.2 Structural Features
Both calpain-1 and -2 are heterodimers, composed of a common 28 kDa regulatory subunit (CAPNS1 /30K) and two distinct 80 kDa catalytic subunits (CAPN1/μCL and CAPN2/mCL, respectively). The catalytic and regulatory subunits of the conventional calpains are divided into four and two region/domains, respectively (see Fig. 12.1b). The protease (CysPc ) domain is the catalytic site containing the cysteine, histidine, asparagine triad (Saido et al. 1994). In the absence of Ca2+, CysPc domain is divided into protease core domains PC1 and PC2 , which are folded into one domain upon Ca2+ binding (Hanna et al. 2008; Hosfield et al. 1999; Strobl et al. 2000). Domains PC1, PC2, CBSW, PEF(L) and PEF(S) bind at least one Ca2+ with varying affinities (Moldoveanu et al. 2002, 2003; Tompa et al. 2001). An extensive knowledge of the structures of the inactive Ca2+-free and the active (Ca2+ and calpastatin bound) forms of calpain-2 is available, and has provided useful insights into the mechanism for the Ca2+-dependent activation of the enzyme but the mechanism by which the protease is regulated in vivo is still not clear, in spite of many suggestions (Hanna et al. 2008).
The classical calpains normally exist as inactive proteases because their catalytic triad residues are not properly aligned in the Ca2+-free form of the protein. It has been deduced from the crystal structures that upon binding of Ca2+ to specific penta-EF-hands in PEF domains and other non-EF-hand sites within PC1, PC2, and CBSW domains, multiple conformational changes occur within the protein to align the catalytic machinery of the active site (Hosfield et al. 1999, 2004; Strobl et al. 2000; Moldoveanu et al. 2002). Furthermore, site-directed mutagenesis studies have allowed the identification of multiple intramolecular sites in the NH2-terminal peptide (residues 1–20) and the domain CBSW-PEF(L) linker peptide (“transducer”) that regulate the response of calpain to Ca2+ binding to CysPc, CBSW, PEF(L) and PEF(S) domains (Moldoveanu et al. 2002; Hosfield et al. 2004; Bozoky et al. 2005). The exact sequence of the steps involved in the relay of the Ca2+ message is however unknown. On activation, calpain autolyzes by truncating the N-terminal portion of the large subunit and most of Gly-rich (GR ) domain of the small subunit. The activated form appears to have greater calcium sensitivity (Hathaway et al. 1982; Imajoh et al. 1986; Suzuki et al. 1981).
An endogenous proteinaceous inhibitor of calpain-1 and -2, called calpastatin , was discovered in the late 1970s (Dayton et al. 1976; Suzuki and Murachi 1977; Nishiura et al. 1978; Takahashi-Nakamura et al. 1981). Among the other calpain homologues, CAPN8 /nCL-2 and CAPN9 /nCL-4 , but not CAPN3 /p94 , are also inhibited by calpastatin. The primary structure of calpastatin consists of a non-inhibitory L-domain and four repeating (1–4) inhibitory domains, each having an independent inhibitory activity against calpain (Emori et al. 1988) (see Fig. 12.1c). The repeating inhibitory domains have highly conserved internal subdomains A, B, and C, with subdomain B representing the calpain inhibitory moiety. A 27-mer synthetic peptide corresponding to subdomain 1B of human calpastatin has been found to exhibit potent and specific inhibition toward calpain-1 and -2 in vitro (Maki et al. 1989; Betts et al. 2003; Betts and Anagli 2004; Fiorino et al. 2007) and in vivo in a rat model of ischemic stroke (Anagli et al. 2009).
12.2.1.3 Biological Significance
Because the conventional calpains are ubiquitously expressed in vertebrate cells, their function is considered to be fundamental and essential. The physiological role of calpain-1 and -2 is not fully understood, but it has been linked to Ca2+-regulated signaling pathways. Their involvement in the regulation of physiological processes such as apoptosis (Benetti et al. 2001; Danial and Korsmeyer 2004; Gomez-Vicente et al. 2005; Norberg et al. 2008; Sanges et al. 2006), signal transduction (Kishimoto et al. 1983; Noguchi et al. 1997; Pontremoli and Melloni 1988), membrane repair (Mellgren and Huang 2007; Mellgren et al. 2007), modulation of cell motility (Satish et al. 2005; Wells et al. 2005) and synaptic function and memory formation (see review by Liu et al. 2008a) has been described. Calpains are somewhat selective with regard to substrate selection; only 5 % of cellular proteins are degraded or fragmented by calpain-1 and -2 (Wang et al. 1989). A number of substrates have been reported for calpains, but few of them have been linked to physiological functions. These include, focal adhesion kinase (FAK) and ezrin, which are involved in fibroblast cell attachment and mobility (Potter et al. 1998; Huang et al. 2004), and phosphotyrosine phosphatase 1 B (PTP1B), which is involved in platelet aggregation (Kuchay et al. 2007). Possible substrates in the central nervous system (CNS) that might be critical to synaptic function and memory formation include αII- and βII-spectrin , the postsynaptic density-95 (PSD95) scaffolding protein, calcium- and calmodulin-dependent protein kinase II (CaMKII), and the N-methyl-D-aspartate (NMDA)-type and other glutamate receptors (Bi et al. 1998; Wang 2000; Liu et al. 2008b).
12.2.1.4 Pathologic Role of Calpain
Evidence accumulated over the past 15 years incriminates calpain hyper-activation in neural cell death following severe cellular challenge or damage in cerebral ischemia (Hong et al. 1994; Roberts-Lewis et al. 1994; Markgraf et al. 1998; Yokota et al. 1999; Liebetrau et al. 1999; Kambe et al. 2005; Kawamura et al. 2005; Anagli et al. 2009), traumatic brain injury (TBI) (Taft et al. 1992; Nath et al. 1996; Postmantur et al. 1996; Buki et al. 1999, 2003; Newcomb et al. 1999; Kampfl et al. 1997; Pike et al. 1998; Pike et al. 2000; Ringger et al. 2004), spinal cord injury (SCI) (Banik et al. 1997; Ray et al. 1999, 2001a, b, 2011; Schumacher et al. 2000; Ray et al. 2003; Sribnick et al. 2007; Yu et al. 2008; Zhang et al. 2003), sub-arachnoid hemorrhage (SAH) (Germano et al. 2002; Zhang et al. 2000; Wang and Yuen 1994; Lee et al. 1997; Yamaura et al. 1993) cerebral vasospasm (Minami et al. 1992), multiple sclerosis (Shields and Banik 1999; Schaecher et al. 2001, 2002; Benjamins et al. 2003; Nakanishi 2003; Shields et al. 1999), Parkinson’s Disease (Crocker et al. 2003; Ray et al. 2000; Chera et al. 2002; Kim et al. 2003; Samantaray et al. 2008, 2011), Alzheimer’s Disease (Veeranna et al. 2004; Chen and Fernandez 2005; Higuchi et al. 2005; Fifre et al. 2006; Rao et al. 2008; Liu et al. 2011), Huntington’s Disease (Gafni and Ellerby 2002; Gafni et al. 2004; Bizat et al. 2003, 2005), and amyotrophic lateral sclerosis (ALS) (Das et al. 2008; Tradewell and Durham 2010).
Investigation of the neuronal pathobiology following cerebral ischemia (Hong et al. 1994; Chen et al. 1998; Kristian et al. 1998) and TBI (Povlishock et al. 1983, 1992; Povlishock 1992; Maxwell et al. 1997; Meaney et al. 2001) indicates that the majority of injured neurons are not damaged at the time of impact but show progressive degenerative changes over the ensuing hours or days. The initial injurious event triggers a destructive cascade of biochemical events that results in cell death within the core of the injured tissue and which spreads out into surrounding tissue over time. As the initial step, ischemic (Szatkowski and Attwell 1994; Rothman and Olney 1986) or traumatic (Hayes et al. 1992; McIntosh et al. 1998) insult induces massive release of glutamate from damaged synapses which leads to activation of glutamate receptor-associated and voltage-dependent calcium channels. Such activation induces influx of calcium ions into the neuron (Fineman et al. 1993; Pettus et al. 1994; Pettus and Povlishock 1996) and release of calcium ions from intracellular stores (Berridge 1993) (see Fig. 12.2). Loss of intracellular calcium homeostasis contributes to cell death by (1) activating various enzymes including proteases, kinases, phosphatases, and phospholipases (Siesjo et al. 1989; Wieloch et al. 1991; Morioka et al. 1992; Verity 1992; Nishida et al. 1994; Wang and Yuen 1994; Dash et al. 1995), (2) induction of free radical release (Kontos 1989; Bading et al. 1993; Hall 1998), and (3) mediating detrimental changes in gene expression (Bading et al. 1993; Rink et al. 1995). Integral to the mechanism of calcium-mediated neuronal degeneration in both ischemic (Yamashima 2000; Rami 2003) and traumatic (Kampfl et al. 1997; Newcomb et al. 1997; Posmantur et al. 1997; Saatman et al. 1996a; Wang 2000) brain injury is the pathological activation of calpain-1 (CAPN1 /μCL + CAPNS1 /30K ) and calpain-2 (CAPN2 /mCL + CAPNS1/30K), which results in proteolytic destruction of many cellular proteins including receptor proteins, calmodulin binding proteins, signal transduction enzymes, transcription factors, and cytoskeletal proteins (Wang and Yuen 1997, 1999). Such uncontrolled proteolysis can result in disruption of axonal transport and structural collapse culminating in oncotic and/or apoptotic cell death (Buki et al. 2000; Medana and Esiri 2003) (see Fig. 12.2).

Conventional calpains in disease. (a) Schematic of the destructive Ca2+-mediated cascade that results in progressive cell damage following traumatic or ischemic brain injury. Initial injury induces massive glutamate release from damaged synapses leading to hyper-activation of NMDA and AMP/kainite receptors (NMDA-R and AMPA/KA-R) leading to influx of calcium and sodium ions. Ca2+ ions can also enter the cell through sodium-activated voltage-sensitive calcium channels (VSCC), via leakage through damaged cell membranes, and they can be released from internal stores. The resulting increase in intracellular Ca2+ levels activates a number of Ca2+-sensitive proteases, including calpain. Once activated, calpain proteolyses critical cellular proteins, including the cytoskeletal protein αII-spectrin, resulting in compromise of the cell membrane and cell death. (b) Calpain as a therapeutic target for ameliorating brain injury. Calpain inhibitors, such as SNJ-1945 or B27-HYD, can block calpain hyper-activation following injury, thereby shutting down the destructive cascade and preserving cell integrity. Thus, calpain inhibitors are neuroprotective
12.2.1.5 Conventional Calpain-Generated Biomarkers of CNS Injury
During brain injury , neural proteins or their breakdown products generated by calpains (calpain-1 and -2) are released into the extracellular environment and eventually reach the cerebrospinal fluid (CSF) in relatively high concentration (Wang et al. 2005). In due time the proteins reach the blood stream either via the compromised blood brain barrier (BBB) or via filtration of the CSF (see Fig. 12.3). Clearance and half life of the biomarkers contribute to the final concentration that can be measured in the blood. The CSF volume of an adult human (CSF 125–150 mL) is about 30- to 40-fold less than the blood volume (4.5–5 L) which explains why the brain biomarker concentration is significantly higher in the CSF samples versus blood samples and makes the former valuable for drug development. Enabled by recent technological advances in proteomics, novel brain injury biomarkers that have elevated levels in biofluid s such as cerebrospinal fluid or blood after traumatic brain injury have been discovered (Vitzthum et al. 2005; Kobeissy et al. 2006; Liu et al. 2006; Mondello et al. 2011). The most well-studied calpain target is the cytoskeletal structural protein αII-spectrin which is cleaved by calpain into signature breakdown products of 150 kDa (SBDP150) and 145 kDa (SBDP145) (Saido et al. 1993; Nath et al. 1996; Wang et al. 1998; Pike et al. 1998; Pike et al. 2000; Zhao et al. 1999; Newcomb-Fernandez et al. 2001). Calpain-generated neural protein breakdown products (BDPs) such as SBDP150, SBDP145, c-Tau, PARP, as well as MAP2, GFAP and CRMP-2 BDPs can be used to monitor acute necrotic/oncotic neural cell death and diagnose brain injury severity and progression (Siman and Noszek 1988; Siman et al. 1989, 2004; Pike et al. 2001, 2003; Ringger et al. 2004; Liu et al. 2006, 2008a, b, 2011; Anagli et al. 2009; Zhang et al. 2009). In addition, sensitive and selective sandwich ELISAs for such biomarkers have allowed quantification of the extent of brain injury in animal studies and clinical studies of brain injury alike (Liu et al. 2010; McGinn et al. 2009; Mondello et al. 2010; Siman et al. 2009).

Generation and detection of brain injury biomarkers
12.2.1.6 Calpain-Target-Based Theranostics for CNS Injury
Although brain (ischemic and traumatic) and spinal cord injury is a significant medical crisis, there are no FDA-approved therapies that have been demonstrated to improve functional outcomes. This is the case despite a large number of clinical trials with promising animal model efficacy data. Major pharmaceutical companies and biotech companies have been trying for years to tackle acute brain injury without success. New therapeutic development traditionally has an extremely high triage rate because more than 90 % of drugs that advance to Phase I clinical trials fail. Some argue such extreme loss can be overcome by guiding all new therapeutic development and clinical trials with a disease-relevant diagnostic test. Discovery of translational biomarker s (from animal studies to clinical trials) might help to finally deliver the long sought after clinical trial success.
