
Abstract
Chronic pain, both inflammatory and neuropathic, is a debilitating condition in which the pain experience persists after the painful stimulus has resolved. The efficacy of current treatment strategies using opioids, NSAIDS and anticonvulsants is limited by the extensive side effects observed in patients, underlining the necessity for novel therapeutic targets. Preclinical models of chronic pain have recently provided evidence for a critical role played by glial cells in the mechanisms underlying the chronicity of pain, both at the site of damage in the periphery and in the dorsal horn of the spinal cord. Here microglia and astrocytes respond to the increased input from the periphery and change morphology, increase in number and release pro-nociceptive mediators such as ATP, cytokines and chemokines. These gliotransmitters can sensitise neurons by activation of their cognate receptors thereby contributing to central sensitization which is fundamental for the generation of allodynia, hyperalgesia and spontaneous pain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Glia
- Microglia
- Astrocytes
- Neuropathic pain
- Inflammatory pain
- Spinal cord
- CX3CL1/R1
- IL-1β
- TNF
- Rheumatoid arthritis
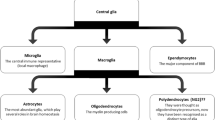
1 Origin and Function of Glia
The term neuroglia was coined to describe the interstitial substance surrounding neurons of the CNS (central nervous system) and was later determined to consist of distinct neuroglial cells. The name astrocyte was introduced to describe ‘star-shaped’ neuroglial cells which were observed to form the supportive system of the CNS. A third cellular element of the CNS was acknowledged in 1913 when improved staining techniques for neuroglial cells led to the recognition of non-neuronal cells that were distinct from astrocytes. During the 1920s del Rio-Hortega employed silver carbon staining and light microscopy to visualise a population of cells that appeared different from the macroglia (oligodendrocytes and astrocytes) that had previously been described in the CNS. These cells, termed microglia, were thought to arise either from CNS invasion of blood mononuclear (monocytic) cells or mesodermal pial elements (Del Rio-Hortega 1932, 2012a, b; Kettenmann et al. 2011; Prinz and Mildner 2011). Whilst alternatives for the origin of microglia have been hypothesised, evidence to support the hypothesis that these cells are of monocytic lineage was later strengthened using autoradiography. Leblond and colleagues demonstrated that microglia possess specific monocytic characteristics, specifically the ability to transform from an amoeboid to a ramified cell (Imamoto and Leblond 1978; Ling et al. 1980). The monocytic/myeloid origins of microglia has now been confirmed conclusively by the absence of microglia from the CNS of a genetically altered mouse strain that lack PU.1, a key transcription factor in the control of myeloid cell differentiation (McKercher et al. 1996; Beers et al. 2006).
Microglia appear at an early stage during embryogenesis where they originate from macrophages in the foetal yolk sac that migrate into the CNS (Saijo and Glass 2011); evidence from postnatal fluorescently labelled cells demonstrates that in this phase of development, microglia are capable of differentiating from monocytes entering the CNS from the circulation (Perry et al. 1985). Conversely, in adult rodents, circulating monocytes only enter the CNS under conditions where there is disruption to the blood-brain barrier (BBB) (King et al. 2009); however, as demonstrated by the use of immune-irradiated mice, the CNS colonisation by these cells is transient and does not contribute to the resident microglial cell pool (Ajami et al. 2011). Rather the microglial population is maintained locally and independently of circulating monocytes via the proliferation of existing cells (Lawson et al. 1992; Ginhoux et al. 2010).
Microglia are specialised phagocytes of the CNS and constitute 5–20 % of the total glial cell population (Saijo and Glass 2011). Under physiological conditions microglia exist in a ‘resting’ or ‘quiescent’ state; these cells can be distinguished morphologically by their small soma and ramified process that perform immune surveillance of the surrounding area. Additionally they express receptors for complement components (Fcγ receptor of IgG) and exhibit low expression of cell surface antigens (Nimmerjahn et al. 2005). Several mechanisms by which these cells maintain a quiescent state have been proposed, including interaction of microglial CX3CR1 receptor with its neuronal ligand the chemokine CX3CL1 and inhibitory signalling through the microglial cell surface proteins CD172, CD200R and CD45 with their neuronal ligands CD47, CD200 and CD22, respectively (Ransohoff and Perry 2009; Ransohoff and Cardona 2010; Cardona et al. 2006; Saijo and Glass 2011).
Within the healthy CNS, microglia have a number of key roles in addition to their function as immune surveyors. As well as monitoring extrasynaptic regions, microglial processes transiently contact synapses, including presynaptic terminals and perisynaptic clefts (Tremblay et al. 2010). Additionally these cells possess a number of neurotransmitter receptors; combined these attributes allow microglia to monitor synaptic function (Salter and Beggs 2014). Microglial cells are present in the CNS from the early stages of development and due to their phagocytic capacity are able to contribute to the elimination of excess neurons that form as part of normal development (Marin-Teva et al. 2011). However, rather than simply removing waste, microglia can initiate apoptosis of cells via the release of several factors including superoxide ions and TNF (Marin-Teva et al. 2004; Sedel et al. 2004; Salter and Beggs 2014). Additionally, a number of inappropriate synaptic connections are made between neurons during development, and microglia play an important role in the regulation of these contacts via the process of synaptic pruning—the phagocytosis of both pre- and postsynaptic elements in a complement cascade-dependent manner (Salter and Beggs 2014; Stevens et al. 2007; Schafer et al. 2012). Microglia contribute to the maturation of established synapses; the functional properties of synapses develop abnormally in mice deficient in several microglial proteins, including CX3CR1, for example, the absence of which results in increased excitatory neurotransmission (Paolicelli et al. 2011; Salter and Beggs 2014). The involvement of microglia to the maintenance of synaptic plasticity has also been investigated in the adult CNS, where they contribute to both homeostatic and activity-triggered plasticity by releasing a number of mediators such as TNF and TGFβ (Butovsky et al. 2014; Koeglsperger et al. 2013).
Of all of the glial cell populations within the CNS, astrocytes are by far the most abundant. In mammals astrocytogenesis begins in late embryogenesis and continues into the postnatal phase. The origin of astrocytes is likely diverse, varying throughout the stages of development. For example, within the cerebral cortex, astrocytes develop from two distinct sources: from radial glia in the ventricular zone during embryogenesis and from subventricular zone (SVZ) progenitors during the postnatal period (Wang and Bordey 2008). Additionally, studies using in vivo retroviral gene transfer have demonstrated that SVZ progenitors can generate both grey and white matter astrocytes as well as oligodendrocytes (Levison and Goldman 1993, 1997; Levison et al. 1993).
The typical image of an astrocyte is that of a stellate cell; however, astrocytes have a complex and heterogeneous morphology (Wang and Bordey 2008). Whilst the nomenclature is considered outdated by some, astrocytes are typically classified into one of two subtypes, protoplasmic and fibrous, where the former are found throughout the grey matter and possess branches that give rise to uniformly distributed processes and the latter are distributed within the white matter and exhibit long fibrelike processes (Sofroniew and Vinters 2010). Furthermore, the branching processes of protoplasmic astrocytes envelop synapses and have endfeet that encase blood vessels, whilst the endfeet of the unbranched processes of fibrous astrocytes envelop nodes of Ranvier (Wang and Bordey 2008). Immunohistochemically astrocytes are commonly identified by the presence of glial fibrils, the major component of which is glial fibrillary acidic protein (GFAP). Antibodies against S100B, a member of the S100 family of EF-band calcium binding proteins, are also used to identify astrocytes; however, this antigen is only expressed by a subset of mature astrocytes. Thus, its expression is not representative of the astrocyte population as a whole (Baudier et al. 1986; Deloulme et al. 2004; Hachem et al. 2005).
Due to the absence of several biophysical properties such as the ability to generate action potentials under physiological conditions, astrocytes were once described as passive (Steinhauser et al. 1992); however, this term is now rarely used. Whilst the initial function of astrocytes was understood to be as little more than an inert scaffold to neurons, it is now widely accepted that they play a critical role in a vast number of processes within the CNS (Volterra and Meldolesi 2005). The close association of astrocytes with many neuronal elements allows astrocytes to regulate the extraneuronal environment around synapses and provide support and nourishment to these cells (Gao and Ji 2010), for example, by buffering extracellular potassium and glutamate. Additionally astrocytes have been demonstrated to significantly contribute to neuronal survival and maturation (via the synthesis and release of growth and trophic factors such as NGF), synapse formation and the regulation of angiogenesis and are a major source of adhesion and extracellular matrix proteins within the CNS that can both promote and inhibit neurite growth (Wang and Bordey 2008).
2 Acute and Chronic Pain
Pain is a subjective sensory experience associated with actual or potential tissue damage (www.iasp.org). It is always unpleasant and therefore an emotional experience. Acute pain is mediated by multi-synaptic pathways beginning with specialised primary afferent fibres (nociceptors) in the periphery, whose cell bodies lie in the dorsal root ganglia (DRG), via the dorsal horn of the spinal cord to supraspinal sites such as the parabrachial area (PBA), periaqueductal grey (PAG), thalamus and cortex. The nociceptors convert the energy of noxious stimuli into electrical impulses which are transmitted to the dorsal horn of the spinal cord. Within the spinal cord, nociceptive transmission is mediated predominately by glutamate acting upon postsynaptic ionotropic receptors; however, the co-release of substance P and calcitonin gene-related peptide (CGRP) and the subsequent activation of their respective NK1 and CGRP receptors postsynaptically modulates glutamatergic transmission (Go and Yaksh 1987; Cheunsuang and Morris 2000; Malcangio and Bowery 1994; Lever et al. 2003; Oku et al. 1987; Seybold et al. 2003). Modulatory control of acute pain is provided by descending inhibitory and facilitatory pathways from the brain and from inhibitory and excitatory interneurons within the spinal cord (Todd 2010; D’Mello and Dickenson 2008).
