
Abstract
The kidney, adrenal gland, and retroperitoneum are important sites of disease in the pediatric population. Some of the most frequently encountered childhood neoplasms, including Wilms tumor and neuroblastoma, arise in the kidney and adrenal gland, respectively. Many of these tumors are deep-seated but amenable to fine-needle aspiration (FNA). Children with a renal, adrenal, or retroperitoneal malignancy may present at an advanced stage and be too ill to withstand the rigors of surgery. Thus, neoadjuvant chemotherapy may be used to debulk some of these tumors before surgery is performed, and an accurate cytologic diagnosis is imperative to ensure the appropriate management is undertaken. This chapter covers both common and more unusual lesions that may be encountered when looking at an FNA from this area. In addition, urine cytology in children is discussed briefly.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others



Keywords
7.1 Kidney
Renal lesions in children and adolescents can be due to cystic or inflammatory etiologies, in addition to neoplasms. Renal tumors are among the most common solid organ malignancies in children due to the peak incidence of Wilms tumor in this age group. Other kidney tumors, including conventional renal cell carcinoma (RCC) of the clear cell type seen in adults, are rare in children.One of the most important factors to know is the age of the child, as some pediatric renal tumors are seen more often in certain age groups (Table 7.1).
7.1.1 Cysts
Clinical Features
Renal cysts in children and adolescents are due to a variety of causes and can be single or multiple, unilateral or bilateral. Autosomal recessive polycystic kidney disease, which is due to a mutation in the PKHD1 gene on chromosome 6 (fibrocystin protein), is characterized by enlarged, diffusely echogenic kidneys with multiple, bilateral cysts and typically presents in the neonatal period or infancy. In countries with widespread use of prenatal ultrasound, it is increasingly recognized in utero. In contrast, autosomal dominant polycystic kidney disease is due to mutations in genes encoding polycystin-1 (PKD1 on chromosome 16) or polycystin-2 (PKD2 on chromosome 4) and is a late-onset disorder characterized by progressive, bilateral renal enlargement due to multiple cysts. It usually becomes symptomatic after the third decade of life but may also present in adolescents or children. Other diseases associated with renal cysts include, but are not limited to, tuberous sclerosis, von Hippel–Lindau syndrome, Ehlers–Danlos syndrome, Zellweger syndrome, Meckel–Gruber syndrome, Beckwith–Wiedemann syndrome, short rib-polydactyly syndrome, renal dysplasia, and calyceal diverticula [1–5]. Acquired simple cysts, which are the most common cysts encountered in adults, are uncommon in children. Current imaging techniques allow accurate assessment of the risk of malignancy in many cystic renal lesions, precluding the need for fine-needle aspiration (FNA) except in cases with indeterminate findings or for decompression of a benign cyst.
Cytological Features
FNA of a renal cyst usually yields clear or blood-stained fluid with macrophages, cyst-lining cells, and occasional inflammatory cells. Renal tubular cells and Liesegang rings are infrequently observed. Benign cysts of various etiologies cannot be distinguished from each other on FNA.
Triage
Cyst fluid may benefit from liquid-based cytology to decrease obscuring blood in hemorrhagic cyst contents and to increase the ability to find cyst-lining cells, to exclude malignancy.
Differential Diagnosis
Neoplasms that undergo cystic degeneration are the main consideration but usually have greater cellularity and more atypia. Cyst contents with macrophages may also mimic granulomatous infections (mycobacterial or fungal) or the xanthomatous histiocytes of xanthogranulomatous pyelonephritis.
Pitfalls
Misinterpretation of reactive atypia or degenerative changes in cyst-lining cells can lead to a false-positive diagnosis of malignancy. Conversely, a hypocellular specimen with few or no malignant cells can result in a false-negative diagnosis in a cystic neoplasm.
7.1.2 Infections
Clinical Features
Pyelonephritis is caused by a variety of bacterial, viral, fungal, and parasitic organisms. Infection can be blood-borne or ascending and in children less than 6 years of age is associated with vesicoureteral reflux in up to 50 % of cases. The most common causes of pyelonephritis are bacteria, such as E. coli, Proteus mirabilis, Klebsiella spp., Enterobacter spp., Enterococcus faecalis, Staphylococcus spp., and others. Fungal pyelonephritis usually occurs in the context of systemic infection with opportunistic or pathogenic organisms, such as Candida spp, Aspergillus spp, Cryptococcus spp, Histoplasma capsulatum, Coccidioides immitis, and others and, although more common in immunocompromised patients, can also affect otherwise healthy individuals. Viruses, especially BK virus and cytomegalovirus, are associated with clinically significant renal infection in immunocompromised patients, particularly those with renal allografts. Pyelonephritis due to parasitic infiltration of the renal parenchyma is rare but can be seen in schistosomiasis, leishmaniasis, and malaria. In addition, schistosomiasis can cause obstructive uropathy at the level of the bladder and/or ureter with consequent ascending pyelonephritis due to other organisms. Mycobacterial infection of the kidney is usually due to M. tuberculosis. Infection may be due to disseminated infection or be localized within the urinary tract. Xanthogranulomatous pyelonephritis occurs in children, albeit infrequently, and is associated with kidney stones and persistent urinary tract infection [1, 2, 6].
Cytological Features
The cytomorphology of these various infections is similar at whatever body site they occur. In the setting of mycobacterial infection, the aspirates show necrotizing granulomatous inflammation (Fig. 7.1a) Immunosuppressed children may present with an FNA comprised almost entirely of necrosis or a more suppurative picture with a moderate number of neutrophils in a necrotic background. A complication of infection, especially bacterial, is a renal abscess, and the FNA yields purulent material with numerous neutrophils, scattered histiocytes, and debris. Xanthogranulomatous pyelonephritis shows vacuolated histiocytes, acute and chronic inflammatory cells, multinucleated giant cells, cholesterol crystals, and necrotic debris (Fig. 7.1b). Rare cases of aspergillosis involving the kidney with fungal balls eliciting ureteral obstruction have been described [6].

Renal Infections (a, b. Papanicolaou stain, high power; b. H&E stain, high power). Mycobacterial infection in the kidney of a 4-year-old girl showing epithelioid histiocytes (thick arrow) and Langhans-type giant cells (thin arrow) in a necrotic background (a). Xanthogranulomatous pyelonephritis diagnosed by FNA of the kidney in a 6-year-old boy. The aspirates showed acute inflammatory cells, vacuolated histiocytes (arrow), and debris (b).
Triage
A portion of the aspirate should be submitted for microbial cultures and cell block if infection is suspected. Special stains, such as Grocott, AFB, and gram stains can be performed on the cell-block material. In addition, if there is limited inflammatory or cellular material, extra unstained smears can be prepared and used for key special stains, in the event there is scant material on the cell block [7]. In cases in which special stains or culture results are negative but clinical suspicion for mycobacterial infection is high, molecular diagnostic techniques can be performed on the FNA material for identification and subtyping.
Differential Diagnosis
In infectious cases dominated by macrophages, the differential diagnosis includes cyst contents.
Pearls
In case of suspected infection, it is important to preserve fresh material for microbial cultures and antibiotic sensitivities.
7.1.3 Miscellaneous Nonneoplastic Conditions
Amyloidosis, renal infarct, sarcoidosis, extramedullary hematopoiesis, and malakoplakia have all been described, albeit infrequently, in the kidneys of children (Fig. 7.2). The cytomorphology is identical to that seen in the FNA of similar conditions in adult kidneys. FNA is not usually utilized in the diagnosis and management of glomerulonephritis. However, cytology plays an important role in the identification of red cell casts in pediatric urine specimens, which may be a clue in the diagnosis of glomerulonephritis.

Extramedullary hematopoiesis (Papanicolaou stain, high power). Megakaryocytes and immature myeloid precursor cells, consistent with extramedullary hematopoiesis, in a 4-year-old girl who presented with anemia and renal mass.
7.1.4 Renal Neoplasms
Children with renal tumors may present symptomatically, e.g., hematuria, abdominal pain or mass, or the tumor may be an incidental finding. A wide variety of renal neoplasms, both benign and malignant, are described in children. These include Wilms tumor, nephrogenic rests and nephroblastomatosis, cystic partially differentiated nephroblastomatoma, metanephric neoplasm, mesoblastic nephroma, clear cell sarcoma, rhabdoid tumor, renal cell carcinoma (in particular translocation-associated and renal medullary), angiomyolipoma and ossifying renal tumor of infancy.
7.1.4.1 Angiomyolipoma
Clinical Features
These benign tumors are rare and occur more often in adults but can be seen in young adults and children with tuberous sclerosis. Tuberous sclerosis is an autosomal dominant syndrome due to alterations of the TSC1 gene on chromosome 9q34 or the TSC2 gene on chromosome 16p13, which is associated with hamartomas of the brain (subependymal giant cell tumor), cutaneous angiofibromas, rhabdomyomas, lymphangioleiomyomatosis, and angiomyolipomas.
Cytological Features
FNA yields a proliferation of thickened blood vessels, sheets and bundles of perivascular spindle and epithelioid cells, and adipose tissue in varying proportions. Fat globules can be seen in the background. In some cases the epithelioid cells show a striking degree of cytologic atypia and pleomorphism. Most imaging studies can identify adipose tissue within a tumor rendering an FNA diagnosis unnecessary; thus, it is the angiomyolipomas with scant fat that typically present for FNA biopsy (Fig. 7.3).

