
Abstract
Dendritic cells (DCs) are the most potent class of antigen presenting cells and are versatile regulators of antigen-specific immune responses. Because of their seminal role in adaptive immunity, DCs have been the subject of therapeutic applications, including the exploration and development of DC-based vaccines (Steinman and Pope 2002; Steinman and Banchereau 2007). To overcome inefficient systemic antigen delivery by conventional DC-based vaccines, where pathogens or pathogenic antigens are introduced to ex vivo cultured DCs, a novel in vivo and in situ DC-targeting strategy has been developed that employs a DC-receptor (DCR) antibody fused to target antigen and administered with adjuvant (Trumpfheller et al. 2012). To date, various DC-specific endocytic receptors have been tested with success in applying this novel approach in animal models and one clinical trial, including the DCRs DEC-205/CD205, DCIR2, langerin and Cle9A (Jiang et al. 1995; Dudziak et al. 2007; Caminschi et al. 2008). Using the ovalbumin (OVA) model antigen, this DC-targeting strategy demonstrated robust targeting efficacy as well as efficient systemic antigen delivery leading to strong T cell immunity. Based on this and other demonstrations of capability, the vaccine efficacy of DC-targeting strategy was further developed and tested in various viral, bacterial, and cancer disease model systems (Trumpfheller et al. 2006; Do et al. 2010; Wang et al. 2012). This chapter focuses on DEC-205/CD205 and DCIR2 DCR antibodies and prophylaxis against pneumonic plague as a principal case study in the development of improved DC-targeting vaccines. The efficacy of DC-targeting strategy is discussed with regard to: (1) breadth and strength of T cell immunity, (2) DC subsets-specific immune responses and DC subsets targeting, (3) mucosal immunity induction, and (4) protection data against virulent human pathogen. This chapter finishes with discussions of lessons learned in developing DC-targeting strategies against other disease models, and future directions for improvement of DC-targeting vaccines.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Dendritic cells
- DC vaccines
- Dendritic cell targeting
- Endocytic receptors
- DEC-205
- DCIR2
- Conjugated antibodies
- T cell immunity
- B cell immunity
- Mucosal immunity
17.1 Introduction
Vaccines are effective and have long-lived immunity only to the extent that they induce T cell memory directed against appropriate target antigens. T cell memory controls humoral and cell-mediated anamnestic responses, and appropriate targeting insures effectual immune action against the etiological agent of disease. This dual requirement for an efficacious vaccine is achieved by delivering target antigen epitopes which drive successful processing, presentation, and T cell induction. The inclusion of such antigens has been somewhat empirical, though modern biotechnology and recombinant engineering has enabled the rational design of potent new chimeric biomolecules. Several types of vaccines have been described in this book, categorized by antigen source as being natural, modified, engineered, combination, or expressed in vivo; exemplified by whole cell-, live attenuated-, subunit-, conjugate- or DNA vaccines. This chapter describes another category of vaccine differentiated by its strategy of antigen presentation. Dendritic cell vaccines share a targeting strategy in which specific antigen is delivered to dendritic cells (DCs) via antibodies directed against their specific endocytic receptors. DC-targeting vaccines have been under development for nearly a decade and recent advances have greatly improved the DC vaccine strategy. This chapter summarizes the development and use of a recent DC-targeting vaccine platform as tested against disease models of particular importance. We also provide a generalized method for development and application against other antigens.
In spite of past advances in biotechnology and the exponential accumulation of genomic data and biomolecular information, some diseases and pathological conditions are still recalcitrant to effective prophylaxis or therapy. To date, conventional vaccine technologies have been problematic or ineffective against several important infectious diseases (i.e., tuberculosis, malaria, HIV/AIDS) and noninfectious diseases (e.g., cancer). Beginning in 1995 (Hsu et al. 1996; Ridgway 2003), the new vaccine approach of DC-targeting entered clinical testing, and continues to gain interest, particularly in areas of cancer prevention and treatment. Recent progress in the DC-targeting strategy has improved the engineering, manufacture and efficacy of targeting products, and promises to revolutionize vaccination against difficult diseases. The first section describes unique and centrally important features of DCs with regard to vaccine biology, and focuses on conventional features and future challenges of DC-based vaccine development.
17.1.1 Dendritic Cells
We begin with a brief overview of DC immunobiology and description of how DCs are pivotal to this vaccine technology platform. Extensive information on DCs immunobiology is available elsewhere (Banchereau and Steinman 1998; Steinman and Banchereau 2007) to which the reader is guided for detailed and current reviews of background knowledge. Basic research and lessons learned from clinical trials have well established that effective vaccines must induce enhanced cell-mediated immune responses with memory in concert with humoral immunity, and must involve mucosal aspects of protection as related to disease pathogenesis. DCs are very capable of meeting these diverse requirements, as is illustrated throughout this chapter. DCs are specialized antigen presenting cells (APCs) that initiate and control immune responses (Banchereau and Steinman 1998; Steinman and Banchereau 2007). Foremost, DCs are potent T cell stimulators.
Immature DCs, as sentinels have a critical role in establishing adaptive immunity to a newly encountered threat; They are abundant at body surfaces including mucosal areas where most infections occur, and they actively acquire antigens by extending their dendrites through the tight junction interfaces of epithelial cells (Rescigno et al. 2001; Niess et al. 2005). DCs have a specialized endocytic system comprised of various uptake receptors and antigen processing compartments (Trombetta and Mellman 2005), which underlie their central and unique role in innate immunity. After encountering nonself entities, DCs bearing antigen then migrate into local lymphoid organs (Cyster 1999; Randolph et al. 2005), which allows efficient selection of specific clones from the diverse antigen-specific repertoire, often less than one in 105 lymphocytes. This facilitates presentation of processed antigens to CD4+ and CD8+ T cells, glycolipids to NKT cells, as well as native antigens to B cells leading to diverse forms of adaptive immunity (Mellman and Steinman 2001). When a pathogen unsuccessfully infects a DC, microbial proteins are imported to the cytoplasm where they are clipped by proteasomes and then transported to the rough endoplasmic reticulum through TAPs (transporters for antigen presentation). Processed peptides of about nine amino acids in length are loaded onto the antigen-binding cleft of newly synthesized MHC class I molecules, and the MHC I-peptide complexes reach the surface during transit through the Golgi apparatus. This classical endogenous pathway of DC processing leads to activation of antigen-specific CD8+ T cells (Pamer and Cresswell 1998). In contrast, the exogenous pathway begins when proteins are delivered to DCs through phagocytosis or receptor-mediated endocytosis. Within non-lysosomal transport vesicles, the processed peptides, either preloaded or newly loaded onto MHC class II, are then presented to specific CD4+ T cells (Turley et al. 2000). Importantly, the vesicles contain not only MHC II-peptide complexes but also CD86, which is a co-stimulatory molecule that enable DCs to stimulate CD4+ T cells very efficiently (Engering and Pieters 2001). Another pathway and special feature of DCs is “cross-presentation,” which is essential for induction of enhanced T cell immunity in vaccine biology (Albert et al. 1998; Heath et al. 2004). During cross-presentation, peptides from extracellular antigens are presented within the MHC class I complex. This cross-presentation capacity allows DCs to elicit CD8+ CTL (cytotoxic T lymphocytes) reaction without infection or endogenous biosynthesis of proteins in contrast to the “classical endogenous pathway.” For example, proteins derived from immune complexes arising via the Fcγ receptor (see Abbreviations), or from dying cells via phosphatidylserine receptor, or from microbes inactivated by ultraviolet, heat or chemical treatments, each gain access to the cytoplasm of DCs resulting in protein ubiquitination, proteasomal degradation, and routing via TAP-mediated transportation to the surface. Thus, many non-replicating antigens, including protein subunit vaccines and chemically inactivated vaccines, can “cross” over to MHC class I products of DCs and elicit CD8+ T cell responses. This cross-presentation by DCs is critical to the effectiveness of vaccines intended against chronic intracellular infectious diseases and cancers.
In the steady state, or in the absence of immune system perturbation, DCs remain in an immature status, and can actively silence untoward immune activation against self and environmental proteins (Hawiger et al. 2001). This induction of tolerance by DCs is also important in immune-regulation, and is particularly important in developing treatments for transplantation, allergy, or autoimmunity. Therefore, awareness of DC “maturation” and tolerance is necessary to exploit DCs for vaccine development (Reis e Sousa 2006). A spectrum of stimuli has been identified as DC maturation signals. One category important for signaling in the presence of microbes is the toll-like receptor (TLR) (Akira et al. 2001; Medzhitov 2001), and a second is TNF signaling by other lymphocytes including NK, NKT, T, and B cells (Fujii et al. 2004). Although different stimuli can affect DC functioning at different levels, matured DCs generally: reduce antigen capturing and processing, increase the expression of co-stimulatory molecules (i.e., CD40, CD80, CD86), increase production of various cytokines and chemokines, and increase migration to draining lymph nodes (LNs) and the survival of DCs (Banchereau and Steinman 1998; Steinman and Banchereau 2007). These series of processes convert DCs from functioning as “immune silencer” to “immune stimulator” and, therein, bridge innate, and adaptive immunities. This was the subject of the 2011 Nobel Prize in Physiology or Medicine, co-awarded posthumously to Dr. Ralph Steinman for his important discoveries in this area. Recent insights also suggest that different functions by distinct DC subsets are responsible for such various and almost contradictory immune responses as tolerance versus activation (Pulendran et al. 1999; Shortman and Liu 2002; Shortman and Naik 2007).
Based on the cell physiology discussed above, the advantages of utilizing DCs in vaccine biology can be summarized in two aspects: the quantity and the quality of induced immune responses. For example, DCs prime antigen-specific CD4+ T cells efficiently and differentiate them into IFN-γ secreting Th1 type CD4+ T cells. DC-delivered peptides are observed to induce specific immune responses that are 100–1,000 times more efficiently than nonspecifically delivered peptides mixed in Complete Freund’s Adjuvant (CFA) (Hawiger et al. 2001). Th1 helper T cells play an important role in activating macrophages to fight against intracellular bacteria and protozoa. They provide aid to CD8+ T cells in defending against viral infections and cancers, and help antigen-specific B cells to proliferate and produce antibodies as well. In addition, DCs can directly stimulate CD8+ T cells and B cells. In this circumstance, the functional affinity of the CD8+ T cells is improved and antibody secretion and isotype switching are enhanced, including IgA production which is important in mucosal protection. Moreover, DCs can induce T cell memory for both CD4+ and CD8+ responses (Steinman 2007; Steinman and Banchereau 2007). Thus, it is essential to harness the DC’s central and versatile role in controlling immune system function while engineering improvements in vaccine efficacy, potency, and longevity.
17.1.2 Conventional DC-Based Vaccine Approach
Valuable concepts of DC-based vaccine approaches have been successfully demonstrated in numerous experimental animal models. To date, the DC-based vaccine platform has proven to be largely safe and able to induce antigen-specific T cell responses in patients. As discussed further below, Table 17.2 lists clinical vaccine trials using a DC strategy, all but one of which employs the conventional method. The difference between conventional and new DC-based vaccine strategies is described in Sect. 17.1.3 below and illustrated in Fig. 17.1. Here, we first describe the general procedure of conventional DC-based vaccines used in clinical applications with some discussion methods used in isolating DC precursors, antigen pulsing, and ex vivo manipulations, as well as suggested improvements to be considered for future DC-based vaccine approach.

Comparison of conventional and new DC-targeting vaccine strategies. The principal stages and components of dendritic cell-based vaccine strategies are depicted for the conventional (a) and new, improved (b) approaches. Diagrammatic representations of critical components are given in the key below for blood, immature DC, matured DC, DC-endocytic receptor, antigen (red), antigen-conjugated DC-targeting antibody, and antigen-specific lymphocyte. Arrows depict stages that involve medical or laboratory handling of material that is withdrawn from or administered to the patient. Numbered stages occur either ex vivo or in vivo as described
Two general sources of DC precursors have been facilitated for use in the conventional DC strategy, CD34+ proliferating hematopoietic stem cells, and CD14+ monocytes (Banchereau et al. 2001; Steinman and Dhodapkar 2001). Monocyte-derived DCs (Mo-DCs) have been extensively used in clinical applications, partly because they are easier to be obtained and a more homogenous population than CD34+-derived DCs. The methods for isolating DC precursors and their subsequent culturing have improved so that large numbers of homogenous Mo-DCs (i.e., 300–500 million mature DCs per apheresis) can be generated with GM-CSF (granular macrophage colony stimulating factor) and IL-4 to give a product of consistent quality (Berger et al. 2002). While increased yields of homogenous DCs would be ideal for use in clinical trials, better understanding is needed with regard to the specialized immune functions induced by distinct DC subsets.
For introducing or “pulsing” antigens into DCs, various forms of antigen and delivery vehicle have been applied, including transfection of RNAs or immune-dominant peptides, and antibody-coated tumor cells, whole cell lysates, or necrotic cells (Schuler et al. 2003). Using cells or cell lysates does not require identification of antigenic epitopes, so is presumed to save time and effort compared to using immune-dominant peptides or RNAs. However, the latter approaches can be used in patients regardless of tumor type and does not require a preparation of large amount of antigen. Moreover, it might provide DCs with selective antigens leading to enhanced antigen-specific immune responses.
As described in Sect. 17.1.1, activating DCs into “mature” status is critical for immune stimulation. There are several established “maturation cocktails” including TLR ligands (e.g., MPLA, poly IC) or inflammatory cytokines (e.g., TNF-α). Monocyte-conditioned medium (MCM) composed of IL-1β, TNF-α, IL-6, PGE2, or CD40L represents a standard for Mo-DCs stimulation (Steinman and Pope 2002; Schuler et al. 2003), although a validated clinical standard has not been published. Previous studies reported that Mo-DCs became more homogenous in population diversity after MCM treatment, and they were highly viable and migrated well upon chemotactic signals (Schuler et al. 2003). A critical question remains, however, of how to determine whether the ex vivo-manipulated DCs are truly matured functional and in vivo. So far, various protocols to check either up-regulation of surface co-stimulatory molecules (e.g., CD80, CD86, CD83), cytokine production (e.g., IL-12), or in vitro immune responses using a positive control antigen pulsed together with real pathogenic antigens such as tetanus toxoid (TT) or keyhole limpet hemocyanin (KLH), have been applied. For clinical DC-based vaccines, standardized protocols are needed that provide improved functional aspects of DCs.
Although optimized vaccination schedule, dosage, and routes of DC-based vaccine approaches have yet to be determined, previous trials have demonstrated that tumor-specific T cells could be induced by either intradermal (Fong et al. 2001a), subcutaneous (Schuler-Thurner et al. 2002), intranodal (Jonuleit et al. 2001), or intravenous (Fong et al. 2001b) injection of ex vivo-manipulated DCs in patients. Given the facts that efficient migration to lymph nodes of the administered DCs is essential for the induction of antigen-specific T cell immunity, and less than 6 % of the Mo-DCs could reach the nodes (Schuler et al. 2003), it can be surmised that improved delivery should be pursued. Although methods are conceived to overcome such migratory limits, including a direct intranodal injection, manipulation of CCL19/CCR7 interaction, or an increased cell count and schedule frequency of DC administration, they bring additional difficulties which have hampered clinical application depending on cancer types. For example, diminished DC functions due to over-manipulation, or induction of unwanted counter immune responses due to already sensitized immune responses have been observed. Therefore, for improved DC-based vaccine approaches, developers must consider such a way to deliver antigen to as many DCs as possible so that effective B and T cell immunity can be induced in systemic lymphoid tissues including mucosal nodes.
Beginning with the first clinical DC vaccination study in 1995 (Hsu et al. 1996), numerous trials have been launched against various cancer diseases and infectious diseases. To date, there are 220 clinical trials worldwide listed that use some form of DC vaccine intervention, as publically listed by www.clinicaltrials.gov (Table 17.2). The majority of these are centered in the US and Canada, followed by Europe and the Asia/Pacifica region. Most studies are in Phase 1 and/or Phase 2, with 33 listed as Phase 3 at the time of this writing. Nearly, all studies target cancer and neoplasms, though 10 target an infectious disease, all of these being against HIV/AIDS. One study uses the new DC-targeting platform described in this chapter, employing a target antigen-fused anti-DC receptor antibody, called the DEC-205-NY-ESO-1 fusion protein vaccine (Study NCT01522820; Table 17.2, italics). While some studies have generated results, these data are not publically accessible.
Although we still need more data to draw broad conclusions on the efficacy of DC-based vaccine approaches, it is clear that valuable proofs of concept in clinical applications have been collected, and the strategy has demonstrated sufficient potential to warrant further pursuit. The new DC-targeting platform is discussed below.
17.1.3 New DC-Targeting Vaccine Approach
The concept of a novel DC-targeting strategy is depicted in Fig. 17.1, where differentiating principles and improvements are compared to conventional DC-based vaccine approaches. The principal advantages of the new targeting strategy is greater targeting efficiency and greater procedural safety through less handling of material introduced into the patient. The strategy is elegant: antigen is couriered directly via an anti-DC receptor antibody. In brief, the new DC-targeting strategy uses a chimeric conjugated antibody in which heavy chain is fused to antigen by recombinant engineering with a systemic DC maturation stimulus (Trumpfheller et al. 2012). The conjugated antibody targets DCs at various DC-specific endocytic receptors, and the targeted antigens are endocytosed, processed, and presented to antigen-specific T cells by mature DCs. Thus, the new DC-targeting strategy employs natural processes occurring in vivo to achieve antigen loading, DC maturation, enrichment, and presentation. This new strategy circumvents several problems associated with the conventional approach as listed below (see boxed insert). Although both approaches involve a single parenteral product, the conventional approaches require collection of DCs from patient for manipulation and purification before re-introduction, creating more risk to negatively impact quality, safety, and efficacy. Complexities associated with the isolation and culture of DCs, their ex vivo manipulation, and their homing to LNs on reintroduction render the conventional approach generally intractable for analytical measurement and therefore problematic in monitoring for quality control.