Since the discovery of calpain in the mid-1960s (Guroff 1964; Huston and Krebs 1968), scientists have sought methods to inhibit its activity. Early attempts used calcium chelators, but as knowledge of calpain substrate specificity grew, and inhibitors from natural products screening programs became available, more selective inhibitors were designed and synthesized (Aoyagi and Umezawa 1975; Parkes et al. 1985; Crawford 1987; Crawford et al. 1988; Anagli et al. 1991; Angliker et al. 1992; Wikstrom et al. 1992; Fukiage et al. 1997; Schroder et al. 1993; Wang and Yuen 1997; Shirasaki et al. 2005; Pietsch et al. 2010; Donkor 2011). Although synthetic calpain inhibitors have proved efficacious in in vivo models of TBI, SCI and cerebral ischemia they have limited therapeutic value because (1) they inhibit other cysteine and serine proteases, (2) they have poor membrane permeability , and (3) they are readily degraded in vivo (Wang and Yuen 1997; Yuen and Wang 1998). The endogenous calpain inhibitor, calpastatin , is the most potent and selective inhibitor identified to date (Murachi 1989; Maki et al. 1991). Other proteins, including the heavy chains of L- and H-kininogens and α2-macroglobulin, also inhibit calpain (Crawford 1987; Salvesen et al. 1986). However, these high molecular weight inhibitors have limited therapeutic utility due to their poor cell permeability and lack of specificity. Calpain inhibition has the potential to shut down the destructive calcium-mediated cascade, thereby reducing proteolysis of essential cell proteins, resulting in cell preservation and protection (see Fig. 12.4). Previous therapeutic strategies for ameliorating the post-injury calcium-mediated destructive cascade have focused on antagonizing glutamate release, glutamate receptors, or voltage-sensitive calcium channels (Hossman 1988; Siesjo and Bengtsson 1989; Bullock and Fujisawa 1992; McIntosh 1994). These strategies have produced limited success because (1) the glutamate response occurs very rapidly after injury leaving only a narrow window for treatment efficacy, and (2) it is difficult to distinguish which of the various receptor and ion channel subclasses are primarily involved in perpetuating the cytotoxic cascade (Hossman 1988; Saatman et al. 1996b). Treatment aimed at a downstream neuropathological event could provide a longer window of opportunity for effective intervention, and therefore be valuable for a greater number of patients. Calpain antagonists may have a further advantage over glutamate and calcium channel receptor antagonists in that calpain exists predominantly in its inactive proenzyme form under normal physiological conditions, and only becomes significantly activated under pathological conditions. Therefore, it would be reasonable to assume that calpain inhibition would not lead to any untoward adverse events. On the other hand, glutamate and calcium channel receptors play a critical neurotransmitter role in and outside of the CNS, and their inhibition could be expected to have profound side effects. Indeed, although glutamate receptors have proved to provide neuroprotection in animal models of cerebral ischemia, they have also been shown to have significant psychotomimetic effects (Olney et al. 1991). Therefore, calpain has unique characteristics as a therapeutic target for CNS injury (Liu et al. 2008a, b) (see Table 12.1). Taken together, these data suggest that brain injury is in need of a paradigm shift with regards to therapeutic development. Calpain-target-based therapeutics and companion diagnostic biomarkers might be the missing component that helps guide successful clinical trials for brain injury.

“Theranostic Approach” to treat traumatic brain injury with a calpain inhibitor drug
“Theranostics ” represents the convergence between Therapeutics and diagnostics. “Theranostics is the term used to describe the proposed process of diagnostic therapy for individual patients—to test them for possible reaction to taking a new medication and to tailor a treatment for them based on the test results” (Warner 2004). Theranostics encompasses the possible utilization of a wide range of procedures including: predictive medicine, personalized medicine, integrated medicine, pharmaco-diagnostics and Dx/Rx partnering. It has been viewed as the parallel use of new therapy and diagnostic tests for a human disease or disorder so as to facilitate drug development and clinical trials and to achieve optimal clinical outcomes in a population of patients. Importantly, in recognizing the emerging role of theranostic approach, FDA has recently drafted a “Drug-Diagnostic Co-Development Concept Paper” with the goal of setting guidelines for prospective co-development of a drug or biological therapy (drugs) and a device test in a scientifically robust and efficient way. Lastly, another advantage of the theranostic approach is its built-in ability to achieve a post-marketing personalized medicine paradigm (i.e., drug treatment could be tailored according to a patient’s diagnostic biomarker profile over time. Most importantly, as proposed in this review, a novel theranostic approach that combines calpain-generated acute brain injury-tracking biomarkers with potent and selective calpain inhibitor drug candidates could fast-track and improve the chances of successful drug development for CNS injury.
12.3 Unconventional Calpains
12.3.1 Skeletal-Muscle-Specific Calpain
12.3.1.1 Discovery and Nomenclature
In 1989, in the course of the cDNA cloning of human calpain-1 and -2 catalytic large subunits (CAPN1 /μCL and CAPN2 /mCL , respectively), a cDNA encoding a novel molecule that was similar to but distinct from both was discovered (Sorimachi et al. 1989). In contrast to the ubiquitous expression of CAPN1/μCL and CAPN2/mCL, the identified mRNA was predominantly expressed in skeletal muscle , i.e., skeletal muscle-specific calpain was the first tissue-specific calpain to be discovered. The novel calpain catalytic subunit was about 50 % identical in aa sequence to those of CAPN1/μCL and CAPN2/mCL. It had the same basic domain structure as the known calpain catalytic subunits, consisting of four region/domains: the N-terminal specific region (NS ), CysPc , CBSW, and PEF domains (see Fig. 12.1a) (Ono et al. 1999; Sorimachi et al. 2010, 2011a, b; Sorimachi and Suzuki 2001).
Initially, this molecule was called p94 for its putative molecular mass (Sorimachi et al. 1989), because there was no information about its tertiary structure. Since the overall aa sequence of p94 was highly similar to those of CAPN1/μCL and CAPN2/mCL, it was thought likely that an as-yet-unknown regulatory small subunit, similar or identical to CAPNS1 /30K , would associate with p94 to make the active enzyme, which would be called a novel calpain with a new name such as n-calpain. Four years later, another novel calpain homolog was identified, clarifying the need for a method to systematize the names of the calpain-related molecules (Sorimachi et al. 1993a). For this purpose, it was proposed that novel calpains, including as-yet-undiscovered ones, would be numbered as n-calpain-1, n-calpain-2, etc., where ‘n’ stands for novel, and p94 would be the catalytic large subunit of n-calpain-1 (for short, nCL-1). In 1990, the human genes for CAPN1/μCL, CAPN2/mCL, p94/nCL-1, and CAPNS1/30K were identified and named CAPN 1, CAPN2, CAPN3, and CAPN4, respectively (Ohno et al. 1990). In 1995, a defect of CAPN3 was shown responsible for limb-girdle muscular dystrophy (LGMD) type 2A (LGMD2A , also called calpainopathy ), and p94/nCL-1 was termed calpain 3 after the gene name CAPN3 (Richard et al. 1995). As discussed in the conventional calpain nomenclature, the skeletal-muscle-specific calpain should now be called CAPN3 . In this review, CAPN3/p94 is used.
12.3.1.2 Structural Features
The basic domain structure of CAPN3/p94 is identical to those of CAPN1/μCL and CAPN2/mCL, as shown in Fig. 12.1a, b. However, CAPN3/p94 contains three unique regions, NS , IS1 , and IS2 (insertion sequence 1 and 2, respectively) , which are neither found in the other calpains nor related to other peptide sequences in the databases.
The NS is a Pro-rich structure of unknown function that is cut off upon the autolysis of CAPN3/p94 (Hayashi et al. 2008; Ono et al. 2004). CysPc domain, as in other calpain homologs, is the most highly conserved. Site-directed mutagenesis identified that Cys129, His334, Asn358 are essential for CAPN3/p94’s activity. IS1 is inserted into the PC2 domain .
IS2 locates between the CBSW and PEF domains and contains a nuclear-localization signal-like sequence, PXKKKKXKP. Deletion of either IS1 or IS2 suppresses autolysis, and thus stabilizes CAPN3/p94 expression. In addition, the IS2 region is necessary and sufficient for CAPN3/p94’s binding to the N2A region of connectin/titin (Sorimachi et al. 1995).
The CBSW and PEF domains of CAPN3/p94 are highly homologous to those of the conventional calpains, strongly suggesting that these domains bind Ca2+ regardless of the Ca2+-independence of CAPN3/p94’s protease activity in the presence of enough Na+ (see below). Many of the pathogenic mutations found in LGMD2A/calpainopathy patients reside in these domains as well as the CysPc domain (Richard et al. 1999).
A comparison of the CAPN3/p94s of different vertebrates showed that more than half aa residues (aar) are more than 80 % conserved between species (Sorimachi et al. 2011a). These conserved aar include more than 70 % of the missense loci found in LGMD2A/calpainopathy pathogenic mutations (see below and Fig. 12.5). Conservation within the CAPN3/p94-characterizing regions (NS, IS1, and IS2) is low; however, it should be noted that some missense mutations, such as the R49C/H, P319L, and S606L mutations within the NS, IS1, and IS2 regions, respectively, occur relatively frequently (Sorimachi et al. 2011a) (Fig. 12.5). This observation suggests that strict sequence conservation within the CAPN3/p94-characterizing regions is not necessarily required, but a few important aar, usually near the borders of these regions, must be conserved to maintain proper function.

Pathogenic CAPN3 missense mutations and their frequencies in LGMD2A/calpainopathy patients. Frequencies are relative to the maximum value. Black vertical lines indicate aar that are conserved more than 80 % of vertebrates. α (red) and β (green) indicate α-helix and β-strand secondary structures of the corresponding calpain-2 3D structure. See Fig. 12.1 legend for other symbols
12.3.1.3 Proteolytic Activity
The mRNA for CAPN3/p94 is expressed predominantly in skeletal muscle, about ten times more abundant than the those for CAPN1/μCL and CAPN2/mCL. For a long time, however, neither the CAPN3/p94 activity nor the protein itself could be detected in skeletal muscle extract by the isolation procedures used successfully for calpain-1 and -2. Then, expression experiments using mutated CAPN3/p94 began to reveal its unique properties (Baghdiguian et al. 1999; Benayoun et al. 2008; Fukiage et al. 2002; Hayashi et al. 2008; Kramerova et al. 2006, 2008, 2009; Ono et al. 1998, 2004, 2010; Richard et al. 1995; Sorimachi et al. 1993b, 1995; Taveau et al. 2003; Ueda et al. 2001). These studies showed that the CAPN3/p94 protein undergoes extremely rapid autolysis (its half-life in vitro is less than 10 min), indicating that CAPN3/p94 has strong protease activity, which, on the other hand, makes studying it at the protein level very difficult. Moreover, this autodegradation is Na+-dependent in the absence of Ca2+, establishing CAPN3/p94 as the first example of an intracellular Na+-dependent enzyme . Specific inhibitors for calpain-1 and -2, including calpastatin , E-64 , and leupeptin , have little effect on CAPN3/p94’s autolytic activity. The in vitro substrates of CAPN3/p94 include CAPN3/p94 itself, myotonin protein kinase, fodrin , heat-shock protein (HSP) 60 , calpastatin, muscle-specific ankyrin-repeat proteins (MARPs ), and connectin/titin , the gigantic filamentous muscle protein.
Although the precise in vivo mechanism of CAPN3/p94’s regulation remains unclear, it is very likely that the activity is suppressed by the interaction of the enzyme with connectin/titin (Hayashi et al. 2008). The physiological relevance of the Na+-dependency of CAPN3/p94 is still unclear, but its substrate specificity differs depending on whether it is activated by Ca2+ or Na+ (Ono et al. 2010). One of the splicing variants of CAPN3 encodes CAPN3:ex1–5|7–14|17–24 (called p94Δ ), which lacks both IS1 and IS2, and has markedly reduced autolytic activity. Therefore, recombinant p94Δ could be purified relatively stable, and was used for enzyme characterization studies. The results showed that p94Δ has characteristics very similar to those of conventional calpains, except that it proteolyzes, and is not inhibited by, calpastatin (Ono et al. 2004).
The lens-specific splicing variant, CAPN3:ex1B|2–5|7–14|17–24 (called Lp82 ), also shows characteristics very similar to those of conventional calpains (Fukiage et al. 2002). Lp82 shows Ca2+-dependent protease activity against βA3 and αB crystallins, and autolyzes at the N-terminus and between the CBSW and PEF domains. Lp82’s activity is inhibited by E-64 and iodoacetamide (Ueda et al. 2001). Like p94Δ, Lp82 is not inhibited by calpastatin, and proteolyzes it. Some other splicing variants with structures similar to Lp82 are expressed in embryonic skeletal muscles (Fougerousse et al. 2000). These variants show a number of unique features compared with CAPN3/p94, although their physiological functions are not clear (Herasse et al. 1999; Ojima et al. 2007).