Angiomyolipoma in kidney (a, b. Papanicolaou stain, high power). Aspirates from this renal mass in a 12-year-old girl show thick-walled blood vessels (a, thin arrow) and naked nuclei of smooth muscle cells (a, thick arrow) with fat globules (b, arrow).
Triage
The epithelioid cells co-express melanocytic markers (HMB45, MART1) and smooth muscle markers (calponin, muscle specific actin). CD117, hormone receptors (PR, less often ER), and occasionally desmin can also be positive.
Differential Diagnosis
When aspirates contain few or no HMB45-positive epithelioid cells, angiomyolipoma may be mistaken for sampling of normal kidney and surrounding soft tissue. When epithelioid cells predominate and/or have significant cytologic atypia and pleomorphism, differential diagnostic considerations include carcinoma and metastatic melanoma. Soft tissue neoplasms (e.g., lipoma) form part of the differential diagnosis.
Pearls
Positivity for melanocytic markers is characteristic of angiomyolipoma and should not be misinterpreted as evidence of melanoma.
7.1.4.2 Wilms Tumor (Nephroblastoma)
Clinical Features
Wilms tumor is among the most common solid organ tumors of childhood and the most common pediatric renal tumor. It occurs throughout childhood but is usually diagnosed before the age of 5 years and occurs equally in both sexes. Wilms tumor can be sporadic or occur with nephroblastomatosis, or in children with syndromes such as Beckwith–Wiedemann, WAGR (Wilms tumor, Aniridia, Genitourinary abnormalities, and mental Retardation due to chromosome 11p13 (WT-1 gene) deletion), and Denys–Drash syndromes. Children usually present with a painless flank or abdominal mass or abdominal pain. Hematuria, elevated blood pressure, or fever may be observed [1, 4].
Cytological Features
The cytomorphology of Wilms tumor recapitulates the embryologic development of the kidney, represented by blastema, mesenchyme, and epithelium (Figs. 7.4 and 7.5).

Wilms tumor (a. Papanicolaou stain, medium power; b. H&E stain, medium power; c. WT1 immunoperoxidase stain, high power). The aspirates show a blastemal component comprised of small round cells with minimal cytoplasm (a). Bland spindle cells (b) that constitute the mesenchymal component can also be seen. Positive WT1 immunostaining is demonstrated in the blastemal component (c).

Wilms tumor with prominent epithelial component (a. Papanicolaou stain, medium power; b. Diff-Quik stain, medium power). The epithelial component can form tubules (a), and the cells may be attached to a metachromatic, basement membrane-like material (b). Please note the well-defined edges of the epithelial component.
-
Blastemal component: Blastema yields a cellular background of small round blue cells. The cells are small and primitive, about twice the size of a lymphocyte, with a high nuclear cytoplasmic (N/C) ratio and minimal, scanty, fragile cytoplasm. The nuclei have finely granular chromatin with inconspicuous nucleoli. Nuclear molding may be seen. Necrosis may be present in the background (Fig. 7.4).
-
Epithelial component: The epithelial component is best identified on scanning power as aggregates of tumor cells forming ill-defined clusters, tubules, and nests. The epithelial component has well-defined edges. The cells are larger than the background blastemal cells, with more cytoplasm, and may be attached to metachromatic basement membrane-like material. Tubules may demonstrate branching [1]. Occasionally, rosettes are seen; however, unlike neuroblastoma, there is no associated eosinophilic fibrillary neuropil. Glomeruloid-like bodies can be observed. These are made up of tumor cells, not the bland-appearing balls of cells that make up normal glomeruli (Fig. 7.5).
-
Mesenchymal component: The mesenchymal spindle cell component is comprised of bland fibroblast-like cells set in fibrous or metachromatic myxoid stroma. Occasionally, smooth muscle or skeletal muscle differentiation may be seen.
-
One, two, or three components may be present. If only blastema is aspirated, differentiation from other small round cell tumors is based on immunoperoxidase stains. A variety of heterologous elements, such as squamous or mucous epithelium, endocrine cells, neuroglial components, osteoid, and cartilage, can be also present, and such tumors are referred to as teratoid Wilms tumor.
-
Anaplasia: Approximately 4 % of Wilms tumors demonstrate a triad of microscopic features diagnostic of anaplasia. These changes can be focal or diffuse and thus may not be seen in the aspirate due to sampling. The features of anaplasia, which can be seen even under scanning at low power, are as follows: enlarged nuclei (at least three times the size of adjacent tumor nuclei), nuclear hyperchromasia, and abnormal or multipolar mitotic figures. All three features must be present for the diagnosis of anaplasia (Fig. 7.6). Although definitive classification of anaplasia as focal or diffuse is not possible in cytologic preparations, it is important to recognize and comment on the presence of any anaplasia, thereby alerting the oncologist to the possibility of diffuse anaplasia which is associated with a worse prognosis.
Fig. 7.6 
Wilms tumor showing anaplasia (a. Papanicolaou stain, high power; b. Diff-Quik stain, high power). Enlarged and hyperchromatic nuclei are observed in this Wilms tumor with anaplasia.

Triage
Ancillary studies are helpful, particularly if the blastemal component predominates, in order to exclude other small round blue cell tumors. All components of a Wilms tumor may show nuclear positivity for WT1, albeit weak in the stromal component. However, WT1 expression is not seen in all Wilms tumors and is not restricted to Wilms tumors. The blastemal component is also positive for vimentin and may show positivity (±) for neuron-specific enolase (NSE), desmin, cytokeratin, and CD56. The epithelial component is positive for EMA and cytokeratin. Staining of the mesenchymal component varies according to its morphologic appearance and differentiation. For example, cells with skeletal muscle differentiation are usually myogenin positive. Material can also be submitted for cytogenetic studies to demonstrate abnormalities of the Wilms tumor (WT) genes 1 (chromosome 11p13) and 2 (chromosome 11p15). Mutations in TP53 are seen with high frequency in the anaplastic subtype of Wilms tumor. Electron microscopy can also be used to demonstrate epithelial differentiation (such as intercellular junctions, basement membrane-like material, oligocilia, and microvilli) and exclude other small round cell tumors, such as neuroblastoma and rhabdomyosarcoma, by demonstrating a lack of neurosecretory granules and muscle fibrils, respectively.
Differential Diagnosis
The differential diagnosis, particularly for those tumors with a predominant blastemal component or round blue cell morphology, is presented in Table 7.2. Wilms tumor cannot be distinguished cytologically from nephroblastomatosis and nephrogenic rests, which are aggregates of persistent primitive metanephric tissue. When microscopic in size, these aggregates are termed rests and, when large, nephroblastomatosis. As the metanephric tissue is primitive, aspiration of nephroblastomatosis will be indistinguishable from nephroblastoma on cytology. Children with nephroblastomatosis are at a markedly increased risk for the development of nephroblastoma. The diagnosis is made by radiological confirmation of the subcapsular location and diffuse nature of the process. It is noted that 30 % of kidneys containing Wilms tumor have foci of nephroblastomatosis, which also show WT-1 mutations.
Pearls
FNA of a flank mass in a child is usually performed via the flank or posterior approach since the kidney is a retroperitoneal structure. Theoretically, it is possible that an anterior approach may lead to the spread of tumor to the peritoneum and upstage a potentially curable childhood neoplasm.Another tumor in the differential diagnosis, particularly in older patients is metanephric adenoma, which shows positivity for BRAF V600E mutations, and is less likely to have a triphasic morphology.
7.1.4.3 Cystic, Partially Differentiated Nephroblastoma
Clinical Features
This is a rare, cystic neoplasm occurring in early infancy or young children, almost always under the age of two. It has low malignant potential. It is also known as multilocular cystic nephroma, multicystic nephroma, or multilocular cyst. This entity may represent a fully differentiated variant of Wilms tumor. Patients commonly present with a mass lesion or ureteral obstruction. Radiological evaluation reveals a large, solitary, well-circumscribed multilocular cystic mass.
Cytological Features
Aspirates tend to be of low cellularity with variable representation of epithelial, stromal, and nephroblastomatous elements. Benign-appearing epithelial cells are arranged singly and in sheets. Bland spindle cells, with or without skeletal muscle differentiation, are generally present in sparse numbers. The background stroma can be myxoid in nature, or have other mesenchymal components such as cartilage and fat.
Triage
Electron microscopy reveals epithelial cells with long cilia suggestive of collecting tubular cells, while cytogenetics shows nonrandom X-chromosome inactivation. Liquid-based cytology may be of benefit in these cases to concentrate the cells available for evaluation in this cystic neoplasm.
Differential Diagnosis
Benign or hemorrhagic cysts of the kidney or retroperitoneum should be considered.
Pearls
The presence of any solid nodules radiologically favors cystic Wilms tumor rather than cystic, partially differentiated nephroblastoma or cystic nephroma [1].
7.1.4.4 Clear Cell Sarcoma
Clinical Features
This is an uncommon but aggressive childhood renal tumor with a poor prognosis. It comprises approximately 5 % of childhood renal tumors, with a peak incidence at 2–3 years of age, and is seen more frequently in boys [1, 4]. It has a propensity to spread to bones, often skull, as well as to soft tissue, orbit, and brain. Children usually present with an abdominal mass. Abdominal pain, fever, hematuria, and hypertension may also be noted. Radiological imaging shows a renal tumor, usually in the medulla or central region.
Cytological Features
These tumors are well circumscribed and commonly show cystic change. Several variants of this tumor are noted; thus, the cytomorphology varies accordingly (Fig. 7.7). In general, the aspirates are cellular and show two cell types, including stellate or spindled tumor cells (“septal cells”) arranged singly in a myxoid background and polygonal tumor cells (“cord cells”) in clusters with pale cytoplasm. Nuclei are generally bland, round, oval, or reniform with finely granular chromatin and inconspicuous nucleoli. Nuclear grooves, when present, are a useful clue to the diagnosis. Myxoid or mucoid material may be seen in the background [8, 9].