Problems with Conventional DC-Based Vaccine Approaches
-
Difficulty in isolation and expansion of DCs with proper number and quality
-
Inefficacy of the ex vivo cultured DCs in homing to draining LNs and purity
-
Potential problems during ex vivo DC manipulation
-
Limitations of antigen preparation
-
Potential risk to quality (e.g., freezing and thawing cells) and safety due to ex vivo handling and manipulation of material introduced back into the patient
The key of DC-targeting strategy for vaccine biology is harnessing the capacity of DCs as APCs and thus, to maximize a role of DCs immune system, or nature’s adjuvant. In other words: (1) by targeting antigens to endocytic receptors, antigen capture and process is significantly enhanced; (2) by administrating DCs stimulus simultaneously, antigen-bearing DCs become true immune stimulators; and (3) by delivering antigens directly to DCs in vivo in situ, critical challenges on DC isolation, ex vivo-manipulation, or migration to the nodes are overcome.
Due to these influential capabilities, the DC-targeting strategy was developed incrementally and extensively wherein several aspects were considered carefully including, the screening and characterization of DC-specific endocytic receptors, generation of monoclonal antibodies with quality control, selection of antigens of interest and disease models, and efficient immune monitoring system to examine the efficacy of the strategy as therapeutic as well as preventive vaccines. Technology discovery and development information on each of these topics is discussed further in following sections of this chapter. We begin with the rationale to select the DEC-205 endocytic receptor for the DC-targeting strategy and lessons learned from first demonstration using a model ovalbulin antigen (OVA) system (Sect. 17.2). The DC-targeting strategy has been refined using pathogenic disease models, giving expected results, and some surprise observations (Sects. 17.3 and 17.4). Lastly, various efforts for clinical applications (Sect. 17.4) and future directions (Sect. 17.5) are considered and discussed.
17.2 DC-Targeting Strategy: Technology Discovery with OVA Model Antigen
This section describes the rationale for selecting DEC-205 over other DC-specific endocytic receptor as a targeting molecule, and valuable information on the DEC-targeting strategy as potential prophylactic and preventive vaccine approach. Much of this information was gained from early experiments using the ovalbumin model antigen and OVA transgenic mice system.
17.2.1 The DC Receptor for Endocytosis, DEC-205
Dendritic cells express various receptors for absorptive uptake of antigens, and DC subtypes are classified in part by the identity of these antigen uptake receptors. For example, Fcγ receptors (Regnault et al. 1999), and macrophage mannose receptor (MMR) (Sallusto et al. 1995) are shared with other type of cells, while CD205 (DEC-205) (Jiang et al. 1995), CD207 (Langerin) (Valladeau et al. 2000), CD209 (DC-SIGN) (Engering et al. 2002) or BDCA-2 (Dzionek et al. 2001), are more DC-restricted receptors. Among these, DEC-205 has received intense interest for use in the DC-targeting strategy due to the following features. First, this endocytic receptor mediates the efficient processing and presentation of antigens on MHC class I and II products in vivo (Jiang et al. 1995). An early study demonstrated that the DEC-205 receptor targets late endosomes or lysosomes rich in MHC class II products, while the homologous MMR was found in more peripheral endosomes (Mahnke et al. 2000). In addition, DEC-205 showed enhanced surface binding, up-taking, and presentation in comparison of MMR. These properties of DEC-205 were mediated by a membrane-proximal region with a coated pit sequence for up-taking, and a distal region with an EDE triad (Glu-Asp-Glu) for the unusual deeper targeting (Mahnke et al. 2000). Second, DEC-205 is expressed at high levels on lymphoid tissue DCs, and particularly in the T cell areas of lymphoid organs, which might enhance antigen-specific DC/T cell contact (Witmer-Pack et al. 1995). Moreover, monoclonal antibody against DEC-205 (anti- or αDEC-205 mAb) demonstrates a target selectively for DEC-205+ DCs in mice (Hawiger et al. 2001). Based on these properties, any protein joined to the DEC-205+ DCs via αDEC-205 mAb was anticipated to be processed and presented efficiently (now, we define this as DEC-targeting). This capability was demonstrated with greatly enhanced induction of antigen-specific CD4+/CD8+ T cell responses (Hawiger et al. 2001).
Given the fact that less than one in 10,000 naïve T cells are able to respond to a specific antigen in general, its stimulation is difficult to detect by conventional immune assays. To aid development and testing, ovalbumin (OVA) was selected as a model antigen within OVA transgenic mice, such as OT-I (transgenic T cell receptor for MHC class I-restricted OVA peptide, amino acids 257–264, SIINFEKL) or OT-II (transgenic T cell receptor for MHC class II-restricted OVA peptide, amino acids 323–339, ISQAVHAAHAEINEAGR). This experimental system proved to be very useful in the analysis of antigen-specific T cell immunity in early exploration of the DC-targeting strategy. When graded doses of the anti-DEC-205 antibody conjugated to OVA protein (αDEC:OVA) were subcutaneously (s.c.) injected into the mice, OVA antigen was efficiently presented to OVA-specific CD8+/CD4+ T cells in vitro, as well as in vivo by CD11c+ DCs, but not by CD11c− DCs or B cells in the draining lymph nodes. These findings indicated high specificity of the DEC-targeting strategy (Bonifaz et al. 2002). In case of MHC class I presentation by CD11c+ DC after αDEC:OVA targeting, transporter associated with antigen processing (TAP) was required which suggested an exogenous MHC class I pathway (Jung et al. 2002). Interestingly, both efficient targeting and peripheral CD8+ T cell tolerance via presentation on MHC class I were observed under steady state or in the absence of infection (Bonifaz et al. 2002). Briefly, in the absence DC stimuli, as achieved experimentally by omitting anti-CD40 mAb (αCD40), OVA-specific CD8+ T cells first proliferated then died by 12–14 days. T cells were not found in lymph nodes, spleen, bloods, or peripheral tissues such as lung. Moreover, there were no recall T cell responses to rechallenge with OVA plus a strong adjuvant such as CFA, which demonstrated the deletion of the T cells or the induction of peripheral tolerance by DEC-205 receptor-mediated antigen delivery. In contrast, when αDEC:OVA was administered in the presence of DC maturation, such as injected αCD40, OVA-specific CD8+ T cells proliferated, produced IL-2 and IFN-γ, and strongly responded to the rechallenge. This demonstrated two important points: first, as previously known, a “danger signal” is required to induce immune stimulatory function of DCs (Bendelac and Medzhitov 2002; Steinman and Banchereau 2007) (see Sect. 17.1.1 for more information); and second, that DEC-205 provides efficient receptor-mediated antigen processing and presentation for MHC class I in vivo, leading to peripheral tolerance with small amounts (see Sect. 17.2.2 for more information). This latter effect typically requires repeated injections of large amounts of proteins or peptides. DEC-205 might play an efficient role for developing tolerance to harmless environmental proteins and self-proteins on MHC class I by continuously deleting those T cells in vivo. Since injection of only αDEC:OVA neither increased DC numbers nor induced DC maturation, these data proved that a coupled target antigen was required in this DC-targeting strategy (Bonifaz et al. 2002).
Based on these findings, the DC-targeting strategy was proposed as an improved DC-based vaccine approach. The effectiveness and practical utility of this platform rests on unique composition of the active pharmaceutical ingredient, comprising αDEC-205 conjugated to target antigen, and adjuvant, comprising αCD40 to induce strong antigen-specific CD4+/CD8+ T cell immune responses. Demonstrations of this new DEC-targeting strategy in model diseases are described in Sects. 17.3 and 17.4 of this chapter.
17.2.2 Highly Efficient CD4/CD8 T-Cell Immunity
The immunostimulatory efficiency of DC-targeting was found to be very efficient compared to the standard subunit vaccine approach using purified antigen. For example, more than 1,000-fold increased efficiency was observed for the OVA antigen. In escalating dose-response tests of αDEC:OVA with αCD40 (fixed at 25 μg/mouse) injected into the mice, and OVA-specific CD4+/CD8+ T cells were assessed by proliferation and by ELISPOT detection of IFN-γ secretion (Bonifaz et al. 2004). A minimum of 2,500 ng non-targeted soluble OVA was required to elicit OVA-specific CD8+ T cell proliferation, while just 2 ng of the DEC-targeted OVA gave the same response. Thus, comparing DEC-targeting delivery to a nontargeted soluble protein administration 1,000 times and 50 times less amount of antigen, respectively, were required to achieve similar level of OVA-specific CD8+ and CD4+ T cell induction. Beyond efficient T cell proliferation and IFN-γ production, DEC-targeting was also observed to induce functional cytotoxic T lymphocytes (CTLs) (Bonifaz et al. 2004).
Interestingly, the efficacy of DEC-targeting for induction of antigen-specific T cell immune responses was more prominent than other vaccination methods tested. For example, mice were injected s.c. with either: (1) splenic DCs pulsed ex vivo with 10 μg/ml of αDEC:OVA and αCD40, (2) 500 μg of the non-targeted soluble OVA protein in CFA, (3) 50 μg of the non-targeted soluble OVA protein with αCD40, (4) 50 μg of OVA peptide (SIINFEKL) with αCD40, or (5) 50 ng of OVA containing αDEC:OVA with αCD40. Following 7 and 30 days after vaccination, OVA-specific T cell expansion was measured by double staining with CD62L and Kb-SIINFEKL-PE tetramer from lymph nodes and spleens. The data showed that at both days, DEC-targeting was the most effective among the group in the induction of the antigen-specific T cell immune responses. The frequency of OVA-specific CD8+ T cells was ~5 % with 50 ng of the OVA containing αDEC:OVA plus αCD40 treatment at day 7 in spleen, while those of the 50 μg soluble OVA protein (1,000 × molar excess of protein) plus αCD40 group and the 50 μg OVA peptide (467 molar excess of processed peptide) plus αCD40 group were around 1 and 0.2 %, respectively. The frequency of the expanded T cells secreting IFN-γ was the highest in the DEC-targeting group as well. Even after 30 days, about 1.3 % of the OVA-specific CD8+ T cells were detected in the group immunized with the DEC-targeting, and such was not detectable with other vaccination strategies demonstrating the efficacy of the DEC-targeting strategy in the induction of T cell immunity (Bonifaz et al. 2004).
17.2.3 Systemic Antigen Presentation and Durability
It was surmised that the DEC-targeting strategy offered an opportunity for improved efficient systemic antigen delivery given the fact that few ex vivo-manipulated DCs were found in the draining lymph nodes of reinjected patients using the conventional DC-based vaccine approach.
In order to test the rate and the durability of the DEC-targeting antibody in lymphoid tissues, Alexa488-conjugated DEC-targeting antibody was injected s.c. into the mice and tracked at various time points (Bonifaz et al. 2004). As early as 30 min after vaccination, the DEC-targeting antibody was bound not only to the CD11c+ DCs in draining lymph nodes but also to the spleen DCs. At 6 h post injection, ~50 % of the DCs in the draining lymph nodes and 40 % of the DCs in distal lymph nodes were targeted, and as late as 3 days after injection, the DEC-targeting antibody was detected in all sites tested. Considering at least 6 h in transit for carriage of the antigen by peripheral DCs to the draining lymph nodes, and that they would not easily move to other organs (Kamath et al. 2000, 2002) (i.e., distal lymph nodes and spleens), this study demonstrated that the DEC-targeting strategy greatly enhanced systemic antigen delivery to the DCs in vivo.
Unexpectedly, the antigen products delivered by the DEC-targeting antibody, particularly those processed on MHC class I products, persisted for longer periods than the typical half life of many DCs (i.e., less than 2 days) (Kamath et al. 2002; Bonifaz et al. 2004). When OVA antigens were given to the mice in two forms-either targeted via αDEC:OVA or as non-targeted soluble OVA-at different time points (1, 3, 7, 15 days) before transferring the OVA-specific T cells, prolonged presentation was detected in the lymph nodes of the DEC-targeted group even after 15 days. In contrast, minimal presentation was detected in the soluble OVA treated group after 7 days. Although further studies are needed to better understand the destiny and mechanisms of antigen targeting, it appears that the DEC-205 receptor is specialized in its capacity for both processing and presentation of antigen as MHC class I products.
Importantly, DC targeted antigens were able to induce stronger antigen-specific T cell responses with durability as well (Bonifaz et al. 2004). For example, IFN-γ secreting CD8+ T cells were examined 14, 21, 60, or 90 days after vaccinating mice with αDEC:OVA, containing 50 ng OVA protein, plus αCD40. Even after 90 days of a subcutaneous single dose of the DEC-targeting antibody vaccination, strong IFN-γCD8+ T cell responses were detected and efficient CTL activity was still observed. This indicated great efficacy in the formation of effector memory T cells by DEC-targeting.
Taken together, these findings demonstrated several practical attributes of the DEC-targeting strategy as a novel DC-based vaccine platform: (1) fast systemic delivery of antigens to large numbers of DCs in lymphoid tissues with durability; (2) induction of higher frequency of antigen-specific T cell immunity with significantly less antigen; and (3) generation of durable effector memory T cells. These salient features of the DEC-targeting strategy were further demonstrated using a tumor model as shown in the below section.
17.2.4 Vaccine Efficacy
To study the efficacy of the DEC-targeting strategy as a novel DC-based vaccine, mice were injected s.c. with αDEC:OVA or isotype control in the presence and absence of αCD40. Sixty days later, they were challenged with B16 melanoma cells bearing OVA (Bonifaz et al. 2004). Only mice vaccinated with αDEC:OVA plus αCD40 were protected and the protection was mediated primarily by OVA-specific CD8+ T cells with protection by CD4+ T cells to a lesser extent. Testing a therapeutic approach, mice were inoculated with the tumor first (5–10 mm diameter) and subsequently received various forms of OVA antigens, including αDEC:OVA + αCD40, soluble OVA in CFA, ex vivo OVA-pulsed spleen DCs, or PBS/adjuvant alone. The only group to show a therapeutic effect was that treated with the DEC-targeting antibody plus adjuvant. Based on the data showing that primed effector T cells induced by the DEC-targeting strategy emigrated to the lung (Bonifaz et al. 2002), mucosal resistance after a single dose of s.c. vaccination was also tested. Mice were injected either with αDEC:OVA or isotype control plus αCD40, and challenged with intranasal recombinant Vaccinia virus expressing OVA. As evaluated by lung viral titers and weight loss, only αDEC:OVA + αCD40 induced systemic protective immunity including at mucosal surfaces.
In summary, all the data obtained in initial studies with OVA model antigen provided promising aspects of the DEC-targeting strategy as a novel DC-based vaccine approach. In particular, greatly enhanced antigen-specific CD8+ T cell immunity induced by the DEC-targeting strategy drew more attention to its potential application as an efficient vaccine against diseases that require strong cytotoxic killer T cell-mediated immunity such as cancer or viral diseases. To our surprise, the DEC-targeting strategy was more efficient in the induction of antigen-specific responses by CD4+ T cells rather than CD8+ T cells when conjugated with various real pathogen antigens as compared to OVA model antigen. These unexpected results will be explained in detail with a case of plague vaccine development in Sect. 17.3 and with other cases in Sect. 17.4. The ability for cross-presentation by the DEC-targeting strategy will be discussed further in Sect. 17.4 as well.
17.3 DC-Targeting Strategy with a Known Pathogenic Antigen, Yersinia Pestis LcrV
Based on results obtained with the OVA model, our DC-targeting strategy was then investigated in various disease models using real disease-related antigens as an improved DC-based vaccine platform. Infectious disease models in which a beneficial role of cell-mediated immunity (CMI) could be investigated were considered first. Plague, caused by Y. pestis, was chosen for study among other bacterial infectious disease models due to the ongoing inquiry about host immune protection and the importance of plague as a potential biological weapon or bioterrorism threat.
17.3.1 Breadth and Efficiency of T Cell Immunity Induced by DC-Targeting Strategy
Yersinia pestis (Y. pestis) is a Gram-negative bacterium and a etiologic agent of plague or Black Death (Perry and Fetherston 1997) (see also, Chap. 7). Three clinical forms of plague occur in humans depending on the route of bacterial infection. Bubonic plague results from subcutaneous deposition of Y. pestis bacillus via infected flea bite. If untreated, bloodstream contamination may result in septicemic plague or pneumonic plague upon infection of the lungs (Perry and Fetherston 1997). Pneumonic plague produces rapid death and natural disbursement through contaminated respiratory droplets. Aerosolized Y. pestis may also be created unnaturally as a bioweapon, and is ranked in the highest category of potential biological threat agents due to its relative ease of dissemination and deadly contagiousness. The development of an effective pneumonic plague vaccine is important for military preparedness and homeland security (Inglesby et al. 2000).