12.3.1.4 Biological Significance
In 1995, human CAPN3 was identified as a gene responsible for LGMD2A /calpainopathy (Richard et al. 1995). The mutations, including missense point mutations, nonsense mutations, frame-shift mutations, and splice-site mutations, are widely distributed in CAPN3. The absence of any ‘hot-spot’ in CAPN3 makes the diagnosis of LGMD2A/calpainopathy difficult (Sorimachi et al. 2011a). So far, more than 450 unique mutations have been reported in CAPN3. More than half of the CAPN3 pathogenic mutations are point mutations, and the majority of these are missense mutations, which are distributed throughout the protein at more than 175 independent loci among the 821 aar (Sorimachi et al. 2011a) (Fig. 12.5). Then, knock-out (Capn3 −/−) mice were shown to emulate a human LGMD2A/calpainopathy-like phenotype (although less severe), indicating that defects in CAPN3/Capn3 and LGMD2A/calpainopathy have a cause-effect relationship (Kramerova et al. 2004; Richard et al. 2000). This is the first and only example so far of a clear cause-effect relationship between human disease and calpain gene mutations.
The following evidence showed that LGMD2A/calpainopathy is primarily caused by compromised CAPN3/p94 protease activity, rather than by damaged structural properties. First, it was shown that 10 pathogenic missense mutants of CAPN3/p94 all showed a deficient ability to proteolyze fodrin (Ono et al. 1998). Second, transgenic mice expressing a structurally intact but inactive CAPN3/p94:C129S protein showed an accumulation of the inactive protein and myopathic phenotypes similar to LGMD2A/calpainopathy (Tagawa et al. 2000). Finally, CAPN3/p94 knock-in (Capn3 CS/CS) mice , which express CAPN3/p94:C129S in place of wild-type CAPN3, showed a muscular dystrophy phenotype (Ojima et al. 2010). Intriguingly, however, the Capn3 CS/CS mice showed a less severe phenotype than Capn3 −/− mice did, indicating that the proteolytically inactive CAPN3/p94 retains some function. One of possible scenarios is that CAPN3/p94 is a novel structural components of the SR , considering its association with ryanodine receptors (Ojima et al. 2010, 2011). On the other hand, transgenic mice expressing a CAPN3/p94 splicing variant lacking exon 6 (CAPN3:ex1–5|7–24) showed muscle phenotypes that were more severe than those of the knock-out mice (Spencer et al. 2002). These observations indicate that autolytic unstability of CAPN3/p94 is related to a certain aspect of its regulatory mechanism.
Studies using Capn3 CS/CS mice have provided some insights into the molecular mechanisms underlying the pathogenesis of LGMD2A/calpainopathy. First, CAPN3/p94 shows a stretch-dependent distribution. The amount of CAPN3/p94 at the M-line relative to that in the N2A region of myofibrils decreases as the sarcomere lengthens. This change in localization is delayed when CAPN3/p94 is inactive, as in Capn3 CS/CS mouse muscle, and is likely to be one of the molecular mechanisms by which LGMD2A/calpainopathy develops. Second, Capn3 CS/CS mice show an impaired ability to adapt to physical stress, accompanied by a compromised exercise-induced upregulation of MARP2 and HSPs. These findings suggest that the stretch-induced dynamic redistribution of p94, which is dependent on its protease activity, functions in the surveillance of myofibrillar conditions, and that this system is essential for protecting muscle tissue from degeneration, particularly under physical stress conditions (Ojima et al. 2011).
12.3.2 Gastrointestinal-Tract-Specific Calpains
12.3.2.1 Discovery and Nomenclature
In 1993, in the course of cDNA screening for tissue-specific calpains, a cDNA was obtained from rat stomach that encoded a novel molecule similar to but distinct from existing calpain catalytic large subunits, CAPN1/μCL, CAPN2/mCL, and CAPN3/p94 (Sorimachi et al. 1993a). The novel catalytic subunit had the same basic domain structure as these (Sorimachi et al. 2010, 2011a) (see below, Fig. 12.1a). As mentioned above, next to the first tissue-specific calpain, CAPN3/p94, which was called nCL-1 at that time, this stomach-specific calpain was designated nCL-2 , and is now called CAPN8 (Sorimachi et al. 1993a). In 1997, Dear and his colleagues discovered the third novel calpain, CAPN5/hTRA-3, and called it nCL-3 in their database submission. However, nCL-3 was not used in the published report, and they named it Capn5 (Dear et al. 1997). Almost simultaneously, the fourth one was found, which was named nCL-4 ; it is now called CAPN9 (Lee et al. 1998).
Comparative examinations revealed that CAPN8/nCL-2 and CAPN9/nCL-4 show predominant expression in the surface mucus-secreting cells (pit cells ) of the stomach, and that smaller amounts are expressed by the goblet cells in the intestines (Hata et al. 2006; Lee et al. 1998). Thus, these calpains are categorized as gastrointestinal-tract-specific calpains (Hata et al. 2010). Their physiological functions and mode of action were unclear until 2010, when they were shown to form a complex with each other in the stomach and to function in gastric mucosal protection against stress-induced ulcer (Hata et al. 2010). For CAPN8/nCL-2 and CAPN9/nCL-4 to be proteolytically active, they must form a complex; this complex is called G-calpain (G stands for gastric). After calpain-1 and -2, G-calpain was the third mammalian calpain enzyme complex shown to function in vivo, and the first to be composed of two different calpain catalytic subunits.
12.3.2.2 Structural Features
The basic domain structures of both CAPN8/nCL-2 and CAPN9/nCL-4 are identical to those of CAPN1/μCL and CAPN2/mCL, as shown in Fig. 12.1a. In cultured cells, CAPN9/nCL-4 is active only in the presence of CAPNS1/30K, and, therefore, is probably similar to, if not exactly the same as, those of the conventional calpains (CAPN1/μCL or CAPN2/mCL + CAPNS1/30K). CAPN8/nCL-2, however, is active without CAPNS1/30K in vitro, and shows homo-oligomerization via the CBSW domain (see below). Some C2-domain-like (β-sandwich ) structures are known to form homo-oligomers (Jones et al. 1989; Lu et al. 2010), and the CAPN8/nCL-2 homo-oligomer may have a structure similar to one of these.
In contrast, these calpains show rather different characteristics in vivo. Endogenous CAPN8/nCL-2 and CAPN9/nCL-4 in mouse stomach form a hybrid complex, G-calpain , and neither CAPN8/nCL-2 nor CAPN9/nCL-4 forms a stable complex with CAPNS1/30K (Hata et al. 2010). Gel filtration analysis of the endogenous mouse stomach G-calpain showed a molecular mass of about 180,000, indicating that G-calpain may contain other small subunit(s) of up to 20,000 (Hata et al. 2010).
The 3D structural analysis of the active (Ca2+-bound) protease domains of CAPN2/mCL and CAPN9/nCL-4 as well as CAPN1/μCL revealed locally distinct structures. Compared with the structure of CAPN1/μCL, that of CAPN2/mCL has a reversible intrinsic silencing mechanism by Trp106 (Moldoveanu et al. 2002, 2003) (Fig. 12.6). In contrast, the structure of CAPN9/nCL-4 shows that its activity can be auto-inhibited by misalignment of the catalytic triad, mediated by large intra-domain movements (Davis et al. 2007) (Fig. 12.6). These findings indicate that, in addition to the common activation mechanism, each calpain family member evolved molecule-specific one, even though they share high levels of sequence conservation.

Superimposed schematic 3D structures of CysPc domains of the active CAPN1/μCL, CAPN2/mCL, and CAPN9/nCL-4. Schematic 3D ribbon structures of the active (Ca2+-bound) forms of CysPc domains of CAPN1/μCL (blue), CAPN2/mCL (white), and CAPN9/nCL-4 (red); their PC1 domains are superimposed by Deli server (PDB data: 2ARY, 1MDW, and 1ZIV) (Davis et al. 2007; Moldoveanu et al. 2003). The active sites are circled in yellow. PC1 subdomains of the three CysPc domains fit very well (Root-Mean-Square Deviation (RMSD) = 1~4 Å). CAPN1/μCL and CAPN2/mCL show overall fitting with very low RMSD (1.0 Å) including Ca2+, but with one significant difference at Trp116 (CAPN1/μCL) and Trp106 (CAPN2/mCL). On the other hand, the PC2 domains of CAPN1/μCL and CAPN9/nCL-4 are markedly displaced. Note that, in the CAPN9/nCL-4 structure, His254 and Asn278 are too far away from Cys97 to form an active triad
12.3.2.3 Activity and Substrates
Of the human calpains, CAPN8/nCL-2 shows the highest similarity (~60 % identity in aa sequence) to CAPN2/mCL. However, unlike calpain-2 (CAPN2/mCL + CAPNS1/30K), recombinant CAPN8/nCL-2 expressed in Escherichia coli exhibits Ca2+-dependent activity without CAPNS1/30K, and forms a homodimer~oligomer via its CBSW domain in vitro (Hata et al. 2007). Recombinant mouse CAPN8/nCL-2 shows half-maximal activity at approximately 0.3 mM, which is similar to calpain-2 (Hata et al. 2007). Its autolytic activity in the presence of Ca2+ is very rapid, with more than 80 % of the protein being cleaved within 30 s (Hata et al. 2007). Several autolytic sites have been identified in the N-terminal anchor helix region, and in the CBSW and PEF domains (Hata et al. 2007). Although most of these features are unique to CAPN8/nCL-2, their physiological relevance remains unclear. On the other hand, recombinant human CAPN9/nCL-4 requires CAPNS1/30K for its activity in vitro, and the activity of this complex is Ca2+-dependent, with half-maximal activity at approximately 0.1 mM Ca2+ under the optimal condition (Lee et al. 1999). The activity of both CAPN9/nCL-4 and CAPN8/nCL-2 is inhibited by calpastatin and other cysteine protease inhibitors, similar to the conventional calpains (Hata et al. 2007; Lee et al. 1999).
The β-subunit (β-COP ) of the coatomer complex, which is involved in retrograde membrane trafficking from the Golgi to the endoplasmic reticulum, is expressed in the stomach pit cells, and was shown to be proteolyzed by CAPN8/nCL-2 in vitro (Hata et al. 2006). Furthermore, β-COP and CAPN8/nCL-2 co-expressed in COS7 cells co-localize to the Golgi, and Ca2+-ionophore stimulation causes limited proteolysis of β-COP near the linker region after Ser528 (as in NP_057535). This cleavage probably results in the dissociation of β-COP from the Golgi, strongly suggesting that CAPN8/nCL-2 is involved in the membrane trafficking of mucus cells via interactions with coat protein.
12.3.2.4 Biological Significance
Xenopus laevis has a CAPN8/nCL-2 orthologue named xCL-2, which causes severe developmental defects when disrupted (Cao et al. 2001). CAPN9/nCL-4 was reported to be down-regulated in gastric cancer (Yoshikawa et al. 2000), and is involved in the formation of a lumen by breast epithelial cells induced by carcinoembryonic antigen-related cell adhesion molecule 1 (Chen et al. 2010).
In mouse stomach in vivo, the disruption of either mouse Capn8 or Capn9 causes a down-regulation of the other gene product, similar to the effect of Capns1 −/− on the levels of CAPN1/μCL and CAPN2/mCL. This suggests a co-dependency in terms of the stability and functionality of both gene products. This point is supported by the finding that the residual CAPN9/nCL-4 in Capn8 −/− mice does not display autolytic activity, and vice versa (Hata et al. 2010). However, Capn8 −/− and Capn9 −/− mice appear healthy under normal conditions. Nevertheless, they are significantly more susceptible to ethanol-induced gastric ulcers (Hata et al. 2010). CAPN8/nCL-2 knock-in (Capn8 CS/CS) mice expressing a protease-inactive CAPN8/nCL-2:C105S mutant also show stress-induced gastropathy, indicating that CAPN8/nCL-2 and CAPN9/nCL-4 (in the form of G-calpain) mediate key components of gastric mucosal defense (Hata et al. 2010).
Gastric mucosal defense is a complex process involving mucus secretion and the migration of the pit cells that differentiate from stem-cell progenitors. Considering that β-COP is a substrate for G-calpain, it might be involved in the regulation of mucus secretion. On the other hand, the expression of CAPN8/nCL-2 in cranial neural crest cells, where it is involved in cell motility, is regulated by ADAM13, a transmembrane metalloprotease containing a disintegrin domain (Cousin et al. 2011), suggesting that G-calpain is involved in pit cell migration.
In another study on the physiological functions of G-calpain, a single nucleotide polymorphism (SNP ) database search showed that human CAPN8 and CAPN9 contain several SNPs that result in aa substitutions (Hata et al. 2010). In vitro expression experiments showed that the G-calpain variants resulting from these SNPs have compromised proteolytic activity, suggesting that individuals expressing certain CAPN8 and CAPN9 variants may suffer from gastrointestinal dysfunction.
12.3.3 Other Calpains
12.3.3.1 PalB Homologs
PalB was first identified in Emericella (Aspergillus) nidulans as a product of the gene responsible for the fungus ’s adaptation to alkaline conditions (Denison et al. 1995). Subsequently, its orthologs were identified in S. cerevisiae (Rim13/Cpl1) and humans (CAPN7 /PalBH) (Futai et al. 1999; Sorimachi et al. 2011a). E. nidulans, like many other microorganisms, grows over a wide pH range. The palB gene locus was identified as one of the pal genes that show defective alkaline adaptation when disrupted. PalB is involved in the proteolytic activation of the PacC transcription factor, a key regulator of pH-dependent gene expression. Here, we refer to this pH adaptation system as the Pal-PacC pathway .