Clear cell sarcoma of the kidney (a. Papanicolaou stain, medium power; b. Papanicolaou stain, high power). The aspirates show dyscohesive and cohesive clusters of stellate and polygonal cells, in addition to bare nuclei. The nuclei are round to oval with very occasional nuclear grooves (b, arrow).
Triage
On immunocytochemistry, these tumors are positive for vimentin and bcl-2 and negative for WT1, EMA, CD34, S-100, desmin, CD99, and synaptophysin. Ultrastructurally, the tumor cells have a high nuclear-to-cytoplasmic ratio, with thin complex cytoplasmic extensions surrounding extracellular matrix. Intermediate filaments are present in the cytoplasm. Fluorescent in situ hybridization (FISH) studies may show a t(10;17) translocation or a chromosome 14q deletion [10].
Differential Diagnosis
Tumors in the perirenal area that may exhibit nuclear grooves include clear cell sarcoma, renal cell carcinoma, renal medullary carcinoma, Langerhans cell histiocytosis, epithelioid hemangioendothelioma, and metastases to the kidney (e.g., papillary thyroid carcinoma, granulosa cell tumor, and solid pseudopapillary neoplasm of the pancreas). Given the variations in morphology of clear cell sarcoma, a wide variety of tumors enters the differential diagnosis, although, in practice, many of these are unlikely based on age at presentation or other clinical considerations. A summary of the differential diagnostic considerations is listed in Tables 7.2, 7.3, and 7.4.
7.1.4.5 Mesoblastic Nephroma
Clinical Features
Mesoblastic nephroma is the most common renal tumor of infancy. It can be seen within a few days of birth, with most diagnosed before 6 months of age. The vast majority are cured by nephrectomy alone. Age at diagnosis (<3 months old) and adequate surgical resection are the most important favorable prognostic features rather than the presence or absence of a cellular subtype on microscopic evaluation. Patients usually present with an abdominal mass. Radiological studies confirm a kidney tumor, often in the region of the hilum. These tumors are commonly well circumscribed and solid. However, infiltration of adjacent renal parenchyma or perirenal fat, hemorrhage, necrosis, and cystic change can occur, particularly in the cellular variant [1, 4].
Cytological Features
Three main subtypes are described: classic, cellular, and mixed (Fig. 7.8). Smears are generally paucicellular, comprised of spindle cells lying both singly and in cohesive clusters. The proliferating cells can acquire features of fibroblasts, myofibroblasts, or smooth muscle cells. The spindle cells have bland nuclei and scant eosinophilic cytoplasm with ill-defined borders. The nuclei show minimal atypia and coarse chromatin. On the Diff-Quick stain, a small amount of fibromyxoid material can usually be seen. Rarely, heterologous elements such as cartilage may be present. Cellular mesoblastic nephroma is a variant of mesoblastic nephroma showing increased cellularity and atypia. Hemorrhage and necrosis can be observed.

Mesoblastic nephroma (Diff-Quik stain, high power). Cohesive clusters of spindle cells with bland nuclear features and scanty cytoplasm are seen in this FNA in a 40-day-old girl with mesoblastic nephroma.
Triage
On immunoperoxidase staining, mesoblastic nephromas are positive for vimentin, actin, and occasionally WT1. Desmin, CD34, and keratin are negative. Features of fibroblasts or myofibroblasts are seen on electron microscopy. The classic mesoblastic nephroma is diploid, while the cellular variant shows t(12;15) (p13;q25), which results in a fusion of the ETV6 and NTRK3 genes. This is also seen in infantile fibrosarcoma, but is not present in fibromatosis. This supports the analogy between cellular mesoblastic nephroma and infantile fibrosarcoma, and between classic mesoblastic nephroma and infantile fibromatosis.
Differential Diagnosis
Table 7.4 highlights other pediatric renal tumors that can show a predominance of spindled cells.
Pearls
Entrapped or adjacent benign tubules or glomeruli may be aspirated together with the mesoblastic nephroma and should not be over-interpreted as an epithelial component of a Wilms tumor.
7.1.4.6 Rhabdoid Tumor
Clinical Features
This is a rare, but aggressive, childhood tumor of the kidney. Rhabdoid tumors can be renal or extrarenal. The mean age at diagnosis is 1 year, with the vast majority of patients presenting by 2 years of age. Patients commonly present with hematuria, fever, or metastases. Tumors are typically large and circumscribed with extensive hemorrhage and necrosis and almost always involve the medullary region [1, 11].
Cytological Features
The aspirate is usually cellular with tumor cells arranged singly or in clusters. The cell population tends to be a monomorphic population of round, oval, or spindled cells. Occasional multinucleated giant cells or a component of small, round, undifferentiated cells may be seen. The nuclei are characteristically large, single, and eccentric with vesicular chromatin and prominent macronuclei. Naked nuclei are common. The cytoplasm is well-defined and abundant. Variable numbers of characteristic “rhabdoid” inclusions, comprised of large, intracytoplasmic, dense eosinophilic hyaline globules, are present. These abut and displace the nucleus laterally resulting in a plasmacytoid appearance.
Triage
On immunoperoxidase staining, rhabdoid tumor is positive for vimentin, desmin, EMA, and keratin. The tumor cells are negative for INI1 and WT1. However, the whorls of intracytoplasmic filaments that make up the hyaline inclusions can cause nonspecific trapping of antibodies and give a wide range of false-positive results on immunocytochemistry. Cytogenetic studies show deletions or mutations of the SMARCB1 (INI1) gene. The intracytoplasmic inclusion consists of whorls of intermediate filaments on electron microscopy.
Differential Diagnosis
Table 7.3 highlights other pediatric renal tumors that can have large epithelioid cells.
Pearls
Intracytoplasmic inclusions occur in a wide variety of other tumors, including Wilms tumor, mesoblastic nephroma, rhabdomyosarcoma, renal medullary carcinoma, and renal cell carcinoma, and are not specific for rhabdoid tumor. Loss of INI1 nuclear staining has also been reported in renal medullary carcinomas and is therefore not entirely specific for rhabdoid tumors, although these tumors are unlikely to be confused clinically or cytologically [12].
7.1.4.7 Renal Cell Carcinoma (RCC)
Clinical Features
Renal cell carcinoma is the second most common kidney neoplasm in the pediatric population, especially older children and adolescents. Many of these are translocation-associated RCC (approximately 20–54 %), with less common variants including papillary (approximately 20 %), clear cell (approximately 15 %), and chromophobe (approximately 5 %) [11, 13, 14]. Translocation-associated, RCC usually involves the TFE3 gene on chromosome 11 or much less frequently the TFEB gene on chromosome 6. Translocation RCCs have a better prognosis compared with non-translocation RCCs and are sometimes referred to as microphthalmia transcription factor (MiT) family translocation carcinomas [13, 14]. One-third of RCCs are associated with either tuberous sclerosis, neuroblastoma, or other syndromes. Signs and symptoms include abdominal pain, hematuria, and a mass lesion.
Cytological Features
-
Translocation-associated RCC tends to mimic clear cell or papillary renal cell carcinoma. Cells are arranged in nests, trabeculae, and papillary structures. Psammoma bodies can be seen. The cells have a low N/C ratio with abundant eosinophilic, granular, or foamy cytoplasm. In some cases, the tumor cell nuclei appear as naked nuclei due to the fragility of the vacuolated cytoplasm (Fig. 7.9a). Metachromatic material and transgressing vessels can also be seen (Fig. 7.9a, b). The nuclei contain granular chromatin and prominent nucleoli. Rare cases may show melanin pigment.
Fig. 7.9 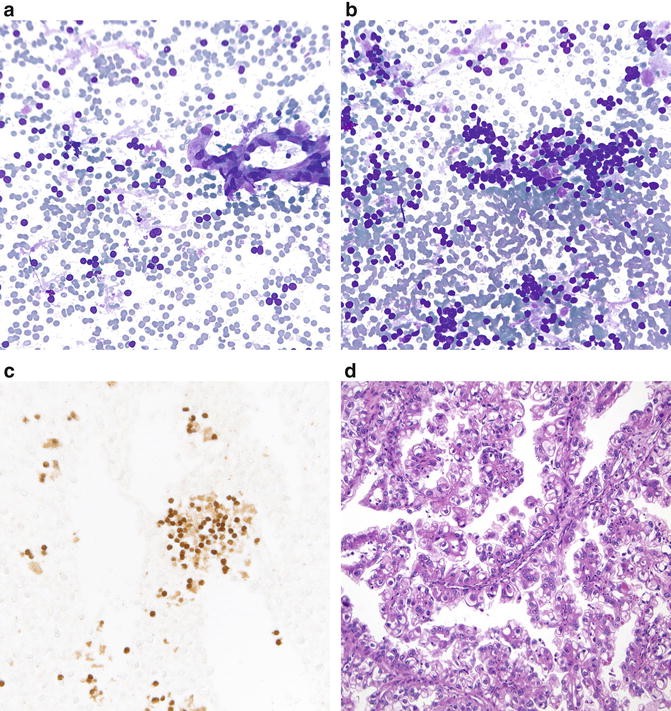
Renal cell carcinoma associated with translocation Xp11.2/TFE gene fusion (a, b. Diff-Quik stain, high power; c. TFE3 immunostain, high power; d. H&E stain, high power). Translocation-associated RCC shows papillary fragments or cohesive clusters of tumor cells with abundant vacuolated cytoplasm that can be stripped off creating naked nuclei (a). There is also usually metachromatic material (b), and transgressing blood vessels (a). A TFE3 immunostain shows nuclear positivity (c). Histological follow-up shows a papillary renal tumor with tumor cells having abundant clear-to-vacuolated cytoplasm (d). (Image courtesy of Dr. Sara Monaco).
-
Papillary RCC can have less cytoplasm that appears basophilic (type 1) or more abundant cytoplasm imparting an eosinophilic or oncocytic appearance (type 2).