There have been numerous efforts to develop human plague vaccines. The only US-licensed plague vaccine (Plague Vaccine USP), a formalin-killed whole cell preparation, was reactogenic, ineffective against pneumonic plague, and ended production in 1999. Live-attenuated plague vaccines such as EV76 are also ineffective against pneumonic plague (Meyer 1970). During pathogenesis, Y. pestis employs a macromolecular needle assembly, called the type III secretion system (T3SS), through which toxic effector proteins are injected into the cytoplasm of host cells. These effector proteins disrupt cell signaling pathways critical for phagocytosis, cytokine release, and normal immune-regulation, resulting in paralysis or induction of apoptosis (Ramamurthi and Schneewind 2002). Macrophages and dendritic cells are early target cells during infection (Marketon et al. 2005). Among numerous molecules explored as protective antigens for vaccine development, the low-calcium response virulence protein, LcrV (or V) and the capsular antigen fraction 1, Caf1 (or F1), have received much focus due largely to their immunogenicity. F1 capsule exerts anti-phagocytic activity on macrophages (Du et al. 2002). As a purified subunit antigen, F1 elicits strong antibody responses in animals and humans (Meyer et al. 1974; Andrews et al. 1996). F1-directed humoral immunity is protective against bubonic plague in animal models caused by Y. pestis strains expressing F1, but ineffective against engineered F1-negative strains or pneumonic plague models (Meyer et al. 1974; Heath et al. 1998). LcrV is multifunctional, comprising the structural tip of the T3SS injectosome (Brubaker 1991; Ramamurthi and Schneewind 2002), regulating the genetic expression and translocation of other Y. pestis effector molecules (Yops) into host cells upon contact (Brubaker 1991), and exerting immunosuppression, including the inhibition of phagocytosis, up-regulation of anti-inflammatory IL-10 (Sing et al. 2002), and down regulation of TNF-gamma (Schmidt et al. 1999). While LcrV antigen alone elicits protective immunity in animal models against infection by either F1-positive or F1-negative strains, combination of LcrV with the F1 antigen provides better protection than either subunit vaccine alone, and protects against pneumonic plague (Andrews et al. 1996; Powell et al. 2005). Many studies with subunit vaccines composed of F1 and LcrV proteins bound to alhydrogel adjuvant showed protection in small animal models of pneumonic plague and indicated that efficacy results mainly from antibody responses (Williamson et al. 1995; Anderson et al. 1996; Heath et al. 1998; Powell et al. 2005). However, aerosol infection studies with nonhuman primates (NHP) have shown inconsistent protection by the F1/V vaccines. While full protective immunity has been achieved for the cynomolgus monkey (Macaca fascicularis), F1/V vaccines have so far failed to elicit demonstrable protection in the African green monkey (Chlorocebus aethiops) (Davis et al. 1996; Overheim et al. 2012a, b; Pitt 2004).
Based on this discrepancy in NHP protection, we proposed that the induction of cellular immunity, and the recruitment of DCs in particular, might also be important for an improved pneumonic plague vaccine (Do et al. 2008, 2010) Our proposal was based on the following reasoning. Helper T cells are known to contribute to antibody-based immunity. The CD4+ T cells can secrete high levels of Th1 cytokines (e.g., IFN-γ) that activate macrophages to kill intracellular pathogens, and Y. pestis is a facultative intracellular pathogen. In addition, CD4+ T cells are cytolytic on cells bearing MHC class II-targets (Bevan 2004). This helps to explain why mice treated with only exogenous IFN-γ and TNF-α are protected against intravenous challenge by LcrV+ Y. pestis (KIM strain, ten median lethal doses), which inhibits Y. pestis growth in vivo (Nakajima and Brubaker 1993). Likewise, IFN-γ induces higher antigen-specific systemic immune responses when co-encapsulated with LcrV antigen (Griffin et al. 1998). Moreover, Stat-4 deficient mice, which produce low levels of IFN-γ, are poorly protected from Y. pestis strain GB despite producing levels of serum antibody as high as wild type controls (Elvin and Williamson 2004). Previous studies by Smiley and colleagues demonstrated that adoptively transferred Y. pestis-primed T cells could protect naïve B cell-deficient mice (μMT−/−) against intranasal challenge of attenuated Y. pestis strain KIM D27 (Parent et al. 2005), and T cells, IFN-γ, TNF-α, and NOS (nitric oxide synthase (2) play important roles in such protection (Parent et al. 2005, 2006). In a subsequent study, they demonstrate that the TNF-α and IFN-γ cytokines contribute to protection against pneumonic plague mediated by anti-F1 and anti-LcrV IgGs in the mouse model (Lin et al. 2010). These data demonstrated a role for Th1 type cellular immunity for an effective immune protection against plague. An additional role for Th17 in protection against pulmonary infection was suggested using B cell-deficient mice primed and boosted with a live attenuated Y. pestis strain D27-pLpxL (Lin et al. 2011a). Finally, a dominant CD8 T cell epitope in another Yesinia virulence protein, YopE, was recently found to confer protection against pneumonic plague (Lin et al. 2011b). Taken together, an effective plague vaccine may need to prime not only humoral immunity but also strong Th1 or Th1/Th17 type cellular immunity. However, the F1/V subunit vaccine in the presence of alhydrogel is known to induce poor cellular immunity.
Although the above-mentioned studies demonstrate participation by Th1 type cellular immunity in protecting mice against plague, these approaches have limitations for clinical application, the foremost concern being extrapolation from the mouse model to human disease. Mouse immunology is not wholly predictive of human immune protection (Mestas and Hughes 2004), and pulmonary anatomical differences confound direct comparisons for pathogenesis and treatment of pneumonic diseases (Bates and Irvin 2003). Another concern is the pathogen model and whether the strain and route of infection represent the actual disease condition intended for prophylaxis. For example, Y. pestis-specific T cells from mice are derived from in vivo/in vitro stimulation with attenuated whole organism, such as pgm- (pigmentation negative strain), which have been already reported to be inefficient against pneumonic plague in humans (Meyer 1970). Mouse challenge experiments performed using an attenuated Y. pestis strains in place of a virulent strain, such as CO92, potentially misrepresent the efficacy of the vaccine regimen. Therefore, it is a necessary to design a vaccine that could induce strong T cell immunity with clinical relevance and confer protection against pneumonic plague by a virulent strain.
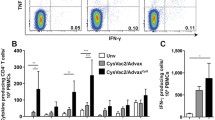
In order to induce broad and efficient T cell immunity against Y. pestis, we developed a DEC-targeting strategy that delivered LcrV protein directly DCs in situ. Full length LcrV sequence was first codon-optimized to improve expression in mammalian transfection system and then cloned in-frame into the heavy chain of anti-mouse DEC-205 mAb (αDEC:LcrV). When mice were immunized subcutaneously with a single dose of this targeted LcrV protein in the presence of poly IC and anti-CD40 (αCD40) as adjuvants, we observed LcrV-specific CD4+ T cell immunity measured by intracellular cytokine staining (ICS) for IFN-γ in three different mouse strains or C57BL/6 (H-2b), BALB/c (H-2d), and C3H/HeJ (H-2k) (Do et al. 2008). The breadth of T cell immunity induced by the DEC-targeting strategy was demonstrated by observation of two (C57BL/6 and C3H/HeJ) and three (BALB/c) distinct CD4+ T cell mimetopes in each haplotype. Induction of broad T cell immunity is critical in considering known human HLA polymorphism as well as immune evasion exploited by the Yersinia bacteria. We also observed similar breadth of T cell immunity via an intranasal (i.n.) administration of the DEC-targeted LcrV at mucosal surfaces indicating efficient DC-targeting strategy regardless of routes and target organs (Do et al. 2012) (see Sect. 17.3.4 for more information). The high efficacy of DEC-targeting strategy in the induction of T cell immunity is further supported by the fact that a single and far smaller relative dose of administration is sufficient to induce efficient T cell immunity (see Sect. 17.3.2 for more information). This contrasts with standard protein immunization which requires high concentrations of proteins or peptides, often requiring repeated injections based on strong standard CFA adjuvant to induce detectable CD4+ T cell responses. Furthermore, the DEC-targeted protein generates multifunctional T cells secreting different cytokines such as IFN-γ, TNF-α, or IL-2 at the same time with a higher frequencies when compared to a non-targeted standard protein vaccine suggesting the quality of T cells induced by DC-targeting strategy (Flynn et al. 2011). We observed that LcrV-specific T cells primed by DEC-targeted protein produced IFN-γ, TNF-α, or IL-2, and it will be interesting to confirm whether these are also multifunctional T cells as found in previous studies.
17.3.2 Efficacy: Targeting Versus Non-Targeting
Lowering the effective dosage of a vaccine is an important issue for global health, including in preparation against a bio-terrorism or bio-warfare threat. In comparison to non-targeted soluble protein vaccine, DC-targeting strategy increases the efficiency of inducing LcrV-specific CD4+ T cell responses greatly. For example, 0.1 μg of the conjugated antibody (αDEC:LcrV) per mouse induced an IFN-γ+CD4+ T cell response comprising ~0.2 % LcrV-specificity, while non-targeted soluble LcrV protein required at least 10 μg/mouse to induce detectable antigen-specific T cells responses by ICS (Do et al. 2008). Considering the actual LcrV protein amount between these two, in that the conjugated antibody is about one-third of LcrV protein (i.e., 0.1 μg αDEC:LcrV contains 0.03 μg of LcrV), the DC-targeting platform increased the efficiency of antigen-specific T cell induction more than 300-fold. Interestingly, even in using different adjuvants, DC-targeting strategy showed improved the efficiency of T cell induction. Thus, the frequency of LcrV-specific IFN-γ secreting CD4+ T cells induced by 100 μg/mouse of the non-targeted soluble LcrV protein with a standard CFA adjuvant was less than the half of that induced by 10 μg of the DEC-targeted LcrV protein in the presence of poly IC and αCD40 as adjuvants (Do et al. 2008). In general, a range of 1–5 μg of the conjugated DC targeted antibodies per mouse has been used with success in various disease models. When we tested LcrV peptide doses during a re-stimulation assay, we observed that most T cells responded to antigen at 0.2 μM peptide or higher, which also indicated efficient priming in vivo via a targeting strategy. Not only a parenteral but also an intranasal administration of the DC-targeting strategy increased the efficiency of the T cell immunity when compared to non-targeted soluble protein. Previously, we showed that at least 10 × more non-targeted soluble LcrV is required to achieve a similar pulmonary T cell response compared to the DEC-targeting antibody following a prime and boost administered intranasally (Do et al. 2012). Moreover, while the number of IL-2+ or TNF-α+ pulmonary CD4+ T cells reaches a plateau when mice are immunized with 3 and 30 μg of non-targeted soluble LcrV, this number significantly increases in mice immunized with 0.3 and 3 μg of the DEC-targeted LcrV, demonstrating again the value of antigen targeting for increasing the efficiency of inducing LcrV-specific cellular immunity (Do et al. 2012). In addition to quantity comparisons, future research should examine the improved quality of T cells induced by DC-targeting strategy, including frequency and features of LcrV-specific memory T cells.
17.3.3 DC Subsets Targeting and B Cell Immunity
The significance of antibody responses in protection against plague is well established (Williamson et al. 1995). Regarding humoral immunity induced by DC-targeting strategy, a previous study showed that 10 μg of the DEC-targeted LcrV protein with poly IC plus αCD40 as adjuvants induced antibody responses comparable to 10 μg of the F1-V subunit vaccine with alhydrogel (Do et al. 2008). Although IgG1 and IgG2b isotypes were predominant in both the DEC-targeted and the F1-V subunit vaccine-treated group as described, the Th1-dependent IgG2a and IgG2c (C57BL/6) isotypes were observed only after the DEC-targeted LcrV immunization. This reflects the influence of Th1 cellular immunity induced by DC-targeting strategy.
In the beginning, two adjuvants were used in the DEC-targeting, combining both poly IC (50 μg) and αCD40 (25 μg) in a single dose of protein vaccine administration. Later, poly IC (50 μg) alone as an adjuvant with a prime and boost strategy was explored since this is a clinically more feasible approach (see Sect. 17.4.2 for more information). As before, broad and efficient LcrV-specific Th1 CD4+ T cell responses were observed when LcrV protein was targeted to DEC-205+ DCs in the presence of poly IC alone in a prime and boost strategy (Do et al. 2010). However, under these conditions, the antibody titers were less in comparison to standard F1-V subunit vaccine administration. For example, priming and boosting a mouse with 10 μg of αDEC:LcrV plus poly IC with 4–6 week separation generated tenfold lower titer when compared to those from priming and boosting a mouse with 10 μg of the F1-V subunit vaccine on alhydrogel. Nevertheless, Th1-dependent isotypes were still observed only in the DEC-targeted group as shown previously (Do et al. 2010).
In order to extend the DC-targeting strategy in the development of an improved pneumonic plague vaccine, LcrV protein was targeted to a new DC subset other than DEC-205+ DCs, and then cellular and humoral immune responses compared. Distinct DC subsets have been characterized and, in the case of mice, there are two major myeloid DC subsets in spleen, based on the expression of CD8α and anatomic location (Shortman and Liu 2002). The DEC-205+ DC subset expresses CD8α and the endocytic receptor, DEC-205, and is located in T cell zone within lymphoid organs. The DCIR2+ DC subset does not express CD8α, but expresses another distinct uptake receptor, DCIR2, which can be recognized by anti-DCIR2 mAb. This subset is enriched in the bridging regions of the marginal zone (Dudziak et al. 2007). Previous studies showed that DEC-205+ DCs are involved in three main activities: (1) uptake of dying cells (Iyoda et al. 2002); (2) resistance against certain viral infection (Allan et al. 2003); as well as, (3) cross-presentation (den Haan et al. 2000). For DEC-205+ DCs, non-replicating antigens can be presented by MHC I, leading to CTL development, while DCIR2+ DCs more rapidly form peptide MHC II complexes (Dudziak et al. 2007). Differences were observed between targeting of these two DC subtypes.
When LcrV was targeted to DCIR2+ DCs via conjugated antibody in the presence of poly IC alone as an adjuvant, lower frequency of IFN-γ secreting T cells, but higher frequency of IL-4, IL-10, IL-13, or IL-5 secreting CD4+ T cells were observed when compared to the DEC-205+ DC targeted group. Moreover, about tenfold higher LcrV-specific antibody titers were observed in DCIR2+ DC targeted group as compared to those induced by DEC-205+ DC targeted group (Do et al. 2010). Thus, overall titers generated via prime-and-boosted with αDCIR2:LcrV plus poly IC adjuvant were comparable to those generated by standard prime-and-boost with the F1-V subunit vaccine on alhydrogel. Immunoglobulin isotypes IgG1 and IgG2b predominated, and again Th1-depdent IgG2a isotypes were highly evident in each of the DEC- and DCIR2-targeted groups, while detection was difficult in the F1-V subunit vaccinated group. Although a previous report showed distinct pathways of T cell differentiation by distinct DC subsets under in vitro condition (Maldonado-Lopez et al. 1999), our study was the first to demonstrate differences in differentiation of CD4+ T cell subsets in vivo leading to different antibody responses by distinct DC subsets. The functional aspects of the increased antibody titers induced by DCIR2+ DC targeted LcrV proteins are further described in the Sect. 17.3.5.
17.3.4 Mucosal Immunity
The value of applying the DEC-targeting strategy as a novel mucosal vaccine against pneumonic plague has been tested. In contrast to pneumonias caused by other Gram-negative bacteria, such as Klebsiella, Y. pestis pathogenesis shows two distinct stages marked by a 48 h delay in recruitment of inflammatory cells to the lungs and appearance of proinflammatory cytokines and chemokines, indicating evasion, and/or suppression of host innate immune responses (Lathem et al. 2005; Bubeck et al. 2007). An adequately designed pneumonic plague vaccine should therefore prevent this delayed proinflammatory response so as to aid bacterial clearance before systemic dissemination. Various approaches have been examined to enhance mucosal immunity, including different immunization routes and adjuvants. For example, heterologous prime/boost strategies and intratracheal/intranasal administration of vaccines each enhanced mucosal immunity as compared to standard routes of administration (Eyles et al. 1998, 2000; Baca-Estrada et al. 2000; Reed and Martinez 2006). Cholera toxin B subunit, proteosome-based adjuvant (ProtollinTM), and the Toll-like receptor five agonist flagellin have all been tested in mucosal co-delivery with various F1/V subunit vaccines (Eyles et al. 1998; Honko et al. 2006; Jones et al. 2006). Antigens co-encapsulated in microspheres or formulated in liposomes induced superior humoral immunity at mucosal surfaces compared to free antigen delivery. In these cases, survival correlated with increased antibody titers in serum and lung washes (Eyles et al. 1998, 2000; Jones et al. 2006). Furthermore, intratracheal loading of anti-F1 or anti-V antibodies was sufficient to protect mice against aerosolized Y. pestis GB strain challenge (Hill et al. 2006), underscoring the correlation between high-antibody level and protection against Y. pestis. Clinical studies, however, are needed to show the induction of antigen-specific humoral and cellular immunity at mucosal surfaces with assay of correlates for protective efficacy.