Acute pain is a vital protective mechanism that persists only for the duration of tissue damage or the presence of a noxious stimulus. Evidence for the importance of acute pain is provided by individuals expressing the rare phenotype of insensitivity to pain; these patients experience regular inadvertent self-mutilation through injury and have a significantly lower than average life expectancy (Cox et al. 2006; Verpoorten et al. 2006).
Commonly pain outlives its usefulness and becomes chronic pain, which is profoundly different from acute pain, as it is the result of plastic changes within the pain pathways. Chronic pain is a debilitating condition lasting longer than 3 months from the noxious stimuli and causes pain to be perceived as out of proportion to the initial inciting injury. Chronic pain can be classified into different types depending on its cause: inflammatory pain is commonly due to tissue inflammation as observed in arthritis, and neuropathic pain arises upon injury to the nervous system.
2.1 Inflammatory and Neuropathic Pain
In both cases a combination of mechanisms results in augmented nociceptive transmission due to peripheral and central sensitisation. Neuropathic pain is that arising from a lesion to the PNS (peripheral nervous system) or CNS as a consequence of physical trauma or disease pathogenesis. Chronic inflammatory pain is typically associated with inflammatory diseases such as arthritis, where the presence of algogenic mediators results in nociceptor sensitisation in the affected tissue, such as the joints in the case of arthritis. Chronic pain is characterised by the presence of a range of symptoms including hyperalgesia (increased sensitivity to noxious stimuli), allodnyia (a painful response to a previously innocuous stimuli) and spontaneous pain (Woolf and Mannion 1999). The treatment of chronic pain is a substantial problem within the clinic as many patients do not respond adequately to available analgesics. The difficulty in treating chronic pain conditions is thought to be a consequence of the heterogeneity of the molecular mechanisms underlying the development and maintenance of chronic pain, many of which are not well understood.
The mechanisms by which these maladaptive pain states arise can be categorised into peripheral sensitisation (changes in peripheral nerves) and central sensitisation (including immune responses in the spinal cord). Peripheral sensitisation due to injury to a localised area leads to primary hyperalgesia and results in increased nociceptive transmission from the periphery which causes changes in dorsal horn neuron activation. Dorsal horn neurons exhibit reduced thresholds to noxious stimuli applied to the periphery, de novo excitation by previously innocuous stimuli, expansion of their receptive fields and increased spontaneous activity. These changes increase excitability of CNS neurons and result in central sensitisation, which is fundamental for the generation of allodynia, secondary hyperalgesia and spontaneous pain (Kuner 2010; Latremoliere and Woolf 2009; Sandkuhler 2009). The resulting increase in glutamate released from central terminals of sensitised neurons trigger phosphorylation of NMDA receptors and post-translational modifications of neuronal proteins. Glial cells within the spinal cord respond to the enhanced nociceptive input by proliferating and switching to a responsive state whereby they release mediators which contribute to dorsal horn mechanisms of chronic pain (Clark and Malcangio 2012; Old and Malcangio 2012). Enhanced activity within the spinal cord triggers cortical and subcortical structures to facilitate excitation and signalling and constitutes higher centre modulation (D’Mello and Dickenson 2008).
Here we will describe and define some of the key mechanisms by which microglia and astrocyte responses in the dorsal horn contribute to the development and maintenance of chronic neuropathic and inflammatory pain states, with a specific focus on chemokine/cytokine signalling and second messenger activation.
3 Spinal Glia Changes in Models of Neuropathic Pain
3.1 Microglial Responses to Injury or Insult
Microglia are the resident immune cells of the CNS and as such respond to pathological insult or tissue injury and subsequent release of mediators from damaged cells. These cells respond to a number of mediators that are increased in the dorsal horn following peripheral injury. They express receptors for the neurotransmitters released from the central terminals of primary afferents: glutamate NMDA and AMPA receptors, the SP receptor NK1 and the CGRP receptor, TrkB receptors for brain derived neurotrophic factor (BDNF) and purinergic receptors including P2X7 and P2X4, many of which are upregulated following injury to the periphery (Rasley et al. 2002; Pezet et al. 2002; McMahon and Malcangio 2009; Ransohoff and Perry 2009).
Microglia respond quickly to an insult by proliferating to expand their population; this process is often termed microgliosis. An insult need not be central in nature, for example it could be the result of a CNS infection or spinal cord injury. In rodent peripheral nerve injury models of neuropathic pain, microglia within the dorsal horn of the spinal cord respond swiftly to augmented primary afferent fibre input. This phenotypic shift is often referred to as the ‘activation’ of microglia. Input from the periphery is vital for this process; blockade of peripheral nerve conduction by the application of a local anaesthetic prevents nerve injury-induced microglial response (Wen et al. 2007; Hathway et al. 2009; Suter et al. 2009). In the region of the dorsal horn in which the injured primary afferents terminate, microglia increase in their numbers and appear more amoeboid, with a hypertrophied soma and thick retracted processes. Additionally they alter their expression of cell surface antigens that play a critical role in immune responses; for example, MHC class II is increased allowing the presentation of antigens by microglia (McMahon and Malcangio 2009). Activated microglia also synthesise and release a variety of pro-nociceptive mediators into the extracellular environment, including cytokines (e.g. TNF and IL-1β), chemokines (e.g. CCL2), reactive oxygen species (ROS) and nitric oxide (NO). Prominent signalling pathways in the development of neuropathic pain are the CX3CR1/L1 loop (discussed below) and the purinergic pathway of microglial activation and subsequent BDNF release. ATP is known to stimulate microglia both in vivo and in vitro via purinergic receptors on the surface membrane of the cells (Honda et al. 2001; Tsuda et al. 2003, 2012; Davalos et al. 2005). Microglial P2X4 is of particular importance for the development of neuropathic pain; it is upregulated in microglia as early as 24 hours after peripheral nerve injury, and the pharmacological inhibition of this receptor transiently attenuates mechanical allodynia in rodents. Furthermore, the intrathecal administration of ATP-treated microglia in naive mice results in the development of mechanical hypersensitivity (Tsuda et al. 2003; Coull et al. 2005). Current evidence indicated that downstream effects of the activation of microglial purinergic receptors are mediated by the release of BDNF via the phosphorylation of p38 MAPK; the administration of an inhibitor of the BDNF receptor, TrkB, prevents the behavioural changes observed following the administration of activated microglia (Coull et al. 2005), and a p38 MAPK inhibitor is able to prevent release of BDNF from microglia (Trang et al. 2009).
The temporal profile of microglial activation within the spinal cord is well documented in models of neuropathic pain. Real-time PCR of spinal cord tissue extracts demonstrates alterations in the levels of microglia-specific mRNA transcripts (e.g. TLR4 and CD14) within hours of injury (Tanga et al. 2004), and the phosphorylation of the microglial MAPK p38 is evident within minutes from peripheral nerve injury (Svensson et al. 2005b; Clark et al. 2007b). Morphological changes (the presence of amoeboid cells and increased immunoreactivity for the microglial marker Ox42) have been observed immunohistochemically as early as 3 days after nerve injury and are maintained for at least 50 days post-surgery (Clark et al. 2007a). Furthermore, the temporal profile of microglial activation is concomitant to the development of behavioural hypersensitivity (Clark et al. 2007b; Zhang et al. 2007; Peters et al. 2007) and the intrathecal administration of compounds that inhibit glial activation to neuropathic rodents is able to attenuate pain behaviours (Clark et al. 2007a; Tawfik et al. 2007), demonstrating that the activation of these cells does indeed contribute to aberrant pain signalling.
