
Abstract
Neurologic complications of cancer and cancer therapy are common and can arise rapidly in pediatric oncology patients. Some of these complications can present as emergent situations that require rapid diagnosis and treatment; familiarity with these conditions and a low index of suspicion are absolutely necessary in caring for such patients. The emergencies of spinal cord compression, acute alteration in mental status, increased intracranial pressure and stroke are addressed in this chapter. Specific attention is given to the acute neurotoxic effects of common chemotherapeutic agents. In addition to background regarding possible etiologies that are unique to the child with cancer, the evidence bases for diagnostic steps and management are discussed. This chapter provides the necessary framework to recognize potential neurologic emergencies in pediatric oncology patients and highlights when to utilize a multidisciplinary team approach including pediatric oncologists and neuro-oncologists, intensivists, neurologists, radiation oncologists, and neurosurgeons to provide optimal care.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Acute Lymphoblastic Leukemia
- Cerebral Perfusion Pressure
- Cord Compression
- Hemorrhagic Stroke
- Malignant Peripheral Nerve Sheath Tumor
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 Introduction
Neurologic complications of cancer and cancer therapy are common and can arise rapidly in pediatric oncology patients. Some of these complications can present as emergent situations that require rapid diagnosis and treatment; familiarity with these conditions and a low index of suspicion are absolutely necessary in caring for such patients. The emergencies of spinal cord compression, acute alteration in mental status, increased intracranial pressure and stroke are addressed in this chapter. Specific attention is given to the acute neurotoxic effects of common chemotherapeutic agents. In addition to background regarding possible etiologies that are unique to the child with cancer, the evidence bases for diagnostic steps and management are discussed and graded (Table 5.1). This chapter provides the necessary framework to recognize potential neurologic emergencies in pediatric oncology patients and highlights when to utilize a multidisciplinary team approach including pediatric oncologists and neuro-oncologists, intensivists, neurologists, radiation oncologists, and neurosurgeons to provide optimal care.
5.2 Spinal Cord Compression
Compression of the spinal cord can be caused by mass effect from either extradural or intradural tumors. Presenting symptoms can initially be nonspecific, and the diagnosis can be missed if the index of suspicion on the part of the examiner is not sufficiently high. Cord compression may herald a new diagnosis of cancer, be a sign of treatment failure or relapse, or indicate a complication of treatment such as a post-lumbar puncture hematoma. In addition to spinal cord compression, this section describes conus medullaris and cauda equina syndromes.
5.2.1 Presentation
Back pain is a presenting symptom in 80–90 % of patients presenting with malignant spinal cord compression (Pollono et al. 2003). In patients old enough to describe their symptoms, this pain is classically radicular, radiating to the legs, sharp, and described as electric. Non-radicular pain may be also reported. However, younger patients may not be able to localize pain or provide any description of its quality.
Weakness is present in similar numbers of patients at presentation, but again can be difficult to elicit in the young child. Increased “clumsiness,” particularly affecting gait, or refusal to walk, can be manifestations of neurologic weakness. In patients who are able to comply with examination, localization of the weakness can help identify the level of compression (Table 5.2). Weakness may initially be flaccid, with subsequent hyperreflexia in extremities distal to the site of compression. Compression at the level of the cauda equina causes absent reflexes due to the peripheral nature of the injury.
Isolated bowel or bladder dysfunction with perineal anesthesia suggests compression of either the conus medullaris or cauda equina, but sympathetic denervation from a higher compression can reproduce symptoms of urinary retention. A history of progressive constipation or abdominal pain with a full bladder should therefore warrant further investigation. Sensory changes (paresthesias or anesthesia) should be carefully investigated to determine patterns of distribution. The presence of any sensory level of such findings is a very concerning sign and can additionally help localize a lesion.
5.2.2 Differential Diagnosis
Cord compression can be the presenting feature of a new malignancy and patients may have either acute or subacute presentations. Of tumors arising from the extradural space, paravertebral Ewing sarcoma is the most frequent malignancy to cause cord compression (Klein et al. 1991; Pollono et al. 2003). Neuroblastoma can also present with cord compression given their origin in the prevertebral sympathetic chain. While less common, primary or metastatic vertebral bone lesions such as osteosarcoma or Langerhans cell histiocytosis have also been associated with cord compression. Pathologic fracture complicating a primary bone lesion may cause the presenting symptoms. Both acute lymphoblastic leukemia (ALL) and acute myelogenous leukemia (AML) can present with extramedullary disease that can cause cord compression. Discrete masses composed of leukemic cells (chloromas or granulocytic sarcomas), are more commonly observed in AML than ALL (Mantadakis et al. 2008; Olcay et al. 2009; Isome et al. 2011). Both Hodgkin and non-Hodgkin lymphoma can similarly lead to cord compression through direct mass effect (Acquaviva et al. 2003; Daley et al. 2003; Gupta et al. 2009).