Triage
On immunoperoxidase staining, TFE3 and CD10 are positive, while vimentin and AE1/AE3 staining is variable. TFE3 should show nuclear positivity (Fig. 7.9c). The negative or focal staining for epithelial markers (e.g., EMA and cytokeratin) and vimentin in a vacuolated epithelioid kidney tumor should raise the possibility of translocation-associated RCC, given that other types of RCC are typically positive for both [1, 14]. Papillary RCC is positive for CK7, which helps to distinguish it from Wilms tumor and metanephric adenoma.
Differential Diagnosis
Tables 7.3 and 7.4 list other pediatric renal tumors in the differential diagnosis.
Pearls
Clear cell sarcoma of the kidney should be distinguished from renal cell carcinoma of the clear cell type and is TFE3 negative.
7.1.4.8 Renal Medullary Carcinoma
Clinical Features
This is an uncommon tumor, most often described in adolescent and young adult African American males with sickle cell trait. It is aggressive with a poor prognosis. Patients present with hematuria, abdominal pain, weight loss, fever, and a mass lesion in the kidney [1, 11].
Cytological Features
The aspirates reveal a marked inflammatory cell infiltrate with intratumoral neutrophils. Tumor cells are seen in loosely cohesive groups or as single cells. The cells have a high N/C ratio, and the cytoplasm is vacuolated. The nuclei are hyperchromatic and irregular in shape with reniform nuclei indented by the cytoplasmic vacuoles. Nuclear grooves and rhabdoid features have also been described. Prominent, sometimes multiple, nucleoli can be seen. A variety of growth patterns can be observed including tubular, solid, yolk sac-like, adenoid cystic-like and micropapillary. Sickled red blood cells may be noted.
Triage
Although there are no definitive immunoperoxidase stains for this renal tumor, a cell block is helpful to confirm positivity for cytokeratin and vimentin. In addition, some renal medullary carcinomas, particularly high-grade variants with rhabdoid features, can have loss of nuclear staining for INI1, as is seen in rhabdoid tumors [12]. A TFE3 stain should be negative.
Differential Diagnosis
Table 7.3 highlights some other tumors to consider in the differential diagnosis.
Pearls
The positivity for cytokeratin and vimentin, and negativity for TFE3, can be helpful to exclude a translocation-associated RCC. In addition, the morphology and clinical history can usually help to exclude a rhabdoid tumor in the cases of renal medullary carcinomas that show loss of nuclear staining for INI1 [12].
7.1.5 Secondary Tumors to the Kidney
Neuroblastoma and non-Hodgkin lymphoma are the most frequently described tumors to metastasize to the kidney in children. Lymphomas are important to recognize, as they usually do not require surgical excision (Fig. 7.10). However, many other childhood tumors (e.g., germ cell tumors) can involve the kidney, either by contiguous spread or from distant metastases. An example would be germ cell tumors metastasizing to the kidney, which can mimic certain primary renal tumors, such as renal medullary carcinoma. Spindle cell tumors, such as synovial sarcoma, can also be metastatic in the kidney and mimic a renal tumor with predominance of spindle cells (Table 7.4) (Fig. 7.11).

Diffuse large B-cell lymphoma in the kidney (Papanicolaou stain, high power). Dyscohesive malignant lymphocytes are seen in this kidney FNA in a 14-year-old girl with a known diagnosis of diffuse large B-cell lymphoma.