Based on background, the efficacy of the DEC-targeting in the induction of LcrV-specific CD4+ T cells at mucosal surfaces was first compared depending on administration routes. A single s.c. administration of the DEC-conjugated LcrV with combined poly IC and αCD40 adjuvants induced LcrV-specific IFN-γ and IL-2 secreting CD4+ T cells at mucosal surfaces, and the frequency of such T cells was increased with an i.n. route of administration (Do et al. 2012). When a prime and a boost strategy in the presence of only poly IC as adjuvant was applied to an intranasal route, again a strong and broad CD4+ T cell response was induced. Three different mouse MHC haplotypes were tested [H-2b(C57BL/6), H-2d(BALB/c), H-2k (C3H/HeJ)], primed and boosted in with αDEC:LcrV or isotype control in the presence of poly IC and lung samples evaluated for antigen-specific IFN-γ production by ICS. This prime-boost study identified two (C57BL/6 and C3H/HeJ) or three (BALB/c) T cell mimetopes that were broad as much as the previously described single dose of αDEC:LcrV + poly IC + αCD40 (Do et al. 2008). These data suggest that antigen processing by lung-associated DCs is similar to that of spleen DCs. Since DEC-205 KO (knock-out) mice produced no detectable antigen-specific CD4+ T cell responses either by parenteral or intranasal routes, it is clear this effect was dependent on the expression of DEC-205 (Do et al. 2012). The mechanism of how DEC-targeting initiates immune responses at mucosal surfaces will be further investigated to better establish the rationale of future mucosal vaccine development. Although previous studies showed that most activated T cells migrated into non lymphoid tissues such as lung, liver, and kidney (Harris et al. 2002; Lefrancois and Puddington 2006), we show here that mucosal priming can induce local cell-mediated immunity. While the effector mechanisms of such protection are undefined, it is evident that enhanced antigen presentation by pulmonary DCs to resident and/or recruited memory—and naive T cells can be beneficial in vaccine design (Harris et al. 2002). Moreover, expression of inflammatory chemokines from effector T cells is critical for the early recruitment of supportive immune cells including immature DCs and natural killer cells (Lefrancois and Puddington 2006).
Different combinations of route in the prime/boost strategy were tested to compare the efficiency of mucosal immunity and found that interestingly priming s.c. and boosting i.n. induced higher frequency of IFN-γ secreting mucosal T cells when compared to s.c. priming and boosting (Do et al. 2012). It might be that an intranasal boost mimics a natural aerosol exposure to Yersinia and, hence, a mucosal boost would be preferable. Priming and boosting intranasally might be the optimal strategy for a vaccine as it induced the strongest antigen-specific cellular immunity at mucosal surfaces. It is advantageous that even after a single parenteral administration, the DEC-targeted protein could prime the immune system sufficiently enough to efficiently recall and activate antigen-specific T cells at mucosal surfaces (Do et al. 2012). Thus, it may be beneficial to develop the DEC-targeted protein vaccine with a mucosal delivery route to enhance T cell immunity at mucosal surfaces.
Regarding humoral immunity at mucosal surfaces, BAL (bronchoalveolar lavage) fluid was collected for comparison of immunoglobulin content (Do et al. 2012). The BAL fluid contained LcrV-specific antibodies from a single s.c. immunization with αDEC:LcrV + poly IC, but prime and boost immunization further enhanced the mucosal antibody responses tenfold. A broad spectrum of IgG isotypes (IgG1, IgG2a, IgG2b) were detected following immunization with αDEC:LcrV, but IgG2a was not found in the BAL fluid from mice immunized with the F1-V subunit vaccine. Intranasal administration of the DEC-targeted LcrV protein increased the BAL antibody titers about 10 to 30-fold relative to the s.c. route. The isotypes of antibody were examined and IgG1 and 2b were consistently predominant while IgG2a isotype was the strongest in the DEC-targeted group. This concurs with other findings that protection against bacterial pneumonias is polarized for the IgG2a isotype (Corbeil et al. 1997; Mills et al. 1998). Interestingly, IgA responses were detected in BAL fluid following i.n. immunization but not with s.c. immunization. This suggests that local immunization is required to promote mucosal-specific antibodies such as IgA. Anti-LcrV IgG antibodies were found in the upper respiratory tract as well as in nasal washes both in the DEC-targeted and the F1-V subunit vaccine-treated group, but IgA responses in nasal washes were detectable only in F1-V plus alhydrogel immunized mice. In addition, after intranasal administration, serum anti-LcrV antibody titers were the highest in the F1-V plus alhydrogel immunized group, demonstrating again the superior efficacy of the F1-V subunit vaccine in the induction of humoral immunity.
17.3.5 Vaccine Efficacy: Challenge with Human Pathogen
For a challenge study using a virulent Y. pestis human pathogen, CO92 strain with intranasal inoculation of up to 25 μl 105 cfu (colony forming units) or ~100 LD50 was used to model the pneumonic form of plague. This is a high dose and a challenging method since Y. pestis CO92 requires special procedures as a CDC Category a agent. In case of the DEC-targeting strategy project, challenge experiments were performed at Public Health Research Institute (PHRI, Newark, NJ), a state of the art, licensed biological containment facility with approved security and safety procedures. All reagents for the vaccination were sent to the PHRI and thus, wherein the entire process from vaccination to challenge and related procedures were performed. In order to check proper priming before the challenge, random blood and spleen samples were submitted to the laboratory where LcrV-specific antibody titers and LcrV-specific CD4+ T cell responses were confirmed by ELISA and ICS, respectively. After confirmation of the priming, intranasal challenge was performed and mice were observed twice a day for 14 days and the survival rate was reported.
The standard F1-V subunit vaccine with alhydrogel treatment was included as a positive control and a PBS treated group was included as a negative control. Various approaches were designed to test individual factors in conferring protection: (1) dosage, (2) targeting versus non-targeting, (3) administration routes, (4) adjuvants (poly IC vs. Ampligen, see more information in Sect. 17.4.2), as well as (5) heterologous priming and boosting strategy. Through all these efforts, the goal was to optimize the targeting strategy for protective efficacy for future clinical trials. The study was designed to answer the critical questions whether to induce combined cellular and humoral immunity, to select a clinically acceptable adjuvant, and to demonstrate protective efficacy against pneumonic plague with a real human pathogen. Relevant findings were reported in the following two published papers.
In one study, the relative protection efficacy between two distinct DC subsets targeting was tested (Do et al. 2010). Groups of mice were primed and boosted intraperitoneally (i.p.) either with DEC- or DCIR2-targeted LcrV in the presence of poly IC at 4–6 weeks interval, then mice were challenged with 100 LD50 Y. pestis CO92 intranasally 6 weeks after the boost. F1-V + alhydrogel protected 100 % and αDCIR2:LcrV + poly IC protected 90 % of the mice, while αDEC:LcrV alone protected only 50 %. Interestingly, 50 % of mice survived after immunization with a single dose of αDCIR2:LcrV plus poly IC, as opposed to 0 % after a single dose of αDEC:LcrV. Similar findings were observed in a duplicate experiment and there was a clear correlation between protection and serum antibody levels.
In another study, protection efficacy of a mucosal DEC-targeted protein was tested as compared to a subcutaneous route of administration (Do et al. 2012). Mice were primed and boosted with αDEC:LcrV plus poly IC, either via an intranasal route or a subcutaneous route. Subcutaneous immunization with the F1-V subunit vaccine plus alhydrogel was included as a positive control. Six weeks after the boost, mice were inoculated intranasally with 100 LD50 of virulent Y. pestis CO92 strain. As expected, immunization with the F1-V conferred 100 % protection while all mice immunized with PBS did not survive the challenge. Interestingly, an intranasal route of αDEC:LcrV with poly IC protected 56 % of the mice while a subcutaneous route only protected 33 % of the mice. Moreover, a single intranasal priming with αDEC:LcrV with adjuvant protected 33 % while a single subcutaneous showed no impact on survival rate.
From these two studies, we have learned a few lessons in the development of plague vaccine. First, mice might not be the optimal animal model for testing the value of cellular immunity against pneumonic plague since we still find a strong correlation between survival rate and serum antibody titers. In particular, a high challenge dose such as 100 LD50 Y. pestis CO92 strain disguises the benefit of cellular immunity since mice cannot survive without antibodies regardless of cellular immunity induction. Further research using nonhuman primates may help discriminate the role for CMI given the fact that the F1-V subunit vaccine, which induces strong humoral immunity but poor cellular immunity, protects mice and cynomolgus macaques, but so far has failed to adequately protect African green monkeys, regardless of Ab titers measured by ELISA (Overheim et al. 2012a, b; Pitt 2004). Second, there appears to be a benefit by using the mucosal delivery of the DEC-targeted protein vaccine. Our findings show potential roles of enhanced mucosal T and B cell immunity induced by the mucosal delivery of the DEC-targeted protein vaccine when compared to a subcutaneous delivery group since serum antibody titers following vaccination were similar between these two groups (Do et al. 2012) (and unpublished data). A subsequent study will focus on demonstrating a more direct involvement of mucosal T and B cell immunity against pneumonic plague. In addition, further studies to investigate whether a mucosal delivery of DCIR2+ DCs with LcrV protein or a combination therapy of distinct DC subsets targeting (DEC-targeting + DCIR2-targeting) might improve the protection efficacy will provide informative knowledge of the DC-targeting strategy in plague vaccine development.
17.4 Lessons from DC-Targeting Strategy
As described above, the DC-targeting strategy has been under intensive investigation as an improved plague vaccine. Beyond this, the DC-targeting platform has also been applied to various other disease models, including infectious viral disease and cancer, to further explore unique features and efficacy. The potential of DC-targeting strategy as a novel prophylactic approach is described in Sect. 17.4.1. Although these preclinical data obtained from animal models provide background and rationale for future clinical applications, we understand there remains a large gap between experimental conditions and actual human disease. Therefore, it is necessary to make efforts to extend these findings to real human pathogenic situations, as described further in Sect. 17.4.3. In our estimation, such translational science begins with the selection of an adjuvant for the DC-targeting platform that is acceptable and well suited for human use (see Sect. 17.4.2 for more information).
17.4.1 Viral/Cancer Model
HIV/AIDS is a major global infectious disease that could benefit from the enhanced T cell immunity induced by the DC-targeting, or more specifically by the DEC-targeting strategy. Existing HIV/AIDS vaccines include prime and boost strategies with naked plasmid DNA and recombinant Vaccinia-Ankara and adenoviruses (Emini and Koff 2004). Since HIV-specific CD4+ T cells help to produce functional HIV-specific CD8+ T cells (Lichterfeld et al. 2004), and HIV patients with stronger CD4+ T cell immunity have shown better clinical outcomes (Rosenberg et al. 1997), the DEC-targeting strategy has been adapted to explore an improved HIV/AIDS vaccine.
The Gag protein was selected as target for designing the anti-DEC-205 mAb due to previously known protective capacity in rhesus monkey studies as well as in clinical cases (Novitsky et al. 2003; Zuniga et al. 2006; Liu et al. 2009). Thus, gag p24 or gag p41 was conjugated to the DEC-targeting antibody. Initial tests used graded doses of αDEC:p24 or αDEC:p41, immunized in the presence of poly IC and αCD40 as adjuvants, and gag-specific T cell responses were measured by ICS following 6 h re-stimulation with peptide pools. This DEC-targeting method was 100 times more efficient for induction of gag-specific IFN-γ+CD4+ T cell responses as compared to a non-targeted soluble gag protein immunization (Trumpfheller et al. 2006). The DEC-targeting strategy was also superior to induce gag-specific IFN-γ+CD4+ T cell responses in comparison to other immunization strategies such as a recombinant adenovirus-gag vaccine or prime-boost administration with a plasmid DNA vaccine. Similar to the plague vaccine studies (see above Sect. 17.3), the DEC-targeting strategy enhanced the breadth of gag-specific CD4+ T cell responses such that two or three mimetopes were identified in different MHC haplotypes as well. Also verified in HIV model were the induction of long-term memory responses and mucosal resistance through a single s.c. immunization as described previously in OVA model. Briefly, even after 19–30 weeks (4−7.5 months) after a single s.c. immunization of the DEC-targeted gag protein, enhanced effector memory CD4+ T cells were induced which could proliferate and secrete IFN-γ upon re-challenge with reactive peptides. Importantly, neither control isotype nor the plasmid DNA vaccine achieved this response. In addition, mice vaccinated with the DEC-targeting gag protein plus adjuvants showed mucosal resistance against a challenge with recombinant vaccinia-gag, and such protection was ablated in IFN-γ receptor knockout mice or by depleting CD4+ T cells (Trumpfheller et al. 2006).
This DEC-targeting strategy was further examined to discern the potential of using only poly IC as an adjuvant (Trumpfheller et al. 2008). Finding an optimal adjuvant with features, such as efficient immune stimulation while also being suitable for human use, is important in developing an improved vaccine (see Sect. 17.4.2 for more information). Thus, poly IC without αCD40 was tested independently since αCD40 is not clinically feasible. As a single s.c. or i.p. immunization, the DEC-targeting antibody with poly IC alone was found not to induce detectable antigen-specific IFN-γ secreting T cell responses. However, when applied in a 4 week prime-and-boost strategy, poly IC alone (50 μg/mouse/injection) induced ~0.5–6 % of gag-specific IFN-γ+CD4+ T cell responses (Trumpfheller et al. 2008). Furthermore, the higher frequency of three cytokine (i.e., IFN-γ, IL-2, and TNF-α) producing gag-specific CD4+ T cells was observed with this strategy as opposed to one or two cytokine producing CD4+ T cells, and these T cells remained stable for 2–7 weeks after the boost. When vaccinia-gag was challenged via airway, the prime-and-boost DEC-targeting + poly IC again provided reasonable protection (Trumpfheller et al. 2008). Taken together, this study demonstrated the efficacy of the DEC-targeting strategy for the development of a novel therapeutic approach in HIV/AIDS vaccine through enhanced CD4+ T cell immunity. In addition, these findings clearly showed the potential of poly IC as sole adjuvant in the DEC-targeting for clinically feasible application, specifically in a prime-boost regimen. This work therefore sets the stage for future development of various DEC-targeting strategies. Detailed data on the DEC-targeted gag protein in nonhuman primates and preclinical studies will be given in Sects. 17. 4.2 and 17.4.3.
Cancer is a complicated and challenging disease for developing a vaccine. Various factors must be considered together in order to achieve desired clinical outcomes, such as breaking the “self” tolerance of tumor antigens, overcoming the barriers of immune checkpoints, facilitating the accessibility of immune cells to the tumor beds, while at the same time homing treatment to the lymphoid tissues, and blocking tumor evasion mechanisms. Since successful cancer vaccination must satisfy these many constraints, it is reasonable strategy to combine more than one approach. The DEC-targeting strategy has been examined to in the aim of solving any of these questions. Given the fact that there are fewer cancer phase trials applying T cell immunity compared to antibody-based immunity (i.e., HerceptinTM and RituxanTM), the demonstrated ability of the DEC-targeting to induce efficient T cell immunity makes it ready for application in the clinic. Ongoing efforts are described in below Sect. 17.4.3.
To enhance immunogenicity against tumor antigens, specific antigens are directly delivered to DCs in vivo via a DEC-targeting antibody. Survivin, mesothelin, or human epidermal growth factor receptor (HER2), were examined in three separate studies as tumor antigens targeted to the DCs. Survivin is non-mutated self-antigen which is expresses in thymus and regulates early thymocyte development (Okada et al. 2004). It shows anti-apoptotic activity, and overexpression of survivin is usually found in fast-growing transformed cell lines as well as in human cancer cells (Tamm et al. 1998; Islam et al. 2000; Grossman et al. 2001). Survivin enforces that the cell cycle to pass G2/M phase checkpoint, leading to oncogenesis (Li et al. 1998). There are correlations between survivin overexpression and unfavorable clinic outcomes (Islam et al. 2000; Altieri 2003; Blanc-Brude et al. 2003). Enhanced survivin-specific CD4+ T cell immunity was reported in the DEC-targeting, especially when human survivin was conjugated to the mouse DEC-targeting antibody (Charalambous et al. 2006). Such a xenogeneic form of tumor antigen could enhance CD4+ T cell immunity and, moreover, depletion of regulatory T cells (Treg), further enhanced anti-tumor immunity (Charalambous et al. 2006). The significance of this study is the demonstration of strong CD4+ T cell immunity to non-mutated self-antigen as induced by the DEC-targeting, which is otherwise rarely observed in other approaches.
Mesothelin is also found to be over-expressed in various human cancers, specifically, lung and pancreatic adenocarcinomas and ovarian cancers (Chang and Pastan 1996; Argani et al. 2001; Ho et al. 2007). Although biological functions of mesothelin need further study, it is known as a tumor differentiation antigen. Interestingly, it shows limited expression in normal tissues while being over-expressed in cancers as a non-mutated protein. For this reason, it has been useful therapeutic target in cancer immunotherapy (Bera and Pastan 2000; Hassan and Ho 2008). When pancreatic cancer patients were vaccinated with GM-CSF transduced allogenic pancreatic tumor cell lines, mesothelin was cross-presented, leading to the induction of cytotoxic T cell immunity (Thomas et al. 2004). This finding showed that our immune system could break the tolerance of “self” tumor antigens and induce anti-tumor immunity. In order to enhance such anti-tumor immunity, human mesothelin was conjugated to the mouse DEC-targeting antibody, and improved mesothelin-specific CD4+ T cell immunity was observed (Wang et al. 2009). Moreover, antigen-specific IFN-γ+CD8+ T cell immune responses were observed by utilizing an ex vivo proliferating assay instead of the conventional (ICS) after 6 h re-stimulation with reactive peptides (Wang et al. 2009). The cross-presentation of proteins targeted via anti-DEC-205 fusion antibody is further described in Sect. 17.4.3.