Intraspinal tumors in children (Fig. 5.1) are most commonly gliomas or ependymomas (Huisman 2009; Benesch et al. 2010). Drop metastases from posterior fossa tumors, most notably primitive neuroectodermal tumors (PNET; medulloblastoma), can also cause cord compression. Patients with neurocutaneous syndromes such as neurofibromatosis type-1 (NF-1) and NF-2, von Hippel-Lindau disease, and tuberous sclerosis warrant special consideration when presenting with signs of spinal cord compression given their propensity for various intraspinal tumors such as malignant peripheral nerve sheath tumor, ependymoma, hemangioblastoma and astrocytoma, respectively.

Intraspinal glioma. This 10-year-old patient presented with several weeks of progressive bilateral lower extremity weakness. The T1-weighted gadolinium-enhanced MRI shown in Panel a demonstrates a cystic mass (“M”) in the upper thoracic spinal cord. The T2-weighted image in Panel b highlights the development of syrinx (“S”), seen as bright cerebrospinal fluid in this sequence, cranial to the mass. Pathology from a biopsy specimen taken at the time of diagnosis was consistent with pilocytic astrocytoma
In the patient with known tumor, new symptoms of cord compression should raise concern for development of metastatic disease. Patients who have recently undergone a lumbar puncture, particularly in the setting of low platelets or coagulopathy, are at risk for an epidural hematoma that may require emergent evacuation. A potential mimic of cord compression in the pediatric oncology patient is acute onset transverse myelitis. This entity can present with symptoms of weakness, bowel and bladder dysfunction, and a sensory level; magnetic resonance imaging (MRI) may demonstrate inflammatory changes within the cord, but no compressive mass. Transverse myelitis has been reported in patients following either intrathecal or high-dose intravenous cytarabine (Schwenn et al. 1991).
5.2.3 Management
The first step in managing a patient with suspected cord compression is involvement of a multidisciplinary team including neurosurgery, neurology, oncology, intensivists and radiation oncology. Frequent monitoring for progression of symptoms is vital, as this can rapidly change the therapeutic plan. Initial steps should be aimed at verifying the diagnosis and attempting to alleviate compressive symptoms medically while a more definitive treatment plan is formulated.
5.2.3.1 Imaging
MRI of the spine can help narrow the differential diagnosis, allow for surgical planning and identify potential sites of multifocal disease. The entire spine should be imaged with pre- and post-gadolinium contrast to maximize yield. Brain imaging may also be warranted if the lesions are suspicious for drop metastases or if the patient presents with signs or symptoms of increased intracranial pressure such as headache, emesis, hypertension with bradycardia, or cranial nerve palsies.
5.2.3.2 Corticosteroids
Steroid treatment is employed to relieve any vasogenic edema that may be contributing to cord compression. In the case of ALL and lymphoma, corticosteroids also have a direct antitumor effect that may benefit the patient in the short term, but ultimately make diagnosis of the underlying malignancy much more challenging. If a hematopoietic malignancy is likely, steroids should be given only when urgent diagnostic procedures have been scheduled (e.g., bone marrow aspiration and biopsy, lymph node excisional biopsy).
Outside of this scenario, the benefits of steroids are thought to outweigh their risks and are routinely used when cord compression is diagnosed, although clinical trials in children are lacking. In adults there is no evidence supporting high-dose dexamethasone; an initial dose of 1–2 mg/kg (max 10 mg) followed by 0.25–0.5 mg/kg (max 4 mg) every 6 h can be considered while awaiting further therapy (Kaal and Vecht 2004; Loblaw et al. 2005; George et al. 2008).
5.2.3.3 Surgery
Surgery in spinal cord compression can be utilized for tumor debulking with spinal cord stabilization or for diagnostic biopsy in the highly chemotherapy- or radiotherapy-sensitive tumor. Prospective studies in adult patients with cord compression have favored early laminectomy and debulking over radiation therapy alone although more recent retrospective studies have called this into question (Patchell et al. 2005; Rades et al. 2010). Laminectomy in the young child can lead to spinal deformities requiring future surgical intervention and therefore pediatric retrospective studies argue against an early surgical approach (Parikh and Crawford 2003). Recovery from surgery can also lead to delays in instituting definitive chemoradiotherapy.