Metastatic synovial sarcoma in the kidney (Papanicolaou stain, medium power). The cellular aspirates from this metastasis show dyscohesive single cells and clusters of oval spindle cells, some with comma-shaped nuclei.
7.2 Adrenal Gland
There are two adrenal glands situated in the retroperitoneum in close proximity to each of the kidneys. The adrenal gland has a cortex and medulla. The cortex secretes glucocorticoids, mineralocorticoids, and androgens, while the medulla produces catecholamines. Adrenal disorders in children include congenital abnormalities, hyper- and hypoplasia, cysts, and inflammatory lesions and tumors, both benign and malignant. Adrenal “incidentaloma ” refers to adrenal masses over 1 cm in size detected, unexpectedly, on radiological imaging. These may be functional (i.e., secrete adrenocortical or medullary hormones) or nonfunctional [1, 4].
7.2.1 Congenital Adrenal Lesions
Congenital adrenal lesions include congenital adrenal hyperplasia, an autosomal recessive disorder, usually due to mutations on chromosome 6p21, in addition to adrenal cytomegaly, adrenal hypoplasia, and storage diseases. Diagnosis, including neonatal screening, is typically based on biochemical assay not FNA biopsy. Some disorders are also associated with adrenal abnormalities, such as enlargement, which may raise concern for malignancy. One such example is Beckwith–Wiedemann syndrome, which is sometimes associated with adrenal enlargement and demonstrates large pleomorphic cells with cytoplasmic nuclear inclusions.
7.2.2 Adrenal Cysts
Adrenal cysts are not often seen in children. Symptoms of adrenal cysts include abdominal pain and palpable mass but are often asymptomatic. Adrenal cysts are occasionally functional. Several types of adrenal cysts are described:
-
Endothelial or simple cysts: These are usually less than 2 cm in size and lined by flattened endothelial cells. Aspiration yields a clear or milky fluid.
-
Pseudocysts: These are unilocular cysts, the cyst lining being fibrous tissue without identifiable epithelial cells. These mainly occur in children following neonatal adrenal infarction but usually resolve spontaneously within a few months. Pseudocysts may also arise in adrenal neoplasms following necrosis and hemorrhage.
-
Epithelial cysts: These are lined by epithelium and may represent retention cysts, cystic adenomas, or embryonal cysts.
-
Parasitic cysts: These are usually due to echinococcal (hydatid) disease.
7.2.3 Adrenal Infections
Various infections of the adrenal gland have been described. These include viral (e.g., herpes family of viruses), bacterial (e.g., staphylococcal spp, mycobacterial), fungal (e.g., histoplasmosis, cryptococcosis), and parasitic (e.g., strongyloidiasis, echinococcosis, amebiasis). The cytomorphology of these infections, and host response, is similar to that found elsewhere in the body. The most frequently encountered infection of the adrenal gland in children is bacterial.
7.2.4 Adrenal Myelolipoma
Clinical Features
This benign neoplasm is usually noted in older adults but has, on occasion, been described in children [15]. These are benign and can be managed conservatively without surgery, unless symptomatic.
Cytological Features
FNA demonstrates mature adipose tissue together with hematopoietic elements that may include erythroid and myeloid precursor cells and megakaryocytes. Large neoplasms can exhibit necrosis, calcification, and hemorrhage.
Differential Diagnosis
The differential diagnosis includes angiomyolipoma (fat cells, smooth muscle, thick-walled vessels, no hematopoietic precursor cells, HMB45-positive epithelioid cells), extramedullary hematopoiesis (lack of adipose tissue), teratoma (mature and immature elements of cell types other than erythroid and myeloid precursor cells), and liposarcoma (lipoblasts, no hematopoietic precursor cells).
Pearls
Megakaryocytes can mimic multinucleated malignant cells or giant cells, leading to a misdiagnosis of malignancy or granulomatous inflammation, respectively.
7.2.5 Adrenal Cortical Neoplasms
Clinical Features
Adrenocortical neoplasms are rare in children, comprising approximately 0.2 % of all pediatric neoplasms. Adrenal cortical neoplasms are encountered more frequently in children with Beckwith–Wiedemann and Li–Fraumeni syndromes. Most occur in children under the age of 8 years with a female preponderance. Many adrenal cortical tumors in childhood are functional, the most common presentation being virilization. Less common manifestations are Cushing syndrome and feminization [1, 4].
Cytological Features
Adrenal cortical hyperplasia and adenoma show identical cytomorphology. However, hyperplasia is bilateral, while adenoma usually presents as a unilateral, solitary mass. The background can be clean or foamy, without necrosis (Fig. 7.12). FNA yields a cellular specimen comprising naked nuclei and cells with low N/C ratio and abundant vacuolated cytoplasm lying singly and in loosely cohesive clusters. The nuclei are round with regular nuclear outlines, finely granular chromatin, and inconspicuous nucleoli. Occasional large, pleomorphic nuclei (so-called endocrine atypia) may be found. Adrenal cortical carcinomas tend to be unilateral and large at time of presentation. Necrosis, hemorrhage and calcification are noted in the background. Arborizing blood vessels are described. Compared to adrenal cortical adenoma, adrenal cortical carcinoma demonstrates cells that have denser, rather than finely vacuolated, cytoplasm and a less frothy background. The nuclei exhibit somewhat more pleomorphism. Fewer naked nuclei are encountered. Cells are arranged singly and in loosely cohesive groups. The cells are cuboidal, polygonal, or plasmacytoid in shape with central to eccentrically situated nuclei and variable amounts of cytoplasm. Intranuclear cytoplasmic inclusions, irregular nuclear membranes, granular chromatin, and nucleoli and mitotic figures may all be seen.

Adrenal cortical hyperplasia (Diff-Quik stain, low power). The aspirates from adrenal cortex typically have this abundantly vacuolated background imparting a “foamy” appearance.
Triage
On immunoperoxidase staining, both adenomas and carcinoma are positive for inhibin, Melan A, calretinin, and vimentin. Synaptophysin is variably positive, while chromogranin is negative. Whorls of smooth endoplasmic reticulum are seen on electron microscopy.
Differential Diagnosis
See Table 7.3. Of note, prominent cytoplasmic vacuolization is typically seen with RCC and some Burkitt lymphomas; however, in adrenal lesions, the vacuoles are more often seen in the background.
Pearls
It may not be possible, on cytomorphology and immunostaining, to distinguish adrenocortical hyperplasia, adenoma, and carcinoma. The term “adrenal cortical lesion/neoplasm” should then be used. The presence of metastatic disease is indicative of a carcinoma.Chromogranin is more helpful than synaptophysin for distinguishing an adrenal cortical tumor from pheochromocytoma, as both adrenal cortex and pheochromocytoma can be synaptophysin positive, whereas adrenal cortex is usually negative for chromogranin.Proliferation index, Ki-67, is much higher in adrenal cortical carcinoma, as compared to adenoma.Loss of heterozygosity on chromosome 11p is seen in most pediatric adrenal cortical carcinomas.
7.2.6 Pheochromocytoma
Clinical Features
Pheochromocytomas are infrequent in children but are the most common pediatric endocrine neoplasms. They arise from the chromaffin cells of the adrenal medulla (intra-adrenal paraganglioma) or extra-adrenal sympathetic or parasympathetic paraganglia (extra-adrenal paraganglioma). Many tumors secrete catecholamines and their metabolites. Compared with those in adults, pheochromocytomas in children are more likely to be familial, extra-adrenal, bilateral, and multifocal. Pheochromocytomas are associated with multiple endocrine neoplasia (MEN) type 2, von Hippel–Lindau syndrome, neurofibromatosis type 1, and familial paraganglioma syndrome. The average age of presentation in children is 11 years, and there is a male preponderance. Some children with pheochromocytoma are asymptomatic, while others present with hypertension, palpitations, headache, excessive sweating, pallor, and anxiety [1, 4].
Cytological Features
Blood, necrosis, or cystic degeneration can be seen in the background. The cells are seen as single cells, loose clusters, syncytia, and occasional rosettes. They are round to spindled with variable amounts of granular cytoplasm. Red cytoplasmic granules may be seen on the Diff-Quik stain. The nuclei are central or eccentrically located with granular chromatin. Multinucleation, intranuclear inclusions, and mitotic figures may be seen. Occasional nuclear pleomorphism is described; however, even in neoplasms showing very mild nuclear pleomorphism, occasional larger hyperchromatic nuclei can be noted (“endocrine atypia”). Ganglion cells that are polygonal in shape with abundant cytoplasm and prominent nucleoli may be encountered (Fig. 7.13). Amyloid is sometimes seen.

Adrenal pheochromocytoma (a. Papanicolaou stain, medium power; b. Diff-Quik stain, high power; c. H&E stain, medium power; d. Synaptophysin immunoperoxidase stain, high power). FNAs of pheochromocytoma show loose clusters and single cells with abundant cytoplasm (a, b), recapitulating the histomorphology (c). Occasional larger nuclei are present. A solid arrangement of polygonal to spindle cells with abundant, eosinophilic cytoplasm can also be seen. Positive staining for synaptophysin within tumor cells is shown (d).
Triage
On immunoperoxidase stains, pheochromocytomas are positive for synaptophysin and chromogranin, while negative for inhibin, Melan A, and calretinin. There is variable vimentin staining. GATA3 is also positive in these tumors. Membrane-bound secretory granules are noted on electron microscopy. Biochemical tests include urinary and plasma catecholamines and metabolites.
Differential Diagnosis
Table 7.3 lists the differential diagnosis.
Pearls
FNA is contraindicated if a pheochromocytoma is suspected due to the risk of hypertensive crisis or hemorrhage. However, the complication rate is rare in those patients who have had an inadvertent FNA of a pheochromocytoma. The biologic behavior of a pheochromocytoma cannot be determined on cytomorphology.
7.2.7 Neuroblastic Tumors
Clinical Features
Neuroblastoma is the most frequently encountered extracranial, solid malignancy in childhood. It develops from neural crest cells in autonomic ganglia or paraganglia along the sympathetic chain, with 35 % arising in the adrenal medulla, 35 % in the extra-adrenal retroperitoneum, and 20 % in the posterior mediastinum. Other sites include cervical, paravertebral and pelvic areas. Given that the primary mass is rarely aspirated, the most common aspirates are from metastatic neuroblastoma, which commonly involves the skin, liver, bone marrow, or lymph nodes. Neuroblastoma is most often seen in children under the age of 4 years, and congenital neuroblastoma is well described. Neuroblastoma is associated with neurofibromatosis type 1, Hirschsprung disease, and Beckwith–Wiedemann syndrome. The prognosis varies greatly, depending on a number of factors including age, mitotic-karyorrhectic index (MKI), histopathologic classification (e.g., favorable vs unfavorable histology), stage of disease, MYCN status, and ploidy (Table 7.5). Presentation depends on the location of the tumor (e.g., mass lesion at primary tumor site), presence of secondary deposits (e.g., cutaneous metastases resulting in a “blueberry muffin” appearance, periorbital edema, and hemorrhage), secretion of catecholamines and metabolites (e.g., hypertension), and paraneoplastic phenomena (e.g., cerebellar ataxia). In congenital forms, the baby often presents as a “blueberry muffin” baby with the bluish-red cutaneous lesions and features of hydrops fetalis. A key clinical finding that can suggest a neuroblastic tumor is the presence of elevated urinary metabolites of catecholamines (vanillylmandelic acid, homovanillic acid) [1, 4, 16].
Cytological Features
These tumors yield cellular smears with small round blue cell morphology with rosette formation (Fig. 7.14). Homer Wright rosettes appear as tumor cells surrounding a small area of neuropil. The cells are small but slightly larger than a lymphocyte, have inconspicuous nucleoli, and have scant cytoplasm, which may cause them to have nuclear molding (Fig. 7.14). The background material may reveal variable amounts of neuropil that appears as granular or fibrillary extracellular material that stains pink on Papanicolaou stain. Necrosis and calcification may also be seen. Other features to look for are comma-shaped spindle cells with cytoplasmic processes to indicate Schwannian stroma or large epithelioid polygonal-shaped cells with prominent nucleoli to indicate a gangliocytic component (Fig. 7.15). In general, neuroblastic tumors form a spectrum ranging from tumors with neuroblasts and scant-to-absent Schwannian stroma (neuroblastoma) to tumors with neuroblasts, in addition to ganglion cells and Schwannian stroma (ganglioneuroblastoma), and tumors dominated by Schwannian stroma without neuroblasts (ganglioneuroma). Ganglioneuroma is a fully differentiated, benign, neuroblastic tumor that has Schwann and ganglion cells in a fibromyxoid or collagenous background without neuroblasts, necrosis, or mitotic figures (Fig. 7.15). Neuroblastomas can also be subclassified as undifferentiated, poorly differentiated, or differentiating.