HER2 is a tyrosine kinase growth factor receptor and its overexpression, which is closely associated with unfavorable clinical consequences, is found in 20–40 % of invasive breast carcinoma and about 70 % in ductal carcinoma patients (Slamon et al. 1987; Hynes and Stern 1994). Although improved diagnostic and therapeutic agents are utilized currently (i.e., HerceptinTM), breast cancer still remains a high-mortality cancer in many countries. HER2 is a non-mutated self antigen and anti-HER2 T and B cell immunity has been reported in cancer patients (Disis et al. 1994, 2000). However, the soluble form of HER2 protein used as a vaccine is known to be inefficient in conferring protective immunity (Taylor et al. 1996; Dela Cruz et al. 2003). Various methods targeting HER2 to antigen presenting cells, including DCs, have also been tested. By utilizing B7-1/2 (Sloots et al. 2008), CD11c (Wei et al. 2009), CD40 (Kim et al. 2010), mannose (Thomann et al. 2011), or Fcγ receptor (Zizzari et al. 2011), the targeted HER2 could induce enhanced immunity when compared to non-targeted soluble protein treatments in mouse model. Development of breast cancer vaccine by utilizing the DEC-targeted HER2 protein represents one of these efforts, and its unique features (i.e., breadth, quality, and quantity of antigen-specific T cell immunity) described so far should be emphasized. When HER2 was targeted to the DEC-targeting antibody, plus poly IC as adjuvant, high quality, and the quantity of antigen-specific CD4+ and CD8+ T cells were induced (Wang et al. 2012). In particular, the HER2 specific CD4+ T cells induced by the DEC-targeting could cross-react to Neu, a homologous rat protein. This implied “epitope spreading”, which could be further exploited in protecting from other HER2 negative variants. Moreover, this cross-reaction allowed xeno-primed T cells (i.e., T cells induced by human HER2 targeted delivery) to be tested in transplantable neu-expressing tumor challenge model, where HER2-specific CD8+ T cells played more important role in delaying tumor growth as well as lengthening the survivial rate (Wang et al. 2012). Taken together, this novel approach is expected to be further tested in cancer patients.
17.4.2 Adjuvants
The combined poly IC (polyinosinic:polycytidylic) and αCD40 was used as a combined adjuvant in initial trials of the DEC-targeting strategy (Bonifaz et al. 2004; Charalambous et al. 2006; Trumpfheller et al. 2006; Do et al. 2008). Given that αCD40 cannot be used in clinical setting, due to the concordant induction of massive apoptotic death of immune cells, the efforts to utilize poly IC alone in a prime and boost strategy were previously explored. Poly IC is a synthetic double strand RNA that mimics viral infection and delivers signal through Toll-like receptor (TLR) 3 (Alexopoulou et al. 2001; Kawai and Akira 2007). A main reason to choose poly IC as an adjuvant in the DEC-targeting strategy is that it is clinically acceptable adjuvant, and it has been used in cancer and HIV patients with efficacy by secreting large amount of interferon mimicking viral infection (Krown et al. 1985; Lampkin et al. 1985; Stevenson et al. 1985; Salazar et al. 1996). Another important feature is that poly IC effectively induces cellular immunity. In the mouse model, various TLR ligands such as MALP-2 (for TLR2/6), Pam3Cys (for TLR1/2), LPS (for TLR4), R-848 (for TLR7/8), CpG (for TLR9), or poly IC (for TLR3) were tested as adjuvant in the DEC-targeting antibody immunization (Longhi et al. 2009). The data showed that poly IC was the superior adjuvant in the induction of antigen-specific CD4+ T cell responses. Section 17.5.2 contains more information on the proposed mechanism. The efficacy of poly IC for inducing T cell immunity was further confirmed in non-human primate studies (Stahl-Hennig et al. 2009). Unlike mice, poly IC is not a strong inducer of type I IFN-α in primates and it might be due to its fast degradation by serum nucleases which are known to be more abundant in primates than in rodents (Nordlund et al. 1970). Thus, naked poly IC was examined alongside poly ICLC (poly IC with poly-L-lysine and carboxymethylcellulose), which is 5–10 times more resistant to RNAase hydrolysis in primate serum and it showed a large amount of type I IFN induction (Levy et al. 1976; Stephen et al. 1977). Briefly, rhesus macaques were immunized s.c. either with KLH or a low dose (10 μg/animal) of poor immunogenic human papillomavirus (HPV)16 capsomers in the presence of either poly IC, poly ICLC, or CpG-C as adjuvant. Although both poly IC and poly ICLC could induce antigen-specific T cell immunity, the highest proliferative responses were observed with 2 mg/animal of poly ICLC treatment (Stahl-Hennig et al. 2009). Moreover, similar dose of CpG-C, a control adjuvant, in similar aqueous solution (not in water-in-oil emulsion) as poly ICLC did not induce HPV16-specific T cell immunity while together they did induce antibody responses (Stahl-Hennig et al. 2009). This data demonstrates that poly IC (or poly ICLC) can be used in primates as an effective adjuvant, particularly for inducing antigen-specific cellular immunity, which might benefit an improved vaccine. In contrast, two well-known approved adjuvants in humans, alum and monophosphoryl lipid A (MPLA), are known to be limited in their capacity to induce cellular immunity.
Recently, interesting results were obtained in healthy volunteers using a subcutaneous route of poly ICLC, contrary to not the intramuscular (i.m.) route as previously employed (Caskey et al. 2011). First, a single s.c. dose of 1.6 mg poly ICLC was well tolerated in phase I studies. Secondly, transcription profiling, using RNAs from whole blood samples obtained from the volunteers before and after the poly ICLC vaccination, showed the up-regulation of genes involved in many different innate immunity pathways, such as IFN-regulated genes, inflammasome-associated genes, NF-κB, DC maturation and antigen-presentation (Caskey et al. 2011). Interestingly, such profiling was very similar to and overlapped with that of attenuated yellow fever viral vaccine YF17D (Caskey et al. 2011), a successful vaccine proven in humans and showing the efficacy of poly IC as a true viral mimic adjuvant.
Poly IC12U (AmpligenTM) is another modified form of poly IC which shows instability in serum, resulting in low toxicity in humans, and thus, has been considered a suitable adjuvant for human vaccines (Thompson et al. 1996; Ichinohe et al. 2007). As mentioned in the nonhuman primate study, poly IC12U also induced antigen-specific T cell immunity, but it required higher dose (6 mg/animal) to show similar effect as poly ICLC (2 mg/animal) (Stahl-Hennig et al. 2009). However, when it was used as an adjuvant in the DEC-targeting strategy in the development of plague vaccine (see Sect. 17.3 for more information), we observed neither adjuvant activity nor protective efficacy (Do unpublished data).
Poly IC was also tested in a mucosal delivery of the DEC-targeting strategy in the development of plague vaccine (see Sect. 17.3.4 for more information) (Do et al. 2012). Although poly IC had been utilized as a mucosal adjuvant before, most of studies showed an adjuvant effect by poly IC in inducing antibody responses in non-targeted protein vaccine or inactivated viral vaccine (Ichinohe et al. 2005; Partidos et al. 2005). However, in the DEC-targeting strategy, poly IC particularly demonstrated the induction of enhanced cellular and humoral immunity at mucosal surfaces (Do et al. 2012). Moreover, when other adjuvants such as LPS (lipopolysaccharide) from Escherichia coli 055:B5 were used as a mucosal delivery of the DEC-targeting, it showed tenfold weaker activity in inducing Th1 immunity as compared to poly IC. Another frequently used adjuvant, the TLR5 agonist flagellin, did not induce pulmonary cellular immunity, either with the DEC-targeted vaccine or F1-V subunit vaccine treatments (Do et al. 2012) (data not shown).
Beyond dsRNA, TLR4 agonists have also been recently tested as adjuvants in the DEC-targeting strategy (Pantel et al. 2012). LPS is a well-known TLR4 agonist and induces DC maturation as well as strong humoral immune responses (Steinhagen et al. 2011). To reduce toxicity while maintaining immunogenicity of the original LPS, chemically modified versions were developed for use as vaccine adjuvants. MPLA, a low-toxicity derivative of lipid A from Salmonella minnesota R595 lipopolysaccharide, is one derivative that has gained popular use for this purpose (Mata-Haro et al. 2007). MPLA in alum is currently being used as adjuvant in hepatitis B and human papilloma vaccine (Dubensky and Reed 2010). Glucopyranosyl lipid A (GLA) is a new synthetic TLR4 agonist and already demonstrated to be safe during phase I trial with Fluzone influenza vaccine (Coler et al. 2010). A stable oil-in-water emulsion called GLA-SE has shown enhanced antibody responses as well as Th1-type cellular responses in mouse and cynomolgus monkey models (Baldwin et al. 2009; Coler et al. 2010). Interestingly, when GLA-SE was included in the DEC-targeting strategy, it induced DC maturation in vivo as early as 4 h after immunization, demonstrating its utility as a novel adjuvant in the induction of antigen-specific antibody responses and Th1-type cellular immunity (Pantel et al. 2012). Altogether, these findings establish that clinically feasible TLR3 and TLR4 agonists can be incorporated into the DEC-targeting strategy for inducing enhanced antigen-specific humoral and cellular immunity. Continuous efforts on exploring improved adjuvants optimal for the DEC-targeting strategy should be encouraged.
17.4.3 Pre-clinical Study
Various efforts have been applied to extend the DEC-targeting strategy into clinical testing. Some of the previous studies utilized human instead of mouse antigen in the DEC-targeting strategy, and such targeted xenogenic protein enhanced overall immunogenicity. Broad human antigen-specific CD4+ T cell immunity on various MHC haplotypes was observed with human antigen peptides pools as well (Charalambous et al. 2006; Wang et al. 2009, 2012).
Anti-human DEC-205 targeting antibody was also generated and tested for comparison to anti-mouse DEC-205 targeting antibody (Guo et al. 2000). In contrast to the mouse counterpart, human DEC-205 is a transmembrane protein with 10 C-type lectin domains having 77 % gene sequence homology (Jiang et al. 1995). Anti-human DEC-205 targeting antibody has been utilized in various settings to gauge its potential for clinical application. The DEC-targeting antibody was found to be superior for inducing antigen-specific IFN-γ secreting CD8+ T cell responses when PBMCs (peripheral blood mononuclear cells) or PBMC-derived DCs and T cells from HIV-infected patients were targeted with HIV gag p24 protein through either anti-human DEC:p24 (αhDEC:p24), αhDC-SIGN:p24, αhMMR:p24, or isotype control antibodies (Bozzacco et al. 2007). In addition, broad CD8+ T cell responses were observed with the DEC-targeting approach. Specifically, eight different gag peptides were identified from 11 patient samples after injection of αhDEC:p24 targeted protein (Bozzacco et al. 2007). These data suggest that through DEC-205 targeting, DCs can enhance cross-presentation of HIV gag p24 protein to broad gag-specific CD8+ T cells leading to IFN-γ secretion. Considering naturally high polymorphic MHC products in human antigen presentation, these results underscore the efficiency and the potential of the DEC-targeting strategy as a novel protein-based vaccine in HIV/AIDS.
In further studies, two novel mouse systems were utilized in demonstrating human DEC-205 targeting strategy as an effective vaccine approach. First, human DEC-205 targeted antibody (MG38.2 Ab) (Guo et al. 2000) was conjugated with nuclear antigen one of Epstein-Bar-virus (EBV, EBNA1) and used as a vaccine against both EBV primary infection and EBV-associated malignancies (Gurer et al. 2008). Briefly, 2–5 days old NOD/LtSz-scid IL2Rγnull (NOG) mice were irradiated and injected with CD34+ human hematopoietic stem cells obtained from human fetal liver. Successful reconstitution was confirmed in the mice after 10–12 weeks and the mice were primed and boosted with αhDEC:EBNA1 in the presence of poly IC as an adjuvant. Two months after the DEC-targeting, EBNA1-specific IFN-γ+CD4+ T cells and antibody responses were observed (Gurer et al. 2008). In another study, a transgenic mouse expressing human DEC-205 receptor on mouse CD11chi DCs was generated and cellular and humoral immune responses induced by a newly synthesized anti-human DEC-205 targeting antibody (3G9 mAb) conjugated with HIV gag p24 protein were examined (Cheong et al. 2010). Again the DEC-targeting showed enhanced antigen-specific CD4+ T cell responses when compared to non-targeted or DC-SIGN-targeted protein approach, as mentioned above (Bozzacco et al. 2007). In addition, the ability for cross-presentation of the DEC-targeting antibody was confirmed where HIV gag-specific CD8+ T cells from both long-term nonprogressor patient and chronically HIV-infected patient proliferated and secreted significant amount of IFN-γ (Cheong et al. 2010). Thus, efficient cross-presentation was demonstrated in the DEC-targeting delivery of antigen.
This newly synthesized anti-human DEC-205 targeting antibody (3G9 mAb) was examined in nonhuman primates as well (Flynn et al. 2011). A heterologous prime-boost strategy, known to increase immunity when compared to a homologous prime-boost regimen, was investigated via the DEC-targeting strategy. Rhesus macaques were primed s.c. three times either with 60 μg of the non-targeted HIV gag p24 protein or the DEC-targeted gag p24 in the presence of 1 mg of poly ICLC at 0, 8, and 27 weeks. All animals were s.c. boosted with New York vaccinia virus (NYVAC) containing HIV Gag/Pol/Nef (Gomez et al. 2007) at 31 weeks after the protein priming (i.e., at 58 weeks). Priming alone with NYVAC-HIV Gag/Pol/Nef as a control group induced low to undetectable gag-specific T cell responses but showed enhanced cellular immunity after boost. Either non-targeted or DEC-targeted HIV gag p24 protein could induce gag-specific CD4+ and CD8+ T cell responses as well as antibody responses (Flynn et al. 2011). However, two interesting and valuable results could be drawn from this study regarding the DEC-targeting strategy. In comparison to non-targeted immunization, the DEC-targeting approach was more effective in cross-presentation as well as in generating multifunctional CD4+ and CD8+ T cells. In particular, when isolated CD8+ T cells from the animals were examined for proliferative capacity, for multi-cytokine secretion (i.e., IFN-γ, IL-2, and TNF-α), and for long-term memory responses, the DEC-targeting showed enhanced efficacy in all aspects in comparison to non-targeted protein vaccine (Flynn et al. 2011). Such multi-functional T cells correlated with better protection against Mycobacterium tuberculosis, Leishmania major, and vaccinia virus in mouse models (Trumpfheller et al. 2006; Darrah et al. 2007; Lindenstrom et al. 2009).
The findings from nonclinical testing including nonhuman primate studies have clearly demonstrated the efficacy of the DEC-targeting as a novel protein vaccine useful for future clinical application. To this objective, a phase I study to evaluate the safety and immunogenicity of the DEC-targeted HIV gag p24 protein with poly ICLC is on-going with 45 healthy volunteers aged 18–60 at The Rockefeller University Hospital (Unpublished data: Personal communication with and oral presentation by Dr. Trumpfheller C., at the 12th International Symposium on Dendritic Cells, October 7–11, 2012, Daegu, Korea). To date, antigen-specific cellular and humoral immune responses with low and transient reactogenicity were reported and additional valuable results are anticipated.
17.5 Summary: Future Direction for DC-Targeting Strategy
In this chapter, we have described the unique features, efficacy, and benefits of the DC-targeting vaccine strategy in various disease models and summarized data that demonstrate it to be an efficient and novel vaccine approach, ready for translation to clinical testing for treating human disease. Beyond the examples introduced in Sect. 17.4.3 as preclinical efforts, the following comments highlight important areas of focus for future investigation in further development of DC-targeting as a viable human vaccine platform.
17.5.1 Human DC Subsets and Targeting
Human DC subsets and mouse DC subsets are not identical (Palucka et al. 2010). This means that a better understanding of human DC subsets is required to extend the DC-targeting strategy into clinical application. Instead of addressing the diversity and biology of human DC subsets in detail, we instead focus here on DEC-205+ DCs which are the human equivalent of mouse CD8α+ DCs (Palucka et al. 2010; Villadangos and Shortman 2010). Previous studies suggested that CD141+ human DCs (also known as BDCA3+ DCs) might be the counterpart of mouse CD8α+ DCs based on four similarities: (1) the expression of chemokine receptor XCR1 (Bachem et al. 2010); (2) the expression of transcription factors Batf3, and IRF-8, and a lack of IRF-4; (3) the expression of TLR3, but not TLR7 and the secretion of IL-12 upon activating through TLR3; and (4) the capacity of phagocytosis of dead cells and cross-presentation (Jongbloed et al. 2010; Poulin et al. 2010). However, differences still exist. For example, CD141+ DCs do not express TLR9 although CD8α+ DCs do, and different signaling is required for IL-12 production from CD141+ DCs as compared to mouse CD8α+ DCs (Jongbloed et al. 2010; Poulin et al. 2010). In addition, the ability of DC cross-presentation in mouse is known to be strictly limited to CD8α+ DCs (Hildner et al. 2008; Lin et al. 2008), but human DC subsets employ more than just CD141+ DCs for cross-presentation (Schnorrer et al. 2006; Segura et al. 2009). The hurdles of characterizing human equivalents of mouse DC subsets arise not only from species intrinsic discrepancy, i.e., anatomical differences and developmental process so that few markers are shared between human and mouse DCs, but also from a paucity of relevant human lymphoid DC supply, caused by ethical and logistical difficulties faced over the past decade.