An early case series from St. Jude Children’s Research Hospital suggested that initial debulking is warranted for sarcomas, whereas biopsy with chemotherapy or radiotherapy should be considered for patients with neuroblastoma, germ cell tumors and lymphomas (Klein et al. 1991). Patients presenting with rapid evolution of symptoms or complete loss of motor function were treated with urgent surgical decompression regardless of tumor histology. Smaller case series have supported the use of initial chemotherapy in neuroblastoma, Ewing sarcoma and germ cell tumors (Hayes et al. 1984). A study of 76 children with neuroblastoma from Italy found no difference in the success rates of chemotherapy, radiotherapy or surgical debulking in improvement of neurological signs, but noted that patients receiving radiotherapy or surgical debulking all went on to receive additional therapy for cord compression, whereas chemotherapy patients did not (De Bernardi et al. 2001). A follow-up study from this same group also noted a higher incidence of spinal deformities in the group receiving surgical intervention with a trend towards worse neurologic outcomes. As with the St. Jude data, patients with rapidly progressive symptoms or severe presentations were all treated with emergent surgery, making that group much higher risk than those treated with chemotherapy or radiotherapy (Angelini et al. 2011). A more contemporary study of 122 patients did not find any difference in spinal outcomes in patients treated with chemotherapy or surgery which the authors attribute to improvement in surgical technique (Simon et al. 2012).
In summary, each case of cord compression warrants individual consideration before a decision to proceed with debulking or biopsy can be made. Rapid progression of symptoms and total plegia are indications for emergent surgical management. Spinal column instability, which warrants surgical intervention, will also often necessitate surgical debulking. If the malignancy is known to be chemotherapy sensitive (i.e., hematopoietic malignancies, neuroblastoma, Ewing sarcoma, germ cell tumors), biopsy either at the site of cord compression or at another more easily accessible site can be considered prior to institution of chemotherapy. The timely availability of these options may also influence the treatment decision.
5.2.3.4 Radiation Therapy
The advantages of external beam radiation therapy include rapid onset of action and minimal invasiveness making radiation therapy an attractive option for patients who are too unstable to be considered surgical candidates, or in the palliative setting. Tumors that are radiosensitive such as hematologic malignancies (i.e., chloromas) and lymphoma may benefit from urgent radiotherapy while leaving additional diagnostic disease sites intact. Extensive metastatic disease may similarly be more amenable to radiation therapy than a surgical approach; when surgery is the primary therapeutic modality, adjunctive radiation may have a role in local control.
A case series from Children’s Hospital of Philadelphia demonstrated the benefit of urgent radiation therapy in combination with chemotherapy in a pediatric population with a variety of tumor types: of 33 patients presenting with cord compression, 55 % demonstrated improvement following chemoradiotherapy and 30 % had stabilization of their neurologic symptoms (Bertsch et al. 1998).
Dose-dependent long-term complications arising from radiation therapy can include future growth complications such as scoliosis as well as risk of secondary malignancy, particularly meningioma (Hoffman and Yock 2009). Stereotactic radiosurgery can deliver a single treatment dose up to 13 Gy to localized tumors without exceeding spinal cord tolerance; additionally, the spinal cord can tolerate high total doses, with a risk of myelopathy of <1 % at 54 Gy in adult patients (Kirkpatrick et al. 2010).
5.2.4 Outcomes
The best predictor of outcome in multiple studies has been the degree of neurologic disability at the time of presentation. One large cohort study suggested that only approximately 50 % of patients with paraplegia at the time of presentation improved despite treatment whereas close to 90 % of patients without paraplegia had improvement in their symptoms (Pollono et al. 2003). Published outcomes data are from small cohort studies with a variety of pathologic diagnoses and treatments, making any broad interpretation difficult. There is a general consensus, however, that outcomes for pediatric cord compression are significantly better than in adult patients.
5.3 Altered Mental Status
For the purposes of this chapter, altered mental status (AMS) includes patients with symptoms of encephalopathy (confusion, somnolence or coma) and those with seizures (which often, though not always, alter the level of consciousness). Although there are often distinct differential diagnoses for encephalopathy and seizure, many conditions unique to the pediatric oncology patient can predispose to either presentation. Additionally, subclinical seizure is a diagnostic consideration in any patient with altered mentation. For these reasons, we suggest a common approach to the differential diagnosis and initial management of such patients.
5.3.1 Presentation
Confusion or somnolence in an oncology patient should prompt a thorough neurologic examination and review of recent medications. Nonresponsive patients require urgent steps to secure the airway, breathing and circulation prior to additional diagnostic interventions. AMS may be due to a diffuse process affecting the brain, such as medication effect or subclinical seizure, or be due to direct involvement of the brainstem, increased intracranial pressure (ICP), or impending herniation. Ruling out the latter by means of neurologic assessment is of utmost importance. Details in the management of increased ICP are discussed in Sect. 5.4.