The Pal-PacC pathway contains palA, palB, palC, palF, palH, palI, and pacC. PalF is a distant homolog of mammalian arrestin and binds to the large cytosolic domain of PalH. The phosphorylation and ubiquitination of PalF under alkaline conditions depend on PalH and PalI, suggesting that pH-sensing is mediated via the interaction between PalF and PalH (similar to that observed for the arrestin receptor, which regulates various processes in mammals, such as photo-sensing). PacC usually adopts an inactive conformation during intramolecular interactions, and is activated by proteolytic removal of its C-terminus. PalB is primarily responsible for the processing of PacC in response to increased pH, which is followed by further proteolysis by the proteasome to yield the active form.
The Pal-PacC pathway is well conserved in yeasts. The yeast calpain, Rim13/Cpl1, was identified as a product of the RIM genes . Like PacC and PalB, Rim101 is proteolytically regulated by Rim13/Cpl1 (Futai et al. 1999), although in this case the proteasome is not involved. Under normal conditions, the environment for Saccharomycetes is rather acidic and this adaptive system (the Rim pathway) functions in near-neutral pH conditions. One example of a biological event in which the Rim pathway plays a role is the infection and invasion of mammalian skin by pathogenic and saprophytic yeasts such as Candida albicans and Yarrowia lipolytica at neutral pH. Disrupting the Rim pathway compromises the pathogenicity of these organisms (Mitchell et al. 2007).
The Pal-PacC and Rim pathways relate to membrane trafficking and involve the ESCRT (endosomal sorting complex required for transport) and Vps (vacuolar protein sorting) proteins. The Pal-PacC and Rim pathways are the first examples of genetic studies that are being used to thoroughly elucidate the molecular components and mechanisms underlying calpain-mediated systems (Hayashi et al. 2005; Rodriguez-Galan et al. 2009). These pathways also exemplify the fact that calpains function as modulator proteases , affecting the function of their substrates through limited proteolysis. Theoretical extension of the knowledge of these pathways is anticipated to provide important keys to understanding other calpain systems, especially those that involve CAPN7 /PalBH (Osako et al. 2010).
12.3.3.2 The TRA-3 Homologs
TRA-3 /CLP-5 was first identified as the product of one of the genes involved in the sex determination cascade of C. elegans . The Ca2+-dependent protease activity of TRA-3/CLP-5 is necessary for the processing of TRA-2A, which is required for female development in XX hermaphrodites . On another front, both TRA-3/CLP-5 and CLP-1 are components of a neuronal necrotic death cascade, which acts upstream of the aspartic proteases, ASP-3 and ASP-4, orthologs for mammalian cathepsins D and E (Syntichaki et al. 2002). In addition, an SNP in tra-3 is reported to be involved in nematode body-size determination (Kammenga et al. 2007).
Mammals have two TRA-3 orthologs, CAPN5 /hTRA-3 and CAPN6 (Sorimachi et al. 2011a). CAPN5/hTRA-3 has Ca2+-dependent autolytic activity and is sensitive to several calpain inhibitors (Waghray et al. 2004). CAPN5 is expressed at varying levels in almost all tissues. Analysis of Capn5 −/− mice shows that CAPN5/hTRA-3 is expressed by a subset of T cells, but is not required for development. SNPs within CAPN5/hTRA-3 are associated with polycystic ovary syndrome, diastolic blood pressure, and cholesterol levels (Saez et al. 2007), and expression of both Capn5 and Capn2 is upregulated in caerulein-induced acute pancreatitis in mice (Nakada et al. 2010).
CAPN6 proteins expressed in eutherians (placental mammals) and schistosomes have a naturally occurring aa substitution at the most important residue in the active site triad (Cys->Lys in humans); strongly suggesting that these CAPN6 proteins have no proteolytic activity. Interestingly, CAPN6 proteins expressed in marsupialia and birds retain all the active-site residues. Moreover, frogs and fish express three TRA-3 homologs, all of which retain the active-site residues (Sorimachi et al. 2011a). Mammalian CAPN6 is predominantly expressed in embryonic muscles, placenta, and in several cultured cell lines. CAPN6 is involved in regulating microtubule dynamics (Tonami et al. 2007) and motility (Tonami et al. 2011) in cellulo, although the in vivo physiological functions of CAPN6 are still unclear (Tonami et al. 2013).
12.3.3.3 The CAPN10 Homologs
A large-scale genetic association study identified an SNP within intron 3 of CAPN10 that is linked to susceptibility to non-insulin-dependent diabetes mellitus (NIDDM , or type 2 diabetes) (Horikawa et al. 2000), although there is no clear molecular explanation for the reason. Capn10 is also a candidate gene responsible for the NIDDM phenotype in the Otsuka Long-Evans Tokushima Fatty (OLETF) rat. Quantitative trait locus (QTL ) analyses using Capn10 −/− mice and two other strains with low and high obesity phenotypes (LG/J and SM/J) show that Capn10 is a component of the obesity QTL, Adip1. This indicates that CAPN10 is involved in obesity in mice (Cheverud et al. 2010).
Studies using Capn10−/− or CAPN10 -overexpressing mice suggest that CAPN10 is involved in type 2 ryanodine receptor-mediated apoptosis (Johnson et al. 2004). Although phenotype of Capn10−/− mice was not described, they are not embryonic lethal. CAPN10 is ubiquitously distributed and its cellular localization is dynamic. When localized in the mitochondria, it mediates mitochondrial dysfunction by cleaving Complex I subunits and promotes mitochondrial permeability transition (Arrington et al. 2006). CAPN10 is also involved in GLUT4 vesicle translocation during insulin-stimulated glucose uptake in adipocytes (Paul et al. 2003).
12.3.3.4 The SOL Homologs
The first genetic calpain study identified the Drosophila gene, small optic lobes (sol) , in 1991 (Delaney et al. 1991). A mutation in sol results in the absence of certain classes of columnar neurons from the optic lobes, the differentiation and/or survival of which is dependent on the protease activity of SOL. SOL comprises an N-terminal six Zn-finger motifs and C-terminal CysPc domain (Delaney et al. 1991; Sorimachi et al. 2011a). Unfortunately, there are no other reports analyzing the molecular mechanisms involved in this process. Mammals express a single SOL ortholog, CAPN15 /SOLH , but its physiological role remains unclear. In parallel with the PalB subfamily, the SOL subfamily (including their mammalian homologs) is a group of evolutionarily interesting molecules that warrant further study.
12.3.3.5 Phytocalpains
A plant calpain, phytocalpain, was first identified from the sugarcane expressed sequence tag database (Correa et al. 2001). Subsequently, many other phytocalpains were identified from dicotyledons, monocotyledons, and gymnospermae. A genetic study identified phytocalpain as the defective kernel 1 (dek1) gene product, DEK1 , which is required for aleurone cell development in the maize endosperm (Lid et al. 2002).
DEK1 contains a potential signal peptide sequence followed by possible three 7-trans-membrane (TM) regions , and CysPc and CBSW domains (Lid et al. 2002; Sorimachi et al. 2011a). Although the CysPc domain of DEK1 is divergent from mammalian calpains, the C-terminal CBSW domain is significantly similar to that of classical calpains such as CAPN1 /μCL , suggesting that DEK1 consists of evolutionarily-different modules. Recombinant maize DEK1 CysPc-CBSW fragment show Ca2+-activated caseinolytic activity, depending on the predicted active site Cys residue. DEK1 is the only calpain homolog in the Arabidopsis genome, and it is important for regulating growth in this plant. Although the full-length DEK1 protein localizes to membranes, intramolecular autolytic cleavage releases the CysPc-CBSW domains into the cytoplasm. These domains are sufficient to fully complement dek1 mutants (Tian et al. 2007).
12.3.3.6 Other Calpain Members
Leishmania and Trypanosoma each expresses around 20 calpain homologs, which likely contribute to cell morphogenesis, drug resistance, and stress-response mechanisms (Olego-Fernandez et al. 2009; Sangenito et al. 2009). Some trypanosome calpains have N-terminal domains with weak similarity to calpastatin. As described above, some calpain homologs show substitutions in one or more of the well-conserved active-site triad residues. This non-proteolytic family of calpain homologs includes eutherian and schistosome CAPN6 , several of the schistosome and nematode calpains, insect CALPC , and all the Trypanosoma homologs (Croall and Ersfeld 2007; Sorimachi et al. 2011a). The evolutionary background for the occurrence of these calpain species is quite interesting, and elucidating their physiological functions will expand our knowledge regarding the functions of the calpain superfamily, e.g., possible non-proteolytic functions (Ojima et al. 2011).
12.4 Concluding Remarks
Here we reviewed pathological and biological situations where involvement of calpains has gained great interest from multiple angles. For example, conventional calpain s (calpain-1 and -2) are involved in neurodegeneration, as well as producing several well documented proteolytic biomarkers that can help track brain injury progress, severity and as a theranostic tool for drug development. In addition, other calpains such as tissue-specific calpains (CAPN3 in skeletal-muscle, and CAPN8 and CAPN9 in gastrointestinal tract) and calpain homologs originally identified in non-mammals represent various physiological, cellular and biochemical pathways.
Although many studies suggest that activation of calpain, especially the conventional ones, is involved in pathophysiological processes as an exacerbating factor, it is absolutely false to recognize calpain as an existence for making diseases worse. Genetic manipulations causing constitutive and conditional disruption of Capn2 in mice result in embryonic lethality and cardiac hypertrophy , respectively. In other words, calpain could be a two-edged sword in that its activity is indispensable for certain aspects of life, yet, its regulation gets somehow fragile within the cells undergoing pathogenic chaos.
Therefore, in addition to investing our efforts in developing novel theranostic approaches aimed at the conventional calpains as useful promising targets for biomarkers and inhibitory drugs, further application fields should be simultaneously pursued with the view that some pathological contexts may be related to insufficient conventional calpain activity. In these cases, appropriate calpain activators, rather than inhibitors, will be candidate drug targets to be developed. In fact, genetic inactivation of tissue-specific calpains causes disease states, indicating that activators/stabilizers for these calpains, if developed, may be a drug for these tissue-specific diseases such as muscular dystrophy and gastropathy .
To date, only the conventional calpains’ over-activation has been associated with diseases. It is, however, equally possible that an inappropriate activation of other calpains including tissue-specific ones is laterally or centrally linked to disease aggravation. In any case, calpain protease research is a rich area for both basic research as well as implicational research in biomedical science. The open question is how to extract and profile its activity with a resolution that is consistently competent for both normal and disease conditions.