3.2 Astrocytic Responses to Injury or Insult
Due to their many functions in the maintenance of CNS homeostasis, under physiological conditions astrocytes are often referred to as ‘active’. Following a change in their environment, such as that occurring in the spinal cord in models of chronic pain, these cells shift to a ‘reactive’ phenotype; astrocytes undergo hypertrophy and increase their expression of a number of cellular proteins including GFAP, S100β and vimentin, which are consequently often used as markers of astrocyte reactivity (Ridet et al. 1997; Pekny and Nilsson 2005). This process is referred to as astrogliosis and has been demonstrated in a number of surgical models of neuropathic pain (Garrison et al. 1991; Colburn et al. 1999; Sweitzer et al. 1999). Astrocyte reactivity can occur through the activation of several pathways; these cells express the receptors for the neurotransmitters NMDA, SP and CGRP (Porter and McCarthy 1997) and thus respond to increased transmitter release in the dorsal horn of the spinal cord subsequent to peripheral nerve injury. In addition, astrocytes possess a variety of cytokine and chemokine receptors that can be activated by their ligands released from microglia; key examples include IL-18/IL-18R, TNF/TNFR and CCL2/CCR2 (Miyoshi et al. 2008; Gao et al. 2009, 2010b). One of the astrocytic changes most likely to be responsible for the contribution of these cells to the maintenance of pain is a decrease in the glutamate transporters GLT1 and GLAST. These transporters function to regulate extracellular glutamate concentrations at non-toxic levels. A decrease in their expression or function results in an elevation in the concentration of spinal extracellular glutamate which can elicit nociceptive hypersensitivity via the activation of NMDA and AMPA receptors (Liaw et al. 2005; Weng et al. 2006). Recent evidence indicates that astrocytes, like microglia, play a critical role in aberrant pain signalling, as the administration of glial inhibitors, such as fluorocitrate, attenuates pain behaviours in rodents (Milligan et al. 2003; Watkins et al. 1997; Okada-Ogawa et al. 2009). Interestingly, the intrathecal administration of reactive astrocytes, briefly incubated with TNF, induces the development of mechanical allodynia in uninjured mice (Gao et al. 2010b). As with microglia, the temporal profile of astrocyte activation has been assessed; astrogliosis becomes apparent after the microglial response, several days after peripheral nerve injury, and lasts for over 4 weeks post-injury (Colburn et al. 1999; Tanga et al. 2004; Romero-Sandoval et al. 2008). Additionally genetically altered mice that are deficient in GFAP develop a shorter lasting mechanical allodynia than their wild-type counterparts. Finally the administration of a GFAP antisense mRNA that prevents translation of GFAP reverses established mechanical allodynia when administered to rats 6 weeks following a peripheral nerve injury (Kim et al. 2009). Together these data demonstrate a role for astrocytes in the maintenance of neuropathic pain.
3.3 CX3CL1, CX3CR1 and Cathepsin S
CX3CL1, also known as fractalkine, is the only member of the CX3C family of cytokines. The cx3cl1 gene was originally described as abundant in the brain and heart but present in all tissues assessed, except peripheral blood leukocytes (Pan et al. 1997). Recent evidence obtained from a series of in situ hybridisation and immunohistochemical studies has determined that neurons are the principle source of CX3CL1 in the CNS (Hughes et al. 2002; Nishiyori et al. 1998; Tarozzo et al. 2003). Additionally, the development of a transgenic mouse expressing a red fluorescent reporter under the control of the CX3CL1 promoter has demonstrated that within the spinal cord CX3CL1 is expressed in dorsal horn neurons (Kim et al. 2011). Both the CX3CL1 protein and mRNA are expressed here, but the protein is not upregulated following peripheral nerve injury (Verge et al. 2004; Clark et al. 2009; Lindia et al. 2005). Furthermore, it has been demonstrated that cytoplasmic glutamate-containing vesicles are present within CX3CL1-positive neurons, indicating that CX3CL1 is expressed within excitatory neurons (Tong et al. 2000).
First described as a potent chemoattractant of T-cells and monocytes in the late 1990s (Bazan et al. 1997; Pan et al. 1997), CX3CL1 exists in two forms. As a membrane-tethered protein it plays a critical role in the firm adhesion of leukocytes to the endothelium to facilitate transmigration (Imai et al. 1997; Fong et al. 1998; Corcione et al. 2009). Soluble CX3CL1 is produced by metalloproteases (ADAM10/17) or protease (Cathepsin S) -mediated cleavage of membrane-bound CX3CL1. Whilst ADAM 10 is responsible for constitutive shedding of CX3CL1, ADAM 17 facilitates inducible shedding of this protein (Bazan et al. 1997; Hundhausen et al. 2003, 2007; Clark et al. 2011). Cathepsin S, on the other hand, is expressed by antigen-presenting cells such as microglia where its release is dependent on the activation of the purinergic receptor P2X7 (Clark et al. 2010) and is enhanced by pro-inflammatory mediators (Liuzzo et al. 1999a, b). CX3CL1 exerts its biological effects by binding to CX3CR1 for which it is the only ligand. Expression analysis of CX3CR1 has demonstrated its mRNA to be abundant in the spleen and peripheral blood leukocytes as well as the brain. Furthermore, the expression of CX3CR1 mRNA is tenfold higher in cultured microglial than whole brain samples, suggesting the receptor is of microglial origin. FACS analysis and calcium imaging of microglia have demonstrated that these cells possess the functional CX3CR1 protein as well as the mRNA (Harrison et al. 1998). This expression profile of CX3CR1 has been confirmed conclusively by the development of a transgenic mouse in which the CX3CR1 contains a green fluorescent protein reporter allowing visualisation of the transcribed protein; here, in the mouse, CX3CR1 protein was observed in microglia throughout the CNS but was not present in astrocytes or oligodendrocytes (Jung et al. 2000).
Within the CNS the CX3CL1–CX3CR1 interaction contributes the maintenance of a quiescent phenotype in microglia and suppresses the release of pro-inflammatory mediators (Mizuno et al. 2003; Lyons et al. 2009). As such, under these conditions, this protein–protein relationship is thought to be neuroprotective. In the context of chronic pain, several observations support a pro-nociceptive role of CX3CL1. The administration of the soluble chemokine domain of CX3CL1 into the intrathecal space at the lumbar level is pro-nociceptive and causes otherwise naive animals to exhibit nocifensive behaviours (Zhuang et al. 2007; Clark et al. 2007b; Milligan et al. 2004, 2005a). Consistently, the intrathecal administration of anti-CX3CL1 antibodies to neuropathic rodents attenuates pain-related behaviour (Clark et al. 2007b). Interestingly, the CSF levels of CX3CL1 increase in neuropathic animals compared to sham controls (Clark et al. 2007b, 2009). CX3CR1 exhibits similar pro-nociceptive attributes under aberrant pain conditions. Enhanced expression of the protein within the dorsal horn of the spinal cord is associated with microgliosis following peripheral nerve injury (Zhuang et al. 2007; Staniland et al. 2010). One mechanism described is IL-6 dependent; IL-6 mRNA and protein expression is induced in neurons following the peripheral nerve injury (Arruda et al. 1998; Lee et al. 2009), and prophylactic treatment with an IL-6 neutralising antibody prevents increased CX3CR1 expression, whilst, conversely, the administration of recombinant IL-6 significantly augments CX3CR1 expression (Lee et al. 2010). Supporting a pro-nociceptive role of CX3CR1 and a critical role for CX3CL1–CX3CR1 interaction in the development of pathological pain responses, CX3CR1-deficient mice do not develop hyperalgesia and/or allodynia in models of nerve injury and exhibit reduced microgliosis when compared to their wild-type littermate controls (Staniland et al. 2010). Similarly, the intrathecal administration of an anti-CX3CR1 antibody attenuates both the behavioural and microglial responses to injury (Zhuang et al. 2007; Milligan et al. 2004, 2005a). Within spinal cord microglia activation of CX3CR1 by CX3CL1 results in increased intracellular calcium concentrations (Harrison et al. 1998), the phosphorylation of p38 MAPK and subsequent release of pro-nociceptive molecules such as IL-6, NO and IL-1β (Zhuang et al. 2007; Clark et al. 2007b).
3.4 TNF and TNFR
TNF, previously known as TNFα, is a small pro-inflammatory cytokine first described in activated macrophages as a molecule with tumour-regression activity (Carswell et al. 1975). TNF belongs to a superfamily of ligand/receptor proteins that share a structural motif — the TNF homology domain. The TNF receptors are the other members of this family of proteins; two have been identified and are either constitutively expressed (TNFR1/p55-R) or inducible (TNFR2/p75-R) (Bodmer et al. 2002; Leung and Cahill 2010). Under physiological conditions TNF is expressed at very low levels in the spinal cord; however, it is rapidly upregulated in both microglia and astrocytes (glia) and neurons following peripheral injury (DeLeo et al. 1997; Ohtori et al. 2004; Hao et al. 2007; Youn et al. 2008). Similarly both TNFR1 and TNFR2 are expressed within glia and neurons in the spinal cord (Gruber-Schoffnegger et al. 2013; Ohtori et al. 2004; Hao et al. 2007). The use of receptor-specific protein ligands has demonstrated that it is TNFR1 activation in the spinal cord that is responsible for the pro-nociceptive effects of TNF under steady-state conditions; TNFR2, on the other hand, contributes to the pro-nociceptive effects of TNF under chronic pain states following its upregulation after nerve injury (Liu et al. 2007; Clark et al. 2013).