5.3.2 Initial Management
The first goal in a seizing patient is to stop the seizures. Benzodiazepines are often used as first-line medications in this setting. Lorazepam can be given intravenously if access is available; otherwise intranasal, buccal, or rectal formulations of diazepam or lorazepam can be employed. It is important to anticipate and treat the adverse effects of these medications including respiratory depression or hypotension, as repeated dosing may be required to halt seizure activity. Failure to respond to benzodiazepines (or recurrent seizures after initial response) should prompt escalation to additional antiepileptics such as phenobarbital or fosphenytoin. Levetiracetam is another antiepileptic that has the advantage of not inducing hepatic enzyme activity and thus has fewer interactions with chemotherapy. Seizures refractory to these interventions can be treated with intravenous loading with either valproic acid or levetiracetam and necessitate urgent determination of the underlying etiology.
Initial diagnostic workup should include serum chemistries, especially sodium, calcium, magnesium, phosphorus, and glucose, and head imaging. CT is often the easiest imaging to obtain in this setting and can rule out emergent, life-threatening causes of seizure. If there are concerns for cerebrovascular accident, a stroke-protocol MRI with diffusion-weighted imaging or magnetic resonance angiography should be considered. The seizing oncology patient should be managed in consultation with neurology; early neurosurgical involvement is also imperative for patients with known or suspected intracranial processes.
Continuous electroencephalography (EEG) monitoring should be strongly considered for any patient with persistent AMS or concerns for subclinical seizure. Lumbar puncture and MRI may be indicated when the patient is stabilized to look for evidence of progressive malignancy, infection or demyelination. Further treatment is dependent on the results of these initial diagnostic studies, as discussed below.
5.3.3 Differential Diagnosis
The differential diagnosis of seizure or AMS can be broadly divided into structural causes, including primary CNS or metastatic disease; metabolic derangements resulting in hyponatremia, hypocalcemia or hypoglycemia; infectious complications; toxic effects of chemotherapy; or stroke (ischemic or hemorrhagic). Posterior reversible encephalopathy syndrome (PRES) can also present with seizure. Seizures secondary to neurologic emergencies such as stroke are discussed in greater detail in Sect. 5.5.
5.3.3.1 Structural
Primary CNS tumors can directly lead to seizures. Oligodendrogliomas and gangliogliomas are common causes of brain tumor-induced seizure, though other low-grade gliomas and dysembryoblastic neuroepithelial tumors (DNETs) can be associated with seizures as well (Ogiwara et al. 2010). Such low-grade lesions may be surgically resected to provide relief from symptomatic seizures. Extracranial tumors can invade the CNS either by direct invasion, as with parameningeal rhabdomyosarcoma, or by hematogenous metastasis, as in Ewing sarcoma (the most common cause of pediatric CNS parenchymal metastasis), extracranial germ cell tumors, and leukemias.
Intracranial hemorrhage is another consideration in the seizing patient. Intratumoral hemorrhage has been variably reported in adults receiving the monoclonal antibody bevacizumab for primary CNS tumors (Seet et al. 2011; Khasraw et al. 2012). Certain primary childhood CNS tumors have a high proclivity for spontaneous intratumoral hemorrhage due to their intrinsic high vascularity; notorious examples are primary CNS choriocarcinoma, choroid plexus carcinoma and malignant gliomas. Derangement of the hemostatic system as seen in acute promyelocytic leukemia or after asparaginase therapy can also lead to intracranial hemorrhage in a patient without CNS disease.
Patients who have undergone resection of CNS tumors may develop postoperative seizures. No clear consensus exists regarding the use of prophylactic antiepileptic therapy in the postoperative setting. The American Academy of Neurology recommends tapering antiepileptic drugs (AEDs) within the first postoperative week, though it does not make a recommendation regarding AED initiation due to a lack of evidence even in the adult setting (Glantz et al. 2000). A retrospective study of 223 patients from Children’s Hospital of Philadelphia found that age younger than 2 years, supratentorial location of tumor and postoperative hyponatremia were the only independent predictors of postoperative seizure (Hardesty et al. 2011). Of the 229 operations reviewed, 7.4 % of patients seized and only 4.4 % received routine postoperative AEDs (Hardesty et al. 2011). Due to insufficient evidence, the use of prophylactic AEDs should be determined under consultation with the patient’s neurosurgeon and intensive care team. Given the low incidence of unprovoked postoperative seizure, workup of new seizures following tumor resection is warranted to exclude alternative etiologies.
5.3.3.2 Metabolic
The metabolic causes of AMS or seizure in pediatric oncology are similar to other pediatric patients: hyponatremia, hypoglycemia, and, less frequently, hypocalcemia secondary to hyperphosphatemia in severe tumor lysis syndrome (discussed in Chap. 3).