Previously, human DC subsets principally in lymphoid organs were investigated (Granelli-Piperno et al. 2005). This aligned with our interest in using DEC-205+ DCs for future DC-targeting strategy vaccines. In addition, these cells are found to be abundant in T-cell areas of human lymph nodes and they co-express CD11c as well as MHC class II, both critical for effective targeting. In the case of human spleen, another study showed that CD11c+ DCs were located in T cell-, B cell-, and marginal zones, and most were found to be immature by lacking CD86 and CD83 receptors (McIlroy et al. 2001). When we used CD205 (DEC-205) to identify human spleen DCs, we found that most DEC-205+ DCs are CD11c+ and immature, consistent with the prior observation (Pack et al. 2008). Interestingly, in addition to known tropism for white pulp, these DEC-205+ DCs were closely associated with cells displaying the mucosal address in cell adhesion molecule, MAdCAM. Moreover, human spleen DEC-205+ DCs were found both in T cell- and non-T cell areas, unlike mouse splenic counterparts found only in T-cell areas. This unexpected finding warrants further investigation to define the natural localization of DEC-205+ DCs. Such differences underlie the need to perform efficacy studies of DEC-targeting strategy using human DC subsets, and findings will guide the design of a fully human DC-targeting strategy. This information is critical since DCs exist in distinct functional states depending on activation/maturation signals derived from microenvironments, such as infectious microbes, tissue-derived cytokines, and other immune cells, which controls the target, timing, and quality of adaptive immunity. Multiple parameters should be considered in designing human DC-targeting strategy, including the desired type of immune protection to be induced (cellular vs. humoral immunity), the tissue distribution and receptors of various DC subsets, and activation signals based on TLRs expressed for optimal choice of adjuvant.
17.5.2 Mechanism
While ample publications show the efficacy of the DEC-targeting strategy in various disease models, few reports address the basic mechanisms at work in DC-targeting. Two studies in particular are worth noting here as leads for further investigation to uncover downstream mechanisms underlying an improved DC-targeting strategy.
The first study investigated how poly IC works as an effective adjuvant for DEC-targeting. As shown in Sect. 17.4.2, poly IC was found to be a superior adjuvant for DEC-targeting in inducing Th1 type cellular immunity as compared to other known TLR ligands (Longhi et al. 2009). Poly IC was shown to be the most effective inducer of type I interferon (IFN) through mda5 (melanoma differentiation-associated protein 5) and TLR3. A blocking experiment showed that type I IFN was in fact essential for DC maturation and CD4+ T cell development. Specifically, type I IFN acted in vivo directly on DCs, leading to up-regulation of DC co-stimulatory molecules, and DCs exposed to type I IFN became better inducers of mixed lymphocyte reactions. In addition, poly IC induced Th1 CD4+ T cell responses primarily through type I IFN, but interestingly type II IFN (IFN-γ) or IL-12 were not necessary. Lastly, bone-marrow chimera experiments demonstrated that the main in vivo sources of type I IFN were nonhematopoietic radio-resistant cells as well as monocytes and DEC-205+ DCs, and the adjuvant effect of poly IC was more prominent when systemic rather than local inflammatory responses were induced (Longhi et al. 2009).
The second study examined the influence of DC subsets on antigen processing. DCs express different antigen uptake receptors, and, to date, we utilized DEC-205 receptor for CD8α+ DC-targeting and DCIR2 receptor for CD8α− DC-targeting. Regarding the OVA model antigen, a previous study demonstrated enhanced MHC class II presentation by targeting DCIR2+ rather than DEC+ DCs (Dudziak et al. 2007). This difference in antigen processing was shown to be intrinsic to the DC subsets, and not due to differences in receptors targeted by anti-DEC-205 or 33D1 (DCIR2) mAbs. The data showed that the enhanced antigen presentation on MHC class II by DCIR2+ DCs was associated with increased expression of proteins involved in MHC class II processing, such as cathepsin (Dudziak et al. 2007). Moreover, as shown in Sect. 17.3.3, when real human pathogenic antigen (Y. pestis LcrV) was utilized in distinct DC subsets targeting, enhanced humoral immunity was observed with targeting DCIR2+ DC (Do et al. 2010). An extensive follow-on study is under way to reveal downstream mechanisms on which distinct DC subsets induce different type of adaptive immunity (Do, manuscript in preparation).
While these studies are a useful beginning, further investigation of various aspects, including at molecular levels, are required for improved DC-targeting strategy as a novel vaccine approach.
17.5.3 Construct Limitation and Antigen Selection
The DC-targeting strategy highlighted in this chapter uses a recombinant conjugated antibody construct. This genetically engineered antibody conjugate has distinct advantages as already described above. However, important aspects of this technology require case-by-case characterization to support clinical application. First and foremost is the necessity of identifying an effective target antigen. This is a challenge not only for the DC-targeting strategy field but also for general vaccine development, especially for vaccines of noninfectious diseases. Second is the actuality that recombinant construct will not always function as intended. Not every construct of the conjugated antibody will be expressed well and successfully induce immunity. Molecular properties such as size, conformation, solubility, and stability of the recombinant antigen-antibody are not yet well predicted by modeling, thus creating need for empirical testing to determine yield and efficacy of the recombinant conjugated antibody. To date, the 293 T cell transient transfection systems has been employed with good result for such engineering (see Sect. 17.6 for more information), but unknown biases may be revealed through exploration of other expression systems. Although these hurdles need to be addressed for improvement of the technology, we have established the great utility of recombinant antibody-antigen vehicle for the DC-targeting vaccine platform. Further applications, for example, combining with other drug delivery systems such as nanoparticles or developing novel technique for efficient screening system of newly synthesized antigen-antibody construct should be encouraged for refining the DC-targeting strategy.
17.6 Generalized Method
Following are general guidelines for creating a DC-targeting strategy to any antigen of interest. The reader is advised to inspect corresponding sections of this chapter and references therein for greater details at each stage of method engineering and testing.
17.6.1 Construct of Antigen-Conjugated DC-Targeting Antibody
The full length coding region of the antigen gene is amplified and then cloned into the COOH terminus of heavy chain of anti-DEC-205 mAb, anti-DCIR2 mAb, or an isotype control mAb. Each construct is transiently transfected into 293 T cells and the expressed conjugated antibody is purified by protein G affinity chromatography. The purified, recombinantly conjugated antibody is then characterized for structure and quality by SDS-PAGE and Western Blotting, and for binding affinity and specificity by FACS (fluorescence activated cell sorting) using Chinese hamster ovary (CHO) cells which have been stably transfected with respective receptors (i.e., DEC-205/DCIR2). It is important to insure that all reagents are free of endotoxin.
17.6.2 Immunization
Each conjugated antibody (5–10 μg/mouse or indicated dosage), with or without a stimulus for DC maturation, is administered subcutaneously (s.c.), intraperitoneally (i.p.), or intranasally (i.n.) depending on the study. DC stimulating reagents that we have found to be successful include anti-CD40, poly IC, or a combination of anti-CD40 and poly IC. In case of using poly IC alone as an adjuvant, a prime and boost strategy is necessary.
17.6.3 Verification of Antigen-Specific T Cell Induction
Antigen-specific T cell responses are analyzed by either ICS, ELISPOT (enzyme-linked immunosorbent spot assay), or by CFSE (Carboxyfluorescein succinimidyl ester)-proliferation assay. Briefly, bulk splenocytes are stimulated with reactive peptide pools (2 μg/ml or indicated concentration) or control medium in the presence of a co-stimulatory anti-CD28 mAb for 6 h. Brefeldin A is added for last 4–5 h to accumulate intracellular cytokines. After stimulation, cells are collected, washed, and stained with various surface antibodies (i.e., CD3/CD4/CD8). Following fixation and permeabilization, cells are incubated with various anti-cytokine antibodies (i.e., IFN-γ, IL-2, TNF-α). Binding specificities are collected by flow cytometer and analyzed (i.e., FlowJo software).
For ELISPOT assays, specialized ELISPOT plates are coated overnight with purified capturing various anti-cytokine antibodies (i.e., IFN-γ, IL-2, TNF-α). The plates are washed, blocked, and then incubated with sorted T cells (CD4+ or CD8+), purified spleen CD11c+ DCs, and in the presence or absence of reactive peptides. After 48 h, the plates are washed and incubated with proper detecting anti-cytokine antibodies and then the spots are visualized and counted.
For CFSE-proliferation assay, bulk splenocytes are labeled with CFSE and the labeled cells are stimulated with or without reactive peptides for 4 days. This can be combined with ICS for the last 6 h of culture. Data are collected by flow cytometer and analyzed.
17.6.4 Verification of Antigen-Specific B Cell Induction
Antigen-specific antibody responses are measured by either ELISA or ELISPOT. Briefly, specialized ELISA plates are coated with respective antigens overnight. Plates are washed, blocked, and placed with serial diluted samples such as serum or bronchoalveolar lavage fluid. Various secondary detecting antibodies (i.e., IgG, IgA) are added and visualized by spectrophotometer and titers are calculated.
For antibody-secreting cell detection by ELISPOT, specialized ELISPOT plates are coated with antigens of interest overnight. The plates are washed, blocked, and then placed with serial diluted bulk splenocytes or bone-marrow cells. After 6 h, the plates are washed and incubated with respective secondary detecting antibodies (i.e., IgG, IgA) and the spots are visualized and counted.
17.6.5 Vaccine Efficacy Tests
The efficacy of conjugated Abs can be evaluated either as a therapeutic or a prophylactic vaccine. Protocol will be different depending on disease models. For example, as a therapeutic vaccine in cancer model, mice are first inoculated with cancers and then treated with various vaccine regimens. In contrast, as a prophylactic vaccine in the cancer model, mice are first injected with various vaccine regimens and then they are challenged with tumors. In either case, tumor volumes, and survival rates are measured and the efficacy is interpreted. The nature of pathogenesis and historical vaccination will determine the study methods to be used measuring effectiveness against a targeted infectious disease (for example, see Sect. 17.3 on vaccination against plague).
17.7 Considerations for Analytical Testing
Nonclinical analytical testing and clinical studies document that a biological drug product meets prescribed specifications for safety, purity and potency, as is required by 21 CFR parts 600.3(s), 601 and 312, and ICH Q5C. However, conventional DC-targeting vaccines are classified as medical device and have somewhat relaxed requirements. Table 17.1 lists analytical tests that have been used in demonstrating intended attributes of DC vaccines as compared to those used for conventional DC vaccines, which require more testing.
Abbreviations
- AIDS/HIV:
-
Acquired immune deficiency syndrome and human immunodeficiency virus disease
- APCs:
-
Antigen presenting cells
- BAL:
-
Bronchoalveolar lavage
- CD40L:
-
CD40 ligand
- CFA:
-
Complete freund’s adjuvant
- CFSE:
-
Carboxyfluorescein succinimidyl ester
- CFU:
-
Colony forming unit
- CHO:
-
Chinese hamster ovary
- CMI:
-
Cell-mediated immunity
- CTL:
-
Cytotoxic T lymphocytes
- DCs:
-
Dendritic cells
- DCR:
-
DC-receptor
- DC-SIGN/CD209:
-
Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin
- DEC-205/CD205:
-
Dendritic and epithelial cells 205 kDa
- EBNA-1:
-
Nuclear antigen 1 of Epstein-Bar-Virus
- EBV:
-
Epstein Bar Virus
- EDE:
-
Glu-Asp-Glu
- ELISA:
-
Enzyme-linked immunosorbent assay
- ELISPOT:
-
Enzyme-linked immunosorbent spot assay
- F1:
-
Capsular antigen fraction 1
- FACS:
-
Fluorescence activated cell sorting
- Fcγ:
-
Cell-surface receptors that bind the Fc portion of IgG
- GLA:
-
Glucopyranosyl lipid A
- GM-CSF:
-
Granular macrophage colony stimulating factor
- HER2:
-
Human epidermal growth factor receptor
- HPV:
-
Human papilloma virus
- ICS:
-
Intracellular cytokine staining
- IFN:
-
Interferon
- I.M.:
-
Intramascular
- I.N.:
-
Intranasal
- I.P.:
-
Intraperitoneally
- KLH:
-
Keyhole limpet hemocyanin
- KO:
-
Knock-out
- LcrV:
-
Low-calcium response virulence protein
- LD50 :
-
Lethal dose, 50 %
- LNs:
-
Lymph nodes
- LPS:
-
Lipopolysaccharide
- mAb:
-
Monoclonal antibody
- MAdCAM:
-
Mucosal addressin cell adhesion molecule
- MALP-2:
-
Macrophage-activating lipopeptide-2
- MCM:
-
Monocyte-conditioned medium
- MDA5:
-
Melanoma differentiation-associated protein 5
- MMR:
-
Macrophage mannose receptor
- Mo-DCs:
-
Monocyte-derived DCs
- MPLA:
-
Monophosphoryl lipid A
- NHP:
-
Nonhuman primates
- NK:
-
Natural killer
- NKT:
-
Natural killer T
- NOG:
-
NOD/LtSz-scid IL2Rγnull
- NOS2:
-
Nitric oxide synthase 2
- NYVAC:
-
New York vaccinia virus
- OVA:
-
Ovalbumin
- PBMC:
-
Peripheral blood mononuclear cell
- PGE2:
-
Prostaglandin E2
- Pgm-:
-
Pigmentation negative strain
- PHRI:
-
Public health research institute
- Poly IC:
-
Polyinosinic: polycytidylic acid
- Poly ICLC:
-
Poly IC with poly-L-lysine and carboxymethyl cellulose
- S.C.:
-
Subcutaneously
- SDS PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- STAT:
-
Signal transducer and activator of transcription
- T3SS:
-
Type III secretion system
- TAPs:
-
Transporters for antigen presentation
- TLR:
-
Toll-like receptors
- TNF:
-
Tumor necrosis factor
- Treg:
-
Regulatory T cell
- TT:
-
Tetanus toxoid
- Y. pestis:
-
Yersinia pestis
- Yops:
-
Y. pestis effector molecules
References
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2:675–680
Albert ML, Sauter B, Bhardwaj N (1998) Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 392:86–89
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738
Allan RS, Smith CM, Belz GT, van Lint AL, Wakim LM, Heath WR, Carbone FR (2003) Epidermal viral immunity induced by CD8alpha + dendritic cells but not by Langerhans cells. Science 301:1925–1928
Altieri DC (2003) Validating survivin as a cancer therapeutic target. Nat Rev Cancer 3:46–54
Anderson GW Jr, Leary SE, Williamson ED, Titball RW, Welkos SL, Worsham PL, Friedlander AM (1996) Recombinant V antigen protects mice against pneumonic and bubonic plague caused by F1-capsule-positive and -negative strains of Yersinia pestis. Infect Immun 64:4580–4585
Andrews GP, Heath DG, Anderson GW Jr, Welkos SL, Friedlander AM (1996) Fraction 1 capsular antigen (F1) purification from Yersinia pestis CO92 and from an Escherichia coli recombinant strain and efficacy against lethal plague challenge. Infect Immun 64:2180–2187
Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, Murugesan SR, Leach SD, Jaffee E, Yeo CJ, Cameron JL, Kern SE, Hruban RH (2001) Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res 7:3862–3868
Baca-Estrada ME, Foldvari MM, Snider MM, Harding KK, Kournikakis BB, Babiuk LA, Griebel PP (2000) Intranasal immunization with liposome-formulated Yersinia pestis vaccine enhances mucosal immune responses. Vaccine 18:2203–2211
Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW, Henn V, Kloetzel PM, Gurka S, Kroczek RA (2010) Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 207:1273–1281
Baldwin SL, Shaverdian N, Goto Y, Duthie MS, Raman VS, Evers T, Mompoint F, Vedvick TS, Bertholet S, Coler RN, Reed SG (2009) Enhanced humoral and Type 1 cellular immune responses with Fluzone adjuvanted with a synthetic TLR4 agonist formulated in an emulsion. Vaccine 27:5956–5963
Banchereau J, Schuler-Thurner B, Palucka AK, Schuler G (2001) Dendritic cells as vectors for therapy. Cell 106:271–274