In the context of peripheral nerve injury models of neuropathic pain, TNF is detectable in the periphery at the site of injury where it is upregulated and exerts pro-nociceptive functions (George et al. 1999; Shubayev and Myers 2000; Sommer and Schafers 1998; Leung and Cahill 2010). Within the spinal cord TNF exerts its pro-nociceptive effects via actions on both neurons and glia. The spinal administration of TNF is associated with the development of mechanical allodynia and thermal hypersensitivity (Youn et al. 2008; Zhang et al. 2011). Additionally the application of exogenous TNF to spinal cord slices induces rapid modulation of Aδ- and C-fibre (nociceptive-fibre)-mediated neurotransmission (Youn et al. 2008), enhances dorsal horn neuronal responses to C-fibre stimulation (Reeve et al. 2000) and augments excitatory neurotransmission in lamina II neurons in a predominantly TNFR1-dependent manner (Zhang et al. 2011). Additionally long-term potentiation (LTP) induced by stimulation of the sciatic nerve is attenuated in TNFR knock-out mice (Park et al. 2011). These effects may not be mediated directly by the activation of neuronal TNF receptors, but rather indirectly by the activation of receptors located on glial cells within the dorsal horn (Gruber-Schoffnegger et al. 2013). Indeed, TNF is also able to modulate glial cell activation within the spinal cord as blockade of TNF signalling is associated with a reduction in glial cell activity in models of peripheral nerve injury (Svensson et al. 2005b; Nadeau et al. 2011). Via P2X7 signalling ATP is able to induce TNF production in microglia by a P38 MAPK-dependent pathway (Suzuki et al. 2004; Lister et al. 2007); in rats with a peripheral nerve injury, concomitant increases of TNF and p-P38 are observed in the spinal cord, and blockade of TNF suppresses P38 phosphorylation and microgliosis (Schafers et al. 2003; Marchand et al. 2009). Evidence from cultured astrocytes demonstrates that TNF transiently activates JNK (Gao et al. 2009), a mitogen-activated kinase (MAPK) associated with the synthesis and release of pro-nociceptive mediators (Ji et al. 2009). In particular this pathway is associated with the upregulation of astrocytic CCL2; CCL2 upregulation by TNF is dose dependently inhibited by a JNK inhibitor (Gao et al. 2009). Similarly, the spinal injection of TNF produces JNK-dependent behavioural hypersensitivity, whilst the administration of a JNK inhibitor to nerve-injured animals suppresses pain-associated behaviour (Gao et al. 2009). Peripheral nerve injury induces a slow but persistent activation of JNK MAPK particularly within astrocytes in the spinal cord. Whilst the administration of a specific JNK inhibitor has no effect on acute pain responses, it potently prevents and reverses nerve injury-induced mechanical allodynia in rodents (Zhuang et al. 2006).
3.5 IL-1β and IL-1R
Interleukin-1β (IL-1β) is a small pro-inflammatory cytokine first described as a pyrogenic factor released from leukocytes (Dinarello 2007), and was one of the first cytokines to be implicated in the mechanisms underlying enhanced nociception in rodents following peripheral nerve injury (Clark et al. 2013). IL-1β, a 17.5 kDa protein, is part of the well-characterised IL-1 family of proteins that also includes IL-1α, the IL-1 receptor (IL-1R1), a decoy receptor (IL-1R2) and the endogenous receptor antagonist (IL-1ra). IL-1ra is structurally similar to IL-1β and binds to IL-1R1 with similar affinity; however, it lacks the ability to activate the receptor and stimulate downstream signalling. Similarly, IL-1R2 is able to bind IL-1β with comparable affinity to IL-1R1 but lacks the appropriate cytosolic region to activate downstream signalling proteins (Weber et al. 2010; Dunn et al. 2001). Within the CNS IL-1β is expressed at low levels by both neurons and glial cells (Ren and Torres 2009; Clark et al. 2006; Copray et al. 2001; Guo et al. 2007; Sommer and Kress 2004), and the expression of IL-1R1 is equally widespread within the spinal cord (Zhang et al. 2008; Sweitzer et al. 1999; Gruber-Schoffnegger et al. 2013).
Under physiological conditions IL-1β has a number of homeostatic functions such as regulation of sleep and temperature (Dinarello 1996). More importantly, IL-1β is a key regulator of pro-inflammatory responses and the control of innate immune responses to pathogen-associated danger signals [pathogen-associated molecular patterns (PAMPS) and danger-associated molecular patterns (DAMPS)], such as lipopolysaccharide (LPS) and bacterial DNA. The mechanism by which IL-1β is secreted from cells differs from the classical ER–Golgi route of protein secretion due to a lack of a leader sequence (Rubartelli et al. 1990). Rather, IL-1β is synthesised as a larger precursor protein and is cleaved into a mature form; the mechanism behind this has now been elucidated and involves caspase-1, which cleaves IL-1β, and a series of accessory proteins that form a complex known as the inflammasome that provides a platform for the activation of caspase (Martinon et al. 2002; Schroder and Tschopp 2010).
Similarly to TNF, IL-1β is pro-nociceptive when administered centrally; the intrathecal administration of exogenous IL-1β results in thermal and mechanical hypersensitivity (Gruber-Schoffnegger et al. 2013; Reeve et al. 2000; Sung et al. 2004; Kawasaki et al. 2008). Further evidence for a pro-nociceptive effect of IL-1β comes from rodents lacking this protein which exhibit attenuated behavioural responses to peripheral nerve injury (Wolf et al. 2006). Furthermore it has been observed that human patients with a range of painful neuropathies exhibit enhanced CSF levels of IL-1β (Alexander et al. 2005; Backonja et al. 2008). Following peripheral nerve injury, IL-1β is upregulated in the spinal cord where it is expressed primarily by glial cells but also by neurons (Clark et al. 2013). Inhibition of normal IL-1β signalling is anti-nociceptive in animal models of neuropathic pain; intrathecal administration of IL-1ra can both prevent and reverse (Milligan et al. 2005b, 2006) nocifensive behaviours in these animals, as can inhibition of caspase-1 which prevents the proteolytic activation of IL-1β, resulting in decreased secretion of the mature form from microglia (Clark et al. 2006).
IL-1β released from glial cells is able to modulate neuronal activity. This cytokine can facilitate glutamatergic transmission via the phosphorylation of the NMDA receptor (Gruber-Schoffnegger et al. 2013) which enhances behavioural hypersensitivity; treatment with IL-1ra attenuates both the behavioural phenotype and receptor phosphorylation. The mechanism underlying this modulation is complex and calcium dependent (Viviani et al. 2003), and involves several intracellular signalling proteins including PKC, IP3, PLC, PLA2 and src kinases. Inhibitors of these proteins are able to block IL-1β-induced phosphorylation of either the NR1 subunit or NR2A/B subunit of the NMDA receptor in vitro similarly to IL-1ra (Viviani et al. 2003). The application of exogenous IL-1β to spinal cord slices is able to increase AMPA-mediated currents in lamina II neurons (Kawasaki et al. 2008) and induce LTP within lamina I neurons (Gruber-Schoffnegger et al. 2013). Patch clamp studies of IL-1β-treated spinal cord slices demonstrate that IL-1β enhances excitatory neurotransmission whilst attenuating inhibitory neurotransmission (Kawasaki et al. 2008). Overall these data indicate that IL-1β contributes to a potentiation of excitatory transmission and a suppression of inhibitory transmission, which together correlate with the facilitation of behavioural hypersensitivity.
As with many other cytokines, as well as a neuronal effect, IL-1β is thought to contribute to a pro-nociceptive state via the activation of glia. Additionally glia may contribute to the effects of IL-1β on neuronal excitability as these are absent following the inhibition of glial cell function (Gruber-Schoffnegger et al. 2013; Liu et al. 2013).
4 Spinal Glia During Inflammatory Pain
Inflammation is a critical protective mechanism occurring in response to injury, infection or irritation. It is characterised by five defining components: redness, heat, swelling, loss of function and pain. Under physiological conditions inflammation allows removal or repair of damaged tissue following an injury to the organism. Under these circumstances the role of inflammatory pain is protective, limiting the use of the affected area and preventing further damage during the healing process. However, in patients with chronic inflammatory conditions, such as arthritis (see section below), chronic pain hypersensitivity is a common complaint (Walsh and McWilliams 2014). The continued presence of algogenic mediators results in the sensitisation of the peripheral tissues (Schaible et al. 2009). Commonly models which involve the direct administration of an exogenous algogenic substance into the hind-paw of the rodent have been utilised in order to investigate how spinal glial mechanisms contribute to inflammatory pain. However, increasing spinal glial mechanisms are being studied in more clinically relevant models of inflammatory pain, such as arthritis.
An extensive body of evidence supports a role for spinal microglia and astrocytes (glia) in inflammatory pain mechanisms. Peripheral inflammation results in sensitization of spinal neurons (Vazquez et al. 2012; Konig et al. 2014) and glial cell reactivity within the dorsal horn of the spinal cord. Spinal astrogliosis and/or microgliosis have been reported in many inflammatory pain models (Watkins et al. 1997; Sweitzer et al. 1999; Clark et al. 2007a; Raghavendra et al. 2004). Critically, inhibition of glial cell activity during peripheral inflammation reduces pain behaviours substantially (Meller et al. 1994; Watkins et al. 1997; Clark et al. 2007a), suggesting that the activity of these cells in the spinal cord is vital for the full development of inflammatory pain.
A number of the spinal glia mechanisms that have to identified to contribute to neuropathic pain may also play a role in pain that occurs as result of peripheral inflammation. The cytokine/chemokine glial mechanisms described in relation to neuropathic pain above also play a role in inflammatory pain. One such astrocytic mechanism is inflammation-induced phosphorylation of JNK in spinal astrocytes (Gao et al. 2010a). Intraplantar injection of CFA (Gao et al. 2010a) or carrageenan (Bas et al. 2015) induces JNK phosphorylation, and JNK inhibition attenuates inflammatory pain behaviours (Gao et al. 2010a; Bas et al. 2015). This inflammation-induced JNK phosphorylation is regulated by spinal TNF secretion (Bas et al. 2015) as is the case during neuropathic pain (Gao et al. 2010a). One spinal microglial mechanism that regulates inflammatory pain is the phosphorylation of p38 MAPK. Peripheral inflammation induces extensive p38 phosphorylation which is restricted to microglial cells (Svensson et al. 2003a, 2005a). Inhibition of p38 in inflammatory pain models results in attenuation of pain behaviours (Svensson et al. 2003b, 2005a). The activation of many microglial receptors results in intracellular p38 phosphorylation, suggesting that this is a key intracellular signalling pathway during both inflammatory and neuropathic pain. Indeed, it has previously been demonstrated that the microglial CX3CR1 receptor leads to p38 phosphorylation (Clark et al. 2007b) and is critical for the full expression of inflammatory pain (Staniland et al. 2010), as well as neuropathic pain (see above section).