References
Anagli J, Hagmann J, Shaw E (1991) Investigation of the role of calpain as a stimulus–response mediator in human platelets using new synthetic inhibitors. Biochem J 274(Pt 2):497–502
Anagli J, Han Y, Stewart L, Yang D, Movsisyan A, Abounit K, Seyfried D (2009) A novel calpastatin-based inhibitor improves postischemic neurological recovery. Biochem Biophys Res Commun 385(1):94–99
Angliker H, Anagli J, Shaw E (1992) Inactivation of calpain by peptidyl fluoromethylketones. J Med Chem 35(2):216–220
Aoyagi T, Umezawa H (1975) In: Reich E, Rifkin DB, Shaw E (eds) Proteases and biological control. Cold Spring Harbor Laboratory, Cold Spring Harbor, pp 429–454
Arrington DD, Van Vleet TR, Schnellmann RG (2006) Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am J Physiol Cell Physiol 291:C1159–C1171
Bading H, Ginty DD, Greenberg ME (1993) Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260:181–186
Baghdiguian S, Martin M, Richard I, Pons F, Astier C, Bourg N, Hay RT, Chemaly R, Halaby G, Loiselet J, Anderson LV, Lopez de Munain A, Fardeau M, Mangeat P, Beckmann JS, Lefranc G (1999) Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IkappaB alpha/NF-kappaB pathway in limb-girdle muscular dystrophy type 2A. Nat Med 5:503–511
Banik NL, Matzelle DC, Gantt-Wilford G, Osborne A, Hogan EL (1997) Increased calpain content and progressive degradation of neurofilament protein in spinal cord injury. Brain Res 752:301–306
Benayoun B, Baghdiguian S, Lajmanovich A, Bartoli M, Daniele N, Gicquel E, Bourg N, Raynaud F, Pasquier MA, Suel L, Lochmuller H, Lefranc G, Richard I (2008) NF-kappaB-dependent expression of the antiapoptotic factor c-FLIP is regulated by calpain 3, the protein involved in limb-girdle muscular dystrophy type 2A. FASEB J 22:1521–1529
Benetti R, Del Sal G, Monte M, Paroni G, Brancolini C, Schneider C (2001) The death substrate Gas2 binds m-calpain and increases susceptibility to p53-dependent apoptosis. EMBO J 20:2702–2714
Benjamins JA, Nedelkoska L, George EB (2003) Protection of mature oligodendrocytes by inhibitors of caspases and calpains. Neurochem Res 28:143–152
Berridge MJ (1993) Inositol trisphosphate and calcium signalling. Nature 361:315–325
Bertipaglia I, Carafoli E (2007) Calpains and human disease. Subcell Biochem 45:29–53
Betts R, Anagli J (2004) The β- and γ-CH2 of B27-WT’s Leu11 and Ile18 side chains play a direct role in calpain inhibition. Biochemistry 43(9):2596–2604
Betts R, Weinsheimer S, Blouse GE, Anagli J (2003) Structural determinants of the calpain inhibitory activity of calpastatin peptide B27-WT. J Biol Chem 278(10):7800–7809
Bi R, Bi XN, Baudry M (1998) Phosphorylation regulates calpain-regulated truncation of glutamate ionotropic receptors. Brain Res 797:154–158
Bizat N, Hermel JM, Boyer F, Jacquard C, Creminon C, Ouary S, Escartin C, Hantraye P, Kajewski S, Brouillet E (2003) Calpain is a major cell death effector in selective striatal degeneration induced in vivo by 3-nitropropionate: implications for Huntington’s disease. J Neurosci 23:5020–5030
Bizat N, Galas MC, Jacquard C, Boyer F, Hermel JM, Schiffmann SN, Hantraye P, Blum D, Brouillet E (2005) Neuroprotective effect of zVAD against the neurotoxin 3-nitropropionic acid involves inhibition of calpain. Neuropharmacology 49:695–702
Bozoky Z, Alexa A, Tompa P, Friedrich P (2005) Multiple interactions of the ‘transducer’ govern its function in calpain activation by Ca2+. Biochem J 388(Pt 3):741–744
Buki A, Siman R, Trojanowski JQ, Povlishock JT (1999) The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol 58(4):365–375
Buki A, Okonkwo DO, Wang KK, Povlishock JT (2000) Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci 20:2825–2834
Buki A, Farkas O, Doczi T, Povlishock JT (2003) Preinjury administration of the calpain inhibitor MDL-28170 attenuates traumatically induced axonal injury. J Neurotrauma 20(3):261–268
Bullock R, Fujisawa H (1992) The role of glutamate antagonists for the treatment of CNS injury. J Neurotrauma 9(Suppl 2):S443–S462
Busch WA, Stromer MH, Goll DE, Suzuki A (1972) Ca2+-specific removal of Z lines from rabbit skeletal muscle. J Cell Biol 52:367–381
Cao Y, Zhao H, Grunz H (2001) xCL-2 is a novel m-type calpain and disrupts morphogenetic movements during embryogenesis in Xenopus laevis. Dev Growth Differ 43:563–571
Chen M, Fernandez HL (2005) Mu-calpain is functionally required for alpha-processing of Alzheimer’s beta-amyloid precursor protein. Biochem Biophys Res Commun 330:714–721
Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP (1998) Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci 18:4914–4928
Chen CJ, Nguyen T, Shively JE (2010) Role of calpain-9 and PKC-delta in the apoptotic mechanism of lumen formation in CEACAM1 transfected breast epithelial cells. Exp Cell Res 316:638–648
Chera B, Schaecher KE, Rocchini A, Imam SZ, Ray SK, Ali SF, Banik NL (2002) Calpain upregulation and neuron death in spinal cord of MPTP-induced parkinsonism in mice. Ann NY Acad Sci 965:274–280
Cheverud JM, Fawcett GL, Jarvis JP, Norgard EA, Pavlicev M, Pletscher LS, Polonsky KS, Ye H, Bell GI, Semenkovich CF (2010) Calpain-10 is a component of the obesity-related quantitative trait locus Adip1. J Lipid Res 51:907–913
Correa GC, Margis-Pinheiro M, Margis R (2001) Identification, classification and expression pattern analysis of sugarcane cysteine proteinases. Genet Mol Biol 24:275–283
Cousin H, Abbruzzese G, Kerdavid E, Gaultier A, Alfandari D (2011) Translocation of the cytoplasmic domain of ADAM13 to the nucleus is essential for Calpain8-a expression and cranial neural crest cell migration. Dev Cell 20:256–263
Crawford C (1987) Inhibition of chicken calpain II by proteins of the cystatin superfamily and alpha 2-macroglobulin. Biochem J 248(5):89–594
Crawford C, Mason RW, Wikstrom P, Shaw E (1988) The design of peptidyldiazomethane inhibitors to distinguish between the cysteine proteinases calpain II, cathepsin L and cathepsin B. Biochem J 253(3):751–758
Croall DE, DeMartino GN (1991) Calcium-activated neutral protease (calpain) system: structure, function, and regulation. Physiol Rev 71(3):813–847
Croall DE, Ersfeld K (2007) The calpains: modular designs and functional diversity. Genome Biol 8:218
Crocker SJ, Smith PD, Jackson-Lewis V, Lamba WR, Hayley SP, Grimm E, Callaghan SM, Slack RS, Melloni E, Przedborski S, Robertson GS, Anisman H, Merali Z, Park DS (2003) Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J Neurosci 23(10):4081–4091
Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116:205–219
Das A, Guyton MK, Butler JT, Ray SK, Banik NL (2008) Activation of calpain and caspase pathways in demyelination and neurodegeneration in animal model of multiple sclerosis. CNS Neurol Disord Drug Targets 7(3):313–320, Review
Dash PK, Moore AN, Dixon CE (1995) Spatial memory deficits, increased phosphorylation of the transcription factor CREB, and induction of the AP-1 complex following experimental brain injury. J Neurosci 15:2030–2039
Davis TL, Walker JR, Finerty PJ Jr, Mackenzie F, Newman EM, Dhe-Paganon S (2007) The crystal structures of human calpains 1 and 9 imply diverse mechanisms of action and auto-inhibition. J Mol Biol 366:216–229
Dayton WR, Goll DE, Zeece MG, Robson RM, Reville WJ (1976) A Ca2+-activated protease possibly involved in myofribrillar protein turnover. Purification from porcine muscle. Biochemistry 15:2150–2158
Dear N, Matena K, Vingron M, Boehm T (1997) A new subfamily of vertebrate calpains lacking a calmodulin-like domain: implications for calpain regulation and evolution. Genomics 45:175–184
Delaney SJ, Hayward DC, Barleben F, Fischbach KF, Miklos GL (1991) Molecular cloning and analysis of small optic lobes, a structural brain gene of Drosophila melanogaster. Proc Natl Acad Sci U S A 88:7214–7218
Denison SH, Orejas M, Arst HN Jr (1995) Signaling of ambient pH in Aspergillus involves a cysteine protease. J Biol Chem 270:28519–28522
Donkor IO (2011) Calpain inhibitors: a survey of compounds reported in the patent and scientific literature. Expert Opin Ther Pat 21(5):601–636, Review
Emori Y, Kawasaki H, Imajoh S, Minami Y, Suzuki K (1988) All four repeating domains of the endogenous inhibitor of calcium-dependent protease independently retain inhibitory activity. Expression of the cDNA fragments in Escherichia coli. J Biol Chem 263(5):2364–2370
Fifre A, Spoone I, Koziel V, Kriem B, Yen-Potin FT, Bihain BB, Oliver JL, Oster T, Pillot T (2006) Microtubule-associated proteins MAP1A, MAP1B and MAP2 proteolysis during soluble amyloid-beta peptide induced neuronal apoptosis: Synergistic involvement of calpain and caspase-3. J Biol Chem 281(1):229–240
Fineman I, Hovda DA, Smith M, Yoshino A, Becker DP (1993) Concussive brain injury is associated with a prolonged accumulation of calcium: a 45Ca autoradiographic study. Brain Res 624:94–102
Fiorino F, Gil-Parrado S, Assfalg-Machleidt I, Machleidt W, Moroder L (2007) A new cell-permeable calpain inhibitor. J Pept Sci 13(1):70–73
Fougerousse F, Anderson LV, Delezoide AL, Suel L, Durand M, Beckmann JS (2000) Calpain3 expression during human cardiogenesis. Neuromuscul Disord 10:251–256
Fukiage C, Azuma M, Nakamura Y, Tamada Y, Nakamura M, Shearer TR (1997) SJA6017, a newly synthesized peptide aldehyde inhibitor of calpain: amelioration of cataract in cultured rat lenses. Biochim Biophys Acta 1361:304–312
Fukiage C, Nakajima E, Ma H, Azuma M, Shearer TR (2002) Characterization and regulation of lens-specific calpain Lp82. J Biol Chem 277:20678–20685
Futai E, Maeda T, Sorimachi H, Kitamoto K, Ishiura S, Suzuki K (1999) The protease activity of a calpain-like cysteine protease in Saccharomyces cerevisiae is required for alkaline adaptation and sporulation. Mol Gen Genet 260:559–568
Gafni J, Ellerby LM (2002) Calpain activation in Huntington’s disease. J Neurosci 22:4842–4849
Gafni J, Hermel E, Young JE, Wellington CL, Hayden MR, Ellerby LM (2004) Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem 279:20211–20220
Germano A, Costa C, DeFord SM, Angileri FF, Arcadi F, Pike BR, Bramanti P, Bausano B, Zhao X, Day AL, Anderson DK, Hayes RL (2002) Systemic administration of a calpain inhibitor reduces behavioral deficits and blood–brain barrier permeability changes after experimental subarachnoid hemorrhage in the rat. J Neurotrauma 19:887–896
Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83(3):731–801
Gomez-Vicente V, Donovan M, Cotter TG (2005) Multiple death pathways in retina-derived 661W cells following growth factor deprivation: cross-talk between caspases and calpains. Cell Death Differ 12:796–804
Guroff G (1964) A neutral calcium-activated proteinase from the soluble fraction of rat brain. J Biol Chem 239:149–155
Hall ED (1998) Antioxidant pharmacotherapy. In: Ginsberg MD, Bogousslavsky J (eds) Cerebrovascular disease: pathophysiology, diagnosis, and management. Blackwell Science, Malden, MA, pp 710–720
Hanna RA, Campbell RL, Davies PL (2008) Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature 456:409–412
Hata S, Koyama S, Kawahara H, Doi N, Maeda T, Toyama-Sorimachi N, Abe K, Suzuki K, Sorimachi H (2006) Stomach-specific calpain, nCL-2, localizes in mucus cells and proteolyzes the β-subunit of coatomer complex, β-COP. J Biol Chem 281:11214–11224
Hata S, Doi N, Kitamura F, Sorimachi H (2007) Stomach-specific calpain, nCL-2/calpain 8, is active without calpain regulatory subunit and oligomerizes through C2-like domains. J Biol Chem 282:27847–27856
Hata S, Abe M, Suzuki H, Kitamura F, Toyama-Sorimachi N, Abe K, Sakimura K, Sorimachi H (2010) Calpain 8/nCL-2 and calpain 9/nCL-4 constitute an active protease complex, G-calpain, involved in gastric mucosal defense. PLoS Genet 6:e1001040
Hathaway DR, Werth DR, Haeberle JR (1982) Limited proteolysis reduces the Ca2+ requirement of a smooth muscle Ca2+-activated protease. J Biol Chem 257:9072–9077
Hayashi M, Fukuzawa T, Sorimachi H, Maeda T (2005) Constitutive activation of the pH-responsive Rim101 pathway in yeast mutants defective in late steps of the MVB/ESCRT pathway. Mol Cell Biol 25:9478–9490
Hayashi C, Ono Y, Doi N, Kitamura F, Tagami M, Mineki R, Arai T, Taguchi H, Yanagida M, Hirner S, Labeit D, Labeit S, Sorimachi H (2008) Multiple molecular interactions implicate the connectin/titin N2A region as a modulating scaffold for p94/calpain 3 activity in skeletal muscle. J Biol Chem 283:14801–14814