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392:245–252
Bates JH, Irvin CG (2003) Measuring lung function in mice: the phenotyping uncertainty principle. J Appl Physiol 94:1297–1306
Bendelac A, Medzhitov R (2002) Adjuvants of immunity: harnessing innate immunity to promote adaptive immunity. J Exp Med 195:F19–F23
Bera TK, Pastan I (2000) Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol 20:2902–2906
Berger TG, Feuerstein B, Strasser E, Hirsch U, Schreiner D, Schuler G, Schuler-Thurner B (2002) Large-scale generation of mature monocyte-derived dendritic cells for clinical application in cell factories. J Immunol Methods 268:131–140
Bevan MJ (2004) Helping the CD8(+) T-cell response. Nat Rev Immunol 4:595–602
Blanc-Brude OP, Mesri M, Wall NR, Plescia J, Dohi T, Altieri DC (2003) Therapeutic targeting of the survivin pathway in cancer: initiation of mitochondrial apoptosis and suppression of tumor-associated angiogenesis. Clin Cancer Res 9:2683–2692
Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM (2002) Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med 196:1627–1638
Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii S, Soares H, Brimnes MK, Moltedo B, Moran TM, Steinman RM (2004) In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med 199:815–824
Bozzacco L, Trumpfheller C, Siegal FP, Mehandru S, Markowitz M, Carrington M, Nussenzweig MC, Piperno AG, Steinman RM (2007) DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc Natl Acad Sci USA 104:1289–1294
Brubaker RR (1991) The V antigen of yersiniae: an overview. Contrib Microbiol Immunol 12:127–133
Bubeck SS, Cantwell AM, Dube PH (2007) Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect Immun 75:697–705
Caminschi I, Proietto AI, Ahmet F, Kitsoulis S, Shin Teh J, Lo JC, Rizzitelli A, Wu L, Vremec D, van Dommelen SL, Campbell IK, Maraskovsky E, Braley H, Davey GM, Mottram P, van de Velde N, Jensen K, Lew AM, Wright MD, Heath WR, Shortman K, Lahoud MH (2008) The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 112:3264–3273
Caskey M, Lefebvre F, Filali-Mouhim A, Cameron MJ, Goulet JP, Haddad EK, Breton G, Trumpfheller C, Pollak S, Shimeliovich I, Duque-Alarcon A, Pan L, Nelkenbaum A, Salazar AM, Schlesinger SJ, Steinman RM, Sekaly RP (2011) Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med 208:2357–2366
Chang K, Pastan I (1996) Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA 93:136–140
Charalambous A, Oks M, Nchinda G, Yamazaki S, Steinman RM (2006) Dendritic cell targeting of survivin protein in a xenogeneic form elicits strong CD4+ T cell immunity to mouse survivin. J Immunol 177:8410–8421
Cheong C, Choi JH, Vitale L, He LZ, Trumpfheller C, Bozzacco L, Do Y, Nchinda G, Park SH, Dandamudi DB, Shrestha E, Pack M, Lee HW, Keler T, Steinman RM, Park CG (2010) Improved cellular and humoral immune responses in vivo following targeting of HIV Gag to dendritic cells within human anti-human DEC205 monoclonal antibody. Blood 116:3828–3838
Coler RN, Baldwin SL, Shaverdian N, Bertholet S, Reed SJ, Raman VS, Lu X, DeVos J, Hancock K, Katz JM, Vedvick TS, Duthie MS, Clegg CH, Van Hoeven N, Reed SG (2010) A synthetic adjuvant to enhance and expand immune responses to influenza vaccines. PLoS ONE 5:e13677
Corbeil LB, Gogolewski RP, Kacskovics I, Nielsen KH, Corbeil RR, Morrill JL, Greenwood R, Butler JE (1997) Bovine IgG2a antibodies to Haemophilus somnus and allotype expression. Can J Vet Res 61:207–213
Cyster JG (1999) Chemokines and the homing of dendritic cells to the T cell areas of lymphoid organs. J Exp Med 189:447–450
Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ, Hoff ST, Andersen P, Reed SG, Morris SL, Roederer M, Seder RA (2007) Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med 13:843–850
Davis KJ, Fritz DL, Pitt ML, Welkos SL, Worsham PL, Friedlander AM (1996) Pathology of experimental pneumonic plague produced by fraction 1-positive and fraction 1-negative Yersinia pestis in African green monkeys (Cercopithecus aethiops). Arch Pathol Lab Med 120:156–163
Dela Cruz JS, Lau SY, Ramirez EM, De Giovanni C, Forni G, Morrison SL, Penichet ML (2003) Protein vaccination with the HER2/neu extracellular domain plus anti-HER2/neu antibody-cytokine fusion proteins induces a protective anti-HER2/neu immune response in mice. Vaccine 21:1317–1326
den Haan JM, Lehar SM, Bevan MJ (2000) CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med 192:1685–1696
Disis ML, Calenoff E, McLaughlin G, Murphy AE, Chen W, Groner B, Jeschke M, Lydon N, McGlynn E, Livingston RB et al (1994) Existent T-cell and antibody immunity to HER-2/neu protein in patients with breast cancer. Cancer Res 54:16–20
Disis ML, Knutson KL, Schiffman K, Rinn K, McNeel DG (2000) Pre-existent immunity to the HER-2/neu oncogenic protein in patients with HER-2/neu overexpressing breast and ovarian cancer. Breast Cancer Res Treat 62:245–252
Do Y, Didierlaurent AM, Ryu S, Koh H, Park CG, Park S, Perlin DS, Powell BS, Steinman RM (2012) Induction of pulmonary mucosal immune responses with a protein vaccine targeted to the DEC-205/CD205 receptor. Vaccine 30:6359–6367
Do Y, Koh H, Park CG, Dudziak D, Seo P, Mehandru S, Choi JH, Cheong C, Park S, Perlin DS, Powell BS, Steinman RM (2010) Targeting of LcrV virulence protein from Yersinia pestis to dendritic cells protects mice against pneumonic plague. Eur J Immunol 40:2791–2796
Do Y, Park CG, Kang YS, Park SH, Lynch RM, Lee H, Powell BS, Steinman RM (2008) Broad T cell immunity to the LcrV virulence protein is induced by targeted delivery to DEC-205/CD205-positive mouse dendritic cells. Eur J Immunol 38:20–29
Du Y, Rosqvist R, Forsberg A (2002) Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect Immun 70:1453–1460
Dubensky TW Jr, Reed SG (2010) Adjuvants for cancer vaccines. Semin Immunol 22:155–161
Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, Steinman RM, Nussenzweig MC (2007) Differential antigen processing by dendritic cell subsets in vivo. Science 315:107–111
Dzionek A, Sohma Y, Nagafune J, Cella M, Colonna M, Facchetti F, Gunther G, Johnston I, Lanzavecchia A, Nagasaka T, Okada T, Vermi W, Winkels G, Yamamoto T, Zysk M, Yamaguchi Y, Schmitz J (2001) BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J Exp Med 194:1823–1834
Elvin SJ, Williamson ED (2004) Stat 4 but not Stat 6 mediated immune mechanisms are essential in protection against plague. Microb Pathog 37:177–184
Emini EA, Koff WC (2004) AIDS/HIV. Developing an AIDS vaccine: need, uncertainty, hope. Science 304:1913–1914
Engering A, Geijtenbeek TB, van Vliet SJ, Wijers M, van Liempt E, Demaurex N, Lanzavecchia A, Fransen J, Figdor CG, Piguet V, van Kooyk Y (2002) The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol 168:2118–2126
Engering A, Pieters J (2001) Association of distinct tetraspanins with MHC class II molecules at different subcellular locations in human immature dendritic cells. Int Immunol 13:127–134
Eyles JE, Sharp GJ, Williamson ED, Spiers ID, Alpar HO (1998) Intra nasal administration of poly-lactic acid microsphere co-encapsulated Yersinia pestis subunits confers protection from pneumonic plague in the mouse. Vaccine 16:698–707
Eyles JE, Williamson ED, Spiers ID, Alpar HO (2000) Protection studies following bronchopulmonary and intramuscular immunisation with yersinia pestis F1 and V subunit vaccines coencapsulated in biodegradable microspheres: a comparison of efficacy. Vaccine 18:3266–3271
Flynn BJ, Kastenmuller K, Wille-Reece U, Tomaras GD, Alam M, Lindsay RW, Salazar AM, Perdiguero B, Gomez CE, Wagner R, Esteban M, Park CG, Trumpfheller C, Keler T, Pantaleo G, Steinman RM, Seder R (2011) Immunization with HIV Gag targeted to dendritic cells followed by recombinant New York vaccinia virus induces robust T-cell immunity in nonhuman primates. Proc Natl Acad Sci USA 108:7131–7136
Fong L, Brockstedt D, Benike C, Wu L, Engleman EG (2001a) Dendritic cells injected via different routes induce immunity in cancer patients. J Immunol 166:4254–4259
Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, Davis MM, Engleman EG (2001b) Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci U S A 98:8809–8814
Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM (2004) The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med 199:1607–1618
Gomez CE, Najera JL, Jimenez V, Bieler K, Wild J, Kostic L, Heidari S, Chen M, Frachette MJ, Pantaleo G, Wolf H, Liljestrom P, Wagner R, Esteban M (2007) Generation and immunogenicity of novel HIV/AIDS vaccine candidates targeting HIV-1 Env/Gag-Pol-Nef antigens of clade C. Vaccine 25:1969–1992
Granelli-Piperno A, Pritsker A, Pack M, Shimeliovich I, Arrighi JF, Park CG, Trumpfheller C, Piguet V, Moran TM, Steinman RM (2005) Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin/CD209 is abundant on macrophages in the normal human lymph node and is not required for dendritic cell stimulation of the mixed leukocyte reaction. J Immunol 175:4265–4273
Griffin KF, Conway BR, Alpar HO, Williamson ED (1998) Immune responses to V antigen of Yersinia pestis co-encapsulated with IFN-gamma: effect of dose and formulation. Vaccine 16:517–521
Grossman D, Kim PJ, Schechner JS, Altieri DC (2001) Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc Natl Acad Sci USA 98:635–640
Guo M, Gong S, Maric S, Misulovin Z, Pack M, Mahnke K, Nussenzweig MC, Steinman RM (2000) A monoclonal antibody to the DEC-205 endocytosis receptor on human dendritic cells. Hum Immunol 61:729–738
Gurer C, Strowig T, Brilot F, Pack M, Trumpfheller C, Arrey F, Park CG, Steinman RM, Munz C (2008) Targeting the nuclear antigen 1 of Epstein-Barr virus to the human endocytic receptor DEC-205 stimulates protective T-cell responses. Blood 112:1231–1239
Harris NL, Watt V, Ronchese F, Le Gros G (2002) Differential T cell function and fate in lymph node and nonlymphoid tissues. J Exp Med 195:317–326
Hassan R, Ho M (2008) Mesothelin targeted cancer immunotherapy. Eur J Cancer 44:46–53
Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC (2001) Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med 194:769–779
Heath DG, Anderson GW Jr, Mauro JM, Welkos SL, Andrews GP, Adamovicz J, Friedlander AM (1998) Protection against experimental bubonic and pneumonic plague by a recombinant capsular F1-V antigen fusion protein vaccine. Vaccine 16:1131–1137
Heath WR, Belz GT, Behrens GM, Smith CM, Forehan SP, Parish IA, Davey GM, Wilson NS, Carbone FR, Villadangos JA (2004) Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol Rev 199:9–26
Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM (2008) Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322:1097–1100
Hill J, Eyles JE, Elvin SJ, Healey GD, Lukaszewski RA, Titball RW (2006) Administration of antibody to the lung protects mice against pneumonic plague. Infect Immun 74:3068–3070
Ho M, Bera TK, Willingham MC, Onda M, Hassan R, FitzGerald D, Pastan I (2007) Mesothelin expression in human lung cancer. Clin Cancer Res 13:1571–1575
Honko AN, Sriranganathan N, Lees CJ, Mizel SB (2006) Flagellin is an effective adjuvant for immunization against lethal respiratory challenge with Yersinia pestis. Infect Immun 74:1113–1120
Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R (1996) Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med 2:52–58
Hynes NE, Stern DF (1994) The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim Biophys Acta 1198:165–184
Ichinohe T, Kawaguchi A, Tamura S, Takahashi H, Sawa H, Ninomiya A, Imai M, Itamura S, Odagiri T, Tashiro M, Chiba J, Sata T, Kurata T, Hasegawa H (2007) Intranasal immunization with H5N1 vaccine plus Poly I:Poly C12U, a Toll-like receptor agonist, protects mice against homologous and heterologous virus challenge. Microbes Infect 9:1333–1340
Ichinohe T, Watanabe I, Ito S, Fujii H, Moriyama M, Tamura S, Takahashi H, Sawa H, Chiba J, Kurata T, Sata T, Hasegawa H (2005) Synthetic double-stranded RNA poly(I:C) combined with mucosal vaccine protects against influenza virus infection. J Virol 79:2910–2919
Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Koerner JF, Layton M, McDade J, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Schoch-Spana M, Tonat K (2000) Plague as a biological weapon: medical and public health management. Working Group Civilian Biodefense. Jama 283:2281–2290
Islam A, Kageyama H, Takada N, Kawamoto T, Takayasu H, Isogai E, Ohira M, Hashizume K, Kobayashi H, Kaneko Y, Nakagawara A (2000) High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene 19:617–623
Iyoda T, Shimoyama S, Liu K, Omatsu Y, Akiyama Y, Maeda Y, Takahara K, Steinman RM, Inaba K (2002) The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med 195:1289–1302
Jiang W, Swiggard WJ, Heufler C, Peng M, Mirza A, Steinman RM, Nussenzweig MC (1995) The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 375:151–155
Jones T, Adamovicz JJ, Cyr SL, Bolt CR, Bellerose N, Pitt LM, Lowell GH, Burt DS (2006) Intranasal Protollin/F1-V vaccine elicits respiratory and serum antibody responses and protects mice against lethal aerosolized plague infection. Vaccine 24:1625–1632
Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V, Vulink AJ, Hart DN, Radford KJ (2010) Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med 207:1247–1260
Jonuleit H, Giesecke-Tuettenberg A, Tuting T, Thurner-Schuler B, Stuge TB, Paragnik L, Kandemir A, Lee PP, Schuler G, Knop J, Enk AH (2001) A comparison of two types of dendritic cell as adjuvants for the induction of melanoma-specific T-cell responses in humans following intranodal injection. Int J Cancer 93:243–251
Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA (2002) In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17: 211–220
Kamath AT, Henri S, Battye F, Tough DF, Shortman K (2002) Developmental kinetics and lifespan of dendritic cells in mouse lymphoid organs. Blood 100:1734–1741
Kamath AT, Pooley J, O’Keeffe MA, Vremec D, Zhan Y, Lew AM, D’Amico A, Wu L, Tough DF, Shortman K (2000) The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J Immunol 165:6762–6770
Kawai T, Akira S (2007) Antiviral signaling through pattern recognition receptors. J Biochem 141:137–145
Kim YS, Kim YJ, Lee JM, Han SH, Ko HJ, Park HJ, Pereboev A, Nguyen HH, Kang CY (2010) CD40-targeted recombinant adenovirus significantly enhances the efficacy of antitumor vaccines based on dendritic cells and B cells. Hum Gene Ther 21:1697–1706
Krown SE, Kerr D, Stewart WE 2nd, Field AK, Oettgen HF (1985) Phase I trials of poly(I, C) complexes in advanced cancer. J Biol Response Mod 4:640–649
Lampkin BC, Levine AS, Levy H, Krivit W, Hammond D (1985) Phase II trial of a complex polyriboinosinic-polyribocytidylic acid with poly-L-lysine and carboxymethyl cellulose in the treatment of children with acute leukemia and neuroblastoma: a report from the Children’s Cancer Study Group. Cancer Res 45:5904–5909
Lathem WW, Crosby SD, Miller VL, Goldman WE (2005) Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci USA 102:17786–17791
Lefrancois L, Puddington L (2006) Intestinal and pulmonary mucosal T cells: local heroes fight to maintain the status quo. Annu Rev Immunol 24:681–704
Levy HB, London W, Fuccillo DA, Baron S, Rice J (1976) Prophylactic control of simian hemorrhagic fever in monkeys by an interferon inducer, polyriboinosinic-polyribocytidylic acid-poly-L-lysine. J Infect Dis 133(Suppl):A256–A259
Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC (1998) Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 396:580–584
Lichterfeld M, Kaufmann DE, Yu XG, Mui SK, Addo MM, Johnston MN, Cohen D, Robbins GK, Pae E, Alter G, Wurcel A, Stone D, Rosenberg ES, Walker BD, Altfeld M (2004) Loss of HIV-1-specific CD8+ T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1-specific CD4+ T cells. J Exp Med 200:701–712
Lin JS, Kummer LW, Szaba FM, Smiley ST (2011a) IL-17 contributes to cell-mediated defense against pulmonary Yersinia pestis infection. J Immunol 186:1675–1684
Lin JS, Park S, Adamovicz JJ, Hill J, Bliska JB, Cote CK, Perlin DS, Amemiya K, Smiley ST (2010) TNFalpha and IFNgamma contribute to F1/LcrV-targeted immune defense in mouse models of fully virulent pneumonic plague. Vaccine 29:357–362
Lin JS, Szaba FM, Kummer LW, Chromy BA, Smiley ST (2011b) Yersinia pestis YopE contains a dominant CD8 T cell epitope that confers protection in a mouse model of pneumonic plague. J Immunol 187:897–904
Lin ML, Zhan Y, Proietto AI, Prato S, Wu L, Heath WR, Villadangos JA, Lew AM (2008) Selective suicide of cross-presenting CD8+ dendritic cells by cytochrome c injection shows functional heterogeneity within this subset. Proc Natl Acad Sci USA 105:3029–3034