5.3.3.2.1 Iatrogenic Hyponatremia
Hyponatremia severe enough to provoke seizures warrants urgent correction though overly rapid correction can lead to central pontine myelinolysis. The risks of this complication are greater in chronic hyponatremia and in adult patients. A reasonable goal is to raise the sodium by ≤10 mEq/L in the first 24 h and then slowly normalize the serum sodium over the next 48 h. Hypertonic saline should be used in the seizing patient as it allows for more rapid correction than isotonic fluids which can be accomplished by calculation of the sodium deficit and either a subsequent fluid infusion rate or bolus therapy (Adrogue and Madias 2000; Sterns et al. 2009; Moritz and Ayus 2010).
5.3.3.2.2 Syndrome of Inappropriate Antidiuretic Hormone (SIADH) Release
The syndrome of inappropriate antidiuretic hormone (SIADH) secretion leads to dilutional hyponatremia due to inappropriate resorption of free water in the renal collecting tubules. SIADH has a multitude of causes including poorly understood effects of CNS or intrapulmonary lesions as well as secondary to chemotherapeutic agents including vincristine, cyclophosphamide, cisplatin and melphalan (Lim et al. 2010). The diagnosis of SIADH relies on the combined laboratory findings of low serum osmolarity (i.e., <290 mOsm/L) with inappropriately concentrated urine osmolality (i.e., >100 mOsm/kg).
Treatment of SIADH depends on: (1) removing the causative agent when possible; (2) restricting free water intake to ≤urine output (±insensible losses); and (3) correction with hypertonic saline, if indicated. Hypertonic saline is generally avoided in patients with asymptomatic SIADH and in those presenting solely with AMS but may be necessary to control seizures. Free water restriction is difficult in situations where hyperhydration is necessary to avoid chemotherapeutic toxicity as with cyclophosphamide administration. In these situations, using normal saline as opposed to hypotonic fluids and checking serum sodium levels frequently is warranted. Chronic SIADH can be managed with oral urea and fluid restriction (Huang et al. 2006). The oral vasopressin receptor antagonist tolvaptan has been shown to be effective in adult patients with paraneoplastic SIADH, but data on broader use in cancer patients and the safety in pediatrics are not yet available (Kenz et al. 2011).
5.3.3.2.3 Cerebral Salt Wasting
Cerebral salt wasting (CSW) is a controversial diagnosis of unclear etiology; some authors suggest CSW does not exist or is exceedingly rare, while others suggest it is not cerebral in origin (Singh et al. 2002; Rivkees 2008). Nonetheless, CSW is a consideration in the pediatric neuro-oncology patient with hyponatremia and seizures. In reviewing children recovering from craniotomy for tumor resection, Hardesty et al. (2011) found that nearly half the patients with laboratory identified CSW developed postoperative seizures. The main clinical distinction between patients with SIADH and CSW is volume status; CSW represents hypovolemic hyponatremia, while patients with SIADH are either slightly hypervolemic or euvolemic. As in patients with SIADH, CSW patients will have hyponatremia, low serum osmolarity and relatively concentrated urine with high urine sodium excretion. The patient with CSW will often respond to isotonic fluid infusion, whereas normal saline frequently worsens hyponatremia in patients with SIADH. Correction of hypovolemia is the main therapy in CSW. Patients with chronic hyponatremia may require oral supplementation with salt tablets.
5.3.3.3 Infection
Patients receiving immunosuppressive chemotherapy have an increased risk for CNS infection which may present with AMS or seizure. Lumbar puncture is a helpful diagnostic tool in this setting, but should be deferred until the patient has been stabilized and CT imaging performed. Institution of broad antimicrobial coverage should not be delayed to obtain cerebrospinal fluid (CSF); cell counts can be informative in non-cytopenic patients even after sterilization of CSF and polymerase chain reaction (PCR)-based assays for viruses can remain positive after antiviral therapy has been instituted.
Infectious studies should include cultures of blood and CSF. Enterovirus, herpes simplex virus (HSV) and human herpesvirus-6 (HHV6) can be detected by CSF PCR. Culture or direct fluorescence antibody (DFA) testing of oropharyngeal lesions should also be considered. Treatment with high-dose acyclovir should be initiated urgently if viral meningoencephalitis is suspected.
A study of 40 cases of bacterial and fungal meningitis in pediatric oncology patients found that recent neurosurgery or a CNS device was present in the majority of cases (Sommers and Hawkins 1999). Among patients without neurosurgical intervention, those with hematologic malignancies and prolonged neutropenia were at the highest risk for infection.
5.3.3.4 Chemotherapy-Associated Neurotoxicity
Methotrexate, ifosfamide, and cytarabine (Ara-C) are commonly used pediatric chemotherapeutic agents that can provoke seizures and encephalopathy. Transverse myelitis is a peculiar complication described following the combined intravenous and intrathecal administration of cytarabine. Vincristine is most commonly known for its associated peripheral neuropathies, but medical errors in administration have highlighted its extreme neurotoxicity when introduced into the CNS or given at erroneously high doses systemically. Thiotepa, carmustine (BCNU) and busulfan are myeloablative agents used at high doses in stem cell transplant with neurologic dose-limiting toxicities (Papadopoulos et al. 1998). Other less commonly used agents such as nelarabine (a prometabolite of Ara-G) have similarly been implicated in patients developing new encephalopathy or seizures.