Hayes RL, Jenkins LW, Lyeth BG (1992) Neurotransmitter-mediated mechanisms of traumatic brain injury: acetylcholine and excitatory amino acids. J Neurotrauma 9(Suppl 1):S173–S187
Herasse M, Ono Y, Fougerousse F, Kimura E, Stockholm D, Beley C, Montarras D, Pinset C, Sorimachi H, Suzuki K, Beckmann JS, Richard I (1999) Expression and functional characteristics of calpain 3 isoforms generated through tissue-specific transcriptional and posttranscriptional events. Mol Cell Biol 19:4047–4055
Higuchi M, Iwata N, Saido TC (2005) Understanding molecular mechanisms of proteolysis in Alzheimer’s disease: progress toward therapeutic interventions. Biochim Biophys Acta 1751:60–67
Hong SC, Lanzino G, Goto Y, Kang SK, Schottler F, Kassell NF, Lee KS (1994) Calcium-activated proteolysis in rat neocortex induced by transient focal ischemia. Brain Res 661:43–50
Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, del Bosque-Plata L, Horikawa Y, Oda Y, Yoshiuchi I, Colilla S, Polonsky KS, Wei S, Concannon P, Iwasaki N, Schulze J, Baier LJ, Bogardus C, Groop L, Boerwinkle E, Hanis CL, Bell GI (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175
Hosfield CM, Elce JS, Davies PL, Jia Z (1999) Crystal structure of calpain reveals the structural basis for Ca2+-dependent protease activity and a novel mode of enzyme activation. EMBO J 18:6880–6889
Hosfield CM, Elce JS, Jia Z (2004) Activation of calpain by Ca2+: roles of the large subunit N-terminal and domain III-IV linker peptides. J Mol Biol 343(4):1049–1053
Hossman K (1988) In: Kreiglstein J (ed) Pharmacology of cerebral ischemia. CRC, Boco Raton, FL, pp 53–63
Huang C, Jacobson K, Schaller MD (2004) MAP kinases and cell migration. J Cell Sci 117:4619–4628
Huston RB, Krebs EG (1968) Activation of skeletal muscle phosphorylase kinase by Ca2+. II. Identification of the kinase activating factor as a proteolytic enzyme. Biochemistry 7:2116–2122
Imajoh S, Kawasaki H, Suzuki K (1986) Limited autolysis of calcium-activated neutral protease (CANP): reduction of the Ca2+-requirement is due to the NH2-terminal processing of the large subunit. J Biochem 100:633–642
Ishuira S, Murofushi H, Suzuki K, Imahori K (1978) Studies of a calcium-activated neutral protease from chicken skeletal muscle. I. Purification and characterization. J Biochem 84:225–230
Johnson JD, Han Z, Otani K, Ye H, Zhang Y, Wu H, Horikawa Y, Misler S, Bell GI, Polonsky KS (2004) RyR2 and calpain-10 delineate a novel apoptosis pathway in pancreatic islets. J Biol Chem 279:24794–24802
Jones EY, Stuart DI, Walker NP (1989) Structure of tumour necrosis factor. Nature 338:225–228
Kambe A, Yokota M, Saido TC, Satokata I, Fujikawa H, Tabuchi S, Kamitani H, Watanabe T (2005) Spatial resolution of calpain-catalyzed proteolysis in focal cerebral ischemia Brain Res 1040(1–2):36–43
Kammenga JE, Doroszuk A, Riksen JA, Hazendonk E, Spiridon L, Petrescu AJ, Tijsterman M, Plasterk RH, Bakker J (2007) A Caenorhabditis elegans wild type defies the temperature-size rule owing to a single nucleotide polymorphism in tra-3. PLoS Genet 3:e34
Kampfl A, Postmantur RM, Zhao X, Schmutzhard E, Clifton GL, Hayes RL (1997) Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: a review and update. J Neurotrauma 14(3):121–134
Kar NC, Pearson CM (1976) A calcium-activated neutral protease in normal and dystrophic human muscle. Clin Chim Acta 73:293–297
Kawamura M, Nakajima W, Ishida A, Ohmura A, Miura S, Takada G (2005) Calpain inhibitor MDL 28170 protects hypoxic-ischemic brain injury in neonatal rats by inhibition of both apoptosis and necrosis. Brain Res 1037:59–69
Kim SJ, Sung JY, Um JW, Hattori N, Mizuno Y, Tanaka K, Paik SR, Kim J, Chung KC (2003) Parkin cleaves intracellular alpha-synuclein inclusions via the activation of calpain. J Biol Chem 278(43):41890–41899
Kishimoto A, Kajikawa N, Shiota M, Nishizuka Y (1983) Proteolytic activation of calcium-activated, phospholipid-dependent protein kinase by calcium-dependent neutral protease. J Biol Chem 258:1156–1164
Kobeissy FH, Ottens AK, Zhang ZQ, Dave JR, Tortella FC, Hayes RL, Wang KKW (2006) Differential proteomic analysis of traumatic brain injury biomarker study using CAX-PAGE/RPLC-MSMS method. Mol Cell Proteomics 5:1887–1898
Kontos HA (1989) Oxygen radicals in CNS damage. Chem Biol Interact 72:229–255
Kramerova I, Kudryashova E, Tidball JG, Spencer MJ (2004) Null mutation of calpain 3 (p94) in mice causes abnormal sarcomere formation in vivo and in vitro. Hum Mol Genet 13:1373–1388
Kramerova I, Kudryashova E, Wu B, Spencer MJ (2006) Regulation of the M-cadherin-beta-catenin complex by calpain 3 during terminal stages of myogenic differentiation. Mol Cell Biol 26:8437–8447
Kramerova I, Kudryashova E, Wu B, Ottenheijm C, Granzier H, Spencer MJ (2008) Novel role of calpain-3 in the triad-associated protein complex regulating calcium release in skeletal muscle. Hum Mol Genet 17:3271–3280
Kramerova I, Kudryashova E, Wu B, Germain S, Vandenborne K, Romain N, Haller RG, Verity MA, Spencer MJ (2009) Mitochondrial abnormalities, energy deficit and oxidative stress are features of calpain 3 deficiency in skeletal muscle. Hum Mol Genet 18:3194–3205
Kristian T, Gido G, Kuroda S, Schutz A, Siesjo BK (1998) Calcium metabolism of focal and penumbral tissues in rats subjected to transient middle cerebral artery occlusion. Exp Brain Res 120:503–509
Kuchay SM, Kim N, Grunz EA, Fay WP, Chrishti AH (2007) Double knockouts reveal that protein tyrosine phosphatase 1B is a physiological target of calpain-1 in platelets. Mol Cell Biol 27:6038–6052
Lee KS, Yanamoto H, Fergus A, Hong SC, Kang SD, Cappelletto B, Toyoda T, Kassell NF, Bavbek M, Kwan AL (1997) Calcium-activated proteolysis as a therapeutic target in cerebrovascular disease. Ann NY Acad Sci 825:95–103
Lee HJ, Sorimachi H, Jeong SY, Ishiura S, Suzuki K (1998) Molecular cloning and characterization of a novel tissue-specific calpain predominantly expressed in the digestive tract. Biol Chem 379:175–183
Lee HJ, Tomioka S, Kinbara K, Masumoto H, Jeong SY, Sorimachi H, Ishiura S, Suzuki K (1999) Characterization of a human digestive tract-specific calpain, nCL-4, expressed in the baculovirus system. Arch Biochem Biophys 362:22–31
Lid SE, Gruis D, Jung R, Lorentzen JA, Ananiev E, Chamberlin M, Niu X, Meeley R, Nichols S, Olsen OA (2002) The defective kernel 1 (dek1) gene required for aleurone cell development in the endosperm of maize grains encodes a membrane protein of the calpain gene superfamily. Proc Natl Acad Sci U S A 99:5460–5465
Liebetrau M, Stauffer B, Auerswald EA, Gabrijelcic-Geiger D, Fritz H, Zimmermann C, Pfefferkom T, Hamann GF (1999) Increased intracellular calpain detection in experimental focal cerebral ischemia. Neuroreport 10:529–534
Liu MC, Akle V, Zheng WR, Dave JR, Tortella FC, Hayes RL, Wang KKW (2006) Comparing calpain- and caspase-3-degradation patterns in traumatic brain injury by differential proteome analysis. Biochem J 394:715–725
Liu J, Liu MC, Wang KKW (2008a) Physiological and pathological actions of calpains in glutamatergic neurons. Sci Signal 1(23):tr3
Liu J, Liu MC, Wang KKW (2008b) Calpain in the CNS: from synaptic function to neurotoxicity. Sci Signal 1(14):re1
Liu MC, Zheng ZQ, Akinyi L, Oli MW, Zheng WR, Larner SF, Kobeissy F, Papa L, Lu XC, Dave JR, Tortella FC, Hayes RL, Wang KKW (2010) Ubiquitin-C-terminal hydrolase as a novel biomarker for stroke and traumatic brain injury in rats. Eur J Neurosci 31:722–732
Liu MC, Kobeissy F, Zheng W, Zhang Z, Hayes RL, Wang KK (2011) Dual vulnerability of tau to calpains and caspase-3 proteolysis under neurotoxic and neurodegenerative conditions. ASN Neuro 3(1):e00051
Lu J, Zheng J, Liu H, Li J, Chen H, Chen K (2010) Protein profiling analysis of skeletal muscle of a pufferfish, Takifugu rubripes. Mol Biol Rep 37:2141–2147
Maki M, Bagci H, Hamaguchi K, Ueda M, Murachi T, Hatanaka M (1989) Inhibition of calpain by a synthetic oligopeptide corresponding to an exon of the human calpastatin gene. J Biol Chem 264(32):18866–18869
Maki M, Ma H, Takano E, Adachi Y, Lee WJ, Hatanaka M, Murachi T (1991) Calpastatins: biochemical and molecular biological studies. Biomed Biochim Acta 50:509–516
Markgraf CG, Velayo NL, Johnson MP, McCarty DR, Medhi S, Koehl JR, Chmielewski PA, Linnik MD (1998) Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia in rats. Stroke 29:152–158
Maxwell WL, Povlishock JT, Graham DL (1997) A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma 14:419–440
McGinn MJ, Kelley BJ, Akinyi L, Oli MW, Liu MC, Hayes RL, Wang KKW, Povlishock JT (2009) Biochemical, structural and biomarker evidence for calpain-mediated cytoskeletal change following diffuse brain injury uncomplicated by contusion. J Neuropathol Exp Neurol 68(3):241–249
McIntosh TK (1994) Neurochemical sequelae of traumatic brain injury: therapeutic implications. Cerebrovasc Brain Metab Rev 6:109–162
McIntosh TK, Saatman KE, Raghupathi R, Graham DI, Smith DH, Lee VM, Trojanowski JQ (1998) The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol Appl Neurobiol 24:251–267
Meaney DF, Margulies SS, Smith DH (2001) Diffuse axonal injury. J Neurosurg 95:1108–1110
Medana IM, Esiri MM (2003) Axonal damage: a key predictor of outcome in human CNS diseases. Brain 126:515–530
Mellgren RL, Huang X (2007) Fetuin A stabilizes m-calpain and facilitates plasma membrane repair. J Biol Chem 282:35868–35877
Mellgren RL, Zhang W, Miyake K, McNeil PL (2007) Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J Biol Chem 282:2567–2575
Meyer WL, Fischer EH, Krebs EG (1964) Activation of skeletal muscle phosphorylase kinase by Ca2+. Biochemistry 3:1033–1039
Minami N, Tani E, Maeda Y, Yamaura I, Fukami M (1992) Effects of inhibitors of protein kinase C and calpain in experimental delayed cerebral vasospasm. J Neurosurg 76:111–118
Mitchell BM, Wu TG, Jackson BE, Wilhelmus KR (2007) Candida albicans strain-dependent virulence and Rim13p-mediated filamentation in experimental keratomycosis. Invest Ophthalmol Vis Sci 48:774–780
Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, Davies PL (2002) A Ca2+ switch aligns the active site of calpain. Cell 108:649–660
Moldoveanu T, Hosfield CM, Lim D, Jia Z, Davies PL (2003) Calpain silencing by a reversible intrinsic mechanism. Nat Struct Biol 10:371–378
Molinari M, Carafoli E (1997) Calpain: a cytosolic proteinase active at the membranes. J Membr Biol 156(1):1–8
Mondello S, Robicsek SA, Gabrielli A, Brophy GM, Papa L, Tepas J, Robertson C, Buki A, Scharf D, Jixiang M, Akinyi L, Muller U, Wang KK, Hayes RL (2010) αII-spectrin breakdown products (SBDPs): diagnosis and outcome in severe traumatic brain injury patients. J Neurotrauma 27(7):1203–1213
Mondello S, Jeromin A, Streeter J, Hayes RL, Wang KKW (2011) Blood-based diagnostics of traumatic brain injuries. Expert Rev Mol Diagn 11(1):65–78
Morioka M, Fukunaga K, Yasugawa S, Nagahiro S, Ushio Y, Miyamoto E (1992) Regional and temporal alterations in Ca2+/calmodulin-dependent protein kinase II and calcineurin in the hippocampus of rat brain after transient forebrain ischemia. J Neurochem 58:1798–1809
Murachi T (1989) Intracellular regulatory system involving calpain and calpastatin. Biochem Int 18:263–294
Nakada S, Tsuneyama K, Kato I, Tabuchi Y, Takasaki I, Furusawa Y, Kawaguchi H, Fujimoto M, Goto H, Hikiami H, Kondo T, Takano Y, Shimada Y (2010) Identification of candidate genes involved in endogenous protection mechanisms against acute pancreatitis in mice. Biochem Biophys Res Commun 391:1342–1347
Nakanishi H (2003) Microglial functions and proteases. Mol Neurobiol 27:163–176
Nath R, Raser KJ, Stafford D, Hajimohammadreza I, Posner A, Allen H, Talanian RV, Yuen P, Gilbertsen RB, Wang KK (1996) Non-erythroid alpha-spectrin breakdown by calpain and interleukin 1 beta-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J 319(Pt 3):683–690
Nath R, Probert A Jr, McGinnis KM, Wang KK (1998) Evidence of activation of caspase-3 like protease in excitotoxin- and hypoxia/hypoglycemia-injured neurons. J Neurochem 71(1):186–195
Newcomb JK, Kampfl A, Posmantur RM, Zhao X, Pike BR, Liu SJ, Clifton GL, Hayes RL (1997) Immunohistochemical study of calpain-mediated breakdown products to alpha-spectrin following controlled cortical impact injury in the rat. J Neurotrauma 14:369–383