Neuroblastoma (a, c. Papanicolaou stain, high power; b. Diff-Quik stain, high power; d. Synaptophysin immunoperoxidase stain, high power). This FNA from an adrenal neuroblastoma in a 15-month-old boy shows Homer Wright rosettes consisting of neuropil surrounded by tumor cells (a, b). Tumor cells are also seen in a background of granular, pink neuropil (c). Tumor cells show positive synaptophysin immunostaining (d).

Primary adrenal ganglioneuroma (a, b. Papanicolaou stain, high power). Large, polygonal ganglion cells with abundant granular cytoplasm are seen and should not be mistaken for malignancy (a). Spindled Schwann cells with ill-defined cytoplasmic borders and nuclei with inconspicuous nucleoli are also observed (b). No neuroblastic component is evident.
Triage
Neuroblastomas and ganglioneuroblastomas exhibit positive immunostaining for synaptophysin, chromogranin, and CD56. In addition, newer antibodies for NB84 and PGP9.5 are also positive in the neuroblastic cells. Neuroblasts tend to be negative for vimentin, desmin, myogenin, cytokeratin, WT1, CD45, and CD99. MYCN amplification can be demonstrated in some neuroblastomas by fluorescence in situ hybridization and is associated with a poorer prognosis. Neurosecretory granules are noted on electron microscopy. There are increased levels of catecholamines and metabolites on biochemical testing.
Differential Diagnosis
The differential diagnosis includes other small round blue cell tumors when the tumor has a predominance of neuroblastic component (Tables 7.2, 7.3, and 7.4).
Pearl
Neuroblastoma is divided into favorable or unfavorable histology based on the Shimada classification, which requires calculation of MKI. As defined in the Shimada classification, MKI cannot be calculated on cytologic specimens. Given that the Shimada classification is important for risk stratification and determination of therapy, a core or open biopsy is needed for appropriate management of these patients. Similarly, it may not be possible to distinguish between a ganglioneuroblastoma and neuroblastoma on cytomorphology alone, or to exclude a neuroblastic component in ganglioneuromas, so most of these lesions will be biopsied or excised for complete histological evaluation.
7.2.8 Metastases
The adrenal gland may be involved by contiguous spread from a renal, adrenal, or retroperitoneal tumor or metastases. Careful attention to cytomorphology and judicious use of ancillary investigations are required in this regard (Tables 7.2, 7.3 and 7.4).
7.3 Retroperitoneum
The retroperitoneum is a complex area that contains many vital organs , including kidneys, adrenal glands, ureters, bladder, duodenum (excluding first segment), colon (excluding transverse component, sigmoid, and cecum), pancreas (excluding tail), aorta, inferior vena cava, para-aortic lymph nodes, nerve trunks, ganglion plexuses, adipose tissue, and skeletal muscle. The retroperitoneum can be the site of diverse disease processes in children including cysts, infections, and neoplasms, both benign and malignant. The retroperitoneal space is relatively large, and symptoms of disease in this area often present at a late stage.
7.3.1 Cysts
Clinical Features
Retroperitoneal cysts in childhood can be neoplastic or nonneoplastic. Nonneoplastic cysts include pancreatic pseudocyst, lymphocele, urinoma, and hematoma. Hydatid cyst, tailgut cysts, and bronchogenic cysts have also been described [3]. Neoplastic cysts include cystic lymphangioma, cystic teratoma, Müllerian cyst, cystic changes in solid tumors (e.g., neuroblastoma), and benign multicystic peritoneal mesothelioma. Mesenteric, omental, and retroperitoneal cysts are typically endothelial- or mesothelial-lined cysts. They present in young children with abdominal mass, intestinal obstruction, volvulus, and abdominal pain. Benign multicystic peritoneal mesothelioma presents as multiple, non-adherent cysts with abdominal distention or as an incidental finding on laparotomy but is more common in young, reproductive-age women than in children [17].
Cytological Features
The fluid specimens usually show cyst contents with varying quantities of mature lymphocytes, histiocytes, and debris. Occasionally endothelial-lining cells or mesothelial cells will be seen. Mesothelial cells can show marked atypia in the setting of reactive conditions or benign multicystic mesothelioma. Cyst-lining cells are the most helpful for classification of these cystic lesions but are infrequently seen. In the case of bronchogenic cysts, ciliated cells are usually seen [3].
Triage
Liquid-based cytology is usually preferred for cystic lesions, particularly benign hypocellular fluid specimens , to concentrate the cells present.
Differential Diagnosis
The other entities to consider are cystic lesions arising from the ovary, kidney, or other solid organs in this region, that compress or distort other structures, and may be misinterpreted as a primary retroperitoneal mass.
Pearls
A variety of malignancies can undergo cystic degeneration, so ensuring adequate sampling of any solid components is essential in order to exclude malignancy. Given that ovarian and mesothelial-lining cells are positive for WT1, other stains, such as calretinin (for mesothelial origin) and ER or PAX8 (for ovarian origin), can be helpful.
7.3.2 Infections
Most retroperitoneal infections in children are bacterial, the most common infectious agent being Staphylococcus aureus. Other bacteria in this regard include E. coli and other gram-negative enteric bacteria, gastrointestinal anaerobes, Actinomyces, and mycobacteria. Paracoccidioides, Cryptococcus, and Histoplasma are causes of retroperitoneal fungal infections and abscesses, while parasitic sources of infection include hydatid disease. These infections may show a granulomatous or histiocytic response. One example would be xanthogranulomatous pyelonephritis from the kidney showing prominent foamy histiocytes and debris. Viruses may cause para-aortic lymphadenopathy, and the corresponding aspirates will typically show reactive lymphadenopathy with a shift to more immunoblastic cells and occasionally features of viral cytopathic effect. In immunosuppressed children (e.g., transplant, HIV/AIDS), unusual infectious agents may be encountered. The cytomorphology will depend on the type of infection and host response. Ancillary tests (e.g., special stains, PCR, culture) can aid in accurate identification of the causative organism.
7.3.3 Lesions and Tumors of the Retroperitoneum with a Predominant Small Round Cell Pattern
Tumors located in the retroperitoneum that consist predominantly of small round cells are highlighted in Table 7.2 and include extra-adrenal neuroblastoma and ganglioneuroblastoma, desmoplastic small round cell tumor, rhabdomyosarcoma, Ewing sarcoma/PNET, and lymphoid lesions, including reactive lymphadenopathy and lymphoma. It is estimated that 35 % of neuroblastomas and ganglioneuroblastomas occur in the retroperitoneum. Rhabdomyosarcoma can arise in the retroperitoneum. In addition, paratesticular or bladder rhabdomyosarcoma can invade the retroperitoneum. Retroperitoneal and para-aortic lymph nodes may exhibit hyperplasia due to various stimuli, in addition to involvement by lymphoma. Burkitt lymphoma not infrequently involves the mesenteric and retroperitoneal lymph nodes.
7.3.3.1 Desmoplastic Small Round Cell Tumor
Clinical Features
Desmoplastic small round cell tumor (DSRCT) is a rare but aggressive tumor, usually located intra-abdominally and in the pelvis, but it can also be found in the retroperitoneum, thorax, and central nervous system. It usually affects adolescent and young adult males.
Cytological Features
Aspirates from DSRCTs yield small round cells with varying amounts of stromal fragments that stain metachromatically on Diff-Quik preparations. The cells can also be signet-ring type, rhabdoid, epithelioid, or spindled (Figs. 7.16 and 7.17). The background typically shows necrosis, karyorrhexis, and cystic debris. This tumor is described in more detail in Chap. 9 on body fluid cytopathology. On histology, small round cells organized in nests and solid areas are found in a desmoplastic stroma, while on aspirates, the small round blue cell component is more pronounced than the fibrotic component [18].