5 Spinal Glia During Rheumatoid Arthritis Pain
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterised by synovial inflammation and joint destruction. The disease aetiology remains unclear but is thought to comprise a complex interplay between environmental and genetic factors. The clinical signs of RA are accompanied by chronic pain, representing a major unmet clinical need. Despite the availability of disease-modifying agents that reduce the clinical signs of RA, the treatment of chronic pain remains inadequate at present (Kidd et al. 2007; Sokka et al. 2007; Dray 2008; Walsh and McWilliams 2014). The mechanisms of RA pathology have been studied extensively using rodent models. Although pain is common in RA patients, it is only relatively recently that pain has been studied in rodent models that replicate the complex mechanisms of this inflammatory disorder. Whilst inflammatory pain states associated with models of mono-arthritis have been extensively characterised, pain that occurs as a result of more clinically relevant poly-arthritic models has only recently been examined.
A number of poly-arthritic rodent models, all of which mimic some aspect of human RA (Williams 1998; Vincent et al. 2012), have been studied in the context of pain. Critically the activation of glial cells within the spinal cord represents a commonality between these models. Collagen-induced arthritis (CIA) represents the rodent model of RA that is most widely used for pathogenesis studies. In CIA immunisation with type II collagen results in an immune response directed against the joints, which closely resembles many aspects of human RA (Trentham 1982; Williams 2004). In addition, passive transfer of collagen antibody cocktails (collagen antibody-induced arthritis; CAIA) or of serum from the spontaneous arthritic KxB/N mouse is also commonly used to model RA.
The models detailed above have all been identified as inducing pain behaviours in the rodent hind-paw. In the CIA model, DBA/1 mice develop mechanical and thermal hypersensitivity from the onset of arthritis (Inglis et al. 2007). In the rat, CIA-induced mechanical hypersensitivity is present before the onset of inflammation (Clark et al. 2012), mirroring the sensory changes reported in models of other autoimmune disorders such as multiple sclerosis, in which hypersensitivity is present before the onset of clinical scores (Olechowski et al. 2009; Clark and Malcangio 2012). Furthermore, pain associated with the CAIA and K/BxN serum transfer models of experimental arthritis in mice has been recently characterised (Christianson et al. 2010, 2011; Bas et al. 2012). In both models joint inflammation is transient, whereas mechanical hypersensitivity is long-lasting, persisting long after joint inflammation has subsided. Importantly, arthritis-induced hypersensitivity is attenuated by analgesic agents used clinically for the treatment of RA, such as anti-TNF agents and NSAIDs (Inglis et al. 2007; Christianson et al. 2010; Bas et al. 2012; Boettger et al. 2010), suggesting good correlation between the inflammatory pain in these models and pain in RA patients. Interestingly in both the CAIA and K/BxN models, mechanical hypersensitivity after the resolution of joint inflammation is NSAID insensitive, suggesting that the early inflammatory stage of these models is followed by a late phase which is non-inflammatory (Christianson et al. 2010; Bas et al. 2012).
The contribution of CNS changes in these RA models is not fully understood, however, spinal glial cell reactivity has been reported. Astrogliosis is observed in the lumbar dorsal horn of CIA (Inglis et al. 2007; Clark et al. 2012), CAIA (Bas et al. 2012; Agalave et al. 2014) and K/BxN serum transfer treated animals (Christianson et al. 2010, 2011). It is evident that glial cells contribute to arthritis-induced hypersensitivity, as intrathecal administration of the glial inhibitor pentoxifylline is able to reverse late-stage CAIA-induced hypersensitivity (Bas et al. 2012). However, it is currently unclear whether this analgesic effect is due to inhibition of astrocytes and/or microglia cell activity. Intrathecal administration of a JNK inhibitor is also able to reverse late-phase hypersensitivity in the CAIA model (Bas et al. 2012). JNK phosphorylation occurs in spinal astrocytes following peripheral inflammation (Gao et al. 2010a) and peripheral nerve injury (Zhuang et al. 2006). CAIA also enhances spinal JNK phosphorylation, however, the cellular localisation of p-JNK in arthritis is yet to be determined (Bas et al. 2012). Thus, it remains to be fully established whether changes in astrocyte activity contribute to arthritis-induced hypersensitivity.
A role for spinal microglia in arthritis-induced hypersensitivity seems increasingly likely. In the rat CIA model, increased microglial cell activity during mechanical hypersensitivity is observed as phosphorylation of p38 MAPK (p-p38) (Clark et al. 2012). It has recently been demonstrated that spinal inhibition of the microglia protease CatS or the chemokine FKN, which constitutes a key neuron-microglial signalling system during neuropathic pain (see above section), is able to reverse established mechanical hypersensitivity in CIA and reduce microglial p-p38 (Clark et al. 2012). This analgesic effect of CatS or FKN inhibition is independent of alteration in disease progression, with inflammation and clinical score unaffected by treatment (Clark et al. 2012). Increased microglia p-p38 has also been observed in adjuvant-induced arthritis (Boyle et al. 2006). Whilst Boyle and colleagues did not examine pain hypersensitivity in detail in this study, spinal administration of a p38 inhibitor was significantly more effective at reducing joint inflammation than systemic administration of the same compound (Boyle et al. 2006). A similar reduction in inflammation was also evident following intrathecal treatment with the anti-TNF agent etanercept, which attenuated arthritis-induced p-p38 (Boyle et al. 2006), suggesting that spinal microglial mechanisms may also regulate peripheral inflammation under some conditions. Indeed, in RA patients, TNF neutralisation inhibits pain prior to reducing inflammation in the joint (Hess et al. 2011), raising the possibility of a spinal microglia contribution. Spinal TLR-4 also appears to regulate microglial cell activity during both K/BxN serum transfer arthritis and CAIA. In TLR-4 knock-out mice, the late phase of K/BxN mechanical hypersensitivity, and spinal gliosis, is attenuated compared to wild-type mice (Christianson et al. 2011). Interestingly, during CAIA, the damage-associated molecular pattern (DAMP) molecule, extracellular high-mobility group box-1 protein (HMGB1) acting as a TLR-4 ligand, is critical for arthritis-induced hypersensitivity (Agalave et al. 2014). Thus, a number of microglial targets may contribute to hypersensitivity in models of RA and may represent promising therapeutic targets for the treatment of established pain in RA patients.
6 Concluding Remarks
The concept that glial-mediated mechanisms modulate neuronal processing in the spinal cord and contribute to the facilitation of pain signalling has developed considerably in recent years. That a substantial number of pre-clinical studies have flourished in this field reinforces the premise that glial targets may be exploited as novel approaches to the treatment of chronic pain.