5.3.3.4.1 Methotrexate
Acute neurotoxicity has been described within 24 h of intravenous or intrathecal methotrexate administration, consisting of confusion, seizures, or encephalopathy. Methotrexate neurotoxicity is frequently self-resolving and is not related to drug levels (Rubnitz et al. 1998). By contrast, acute neurotoxicity following a methotrexate overdose can be very severe. Symptoms include myelopathy, seizures, encephalopathy and death from progressive necrotizing leukoencephalopathy. Published experience in overdose suggests benefit from high-dose intravenous (IV) leucovorin, IV carboxypeptidase G2 and alkalinization of the urine, with hemodialysis if renal insufficiency develops. If the overdose was administered intrathecally, the addition of CSF lavage or exchange and intrathecal administration of carboxypeptidase G2 have been used with good outcomes (Spiegel et al. 1984; Widemann et al. 2004).
Subacute methotrexate neurotoxicity includes a constellation of neurologic symptoms, often reversible, that occur after either IV or IT methotrexate. Symptoms include severe headache, focal or generalized seizures, AMS ranging from confusion to coma, and sensory and motor findings consistent with cerebrovascular accident. Aphasia is commonly reported (Dufourg et al. 2007). The median time from methotrexate administration to symptom development is consistently reported to be 10 days (Mahoney et al. 1998). The risk of developing methotrexate neurotoxicity increases with combination IV/IT therapy, increasing total cumulative administered methotrexate dose and cranial irradiation (Land et al. 1994; Reddick et al. 2005).
There is no single diagnostic test for methotrexate neurotoxicity; therefore recognizing the clinical scenario is vital to making the diagnosis. MRI can support the diagnosis (Fig. 5.2), as patients have characteristic changes on diffusion-weighted imaging that mimic ischemia (Reddick et al. 2005). However, unlike true ischemic changes, there is rapid normalization within several weeks (Sandoval et al. 2003). MRI findings are frequently multifocal and do not fit vascular distribution patterns. These MRI changes can also be seen in asymptomatic patients receiving methotrexate. Therefore, more extensive investigation to rule out other causes of seizure or AMS is often necessary.

Subacute methotrexate neurotoxicity. This 12-year-old patient with acute lymphoblastic leukemia developed symptoms of aphasia and lethargy approximately 10 days following combined intrathecal and high-dose intravenous (5 g/m2) methotrexate during delayed intensification. The T2-FLAIR MRI shown in Panel a demonstrates the development of ill-defined patches of hyperintensity within the white matter (white arrow). Diffusion-weighted imaging taken concurrently, shown in Panel b, shows restricted diffusion (black arrows). Although the pattern is not consistent with any one vascular territory, infarct is on the differential diagnosis with such findings. Fortunately, the patient’s symptoms resolved over 48 h, and while repeat scans taken 1 month later demonstrate continuation and evolution of the T2-FLAIR changes (Panel c, white arrows), the diffusion changes have now completely resolved (Panel d). The patient has continued to be completely asymptomatic but has not yet been rechallenged with intrathecal methotrexate
As mentioned, the majority of cases of methotrexate neurotoxicity are self-resolving. There are varying amounts of preclinical data and published case series that support the use of aminophylline or dextromethorphan in the acute phase and leucovorin rescue with future IT methotrexate in patients who have developed neurotoxicity in the past (Winick et al. 1992; Drachtman et al. 2002; Inaba et al. 2008). Aminophylline can be considered for patients who have acute obtundation after methotrexate. If toxicity is thought secondary to IT therapy, one can consider postponing the next scheduled IT methotrexate or substituting IT Ara-C and hydrocortisone, though there are very limited data to show this is an effective IT therapy in ALL. Patients can be safely rechallenged with IT methotrexate, either with or without leucovorin or aminophylline rescue, and not redevelop neurotoxicity (Rollins et al. 2004; Inaba et al. 2008). Thus, completely eliminating methotrexate from future therapy is not usually indicated.