Newcomb JK, Pike BR, Zhao X, Banik NL, Hayes RL (1999) Altered calpastatin protein levels following traumatic brain injury. J Neurotrauma 16(1):1–11
Newcomb-Fernandez JK, Zhao X, Pike BR, Wang KK, Kampfl A, Beer R, DeFord SM, Hayes RL (2001) Concurrent assessment of calpain and caspase-3 activation after oxygen-glucose deprivation in primary septo-hippocampal cultures. J Cereb Blood Flow Metab 21(11):1281–1294
Nishida A, Emoto K, Shimizu M, Uozumi T, Yamawaki S (1994) Brain ischemia decreases phosphatidylcholine-phospholipase D but not phosphatidylinositol-phospholipase C in rats. Stroke 25:1247–1251
Nishiura I, Tanaka K, Yamamoto S, Murachi T (1978) The occurence of an inhibitor of Ca2+-dependent neutral protease in rat liver. J Biochem 84(6):1657–1659
Noguchi M, Sarin A, Aman MJ, Najajima H, Shores EW, Henkart PA, Leonard WJ (1997) Functional cleavage of the common cytokine receptor gamma chain (γc) by calpain. Proc Natl Acad Sci U S A 94:11534–11539
Norberg E, Gogvadze V, Ott M, Horn M, Uhlen P, Orrenius S, Zhivotovsky B (2008) An increase in intracellular Ca2+ is required for the activation of mitochondrial calpain to release AIF during cell death. Cell Death Differ 15(12):1857–1864
Ohno S, Emori Y, Imajoh S, Kawasaki H, Kisaragi M, Suzuki K (1984) Evolutionary origin of a calcium-dependent protease by fusion of genes for a thiol protease and a calcium-binding protein? Nature 312(5994):566–570
Ohno S, Minoshima S, Kudoh J, Fukuyama R, Shimizu Y, Ohmi-Imajoh S, Shimizu N, Suzuki K (1990) Four genes for the calpain family locate on four distinct human chromosomes. Cytogenet Cell Genet 53:225–229
Ojima K, Ono Y, Doi N, Yoshioka K, Kawabata Y, Labeit S, Sorimachi H (2007) Myogenic stage, sarcomere length, and protease activity modulate localization of muscle-specific calpain. J Biol Chem 282:14493–14504
Ojima K, Kawabata Y, Nakao H, Nakao K, Doi N, Kitamura F, Ono Y, Hata S, Suzuki H, Kawahara H, Bogomolovas J, Witt C, Ottenheijm C, Labeit S, Granzier H, Toyama-Sorimachi N, Sorimachi M, Suzuki K, Maeda T, Abe K, Aiba A, Sorimachi H (2010) Dynamic distribution of muscle-specific calpain in mice has a key role in physical-stress adaptation and is impaired in muscular dystrophy. J Clin Invest 120:2672–2683
Ojima K, Ono Y, Ottenheijm C, Hata S, Suzuki H, Granzier H, Sorimachi H (2011) Non-proteolytic functions of calpain-3 in sarcoplasmic reticulum in skeletal muscles. J Mol Biol 407:439–449
Olego-Fernandez S, Vaughan S, Shaw MK, Gull K, Ginger ML (2009) Cell morphogenesis of Trypanosoma brucei requires the paralogous, differentially expressed calpain-related proteins CAP5.5 and CAP5.5V. Protist 160:576–590
Olney JW, Labruyere J, Wang G, Wozniak DF, Price MT, Sesma MA (1991) NMDA antagonist neurotoxicity: mechanism and prevention. Science 254:1515–1518
Ono Y, Shimada H, Sorimachi H, Richard I, Saido TC, Beckmann JS, Ishiura S, Suzuki K (1998) Functional defects of a muscle-specific calpain, p94, caused by mutations associated with limb-girdle muscular dystrophy type 2A. J Biol Chem 273:17073–17078
Ono Y, Sorimachi H, Suzuki K (1999) New aspect of the research on limb-girdle muscular dystrophy 2A: a molecular biologic and biochemical approach to pathology. Trends Cardiovasc Med 9:114–118
Ono Y, Kakinuma K, Torii F, Irie A, Nakagawa K, Labeit S, Abe K, Suzuki K, Sorimachi H (2004) Possible regulation of the conventional calpain system by skeletal muscle-specific calpain, p94/calpain 3. J Biol Chem 279:2761–2771
Ono Y, Ojima K, Torii F, Takaya E, Doi N, Nakagawa K, Hata S, Abe K, Sorimachi H (2010) Skeletal muscle-specific calpain is an intracellular Na+-dependent protease. J Biol Chem 285:22986–22998
Osako Y, Maemoto Y, Tanaka R, Suzuki H, Shibata H, Maki M (2010) Autolytic activity of human calpain 7 is enhanced by ESCRT-III-related protein IST1 through MIT-MIM interaction. FEBS J 277:4412–4426
Parkes C, Kembhavi AA, Barrett AJ (1985) Calpain inhibition by peptide epoxides. Biochem J 230:509–516
Paul DS, Harmon AW, Winston CP, Patel YM (2003) Calpain facilitates GLUT4 vesicle translocation during insulin-stimulated glucose uptake in adipocytes. Biochem J 376:625–632
Pettus EH, Povlishock JT (1996) Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res 722:1–11
Pettus EH, Christman CW, Giebel ML, Povlishock JT (1994) Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma 11:507–522
Pietsch M, Chua KC, Abell AD (2010) Calpains: attractive targets for the development of synthetic inhibitors. Curr Top Med Chem 10:270–293
Pike BR, Zhao X, Newcomb JK, Postmantur RM, Wang KK, Hayes RL (1998) Regional calpain and caspase-3 proteolysis of alpha-spectrin after traumatic brain injury. Neuroreport 9(11):2437–2442
Pike BR, Zhao X, Newcomb JK, Glenn CC, Anderson DK, Hayes RL (2000) Stretch injury causes calpain and caspase-3 activation and necrotic and apoptotic cell death in septo-hippocampal cell cultures. J Neurotrauma 17(4):283–298
Pike BR, Flint J, Johnson E, Glenn CC, Dutta S, Wang KKW, Hayes RL (2001) Accumulation of calpain-cleaved non-erythroid αII-spectrin in cerebrospinal fluid after traumatic brain injury in rats. J Neurochem 78:1297–1306
Pike BR, Flint J, Dave JR, Lu XC, Wang KKW, Tortella FC, Hayes RL (2003) Accumulation of calpain and caspase-3 proteolytic fragments of brain-derived αII-spectrin in CSF after middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab 24(1):98–106
Pontremoli S, Melloni E (1988) The role of calpain and protein kinase C in activation of human neutrophils. Prog Clin Biol Res 282:195–208
Posmantur R, Kampfl A, Siman R, Liu J, Zhao X, Clifton GL, Hayes RL (1997) A calpain inhibitor attenuates cortical cytoskeletal protein loss after experimental traumatic brain injury in the rat. Neuroscience 77:875–888
Postmantur RM, Kampfl A, Liu SJ, Heck K, Taft WC, Clifton GL, Hayes RL (1996) Cytoskeletal derangements of cortical neuronal processes three hours after traumatic brain injury in rats: an immunofluorescence study. J Neuropathol Exp Neurol 55(1):68–80
Potter DA, Tirnauer JS, Janssen R, Croall DE, Hughes CN, Fiacco KA, Mier JW, Maki M, Herman IM (1998) Calpain regulates actin remodeling during cell spreading. J Cell Biol 141:647–662
Povlishock JT (1992) Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol 2:1–12
Povlishock JT, Becker DP, Cheng CL, Vaughan GW (1983) Axonal change in minor head injury. J Neuropathol Exp Neurol 42:225–242
Povlishock JT, Erb DE, Astruc J (1992) Axonal response to traumatic brain injury: reactive axonal change, deafferentation, and neuroplasticity. J Neurotrauma 9(Suppl 1):S189–S200
Rami A (2003) Ischemic neuronal death in the rat hippocampus: the calpain-calpastatin-caspase hypothesis. Neurobiol Dis 13:75–88
Rao MV, Mohan PS, Peterhoff CM, Yang DS, Schmidt SD, Stavrides PH, Campbell J, Chen Y, Jiang Y, Paskevich PA, Cataldo AM, Haroutunian V, Nixon RA (2008) Marked calpastatin (CAST) depletion in Alzheimer’s disease accelerates cytoskeleton disruption and neurodegeneraton: neuroprotection by CAST overexpression. J Neurosci 28(47):12241–12254
Ray SK, Shields DC, Saido TC, Matzelle DC, Wilford GG, Hogan EL, Banik NL (1999) Calpain activity and translational expression increased in spinal cord injury. Brain Res 816:375–380
Ray SK, Wilford GG, Ali SF, Banik NL (2000) Calpain upregulation in spinal cords of mice with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson’s disease. Ann NY Acad Sci 914:275–283
Ray SK, Matzelle DD, Wilford GG, Hogan EL, Banik NL (2001a) Cell death in spinal cord injury (SCI) requires de novo protein synthesis. Calpain inhibitor E-64-d provides neuroprotection in SCI lesion and penumbra. Ann NY Acad Sci 939:436–449
Ray SK, Matzelle DD, Wilford GG, Hogan EL, Banik NL (2001b) Inhibition of calpain-mediated apoptosis by E-64 d-reduced immediate early gene (IEG) expression and reactive astrogliosis in the lesion and penumbra following spinal cord injury in rats. Brain Res 916:115–126
Ray SK, Hogan EL, Banik NL (2003) Calpain in the pathophysiology of spinal cord injury: neuroprotection with calpain inhibitors. Brain Res Brain Res Rev 42(2):169–185
Ray SK, Samantaray S, Smith JA, Matzelle DD, Das A, Banik NL (2011) Inhibition of cysteine proteases in acute and chronic spinal cord injury. Neurotherapeutics 8(2):180–186
Reddy MK, Etlinger JD, Rabinowitz M, Fischman DA, Zak R (1975) Removal of Z-lines and alpha-actinin from isolated myofibrils by a calcium-activated neutral protease. J Biol Chem 250:4278–4284
Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C, Pasturaud P, Roudaut C, Hillaire D, Passos-Bueno M-R, Zats M, Tischfield JA, Fardeau M, Jackson CE, Cohen D, Beckmann JS (1995) Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 81:27–40
Richard I, Roudaut C, Saenz A, Pogue R, Grimbergen JE, Anderson LV, Beley C, Cobo AM, de Diego C, Eymard B, Gallano P, Ginjaar HB, Lasa A, Pollitt C, Topaloglu H, Urtizberea JA, de Visser M, van der Kooi A, Bushby K, Bakker E, Lopez de Munain A, Fardeau M, Beckmann JS (1999) Calpainopathy-a survey of mutations and polymorphisms. Am J Hum Genet 64:1524–1540
Richard I, Roudaut C, Marchand S, Baghdiguian S, Herasse M, Stockholm D, Ono Y, Suel L, Bourg N, Sorimachi H, Lefranc G, Fardeau M, Sebille A, Beckmann JS (2000) Loss of calpain 3 proteolytic activity leads to muscular dystrophy and to apoptosis-associated IkappaBalpha/nuclear factor kappaB pathway perturbation in mice. J Cell Biol 151:1583–1590
Ringger NC, O’Steen BE, Brabham JG, Silver X, Pineda JA, Wang KKW, Hayes RL (2004) A novel marker for traumatic brain injury: CSF aII-spectrin breakdown product levels. J Neurotrauma 21:1443–1456
Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK (1995) Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol 147:1575–1583
Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R (1994) Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci 14:3934–3944
Rodriguez-Galan O, Galindo A, Hervas-Aguilar A, Arst HN Jr, Penalva MA (2009) Physiological involvement in pH signaling of Vps24-mediated recruitment of Aspergillus PalB cysteine protease to ESCRT-III. J Biol Chem 284:4404–4412
Rothman SM, Olney JW (1986) Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann Neurol 19:105–111
Saatman KE, Murai H, Bartus RT, Smith DH, Hayward NJ, Perri BR, McIntosh TK (1996a) Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci U S A 93:3428–3433
Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK (1996b) Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol 55:850–860
Saez ME, Martinez-Larrad MT, Ramirez-Lorca R, Gonzalez-Sanchez JL, Zabena C, Martinez-Calatrava MJ, Gonzalez A, Moron FJ, Ruiz A, Serrano-Rios M (2007) Calpain-5 gene variants are associated with diastolic blood pressure and cholesterol levels. BMC Med Genet 8:1
Saido TC, Yokota M, Nagao S, Yamaura I, Tani E, Tsuchiya T, Suzuki K, Kawashima S (1993) Spatial resolution of fodrin proteolysis in postischemic brain. J Biol Chem 268:25239–25243
Saido TC, Sorimachi H, Suzuki K (1994) Calpain: new perspectives in molecular diversity and physiological-pathological involvement. FASEB J 8:814–822
Salvesen G, Parkes C, Abrahamson M, Grubb A, Barrett AJ (1986) Human low-Mr kininogen contains three copies of a cystatin sequence that are divergent in structure and in inhibitory activity for cysteine proteinases. Biochem J 234:429–434
Samantaray S, Ray SK, Banik NL (2008) Calpain as a potential therapeutic target in Parkinson’s disease. CNS Neurol Disord Drug Targets 7(3):305–312
Samantaray S, Knaryan VH, Gal L, Ray SK, Banik NL (2011) Calpain inhibition protected spinal cord motoneurons against 1-methyl-4-phenylpyridinium ion and rotenone. Neuroscience 129:263–274
Sangenito LS, Ennes-Vidal V, Marinho FA, Da Mota FF, Santos AL, D’Avila-Levy CM, Branquinha MH (2009) Arrested growth of Trypanosoma cruzi by the calpain inhibitor MDL28170 and detection of calpain homologues in epimastigote forms. Parasitology 136:433–441
Sanges D, Comitato A, Tammaro R, Marigo V (2006) Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and blocked by calpain inhibitors. Proc Natl Acad Sci U S A 103:17366–17371
Satish L, Blair HC, Glading A, Wells A (2005) Interferon-inducible protein 9 (CXCL11)-induced cell motility in keratinocytes requires calcium flu-dependent activation of mu-calpain. Mol Cell Biol 25(5):1922–1941