Lindenstrom T, Agger EM, Korsholm KS, Darrah PA, Aagaard C, Seder RA, Rosenkrands I, Andersen P (2009) Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J Immunol 182:8047–8055
Liu J, O’Brien KL, Lynch DM, Simmons NL, La Porte A, Riggs AM, Abbink P, Coffey RT, Grandpre LE, Seaman MS, Landucci G, Forthal DN, Montefiori DC, Carville A, Mansfield KG, Havenga MJ, Pau MG, Goudsmit J, Barouch DH (2009) Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature 457:87–91
Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, Salazar AM, Colonna M, Steinman RM (2009) Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med 206:1589–1602
Mahnke K, Guo M, Lee S, Sepulveda H, Swain SL, Nussenzweig M, Steinman RM (2000) The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J Cell Biol 151:673–684
Maldonado-Lopez R, De Smedt T, Michel P, Godfroid J, Pajak B, Heirman C, Thielemans K, Leo O, Urbain J, Moser M (1999) CD8alpha+ and CD8alpha- subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J Exp Med 189:587–592
Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O (2005) Plague bacteria target immune cells during infection. Science 309:1739–1741
Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC (2007) The vaccine adjuvant monophosphoryl lipid a as a TRIF-biased agonist of TLR4. Science 316:1628–1632
McIlroy D, Troadec C, Grassi F, Samri A, Barrou B, Autran B, Debre P, Feuillard J, Hosmalin A (2001) Investigation of human spleen dendritic cell phenotype and distribution reveals evidence of in vivo activation in a subset of organ donors. Blood 97:3470–3477
Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1:135–145
Mellman I, Steinman RM (2001) Dendritic cells: specialized and regulated antigen processing machines. Cell 106:255–258
Mestas J, Hughes CC (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172:2731–2738
Meyer KF (1970) Effectiveness of live or killed plague vaccines in man. Bull World Health Organ 42:653–666
Meyer KF, Hightower JA, McCrumb FR (1974) Plague immunization. VI. Vaccination with the fraction i antigen of Yersinia pestis. J Infect Dis 129(Suppl):S41–S45
Mills KH, Ryan M, Ryan E, Mahon BP (1998) A murine model in which protection correlates with pertussis vaccine efficacy in children reveals complementary roles for humoral and cell-mediated immunity in protection against Bordetella pertussis. Infect Immun 66:594–602
Nakajima R, Brubaker RR (1993) Association between virulence of Yersinia pestis and suppression of gamma interferon and tumor necrosis factor alpha. Infect Immun 61:23–31
Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, Littman DR, Reinecker HC (2005) CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307:254–258
Nordlund JJ, Wolff SM, Levy HB (1970) Inhibition of biologic activity of poly I: poly C by human plasma. Proc Soc Exp Biol Med 133:439–444
Novitsky V, Gilbert P, Peter T, McLane MF, Gaolekwe S, Rybak N, Thior I, Ndung’u T, Marlink R, Lee TH, Essex M (2003) Association between virus-specific T-cell responses and plasma viral load in human immunodeficiency virus type 1 subtype C infection. J Virol 77:882–890
Okada H, Bakal C, Shahinian A, Elia A, Wakeham A, Suh WK, Duncan GS, Ciofani M, Rottapel R, Zuniga-Pflucker JC, Mak TW (2004) Survivin loss in thymocytes triggers p53-mediated growth arrest and p53-independent cell death. J Exp Med 199:399–410
Overheim K, Lemoine S, Gallegos K, Fisher I, Monier A, Schneider C, Valderas M, Wilder J, Russell R, Sherwood R, Leclaire R (2012a) Increased dose of recombinant F1/V plague vaccine results in increased survival in plague-challenged cynomologus macaques. In: ASM biodefense and emerging diseases research meeting, Washington, DC
Overheim K, Lemoine S, Gallegos K, Fisher I, Monier A, Schneider C, Valderas M, Wilder J, Russell R, Sherwood R, Leclaire R (2012b) Increased protection of african green monkeys vaccinated with increasing doses of a recombinant F1/V plague vaccine. In: ASM Biodefense and Emerging Diseases research meeting, Washington, DC
Pack M, Trumpfheller C, Thomas D, Park CG, Granelli-Piperno A, Munz C, Steinman RM (2008) DEC-205/CD205+ dendritic cells are abundant in the white pulp of the human spleen, including the border region between the red and white pulp. Immunology 123:438–446
Palucka K, Banchereau J, Mellman I (2010) Designing vaccines based on biology of human dendritic cell subsets. Immunity 33:464–478
Pamer E, Cresswell P (1998) Mechanisms of MHC class I–restricted antigen processing. Annu Rev Immunol 16:323–358
Pantel A, Cheong C, Dandamudi D, Shrestha E, Mehandru S, Brane L, Ruane D, Teixeira A, Bozzacco L, Steinman RM, Longhi MP (2012) A new synthetic TLR4 agonist, GLA, allows dendritic cells targeted with antigen to elicit Th1 T-cell immunity in vivo. Eur J Immunol 42:101–109
Parent MA, Berggren KN, Kummer LW, Wilhelm LB, Szaba FM, Mullarky IK, Smiley ST (2005) Cell-mediated protection against pulmonary Yersinia pestis infection. Infect Immun 73:7304–7310
Parent MA, Wilhelm LB, Kummer LW, Szaba FM, Mullarky IK, Smiley ST (2006) Gamma interferon, tumor necrosis factor alpha, and nitric oxide synthase 2, key elements of cellular immunity, perform critical protective functions during humoral defense against lethal pulmonary Yersinia pestis infection. Infect Immun 74:3381–3386
Partidos CD, Hoebeke J, Moreau E, Chaloin O, Tunis M, Belliard G, Briand JP, Desgranges C, Muller S (2005) The binding affinity of double-stranded RNA motifs to HIV-1 Tat protein affects transactivation and the neutralizing capacity of anti-Tat antibodies elicited after intranasal immunization. Eur J Immunol 35:1521–1529
Perry RD, Fetherston JD (1997) Yersinia pestis–etiologic agent of plague. Clin Microbiol Rev 10:35–66
Pitt ML (2004) Animal models and correlates of protection for plague vaccines workshop, Gaithersburg, MD. http://www.fda.gov/cber/minutes/workshop-min.htm
Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, Keller AM, Joffre O, Zelenay S, Nye E, Le Moine A, Faure F, Donckier V, Sancho D, Cerundolo V, Bonnet D, Reis e Sousa C (2010) Characterization of human DNGR-1+BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med 207:1261–1271
Powell BS, Andrews GP, Enama JT, Jendrek S, Bolt C, Worsham P, Pullen JK, Ribot W, Hines H, Smith L, Heath DG, Adamovicz JJ (2005) Design and testing for a nontagged F1-V fusion protein as vaccine antigen against bubonic and pneumonic plague. Biotechnol Prog 21:1490–1510
Pulendran B, Smith JL, Caspary G, Brasel K, Pettit D, Maraskovsky E, Maliszewski CR (1999) Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci USA 96:1036–1041
Ramamurthi KS, Schneewind O (2002) Type iii protein secretion in yersinia species. Annu Rev Cell Dev Biol 18:107–133
Randolph GJ, Angeli V, Swartz MA (2005) Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol 5:617–628
Reed DS, Martinez MJ (2006) Respiratory immunity is an important component of protection elicited by subunit vaccination against pneumonic plague. Vaccine 24:2283–2289
Regnault A, Lankar D, Lacabanne V, Rodriguez A, Thery C, Rescigno M, Saito T, Verbeek S, Bonnerot C, Ricciardi-Castagnoli P, Amigorena S (1999) Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med 189:371–380
Reis e Sousa C (2006) Dendritic cells in a mature age. Nat Rev Immunol 6:476–483
Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P (2001) Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol 2:361–367
Ridgway D (2003) The first 1000 dendritic cell vaccinees. Cancer Invest 21:873–886
Rosenberg ES, Billingsley JM, Caliendo AM, Boswell SL, Sax PE, Kalams SA, Walker BD (1997) Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science 278:1447–1450
Salazar AM, Levy HB, Ondra S, Kende M, Scherokman B, Brown D, Mena H, Martin N, Schwab K, Donovan D, Dougherty D, Pulliam M, Ippolito M, Graves M, Brown H, Ommaya A (1996) Long-term treatment of malignant gliomas with intramuscularly administered polyinosinic-polycytidylic acid stabilized with polylysine and carboxymethylcellulose: an open pilot study. Neurosurgery 38: 1096–1103 (Discussion 1103–1094)
Sallusto F, Cella M, Danieli C, Lanzavecchia A (1995) Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med 182:389–400
Schmidt A, Rollinghoff M, Beuscher HU (1999) Suppression of TNF by V antigen of Yersinia spp. involves activated T cells. Eur J Immunol 29:1149–1157
Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, Davey G, Kupresanin F, Li M, Maraskovsky E, Belz GT, Carbone FR, Shortman K, Heath WR, Villadangos JA (2006) The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc Natl Acad Sci USA 103:10729–10734
Schuler-Thurner B, Schultz ES, Berger TG, Weinlich G, Ebner S, Woerl P, Bender A, Feuerstein B, Fritsch PO, Romani N, Schuler G (2002) Rapid induction of tumor-specific type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopreserved, peptide-loaded monocyte-derived dendritic cells. J Exp Med 195:1279–1288
Schuler G, Schuler-Thurner B, Steinman RM (2003) The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol 15:138–147
Segura E, Albiston AL, Wicks IP, Chai SY, Villadangos JA (2009) Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc Natl Acad Sci USA 106:20377–20381
Shortman K, Liu YJ (2002) Mouse and human dendritic cell subtypes. Nat Rev Immunol 2:151–161
Shortman K, Naik SH (2007) Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol 7:19–30
Sing A, Rost D, Tvardovskaia N, Roggenkamp A, Wiedemann A, Kirschning CJ, Aepfelbacher M, Heesemann J (2002) Yersinia V-antigen exploits toll-like receptor 2 and CD14 for interleukin 10-mediated immunosuppression. J Exp Med 196:1017–1024
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235:177–182
Sloots A, Mastini C, Rohrbach F, Weth R, Curcio C, Burkhardt U, Jager E, Forni G, Cavallo F, Wels WS (2008) DNA vaccines targeting tumor antigens to B7 molecules on antigen-presenting cells induce protective antitumor immunity and delay onset of HER-2/Neu-driven mammary carcinoma. Clin Cancer Res 14:6933–6943
Stahl-Hennig C, Eisenblatter M, Jasny E, Rzehak T, Tenner-Racz K, Trumpfheller C, Salazar AM, Uberla K, Nieto K, Kleinschmidt J, Schulte R, Gissmann L, Muller M, Sacher A, Racz P, Steinman RM, Uguccioni M, Ignatius R (2009) Synthetic double-stranded RNAs are adjuvants for the induction of T helper 1 and humoral immune responses to human papillomavirus in rhesus macaques. PLoS Pathog 5:e1000373
Steinhagen F, Kinjo T, Bode C, Klinman DM (2011) TLR-based immune adjuvants. Vaccine 29:3341–3355
Steinman RM (2007) Dendritic cells: understanding immunogenicity. Eur J Immunol 37(Suppl 1):S53–S60
Steinman RM, Banchereau J (2007) Taking dendritic cells into medicine. Nature 449:419–426
Steinman RM, Dhodapkar M (2001) Active immunization against cancer with dendritic cells: the near future. Int J Cancer 94:459–473
Steinman RM, Pope M (2002) Exploiting dendritic cells to improve vaccine efficacy. J Clin Invest 109:1519–1526
Stephen EL, Sammons ML, Pannier WL, Baron S, Spertzel RO, Levy HB (1977) Effect of a nuclease-resistant derivative of polyriboinosinic-polyribocytidylic acid complex on yellow fever in rhesus monkeys (Macaca mulatta). J Infect Dis 136:122–126
Stevenson HC, Abrams PG, Schoenberger CS, Smalley RB, Herberman RB, Foon KA (1985) A phase I evaluation of poly(I, C)-LC in cancer patients. J Biol Response Mod 4:650–655
Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC (1998) IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res 58:5315–5320
Taylor P, Gerder M, Moros Z, Feldmann M (1996) Humoral and cellular responses raised against the human HER2 oncoprotein are cross-reactive with the homologous product of the new proto-oncogene, but do not protect rats against B104 tumors expressing mutated neu. Cancer Immunol Immunother 42:179–184
Thomann JS, Heurtault B, Weidner S, Braye M, Beyrath J, Fournel S, Schuber F, Frisch B (2011) Antitumor activity of liposomal ErbB2/HER2 epitope peptide-based vaccine constructs incorporating TLR agonists and mannose receptor targeting. Biomaterials 32:4574–4583
Thomas AM, Santarsiero LM, Lutz ER, Armstrong TD, Chen YC, Huang LQ, Laheru DA, Goggins M, Hruban RH, Jaffee EM (2004) Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med 200:297–306
Thompson KA, Strayer DR, Salvato PD, Thompson CE, Klimas N, Molavi A, Hamill AK, Zheng Z, Ventura D, Carter WA (1996) Results of a double-blind placebo-controlled study of the double-stranded RNA drug polyI:polyC12U in the treatment of HIV infection. Eur J Clin Microbiol Infect Dis 15:580–587
Trombetta ES, Mellman I (2005) Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol 23:975–1028
Trumpfheller C, Finke JS, Lopez CB, Moran TM, Moltedo B, Soares H, Huang Y, Schlesinger SJ, Park CG, Nussenzweig MC, Granelli-Piperno A, Steinman RM (2006) Intensified and protective CD4+ T cell immunity in mice with anti-dendritic cell HIV gag fusion antibody vaccine. J Exp Med 203:607–617
Trumpfheller C, Longhi MP, Caskey M, Idoyaga J, Bozzacco L, Keler T, Schlesinger SJ, Steinman RM (2012) Dendritic cell-targeted protein vaccines: a novel approach to induce T-cell immunity. J Intern Med 271:183–192
Trumpfheller C, Caskey M, Nchinda G, Longhi MP, Mizenina O, Huang Y, Schlesinger SJ, Colonna M, Steinman RM (2008) The microbial mimic polyIC induces durable and protective CD4+ T cell immunity together with a dendritic cell targeted vaccine. In: Proc Nat Acad Sci.USA (In Press)
Turley SJ, Inaba K, Garrett WS, Ebersold M, Unternaehrer J, Steinman RM, Mellman I (2000) Transport of peptide-MHC class II complexes in developing dendritic cells. Science 288:522–527
Valladeau J, Ravel O, Dezutter-Dambuyant C, Moore K, Kleijmeer M, Liu Y, Duvert-Frances V, Vincent C, Schmitt D, Davoust J, Caux C, Lebecque S, Saeland S (2000) Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity 12:71–81
Villadangos JA, Shortman K (2010) Found in translation: the human equivalent of mouse CD8+ dendritic cells. J Exp Med 207:1131–1134
Wang B, Kuroiwa JM, He LZ, Charalambous A, Keler T, Steinman RM (2009) The human cancer antigen mesothelin is more efficiently presented to the mouse immune system when targeted to the DEC-205/CD205 receptor on dendritic cells. Ann NY Acad Sci 1174:6–17
Wang B, Zaidi N, He LZ, Zhang L, Kuroiwa JM, Keler T, Steinman RM (2012) Targeting of the non-mutated tumor antigen HER2/neu to mature dendritic cells induces an integrated immune response that protects against breast cancer in mice. Breast Cancer Res 14:R39
Wei H, Wang S, Zhang D, Hou S, Qian W, Li B, Guo H, Kou G, He J, Wang H, Guo Y (2009) Targeted delivery of tumor antigens to activated dendritic cells via CD11c molecules induces potent antitumor immunity in mice. Clin Cancer Res 15:4612–4621
Williamson ED, Eley SM, Griffin KF, Green M, Russell P, Leary SE, Oyston PC, Easterbrook T, Reddin KM, Robinson A et al (1995) A new improved sub-unit vaccine for plague: the basis of protection. FEMS Immunol Med Microbiol 12:223–230
Witmer-Pack MD, Swiggard WJ, Mirza A, Inaba K, Steinman RM (1995) Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. II. Expression in situ in lymphoid and nonlymphoid tissues. Cell Immunol 163:157–162
Zizzari IG, Veglia F, Taurino F, Rahimi H, Quaglino E, Belleudi F, Riccardo F, Antonilli M, Napoletano C, Bellati F, Benedetti-Panici P, Torrisi MR, Frati L, Nuti M, Rughetti A (2011) HER2-based recombinant immunogen to target DCs through FcgammaRs for cancer immunotherapy. J Mol Med (Berl) 89:1231–1240
Zuniga R, Lucchetti A, Galvan P, Sanchez S, Sanchez C, Hernandez A, Sanchez H, Frahm N, Linde CH, Hewitt HS, Hildebrand W, Altfeld M, Allen TM, Walker BD, Korber BT, Leitner T, Sanchez J, Brander C (2006) Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J Virol 80:3122–3125
Acknowledgment
We thank the late Dr. Ralph M. Steinman and his past colleagues for their tremendous works on DC-targeting strategy, and Dr. Seongho Ryu from Soonchunhyang Institute of Medi-bio Sciences (SIMS) for a “human body image” in the Fig. 17.1, and J-A Han for assistance with review of clinical data in Table 17.2. This work was supported by funds from the National Institutes of Health (1 R21 AI082331-01, PI. Y. Do). This study was also supported by the National Research Foundation of Korea (NRF) (2013R1A1A2005545 & 2011-0020163) and UNIST research fund (No. 1.120020.01).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Do, Y., Powell, B. (2015). Dendritic Cell Targeting Vaccines. In: Nunnally, B., Turula, V., Sitrin, R. (eds) Vaccine Analysis: Strategies, Principles, and Control. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-45024-6_17
Download citation
DOI: https://doi.org/10.1007/978-3-662-45024-6_17
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-45023-9
Online ISBN: 978-3-662-45024-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)