5.3.3.4.2 Ifosfamide
Patients receiving ifosfamide may develop encephalopathy that can range from sleepiness and confusion to seizures or coma, starting several hours after infusion commencement. The reported frequency of this side effect is as high as 22 % in pediatric patients (Pratt et al. 1986). Although there is no clear dose-dependence, studies have suggested that hypoalbuminemia and renal dysfunction are risk factors (David and Picus 2005). The exact mechanism for ifosfamide-induced encephalopathy (IIE) is unclear although it is thought secondary to high levels of urine glutaric acid which is linked to one particular metabolite of ifosfamide, chloroethylamine (Kupfer et al. 1994). Methylene blue has been reported to successfully treat IIE and may also be effective as prophylaxis through unclear mechanisms, possibly by correcting derangement in mitochondrial flavoproteins (Pelgrims et al. 2000; Hamadani and Awan 2006). A review by Patel (2006) found that a lack of controlled evidence makes the effectiveness of methylene blue unclear. Thiamine has also been reported to be an effective treatment and prophylaxis for IIE through unclear mechanisms and also with a lack of controlled data (Hamadani and Awan 2006).
5.3.3.4.3 Cytarabine
The pyrimidine analog cytarabine is frequently used in the treatment of pediatric leukemia and lymphoma. There is a wide range of employed dosages and at lower doses neurologic toxicity is uncommon. Higher doses, however, are associated with an acute cerebellar syndrome in ≥10 % of patients (Baker et al. 1991). There is a clear dose-dependence of this side effect and it is generally seen at doses ≥3 g/m2/day; spacing administration from twice daily to once daily has been shown to reduce the incidence of cerebellar toxicity (Smith et al. 1997).
The clinical manifestations of cytarabine-induced cerebellar toxicity include the classic cerebellar signs of dysarthria, dysdiadochokinesia and ataxia (Smith et al. 1997). Cerebral dysfunction can also be seen in a subset of cases, with manifestations including encephalopathy, seizures and coma. Histopathologic changes seen in the cerebellar syndrome include loss of Purkinje cells in the cerebellar hemispheres and vermis along with a proliferation of glial cells known as Bergmann’s gliosis (Baker et al. 1991).
Symptoms often resolve following discontinuation of cytarabine; however some patients will have permanent neurologic damage. One large case series of adults and children receiving high-dose cytarabine found that approximately 1 % of patients who developed severe cerebellar symptoms had an irreversible or fatal course (Herzig et al. 1987). Although this same report demonstrated that some patients may be rechallenged without permanent neurologic damage, the decision to proceed with additional cytarabine is a difficult one. Identified risk factors for cerebellar syndrome include dose and timing as discussed above as well as hepatic or renal insufficiency and older age (Herzig et al. 1987; Baker et al. 1991; Rubin et al. 1992). No potential genetic modifiers have been reported in the literature.
IT cytarabine can be administered in its standard formulation or as a slow-release liposomal formulation (DepoCyt). The liposomal formulation appears to have a higher incidence of arachnoiditis; prophylactic corticosteroids may be beneficial in this situation (Glantz et al. 1999). As discussed, concomitant administration of IT and high-dose IV cytarabine is associated with the development of transverse myelitis presenting as bowel and bladder dysfunction with lower extremity weakness and should be avoided (Dunton et al. 1986; Schwenn et al. 1991).
5.3.3.5 Posterior Reversible Encephalopathy Syndrome
Posterior reversible encephalopathy syndrome (PRES, also called RPLS, reversible posterior leukoencephalopathy syndrome) was first described as a syndrome of encephalopathy accompanied by transient subcortical white matter changes on T2-weighted MRI (Hinchey et al. 1996). The clinical manifestations of PRES include headaches, seizures, AMS and cortical blindness. Hypertension is a clear antecedent and most significant risk factor in the development of PRES. Hypertension may be secondary to medications, such as corticosteroids, or concurrent medical conditions such as renal failure from tumor lysis syndrome (Greenwood et al. 2003). Retrospective studies also suggest a higher incidence of PRES during induction chemotherapy, independent of tumor lysis syndrome or hyperleukocytosis (Norman et al. 2007).
Since its initial description, PRES has been recognized to not be limited to the posterior circulation of the brain nor to white matter exclusively nor be fully reversible in all cases (Lucchini et al. 2008). While the initial report postulated a role for immunosuppression in the pathogenesis of PRES, it has been subsequently described in non-immunocompromised patients such as in pregnancy-induced hypertension. Specific immunosuppressive medications, such as tacrolimus and cyclosporine, continue to be implicated in case reports in part due to their effect on blood pressure.
PRES has been noted in pediatric malignancies including ALL, AML, non-Hodgkin lymphoma, and solid tumors including neuroblastoma, osteosarcoma, and Ewing sarcoma. Hypertension was a clearly identified preceding event in the majority of cases. Seizures were the most common clinical manifestation, with AMS, headache and some form of visual disturbance occurring in 30–40 % of cases (de Laat et al. 2011). While the majority of patients had a reversible course, 12 % had persistent neurologic symptoms.
Brain MRI is required to make the diagnosis of PRES. Vasogenic edema most often symmetrically affects the parietal and occipital subcortical white matter. Cortex, basal ganglia, and posterior fossa structures such as cerebellum and brainstem can also be involved.