References
Agalave NM, Larsson M, Abdelmoaty S, Su J, Baharpoor A, Lundback P, Palmblad K, Andersson U, Harris H, Svensson CI (2014) Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 155(9):1802–1813
Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14:1142–1149
Alexander GM, van Rijn MA, van Hilten JJ, Perreault MJ, Schwartzman RJ (2005) Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. Pain 116:213–219
Arruda JL, Colburn RW, Rickman AJ, Rutkowski MD, DeLeo JA (1998) Increase of interleukin-6 mRNA in the spinal cord following peripheral nerve injury in the rat: potential role of IL-6 in neuropathic pain. Brain Res Mol Brain Res 62:228–235
Backonja MM, Coe CL, Muller DA, Schell K (2008) Altered cytokine levels in the blood and cerebrospinal fluid of chronic pain patients. J Neuroimmunol 195:157–163
Bas DB, Su J, Sandor K, Agalave NM, Lundberg J, Codeluppi S, Baharpoor A, Nandakumar KS, Holmdahl R, Svensson CI (2012) Collagen antibody-induced arthritis evokes persistent pain with spinal glial involvement and transient prostaglandin dependency. Arthritis Rheum 64:3886–3896
Bas DB, Abdelmoaty S, Sandor K, Codeluppi S, Fitzsimmons B, Steinauer J, Hua XY, Yaksh TL, Svensson CI (2015) Spinal release of tumour necrosis factor activates c-Jun N-terminal kinase and mediates inflammation-induced hypersensitivity. Eur J Pain 19(2):260–270
Baudier J, Glasser N, Gerard D (1986) Ions binding to S100 proteins. I. Calcium- and zinc-binding properties of bovine brain S100 alpha alpha, S100a (alpha beta), and S100b (beta beta) protein: Zn2+ regulates Ca2+ binding on S100b protein. J Biol Chem 261:8192–8203
Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ (1997) A new class of membrane-bound chemokine with a CX3C motif. Nature 385:640–644
Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH (2006) Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 103:16021–16026
Bodmer JL, Schneider P, Tschopp J (2002) The molecular architecture of the TNF superfamily. Trends Biochem Sci 27:19–26
Boettger MK, Weber K, Grossmann D, Gajda M, Bauer R, Bar KJ, Schulz S, Voss A, Geis C, Brauer R, Schaible HG (2010) Spinal tumor necrosis factor alpha neutralization reduces peripheral inflammation and hyperalgesia and suppresses autonomic responses in experimental arthritis: a role for spinal tumor necrosis factor alpha during induction and maintenance of peripheral inflammation. Arthritis Rheum 62:1308–1318
Boyle DL, Jones TL, Hammaker D, Svensson CI, Rosengren S, Albani S, Sorkin L, Firestein GS (2006) Regulation of peripheral inflammation by spinal p38 MAP kinase in rats. PLoS Med 3:e338
Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL (2014) Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–143
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924
Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A 72:3666–3670
Cheunsuang O, Morris R (2000) Spinal lamina I neurons that express neurokinin 1 receptors: morphological analysis. Neuroscience 97:335–345
Christianson CA, Corr M, Firestein GS, Mobargha A, Yaksh TL, Svensson CI (2010) Characterization of the acute and persistent pain state present in K/BxN serum transfer arthritis. Pain 151:394–403
Christianson CA, Dumlao DS, Stokes JA, Dennis EA, Svensson CI, Corr M, Yaksh TL (2011) Spinal TLR4 mediates the transition to a persistent mechanical hypersensitivity after the resolution of inflammation in serum-transferred arthritis. Pain 152:2881–2891
Clark AK, Malcangio M (2012) Microglial signalling mechanisms: Cathepsin S and Fractalkine. Exp Neurol 234:283–292
Clark AK, D’Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M (2006) Rapid co-release of interleukin 1beta and caspase 1 in spinal cord inflammation. J Neurochem 99:868–880
Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M (2007a) Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain 11:223–230
Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M (2007b) Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A 104:10655–10660
Clark AK, Yip PK, Malcangio M (2009) The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci 29:6945–6954
Clark AK, Staniland AA, Marchand F, Kaan TK, McMahon SB, Malcangio M (2010) P2X7-dependent release of interleukin-1beta and nociception in the spinal cord following lipopolysaccharide. J Neurosci 30:573–582
Clark AK, Staniland AA, Malcangio M (2011) Fractalkine/CX3CR1 signalling in chronic pain and inflammation. Curr Pharm Biotechnol 12:1707–1714
Clark AK, Grist J, Al-Kashi A, Perretti M, Malcangio M (2012) Spinal cathepsin S and fractalkine contribute to chronic pain in the collagen-induced arthritis model. Arthritis Rheum 64:2038–2047
Clark AK, Old EA, Malcangio M (2013) Neuropathic pain and cytokines: current perspectives. J Pain Res 6:803–814
Colburn RW, Rickman AJ, DeLeo JA (1999) The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol 157:289–304
Copray JC, Mantingh I, Brouwer N, Biber K, Kust BM, Liem RS, Huitinga I, Tilders FJ, Van Dam AM, Boddeke HW (2001) Expression of interleukin-1 beta in rat dorsal root ganglia. J Neuroimmunol 118:203–211
Corcione A, Ferretti E, Bertolotto M, Fais F, Raffaghello L, Gregorio A, Tenca C, Ottonello L, Gambini C, Furtado G, Lira S, Pistoia V (2009) CX3CR1 is expressed by human B lymphocytes and mediates [corrected] CX3CL1 driven chemotaxis of tonsil centrocytes. PLoS One 4:e8485
Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438:1017–1021
Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG (2006) An SCN9A channelopathy causes congenital inability to experience pain. Nature 444:894–898
D’Mello R, Dickenson AH (2008) Spinal cord mechanisms of pain. Br J Anaesth 101:8–16
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758
Del Rio-Hortega P (1932) Microglia. In: Penfield W (ed) Cytology and cellular pathology of the nervous system. Hoeber, New York, pp 482–534
Del Rio-Hortega P (2012a) Are the glia with very few processes homologous with Schwann cells? by Pio del Rio-Hortega. 1922. Clin Neuropathol 31:460–462
Del Rio-Hortega P (2012b) Studies on neuroglia: glia with very few processes (oligodendroglia) by PA-o del RA-o-Hortega. 1921. Clin Neuropathol 31:440–459
DeLeo JA, Colburn RW, Rickman AJ (1997) Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Res 759:50–57
Deloulme JC, Raponi E, Gentil BJ, Bertacchi N, Marks A, Labourdette G, Baudier J (2004) Nuclear expression of S100B in oligodendrocyte progenitor cells correlates with differentiation toward the oligodendroglial lineage and modulates oligodendrocytes maturation. Mol Cell Neurosci 27:453–465
Dinarello CA (1996) Biologic basis for interleukin-1 in disease. Blood 87:2095–2147
Dinarello CA (2007) Historical insights into cytokines. Eur J Immunol 37(Suppl 1):S34–S45
Dray A (2008) New horizons in pharmacologic treatment for rheumatic disease pain. Rheum Dis Clin North Am 34:481–505
Dunn E, Sims JE, Nicklin MJ, O’Neill LA (2001) Annotating genes with potential roles in the immune system: six new members of the IL-1 family. Trends Immunol 22:533–536
Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, Patel DD (1998) Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med 188:1413–1419
Gao YJ, Ji RR (2010) Targeting astrocyte signaling for chronic pain. Neurotherapeutics 7:482–493
Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR (2009) JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci 29:4096–4108
Gao YJ, Xu ZZ, Liu YC, Wen YR, Decosterd I, Ji RR (2010a) The c-Jun N-terminal kinase 1 (JNK1) in spinal astrocytes is required for the maintenance of bilateral mechanical allodynia under a persistent inflammatory pain condition. Pain 148:309–319
Gao YJ, Zhang L, Ji RR (2010b) Spinal injection of TNF-alpha-activated astrocytes produces persistent pain symptom mechanical allodynia by releasing monocyte chemoattractant protein-1. Glia 58:1871–1880
Garrison CJ, Dougherty PM, Kajander KC, Carlton SM (1991) Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res 565:1–7
George A, Schmidt C, Weishaupt A, Toyka KV, Sommer C (1999) Serial determination of tumor necrosis factor-alpha content in rat sciatic nerve after chronic constriction injury. Exp Neurol 160:124–132
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845
Go VL, Yaksh TL (1987) Release of substance P from the cat spinal cord. J Physiol 391:141–167
Gruber-Schoffnegger D, Drdla-Schutting R, Honigsperger C, Wunderbaldinger G, Gassner M, Sandkuhler J (2013) Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-alpha and IL-1beta is mediated by glial cells. J Neurosci 33:6540–6551
Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K (2007) Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci 27:6006–6018
Hachem S, Aguirre A, Vives V, Marks A, Gallo V, Legraverend C (2005) Spatial and temporal expression of S100B in cells of oligodendrocyte lineage. Glia 51:81–97
Hao S, Mata M, Glorioso JC, Fink DJ (2007) Gene transfer to interfere with TNFalpha signaling in neuropathic pain. Gene Ther 14:1010–1016
Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L (1998) Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A 95:10896–10901
Hathway GJ, Vega-Avelaira D, Moss A, Ingram R, Fitzgerald M (2009) Brief, low frequency stimulation of rat peripheral C-fibres evokes prolonged microglial-induced central sensitization in adults but not in neonates. Pain 144:110–118
Hess A, Axmann R, Rech J, Finzel S, Heindl C, Kreitz S, Sergeeva M, Saake M, Garcia M, Kollias G, Straub RH, Sporns O, Doerfler A, Brune K, Schett G (2011) Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc Natl Acad Sci U S A 108:3731–3736
Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S (2001) Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci 21:1975–1982
Hughes PM, Botham MS, Frentzel S, Mir A, Perry VH (2002) Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia 37:314–327
Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A (2003) The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 102:1186–1195