Desmoplastic small round cell tumor (a. Papanicolaou stain, medium power; b. Diff-Quik stain, medium power). These aspirates from a retroperitoneal DSRCT show small round cells with very high nuclear-to-cytoplasmic ratios in a necrotic background (a). Small round cells with some attempt at rosette formation are noted (b).

Desmoplastic small round cell tumor in a 7-year-old boy (a. Papanicolaou stain, high power; b. AE1/AE3 stain, high power; c. Desmin stain, high power). Small fragments of collagenous stroma can be noted within a small round blue cell tumor (a). The polyphenotypic nature of this tumor can be demonstrated by both positive AE1/AE3 and desmin staining (b, c).
Triage
Immunoperoxidase stains are helpful to demonstrate co-expression of epithelial markers (EMA, cytokeratin), neural markers (S100, NSE), and mesenchymal markers (vimentin and dot-like perinuclear staining for desmin). The WT1 stain using an antibody directed to the carboxy-terminus also shows nuclear positivity. This tumor demonstrates a translocation t(11;22)(p13;q12) with fusion of the EWSR and WT1 genes.
Differential Diagnosis
See Table 7.2. One of the most difficult tumors to distinguish from DSRCT is blastemal predominant Wilms tumor, given that both tumors show poorly differentiated morphology with immunopositivity for desmin and cytokeratin. Even the dot-like positivity for desmin can be seen in both tumors. Thus, the best way for distinguishing DSRCTs is to demonstrate that the tumor cells have the EWSR1-WT1 rearrangement and show selective immunoreactivity to the WT1 carboxy-terminus. In contrast, Wilms tumors are typically positive with antibodies directed to both the WT1 amino-terminus and carboxy-terminus [19].
Pearls
A variety of tumors in addition to DSRCTs have EWS gene rearrangements that can be detected by FISH using EWS break-apart probes. Thus, morphology and immunophenotype and/or identification of the translocation partner by FISH, RT-PCR, or other molecular methods is necessary for a definitive diagnosis.
7.3.3.2 Ewing Sarcoma/Primitive Neuroectodermal Tumors (EWS/PNET)
Clinical Features
This is a small round blue cell tumor originating in soft tissue, including retroperitoneum, and bone. Despite differences in morphology and immunophenotype which reflect the degree of neural differentiation, EWS and PNET share cytogenetic features and, thus, are considered a single entity. This tumor is seen most frequently in older children and adolescents. The most common presentation is that of a painful mass. Ewing sarcoma typically occurs in long bones, while the soft tissue PNET is usually located in the chest wall, paraspinal region, and head and neck. Very occasionally a PNET can arise in an organ (e.g., kidney, lung) [1, 4, 20].
Cytological Features
The background comprises variable amounts of blood, necrosis, apoptosis, and mitotic figures. A tigroid background, due to intracellular glycogen, is a feature in some tumors. Cells are arranged in sheets, loose and tight clusters, single cells, and occasional rosettes. The cells usually have a high nuclear-to-cytoplasmic ratio with a small amount of cytoplasm. The cytoplasm contains glycogen vacuoles, which are best seen on Diff-Quik-stained material. Occasionally, somewhat larger cells with more abundant cytoplasm are seen, imparting a light (cells with increased cytoplasm) and dark (cells with less cytoplasm or degenerating cells) pattern on aspirates. The nuclei are round to oval with mildly irregular borders and finely granular chromatin. Nucleoli may be inconspicuous, single, or multiple. Nuclear molding may be seen. Neural differentiation, in the form of fibrillary neuropil and rosettes, can be seen in varying quantities. This tumor is discussed in greater detail in Chap. 5 with images.
Triage
Immunocytochemistry is positive for CD99/O13 and FLI-1. Neuroendocrine and broad spectrum epithelial markers are variably positive. Muscle and lymphoid markers are negative. Glycogen can be demonstrated using PAS stains with and without diastase. Several cytogenetic aberrations involving the EWS gene on chromosome 22q12 are described in EWS/PNET, the most common being t(11;22)(q24;q12). Others include t(21;22)(q12q12), t(7;22)(p22;q12), t(17;22)(q12;q12), and t(2;22)(q33;q12).
Differential Diagnosis
See Table 7.2. One of the main entities in the differential diagnosis is undifferentiated (Ewing-like) round cell sarcoma, which typically has cells with paler cytoplasm than in EWS/PNET, more atypia, shows a CIC-DUX4 rearrangement t(4;19), and is WT1 positive.
Pearls
Other tumors that show the EWS (chromosome 22) gene rearrangement include clear cell sarcoma, desmoplastic small round cell tumor, extraskeletal myxoid chondrosarcoma, myoepithelial tumors, and angiomatoid fibrous histiocytoma. Thus, morphology and immunophenotype and/or identification of the translocation partner by FISH, RT-PCR, or other molecular methods is necessary for a definitive diagnosis.
7.3.4 Lesions and Tumors of the Retroperitoneum with a Predominant Spindle Cell Pattern
Various spindle cell soft tissue tumors, including but not limited to those highlighted in Table 7.4, can involve the retroperitoneum in childhood. These include muscle tumors [e.g., rhabdomyosarcoma (Fig. 7.18), leiomyoma, and leiomyosarcoma (Fig. 7.19)], fibrotic lesions (e.g., fibrosarcoma), vascular lesions (e.g., Kaposi sarcoma), neural lesions (e.g., schwannoma, ganglioneuroma), and synovial sarcoma. Soft tissue tumors are described in greater detail in Chap. 5.

Primary retroperitoneal alveolar rhabdomyosarcoma (a, c. Papanicolaou stain, high power; b. Diff-Quik stain, high power; d. H&E stain, medium power). The aspirates show features of a small round cell tumor (a, b), with some cells containing more abundant, dense, orangeophilic cytoplasm (c). The corresponding histologic section is shown (d).

Retroperitoneal smooth muscle tumor in an HIV-positive child (a. Papanicolaou stain, high power; b. H&E stain, medium power). The aspirates show spindle cells with ill-defined cell borders, blunt-ended nuclei, irregular nuclear borders, and prominent nucleoli (a). Fascicles of spindle cells are also observed in the resection specimen (b).
7.3.4.1 Fibroblastic-Myofibroblastic Lesions
Lesions of this type seen in the pediatric retroperitoneum include inflammatory myofibroblastic tumor, solitary fibrous tumor, and fibrosarcoma. Inflammatory myofibroblastic tumor comprises spindle and inflammatory cells, including plasma cells and eosinophils. These tumors demonstrate chromosomal rearrangements of the 2p23 locus. Solitary fibrous tumor presents with uniform spindle cells which show variable CD34 and vimentin positivity. Fibrosarcoma (infantile and adult-type) can be seen in the retroperitoneum. Infantile fibrosarcoma comprises small or large spindle cells that may demonstrate marked pleomorphism in a hemorrhagic or necrotic background. The majority exhibit translocation t(12;15)(p13;q26). The adult type of fibrosarcoma may occasionally be seen in older children and adolescents. This comprises spindle cells in a background of variable amounts of collagen.
7.3.5 Lesions and Tumors of the Retroperitoneum Comprising Cells with Moderate to Abundant Cytoplasm
Angiomyolipoma, PEComa, germ cell tumors, chordoma, and ganglioneuroblastoma may all occur in the retroperitoneum. These lesions consist of cells with fairly abundant cytoplasm. Most germ cell tumors found in the retroperitoneum are metastases, and it is currently controversial whether the entity of primary extragonadal germ cell tumors of the retroperitoneum exists. PEComas arise from the perivascular epithelioid cell and express both muscle and melanocytic markers (Fig. 7.20). There are several tumors that form part of the PEComa family of tumors including angiomyolipoma, sugar tumor, and lymphangioleiomyomatosis. Chordoma and parachordoma are very uncommon in children. Cytologically, physaliferous cells are present in a background of myxoid material. Physaliferous cells have abundant vacuolated cytoplasm and pleomorphic nuclei that can show scalloped nuclear contours. On immunoperoxidase stains, these tumors are positive for S100, EMA, and brachyury. Table 7.3 highlights some of the differential diagnostic considerations.