Schaecher KE, Shields DC, Banik NL (2001) Mechanism of myelin breakdown in experimental demyelination: a putative role for calpain. Neurochem Res 26:731–737
Schaecher K, Rocchini A, Dinkins J, Matzelle DD, Banik NL (2002) Calpain expression and infiltration of activated T cells in experimental allergic encephalomyelitis over time: increased calpain activity begins with onset of disease. J Neuroimmunol 129:1–9
Schroder E, Phillips C, Garman E, Harlos K, Crawford C (1993) X-ray crystallographic structure of a papain-leupeptin complex. FEBS Lett 315:38–42
Schumacher PA, Siman RG, Fehlings MG (2000) Pretreatment with calpain inhibitor CEP-4143 inhibits calpain I activation and cytoskeletal degradation, improves neurological function, and enhances axonal survival after traumatic spinal cord injury. J Neurochem 74:1646–1655
Shields DC, Banik NL (1999) Pathological role of calpain in experimental demyelination. J Neurosci Res 55(5):533–541
Shields DC, Schaecher KE, Saido TC, Banik NL (1999) A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc Natl Acad Sci U S A 96:11486–11491
Shirasaki Y, Miyashita H, Yamaguchi M, Inoue J, Nakamura M (2005) Exploration of orally available calpain inhibitors: peptidyl alpha-ketoamides containing an amphiphile at P3 site. Bioorg Med Chem 13:4473–4484
Siesjo BK, Bengtsson F (1989) Calcium fluxes, calcium antagonists, and calcium-related pathology in brain ischemia, hypoglycemia, and spreading depression: a unifying hypothesis. J Cereb Blood Flow Metab 9:127–140
Siesjo BK, Bengtsson F, Grampp W, Theander S (1989) Calcium, excitotoxins, and neuronal death in the brain. Ann NY Acad Sci 568:234–251
Siman R, Noszek JC (1988) Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron 1:279–287
Siman R, Noszek JC, Kegerise C (1989) Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci 9:1579–1590
Siman R, McIntosh TK, Soltesz KM, Chen Z, Neumar RW, Roberts VL (2004) Proteins released from degenerating neurons are surrogate markers for acute brain damage. Neurobiol Dis 16:311–320
Siman R, Toraskar N, Dang A, McNeil E, McGarvey M, Plaum J, Maloney E, Grady MS (2009) A panel of neuron-enriched proteins as markers for traumatic brain injury in humans. J Neurotrauma 26(11):1867–1877
Sorimachi H, Suzuki K (2001) The structure of calpain. J Biochem 129:653–664
Sorimachi H, Imajoh-Ohmi S, Emori Y, Kawasaki H, Ohno S, Minami Y, Suzuki K (1989) Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-types. Specific expression of the mRNA in skeletal muscle. J Biol Chem 264:20106–20111
Sorimachi H, Ishiura S, Suzuki K (1993a) A novel tissue-specific calpain species expressed predominantly in the stomach comprises two alternative splicing products with and without Ca2+-binding domain. J Biol Chem 268:19476–19482
Sorimachi H, Toyama-Sorimachi N, Saido TC, Kawasaki H, Sugita H, Miyasaka M, Arahata K, Ishiura S, Suzuki K (1993b) Muscle-specific calpain, p94, is degraded by autolysis immediately after translation, resulting in disappearance from muscle. J Biol Chem 268:10593–10605
Sorimachi H, Kinbara K, Kimura S, Takahashi M, Ishiura S, Sasagawa N, Sorimachi N, Shimada H, Tagawa K, Maruyama K, Suzuki K (1995) Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. J Biol Chem 270:31158–31162
Sorimachi H, Hata S, Ono Y (2010) Expanding members and roles of the calpain superfamily and their genetically modified animals. Exp Anim 59:549–566
Sorimachi H, Hata S, Ono Y (2011a) Calpain chronicle – an enzyme family under multidisciplinary characterization. Proc Jpn Acad Ser B Phys Biol Sci 87(6):287–327
Sorimachi H, Hata S, Ono Y (2011b) Impact of genetic insights into calpain biology. J Biochem 150:23–37
Spencer MJ, Guyon JR, Sorimachi H, Potts A, Richard I, Herasse M, Chamberlain J, Dalkilic I, Kunkel LM, Beckmann JS (2002) Stable expression of calpain 3 from a muscle transgene in vivo: immature muscle in transgenic mice suggests a role for calpain 3 in muscle maturation. Proc Natl Acad Sci U S A 99:8874–8879
Sribnick EA, Matzelle DD, Banik NL, Ray SK (2007) Direct evidence for calpain involvement in apoptotic death of neurons in spinal cord injury in rats and neuroprotection with calpain inhibitor. Neurochem Res 32(12):2210–2216
Strobl S, Fernandez-Catalan C, Braun M, Huber R, Masumoto H, Nakagawa K, Irie A, Sorimachi H, Bourenkow G, Bartunik H, Suzuki K, Bode W (2000) The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proc Natl Acad Sci U S A 97:588–592
Suzuki Y, Murachi T (1977) The occurence of a neutral protease and its inhibitor in rat peritoneal macrophages. J Biochem 82(1):215–220
Suzuki K, Tsuji S, Kubota S, Kimura Y, Imahori K (1981) Limited proteolysisof Ca2+-activated neutral protease (CANP) changes its sensitivity to Ca2+ ions. J Biochem 90:275–278
Syntichaki P, Xu K, Driscoll M, Tavernarakis N (2002) Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 419:939–944
Szatkowski M, Attwell D (1994) Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci 17:359–365
Taft WC, Yang K, Dixon CE, Hayes RL (1992) Microtubule-associated protein 2 levels decrease in hippocampus following traumatic brain injury. J Neurotrauma 9(3):281–290
Tagawa K, Taya C, Hayashi Y, Nakagawa M, Ono Y, Fukuda R, Karasuyama H, Toyama-Sorimachi N, Katsui Y, Hata S, Ishiura S, Nonaka I, Seyama Y, Arahata K, Yonekawa H, Sorimachi H, Suzuki K (2000) Myopathy phenotype of transgenic mice expressing active site-mutated inactive p94 skeletal muscle-specific calpain, the gene product responsible for limb girdle muscular dystrophy type 2A. Hum Mol Genet 9:1393–1402
Takahashi-Nakamura M, Tsuji S, Suzuki K, Imahori K (1981) Purification and characterization of an inhibitor of calcium-activated neutral protease from rabbit skeletal muscle. J Biochem 90(6):1583–1589
Takai Y, Yamamoto M, Inoue M, Kishimoto A, Nishizuka Y (1977) A proenzyme of cyclic nucleotide-independent protein kinase and its activation by calcium-dependent neutral protease from rat liver. Biochem Biophys Res Commun 77:542–550
Taveau M, Bourg N, Sillon G, Roudaut C, Bartoli M, Richard I (2003) Calpain 3 is activated through autolysis within the active site and lyses sarcomeric and sarcolemmal components. Mol Cell Biol 23:9127–9135
Tian Q, Olsen L, Sun B, Lid SE, Brown RC, Lemmon BE, Fosnes K, Gruis DF, Opsahl-Sorteberg HG, Otegui MS, Olsen OA (2007) Subcellular localization and functional domain studies of DEFECTIVE KERNEL1 in maize and Arabidopsis suggest a model for aleurone cell fate specification involving CRINKLY4 and SUPERNUMERARY ALEURONE LAYER1. Plant Cell 19:3127–3145
Tompa P, Emori Y, Sorimachi H, Suzuki K, Friedrich P (2001) Domain III of calpain is a Ca2+-regulated phospholipid-binding domain. Biochem. Biophys Res Commun 280:1333–1339
Tonami K, Kurihara Y, Aburatani H, Uchijima Y, Asano T, Kurihara H (2007) Calpain 6 is involved in microtubule stabilization and cytoskeletal organization. Mol Cell Biol 27:2548–2561
Tonami K, Hata S, Ojima K, Ono Y, Kurihara Y, Amano T, Sato T, Kawamura Y, Kurihara H, Sorimachi H (2013) Calpain-6 Deficiency Promotes Skeletal Muscle Development and Regeneration. PLoS Genet 9:e1003668
Tonami K, Kurihara Y, Arima S, Nishiyama K, Uchijima Y, Asano T, Sorimachi H, Kurihara H (2011) Calpain 6, a microtubule-stabilizing protein, regulates Rac1 activity and cell motility through interaction with GEF-H1. J Cell Sci 124:1214–1223
Tradewell ML, Durham HD (2010) Calpastatin reduces toxicity of SOD1G93A in a culture model of amyotrophic lateral sclerosis. Neuroreport 21(15):976–979
Ueda Y, McCormack AL, Shearer TR, David LL (2001) Purification and characterization of lens specific calpain (Lp82) from bovine lens. Exp Eye Res 73:625–637
Veeranna KT, Boland B, Odrljin T, Mohan P, Basavarajappa BS, Peterhoff C, Cataldo A, Rudnicki A, Amin N, Li BS, Pant HC, Hungund BL, Arancio O, Nixon RA (2004) Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer’s disease. Am J Pathol 165:795–805
Verity MA (1992) Ca(2+)-dependent processes as mediators of neurotoxicity. Neurotoxicology 13:139–147
Vitzthum F, Behrens F, Anderson NL, Shaw JH (2005) Proteomics: from basic research to diagnostic application. A review of requirements and needs. J Proteome Res 4(4):1086–1097
Waghray A, Wang DS, McKinsey D, Hayes RL, Wang KK (2004) Molecular cloning and characterization of rat and human calpain-5. Biochem Biophys Res Commun 324:46–51
Wang KKW (2000) Calpain and caspase: can you tell the difference? Trends Neurosci 23:20–26
Wang KK, Yuen PW (1994) Calpain inhibition: an overview of its therapeutic potential. Trends Pharmacol Sci 15:412–419
Wang KK, Yuen PW (1997) Development and therapeutic potential of calpain inhibitors. Adv Pharmacol 37:117–152
Wang KK, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV, Glantz SB, Morrow JS (1998) Simultaneous degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem 273:22490–22497
Wang KKW, Yuen PW (1999) Calpain substrates, assay methods, regulation and its inhibitory agents. In: Wang KKW, Yuen PW (eds) Calpain: pharmacology and toxicology of a calcium-dependent cellular protease. Taylor and Francis, Philadelphia, PA, pp 77–102
Wang KKW, Villalobo A, Roufogalis BD (1989) Calmodulin-binding proteins as calpain substrates. Biochem J 262:693–706
Wang KKW, Ottens A, Liu MC, Lewis SB, Meegan C, Oli MW, Tortella FC, Hayes RL (2005) Proteomic identification of biomarkers of traumatic brain injury. Expert Rev Proteomics 2(4):603–614
Warner S (2004) The Scientist 18(16):38
Wells A, Huttenlocher A, Lauffenburger DA (2005) Calpain proteases in cell adhesion and motility. Int Rev Cytol 245:1–16
Wieloch T, Cardell M, Bingren H, Zivin J, Saitoh T (1991) Changes in the activity of protein kinase C and the differential subcellular redistribution of its isozymes in the rat striatum during and following transient forebrain ischemia. J Neurochem 56:1227–1235
Wikstrom P, Anagli J, Angliker H, Shaw E (1992) Additional peptidyl diazomethyl ketones, including biotinyl derivatives, which affinity-label calpain and related cysteinyl proteinases. J Enzyme Inhib 6(4):259–269
Yamashima T (2000) Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol 62:273–295
Yamaura I, Tani E, Saido TC, Suzuki K, Minami N, Maeda Y (1993) Calpain-calpastatin system of canine basilar artery in vasospasm. J Neurosurg 79:537–543
Yokota M, Tani E, Tsubuki S, Yamaura I, Nakagaki I, Hori S, Saido TC (1999) Calpain inhibitor entrapped in liposome rescues ischemic neuronal damage. Brain Res 819:8–14
Yoshikawa Y, Mukai H, Hino F, Asada K, Kato I (2000) Isolation of two novel genes, down-regulated in gastric cancer. Jpn J Cancer Res 91:459–463
Yu CG, Joshi A, Geddes JW (2008) Intraspinal MDL28170 microinjection improves functional and pathological outcome following spinal cord injury. J Neurotrauma 25(7):833–840
Yuen PW, Wang KKW (1998) Calpain inhibitors: Novel neuroprotectants and potential anticataract agents. Drugs Future 23:741–749
Zhang SX, Underwood M, Landfield A, Huang FF, Gison S, Geddes JW (2000) Cytoskeletal disruption following contusion injury to the rat spinal cord. J Neuropathol Exp Neurol 59:287–296
Zhang SX, Bondada V, Geddes JW (2003) Evaluation of conditions for calpain inhibition in the rat spinal cord: effective postinjury inhibition with intraspinal MDL28170 microinjection. J Neurotrauma 20:59–67
Zhang Z, Larner S, Liu MC, Zheng W, Hayes RL, Wang KKW (2009) Multiple αII-spectrin breakdown products distinguish calpain and caspase dominated necrotic and apoptotic cell death pathways. Apoptosis 14(11):1289–1298
Zhao X, Pike BR, Newcomb JK, Wang KK, Posmantur RM, Hayes RL (1999) Maitotoxin induces calpain but not caspase-3 activation and necrotic cell death in primary septo-hippocampal cultures. Neurochem Res 24:371–382
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this chapter
Cite this chapter
Anagli, J., Wang, K.K.W., Ono, Y., Sorimachi, H. (2013). Calpains in Health and Disease. In: Brix, K., Stöcker, W. (eds) Proteases: Structure and Function. Springer, Vienna. https://doi.org/10.1007/978-3-7091-0885-7_12
Download citation
DOI: https://doi.org/10.1007/978-3-7091-0885-7_12
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-0884-0
Online ISBN: 978-3-7091-0885-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)