Hundhausen C, Schulte A, Schulz B, Andrzejewski MG, Schwarz N, von Hundelshausen P, Winter U, Paliga K, Reiss K, Saftig P, Weber C, Ludwig A (2007) Regulated shedding of transmembrane chemokines by the disintegrin and metalloproteinase 10 facilitates detachment of adherent leukocytes. J Immunol 178:8064–8072
Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O (1997) Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91:521–530
Imamoto K, Leblond CP (1978) Radioautographic investigation of gliogenesis in the corpus callosum of young rats. II. Origin of microglial cells. J Comp Neurol 180:139–163
Inglis JJ, Notley CA, Essex D, Wilson AW, Feldmann M, Anand P, Williams R (2007) Collagen-induced arthritis as a model of hyperalgesia: functional and cellular analysis of the analgesic actions of tumor necrosis factor blockade. Arthritis Rheum 56:4015–4023
Ji RR, Gereau RW, Malcangio M, Strichartz GR (2009) MAP kinase and pain. Brain Res Rev 60:135–148
Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20:4106–4114
Kawasaki Y, Zhang L, Cheng JK, Ji RR (2008) Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 28:5189–5194
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91:461–553
Kidd BL, Langford RM, Wodehouse T (2007) Arthritis and pain. Current approaches in the treatment of arthritic pain. Arthritis Res Ther 9:214
Kim DS, Figueroa KW, Li KW, Boroujerdi A, Yolo T, Luo ZD (2009) Profiling of dynamically changed gene expression in dorsal root ganglia post peripheral nerve injury and a critical role of injury-induced glial fibrillary acidic protein in maintenance of pain behaviors [corrected]. Pain 143:114–122
Kim KW, Vallon-Eberhard A, Zigmond E, Farache J, Shezen E, Shakhar G, Ludwig A, Lira SA, Jung S (2011) In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood 118:e156–e167
King IL, Dickendesher TL, Segal BM (2009) Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 113:3190–3197
Koeglsperger T, Li S, Brenneis C, Saulnier JL, Mayo L, Carrier Y, Selkoe DJ, Weiner HL (2013) Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-beta1 in the CNS. Glia 61:985–1002
Konig C, Zharsky M, Moller C, Schaible HG, Ebersberger A (2014) Involvement of peripheral and spinal tumor necrosis factor alpha in spinal cord hyperexcitability during knee joint inflammation in rats. Arthritis Rheumatol 66:599–609
Kuner R (2010) Central mechanisms of pathological pain. Nat Med 16:1258–1266
Latremoliere A, Woolf CJ (2009) Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 10:895–926
Lawson LJ, Perry VH, Gordon S (1992) Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48:405–415
Lee KM, Jeon SM, Cho HJ (2009) Tumor necrosis factor receptor 1 induces interleukin-6 upregulation through NF-kappaB in a rat neuropathic pain model. Eur J Pain 13:794–806
Lee KM, Jeon SM, Cho HJ (2010) Interleukin-6 induces microglial CX3CR1 expression in the spinal cord after peripheral nerve injury through the activation of p38 MAPK. Eur J Pain 14:682.e1–e12
Leung L, Cahill CM (2010) TNF-alpha and neuropathic pain—a review. J Neuroinflammation 7:27
Lever IJ, Grant AD, Pezet S, Gerard NP, Brain SD, Malcangio M (2003) Basal and activity-induced release of substance P from primary afferent fibres in NK1 receptor knockout mice: evidence for negative feedback. Neuropharmacology 45:1101–1110
Levison SW, Goldman JE (1993) Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron 10:201–212
Levison SW, Goldman JE (1997) Multipotential and lineage restricted precursors coexist in the mammalian perinatal subventricular zone. J Neurosci Res 48:83–94
Levison SW, Chuang C, Abramson BJ, Goldman JE (1993) The migrational patterns and developmental fates of glial precursors in the rat subventricular zone are temporally regulated. Development 119:611–622
Liaw WJ, Stephens RL Jr, Binns BC, Chu Y, Sepkuty JP, Johns RA, Rothstein JD, Tao YX (2005) Spinal glutamate uptake is critical for maintaining normal sensory transmission in rat spinal cord. Pain 115:60–70
Lindia JA, McGowan E, Jochnowitz N, Abbadie C (2005) Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain 6:434–438
Ling EA, Penney D, Leblond CP (1980) Use of carbon labeling to demonstrate the role of blood monocytes as precursors of the ‘ameboid cells’ present in the corpus callosum of postnatal rats. J Comp Neurol 193:631–657
Lister MF, Sharkey J, Sawatzky DA, Hodgkiss JP, Davidson DJ, Rossi AG, Finlayson K (2007) The role of the purinergic P2X7 receptor in inflammation. J Inflamm (Lond) 4:5
Liu YL, Zhou LJ, Hu NW, Xu JT, Wu CY, Zhang T, Li YY, Liu XG (2007) Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology 52:708–715
Liu T, Jiang CY, Fujita T, Luo SW, Kumamoto E (2013) Enhancement by interleukin-1beta of AMPA and NMDA receptor-mediated currents in adult rat spinal superficial dorsal horn neurons. Mol Pain 9:16
Liuzzo JP, Petanceska SS, Devi LA (1999a) Neurotrophic factors regulate cathepsin S in macrophages and microglia: a role in the degradation of myelin basic protein and amyloid beta peptide. Mol Med 5:334–343
Liuzzo JP, Petanceska SS, Moscatelli D, Devi LA (1999b) Inflammatory mediators regulate cathepsin S in macrophages and microglia: a role in attenuating heparan sulfate interactions. Mol Med 5:320–333
Lyons A, Lynch AM, Downer EJ, Hanley R, O’Sullivan JB, Smith A, Lynch MA (2009) Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem 110:1547–1556
Malcangio M, Bowery NG (1994) Spinal cord SP release and hyperalgesia in monoarthritic rats: involvement of the GABAB receptor system. Br J Pharmacol 113:1561–1566
Marchand F, Tsantoulas C, Singh D, Grist J, Clark AK, Bradbury EJ, McMahon SB (2009) Effects of Etanercept and Minocycline in a rat model of spinal cord injury. Eur J Pain 13:673–681
Marin-Teva JL, Dusart I, Colin C, Gervais A, Van RN, Mallat M (2004) Microglia promote the death of developing Purkinje cells. Neuron 41:535–547
Marin-Teva JL, Cuadros MA, Martin-Oliva D, Navascues J (2011) Microglia and neuronal cell death. Neuron Glia Biol 7:25–40
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10:417–426
McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, Maki RA (1996) Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J 15:5647–5658
McMahon SB, Malcangio M (2009) Current challenges in glia-pain biology. Neuron 64:46–54
Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF (1994) The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology 33:1471–1478
Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR (2003) Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci 23:1026–1040
Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O’Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR (2004) Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci 20:2294–2302
Milligan E, Zapata V, Schoeniger D, Chacur M, Green P, Poole S, Martin D, Maier SF, Watkins LR (2005a) An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur J Neurosci 22:2775–2782
Milligan ED, Langer SJ, Sloane EM, He L, Wieseler-Frank J, O’Connor K, Martin D, Forsayeth JR, Maier SF, Johnson K, Chavez RA, Leinwand LA, Watkins LR (2005b) Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur J Neurosci 21:2136–2148
Milligan ED, Soderquist RG, Malone SM, Mahoney JH, Hughes TS, Langer SJ, Sloane EM, Maier SF, Leinwand LA, Watkins LR, Mahoney MJ (2006) Intrathecal polymer-based interleukin-10 gene delivery for neuropathic pain. Neuron Glia Biol 2:293–308
Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K (2008) Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J Neurosci 28:12775–12787
Mizuno T, Kawanokuchi J, Numata K, Suzumura A (2003) Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res 979:65–70
Nadeau S, Filali M, Zhang J, Kerr BJ, Rivest S, Soulet D, Iwakura Y, de Rivero Vaccari JP, Keane RW, Lacroix S (2011) Functional recovery after peripheral nerve injury is dependent on the pro-inflammatory cytokines IL-1beta and TNF: implications for neuropathic pain. J Neurosci 31:12533–12542
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318
Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A, Satoh M (1998) Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett 429:167–172
Ohtori S, Takahashi K, Moriya H, Myers RR (2004) TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine (Phila Pa 1976) 29:1082–1088
Okada-Ogawa A, Suzuki I, Sessle BJ, Chiang CY, Salter MW, Dostrovsky JO, Tsuboi Y, Kondo M, Kitagawa J, Kobayashi A, Noma N, Imamura Y, Iwata K (2009) Astroglia in medullary dorsal horn (trigeminal spinal subnucleus caudalis) are involved in trigeminal neuropathic pain mechanisms. J Neurosci 29:11161–11171
Oku R, Satoh M, Fujii N, Otaka A, Yajima H, Takagi H (1987) Calcitonin gene-related peptide promotes mechanical nociception by potentiating release of substance P from the spinal dorsal horn in rats. Brain Res 403:350–354
Old EA, Malcangio M (2012) Chemokine mediated neuron-glia communication and aberrant signalling in neuropathic pain states. Curr Opin Pharmacol 12:67–73
Olechowski CJ, Truong JJ, Kerr BJ (2009) Neuropathic pain behaviours in a chronic-relapsing model of experimental autoimmune encephalomyelitis (EAE). Pain 141:156–164
Pan Y, Lloyd C, Zhou H, Dolich S, Deeds J, Gonzalo JA, Vath J, Gosselin M, Ma J, Dussault B, Woolf E, Alperin G, Culpepper J, Gutierrez-Ramos JC, Gearing D (1997) Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature 387:611–617
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458
Park CK, Lu N, Xu ZZ, Liu T, Serhan CN, Ji RR (2011) Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J Neurosci 31:15072–15085
Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50:427–434
Perry VH, Hume DA, Gordon S (1985) Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 15:313–326
Peters CM, Jimenez-Andrade JM, Kuskowski MA, Ghilardi JR, Mantyh PW (2007) An evolving cellular pathology occurs in dorsal root ganglia, peripheral nerve and spinal cord following intravenous administration of paclitaxel in the rat. Brain Res 1168:46–59
Pezet S, Malcangio M, Lever IJ, Perkinton MS, Thompson SW, Williams RJ, McMahon SB (2002) Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol Cell Neurosci 21:684–695