PEComa (a. Diff-Quik stain, medium power; b. Papanicolaou stain, high power). FNA from a renal melanotic PEComa shows cells with abundant vacuolated cytoplasm containing melanin pigment.
7.3.6 Metastases to the Retroperitoneum
Many tumors of childhood can spread to the retroperitoneum, either directly or metastatic, from a more distant site. A thorough clinical history and physical examination, in addition to careful attention to cytomorphology and the results of ancillary studies, is required to make the correct diagnosis.
7.4 Urine Cytology
7.4.1 Urine Collection
Toilet-trained children, depending on age, may be able to provide a urine sample. To collect a urine sample in younger children, the use of urine bags, disposable diapers, urine collection pads, catheterization, or suprapubic aspiration is required. The collection method also depends on the type of test to be performed (e.g., a urine sample for culture and sensitivity should be collected in a way that minimizes contamination). Cleaning the perineum and genital area before collecting urine may help reduce contamination.
7.4.2 Urinary Inflammation
Bacterial infection is the most common infection seen in urine. However, viral, fungal, and parasitic diseases may cause cystitis. In addition, medications, especially chemotherapeutic agents, can cause cystitis, including hemorrhagic cystitis.
-
Viral infection: Cytomegalovirus (CMV) and BK virus (a polyomavirus) are the most common viral infections seen in urine cytology, especially in immunosuppressed children. The cytopathic effect of CMV includes a large cell with a prominent eosinophilic intranuclear inclusion and small intracytoplasmic inclusions. BK virus infection can cause renal transplant rejection. The cytomorphology of BK viral cytopathic effect includes urothelial cells with eccentrically situated nuclei containing a large basophilic intranuclear inclusion that imparts a “smudgy” look to the nucleus. The cells are usually seen as single cells (Fig. 7.21). The diagnosis of BK viruses can be confirmed on PCR, electron microscopy, immunostaining for the SV40 antibody, and tissue biopsy. Herpes simplex infection can be seen in the form of multinucleated cells with nuclear molding and peripheral condensation of chromatin (Fig. 7.21).
Fig. 7.21 
Urine cytology in children (a–d. Papanicolaou stain, high power). Decoy cells due to polyomavirus in the urine from a 17-year-old girl who had undergone renal transplant (a, b). Multinucleated cells with nuclear molding and margination of chromatin due to herpes simplex virus infection in urine from a 19-year-old girl (c). Schistosoma haematobium ovum in urine from a 9-year-old boy (d).
-
Bacterial infection: In children, there may be an association between bacterial infection of the urinary tract, vesicoureteral reflux, and renal scarring. The causative bacteria are most often gram-negative bacteria, especially Escherichia coli. Staphylococcal spp. are associated with bladder instrumentation. Cytologic evaluation reveals a marked acute inflammatory cell infiltrate, red blood cells, reactive urothelial cells, and debris. Bacterial colonies may be noted. Gram stain and culture are ancillary investigations that can be performed.
-
Fungal infection: Fungal cystitis is unusual in children and is usually associated with immunosuppression or bladder instrumentation. Candida spp., Cryptococcus, and Aspergillus are some examples of causative agents. Fungal casts may be identified, in addition to inflammatory cells, blood, and urothelial cells. Special stains (e.g., GMS, PAS) and fungal culture will aid in identification of the fungus.
-
Parasitic infection: Schistosomiasis, especially S. haematobium, is the most important cause of parasitic infection of the bladder. Schistosoma ovum is large and oval in shape, measuring 150 × 50 μm surrounded by a chitinous shell with a terminal spine, as in the case of S. haematobium (Fig. 7.21). Trichomonas vaginalis causes cystitis and urethritis. In urine, they often appear more degenerate and smaller than on cervical cytology, in a background of marked inflammation. Their presence in urine may indicate onset of sexual activity in adolescents or sexual abuse.

7.4.3 Miscellaneous Conditions
Xanthogranulomatous pyelonephritis has been described in children. Urine cytomorphology reveals nonspecific inflammation or blood. Urine culture is usually negative. Lipid storage disorders (e.g., Niemann–Pick disease) have been diagnosed on urine cytology in the presence of cells with numerous small vacuoles, combined with special stains (e.g., PAS, Sudan stains). A high proportion of columnar cells are seen in children with cystitis cystica et glandularis.
7.4.4 Neoplasia
The most common childhood tumor to exfoliate into urine is the rhabdomyosarcoma, especially the embryonal and botryoides subtypes. Presentation includes hematuria, abdominal pain, and recurrent urinary tract infection. Cytologic examination of urine is associated with a low diagnostic yield of tumor cells. When present, the tumor cells exfoliate singly and in small clusters. The cells are often degenerate and smaller than seen on FNA, with high nuclear-to-cytoplasmic ratios. More differentiated cells such as strap cells or cells with cross striations have, very occasionally, been reported in urine. Immunoperoxidase stains such as desmin, myoD1, and myogenin are usually positive. The alveolar subtype will show translocation t(2; 13)(q35;q14) or t(1;13)(p36;q14). Other small round cell tumors that have been described on urine cytology include nephroblastoma, non-Hodgkin lymphoma, and EWS/PNET. Other neoplasms, like inflammatory myofibroblastic tumors, can also occur.
References
Husain AN, Stocker JT, editors. Color atlas of pediatric pathology. New York, NY: Demos Medical Publishing; 2011.
Kini S. Color atlas of differential diagnosis in exfoliation and aspiration cytopathology. 2nd ed. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins; 2011.
Yang D, Jung D, Kim H, Kang J, Kim S, Kim J, Hwang H. Retroperitoneal cystic masses: CT, clinical and pathologic findings and literature review. Radiographics. 2004;24:1353–65.
Stocker J, Dehner L, editors. Pediatric pathology. 2nd ed. Philadelphia, PA: Lippincott, Williams and Wilkins; 2001.
Khalbuss W, Monaco S, Pantanowitz L. Quick compendium of cytopathology. Chicago: ASCP Press; 2013.
Shohab D, Hussain I, Khawaia A, Jamil I, Raia NU, Ahmed F, Akhter S. Primary renal aspergillosis and xanthogranulomatous pyelonephritis in an immune-competent toddler. J Coll Physicians Surg Pak. 2014;24:S101–3.
Silowash R, Monaco SE, Pantanowitz L. Ancillary techniques on direct-smear aspirate slides: a significant evolution for cytopathology techniques. Cancer Cytopathol. 2013;121:670.
Iyer V, Agarwala S, Verma K. Fine needle aspiration cytology of clear cell sarcoma of the kidney: study of eight cases. Diagn Cytopathol. 2005;33:83–9.
Portugal R, Barroca H. Clear cell sarcoma, cellular mesoblastic nephroma and metanephric adenoma: cytological features and differential diagnosis with Wilms tumor. Cytopathology. 2008;19:80–5.
Brownlee NA, Perkins LA, Stewart W, Jackle B, Pettenati MJ, Koty PP, Iskandar SS, Garvin AJ. Recurring translocation (10;17) and deletion (14q) in clear cell sarcoma of the kidney. Arch Pathol Lab Med. 2007;131:446–51.
Sharifah N. Fine needle aspiration cytology characteristics of renal tumors in children. Pathology. 1994;26:359–64.
Cheng JX, Tretiakova M, Gong C, Mandal S, Krausz T, Taxy JB. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol. 2008;21:647–52.
Schinstine M, Filie A, Torres-Cabala C, et al. Fine needle aspiration of Xp11.2 translocation/TFE3 fusion renal cell carcinoma metastatic to the lung: report of a case and review of the literature. Diagn Cytopathol. 2006;34:751–6.
Ramphal R, Pappo A, Zielenska M, Grant R, Ngan BY. Pediatric renal cell carcinoma: clinical, pathologic, and molecular abnormalities associated with the members of the mit transcription factor family. Am J Clin Pathol. 2006;126:349–64.
Barman S, Mandal KC, Mukhopadhyay M. Adrenal myelolipoma: an incidental and rare benign tumor in children. J Indian Assoc Pediatr Surg. 2014;19:236–8.
Davidoff A. Neuroblastoma. Semin Pediatr Surg. 2012;21:2–14.
Durell J, Dagash J, Eradi B, Nour S. Pediatric benign cystic mesothelioma. J Pediatr Adolesc Gynecol. 2016;29:e33–4.
Crapazano J, Cardillo M, Lin O, Zakowski M. Cytology of desmoplastic small round cell tumor. Cancer. 2002;96:21–31.
Arnold MA, Schoenfield L, Limketkai BN, Arnold CA. Diagnostic pitfalls of differentiating desmoplastic small round cell tumor (DSRCT) from Wilms tumor (WT): overlapping morphologic and immunohistochemical features. Am J Surg Pathol. 2014;38:1220–6.
Tsokos M, Alaggio R, Dehner L, Dickman P. Ewing sarcoma/peripheral primitive neuroectodermal tumor and related tumors. Pediatr Dev Pathol. 2012;15(1 Suppl):108–26.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Michelow, P., Dubb, M. (2017). Kidney, Adrenal Gland, and Retroperitoneum. In: Monaco, S., Teot, L. (eds) Pediatric Cytopathology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-53441-0_7
Download citation
DOI: https://doi.org/10.1007/978-3-662-53441-0_7
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-53439-7
Online ISBN: 978-3-662-53441-0
eBook Packages: MedicineMedicine (R0)