Porter JT, McCarthy KD (1997) Astrocytic neurotransmitter receptors in situ and in vivo. Prog Neurobiol 51:439–455
Prinz M, Mildner A (2011) Microglia in the CNS: immigrants from another world. Glia 59:177–187
Raghavendra V, Tanga FY, DeLeo JA (2004) Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci 20:467–473
Ransohoff RM, Cardona AE (2010) The myeloid cells of the central nervous system parenchyma. Nature 468:253–262
Ransohoff RM, Perry VH (2009) Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27:119–145
Rasley A, Bost KL, Olson JK, Miller SD, Marriott I (2002) Expression of functional NK-1 receptors in murine microglia. Glia 37:258–267
Reeve AJ, Patel S, Fox A, Walker K, Urban L (2000) Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain 4:247–257
Ren K, Torres R (2009) Role of interleukin-1beta during pain and inflammation. Brain Res Rev 60:57–64
Ridet JL, Malhotra SK, Privat A, Gage FH (1997) Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 20:570–577
Romero-Sandoval A, Chai N, Nutile-McMenemy N, DeLeo JA (2008) A comparison of spinal Iba1 and GFAP expression in rodent models of acute and chronic pain. Brain Res 1219:116–126
Rubartelli A, Cozzolino F, Talio M, Sitia R (1990) A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J 9:1503–1510
Saijo K, Glass CK (2011) Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 11:775–787
Salter MW, Beggs S (2014) Sublime microglia: expanding roles for the guardians of the CNS. Cell 158:15–24
Sandkuhler J (2009) Models and mechanisms of hyperalgesia and allodynia. Physiol Rev 89:707–758
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705
Schafers M, Svensson CI, Sommer C, Sorkin LS (2003) Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci 23:2517–2521
Schaible HG, Richter F, Ebersberger A, Boettger MK, Vanegas H, Natura G, Vazquez E, von Segond BG (2009) Joint pain. Exp Brain Res 196:153–162
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832
Sedel F, Bechade C, Vyas S, Triller A (2004) Macrophage-derived tumor necrosis factor alpha, an early developmental signal for motoneuron death. J Neurosci 24:2236–2246
Seybold VS, McCarson KE, Mermelstein PG, Groth RD, Abrahams LG (2003) Calcitonin gene-related peptide regulates expression of neurokinin1 receptors by rat spinal neurons. J Neurosci 23:1816–1824
Shubayev VI, Myers RR (2000) Upregulation and interaction of TNFalpha and gelatinases A and B in painful peripheral nerve injury. Brain Res 855:83–89
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35
Sokka T, Kautiainen H, Toloza S, Makinen H, Verstappen SM, Lund HM, Naranjo A, Baecklund E, Herborn G, Rau R, Cazzato M, Gossec L, Skakic V, Gogus F, Sierakowski S, Bresnihan B, Taylor P, McClinton C, Pincus T (2007) QUEST-RA: quantitative clinical assessment of patients with rheumatoid arthritis seen in standard rheumatology care in 15 countries. Ann Rheum Dis 66:1491–1496
Sommer C, Kress M (2004) Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett 361:184–187
Sommer C, Schafers M (1998) Painful mononeuropathy in C57BL/Wld mice with delayed Wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Res 784:154–162
Staniland AA, Clark AK, Wodarski R, Sasso O, Maione F, D’Acquisto F, Malcangio M (2010) Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J Neurochem 114:1143–1157
Steinhauser C, Berger T, Frotscher M, Kettenmann H (1992) Heterogeneity in the membrane current pattern of identified glial cells in the hippocampal slice. Eur J Neurosci 4:472–484
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178
Sung CS, Wen ZH, Chang WK, Ho ST, Tsai SK, Chang YC, Wong CS (2004) Intrathecal interleukin-1beta administration induces thermal hyperalgesia by activating inducible nitric oxide synthase expression in the rat spinal cord. Brain Res 1015:145–153
Suter MR, Berta T, Gao YJ, Decosterd I, Ji RR (2009) Large A-fiber activity is required for microglial proliferation and p38 MAPK activation in the spinal cord: different effects of resiniferatoxin and bupivacaine on spinal microglial changes after spared nerve injury. Mol Pain 5:53
Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y (2004) Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci 24:1–7
Svensson CI, Hua XY, Protter AA, Powell HC, Yaksh TL (2003a) Spinal p38 MAP kinase is necessary for NMDA-induced spinal PGE(2) release and thermal hyperalgesia. Neuroreport 14:1153–1157
Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL (2003b) Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem 86:1534–1544
Svensson CI, Fitzsimmons B, Azizi S, Powell HC, Hua XY, Yaksh TL (2005a) Spinal p38beta isoform mediates tissue injury-induced hyperalgesia and spinal sensitization. J Neurochem 92:1508–1520
Svensson CI, Schafers M, Jones TL, Powell H, Sorkin LS (2005b) Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett 379:209–213
Sweitzer SM, Colburn RW, Rutkowski M, DeLeo JA (1999) Acute peripheral inflammation induces moderate glial activation and spinal IL-1beta expression that correlates with pain behavior in the rat. Brain Res 829:209–221
Tanga FY, Raghavendra V, DeLeo JA (2004) Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int 45:397–407
Tarozzo G, Bortolazzi S, Crochemore C, Chen SC, Lira AS, Abrams JS, Beltramo M (2003) Fractalkine protein localization and gene expression in mouse brain. J Neurosci Res 73:81–88
Tawfik VL, Nutile-McMenemy N, Lacroix-Fralish ML, DeLeo JA (2007) Efficacy of propentofylline, a glial modulating agent, on existing mechanical allodynia following peripheral nerve injury. Brain Behav Immun 21:238–246
Todd AJ (2010) Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 11:823–836
Tong N, Perry SW, Zhang Q, James HJ, Guo H, Brooks A, Bal H, Kinnear SA, Fine S, Epstein LG, Dairaghi D, Schall TJ, Gendelman HE, Dewhurst S, Sharer LR, Gelbard HA (2000) Neuronal fractalkine expression in HIV-1 encephalitis: roles for macrophage recruitment and neuroprotection in the central nervous system. J Immunol 164:1333–1339
Trang T, Beggs S, Wan X, Salter MW (2009) P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci 29:3518–3528
Tremblay ME, Lowery RL, Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8:e1000527
Trentham DE (1982) Collagen arthritis as a relevant model for rheumatoid arthritis. Arthritis Rheum 25:911–916
Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K (2003) P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424:778–783
Tsuda M, Tozaki-Saitoh H, Inoue K (2012) Purinergic system, microglia and neuropathic pain. Curr Opin Pharmacol 12:74–79
Vazquez E, Kahlenbach J, von Segond BG, Konig C, Schaible HG, Ebersberger A (2012) Spinal interleukin-6 is an amplifier of arthritic pain in the rat. Arthritis Rheum 64:2233–2242
Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC (2004) Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci 20:1150–1160
Verpoorten N, Claeys KG, Deprez L, Jacobs A, Van Gerwen V, Lagae L, Arts WF, De Meirleir L, Keymolen K, Ceuterick-de GC, De Jonghe P, Timmerman V, Nelis E (2006) Novel frameshift and splice site mutations in the neurotrophic tyrosine kinase receptor type 1 gene (NTRK1) associated with hereditary sensory neuropathy type IV. Neuromuscul Disord 16:19–25
Vincent TL, Williams RO, Maciewicz R, Silman A, Garside P (2012) Mapping pathogenesis of arthritis through small animal models. Rheumatology (Oxford) 51:1931–1941
Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di LM, Galli CL, Marinovich M (2003) Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 23:8692–8700
Volterra A, Meldolesi J (2005) Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6:626–640
Walsh DA, McWilliams DF (2014) Mechanisms, impact and management of pain in rheumatoid arthritis. Nat Rev Rheumatol 10(10):581–592
Wang DD, Bordey A (2008) The astrocyte odyssey. Prog Neurobiol 86:342–367
Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF (1997) Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain 71:225–235
Weber A, Wasiliew P, Kracht M (2010) Interleukin-1 (IL-1) pathway. Sci Signal 3:cm1
Wen YR, Suter MR, Kawasaki Y, Huang J, Pertin M, Kohno T, Berde CB, Decosterd I, Ji RR (2007) Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology 107:312–321
Weng HR, Chen JH, Cata JP (2006) Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience 138:1351–1360
Williams RO (1998) Rodent models of arthritis: relevance for human disease. Clin Exp Immunol 114:330–332
Williams RO (2004) Collagen-induced arthritis as a model for rheumatoid arthritis. Methods Mol Med 98:207–216
Wolf G, Gabay E, Tal M, Yirmiya R, Shavit Y (2006) Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain 120:315–324
Woolf CJ, Mannion RJ (1999) Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353:1959–1964
Youn DH, Wang H, Jeong SJ (2008) Exogenous tumor necrosis factor-alpha rapidly alters synaptic and sensory transmission in the adult rat spinal cord dorsal horn. J Neurosci Res 86:2867–2875
Zhang J, Shi XQ, Echeverry S, Mogil JS, De KY, Rivest S (2007) Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci 27:12396–12406
Zhang RX, Li A, Liu B, Wang L, Ren K, Zhang H, Berman BM, Lao L (2008) IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain 135:232–239
Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR (2011) TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain 152:419–427
Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR (2006) A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci 26:3551–3560
Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR (2007) Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun 21:642–651
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Old, E.A., Clark, A.K., Malcangio, M. (2015). The Role of Glia in the Spinal Cord in Neuropathic and Inflammatory Pain. In: Schaible, HG. (eds) Pain Control. Handbook of Experimental Pharmacology, vol 227. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-46450-2_8
Download citation
DOI: https://doi.org/10.1007/978-3-662-46450-2_8
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-46449-6
Online ISBN: 978-3-662-46450-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)