Treatment of PRES begins with aborting seizures, if present, and decreasing the patient’s blood pressure using calcium channel blockers such as nifedipine or ß-blockers such as labetalol. Ongoing antiepileptic therapy is often warranted in the acute setting. While many studies suggest that AEDs may be tapered when neuroimaging normalizes, multiple retrospective reviews of pediatric oncology patients with PRES have found high rates of ongoing epilepsy (Morris et al. 2007; de Laat et al. 2011). Chemotherapy is often held while the patient is symptomatic and there are multiple reports of safe resumption of antineoplastic therapy, including any possible offending agents, with no recurrence of symptoms (Morris et al. 2007; Norman et al. 2007; de Laat et al. 2011).
5.3.4 Outcomes
While many of the described causes of seizures are temporary, some patients with childhood cancer will require long-term prophylactic AEDs. Data from the Childhood Cancer Survivor Study showed that 6 % of childhood ALL survivors were identified as having a seizure disorder with at least half developing this as a late effect of therapy (Goldsby et al. 2010). The choice of prophylactic AED is made based on seizure etiology and response to therapy, but drug-drug interactions with chemotherapeutics is an important consideration as well.
In a retrospective study of ALL patients, treatment with phenytoin, phenobarbital or carbamazepine for at least 1 month duration was associated with an increased risk of relapse in patients with B cell, though not T cell, ALL (Relling et al. 2000). The investigators showed that the use of these enzyme-inducing antiepileptic medications led to increased clearance of some chemotherapeutic agents.
Levetiracetam is an AED that is not metabolized through the CYP450 system, making it a safer choice for prolonged therapy. Although considered efficacious without affecting relapse risk, studies in pediatric ALL are minimal (Ruggiero et al. 2010). Regardless of the choice of agent, the practitioner should be aware of the high potential for altered drug metabolism in patients receiving cytochrome P450-inducing or P450-inhibiting AEDs.
5.4 Increased Intracranial Pressure
Increased ICP is due to an increase in the volume of one of three constituents of the intracranial space: blood, CSF or soft tissue. In infants whose fontanels are still open there is a greater capacity for an increase in any one of these compartments prior to the development of symptoms.
Intracranial hypertension can lead to a loss of cerebral perfusion pressure (CPP), defined as the mean arterial pressure less the intracranial pressure (CPP = MAP−ICP). Insufficient CPP will produce secondary ischemia due to a lack of forward blood flow. Even more concerning, intracranial hypertension can lead to herniation syndromes wherein brain parenchyma is pushed downward below the tentorium cerebelli, through the foramen magnum, or transversely across the falx cerebri, in all cases leading to potentially irreversible neurologic damage or death. Early diagnosis, stabilization and institution of therapy appropriate to the cause of increased ICP are essential in preventing these outcomes.
5.4.1 Presentation
Headache is one of the most common, and unfortunately least specific, features that can herald increased ICP. In a retrospective review of 315 pediatric patients with headache who had neuroimaging, headache awakening the patient from sleep and a negative family history for migraine were the strongest individual predictors of a mass lesion (Medina et al. 1997). Confusion and abnormal neurologic examination were also risk factors in that study. Vomiting is another common presenting sign; this is classically worst in the morning, due to increasing ICP while sleeping in the supine position relative to standing. Rapidly increasing ICP may present with a short interval of headache or constitutional symptoms that precedes coma or death due to pressure on the reticular activating system in the midbrain.
In infants, irritability may be the only presenting symptom. Physical examination may reveal a wide, tense fontanel with split sutures. Measuring head circumference should be a standard part of the neurologic examination in an infant, and discordance between circumference and weight or length should prompt closer investigation. Sundowning, a downward gaze preference at rest, may also be seen.
Focal neurologic features of increased ICP depend on the location of the mass or the progression of any herniation. Optic nerve swelling in response to intracranial hypertension can lead to papilledema. However, papilledema may not develop for weeks after the onset of increased ICP and therefore a normal fundoscopic exam does not rule out intracranial hypertension. Cranial nerves III, IV and VI can also be affected leading to paralysis of gaze or complaints of diplopia. Ipsilateral mydriasis with poor pupillary response to light can be seen due to the effect of increased ICP on CN III or from direct impingement in uncal herniation. Finally, a localized mass or ischemia from decreased CPP can lead to focal deficits of strength or sensation. Cushing’s triad, the combination of hypertension, bradycardia, and an irregular respiratory pattern, is a late manifestation of increased ICP and an ominous sign of impending herniation.
5.4.2 Initial Management
The patient with suspected increased ICP should have their airway, breathing and circulation secured before proceeding with diagnostic evaluation. A normal arterial partial pressure of carbon dioxide (PaCO2) will prevent hypercapnea-induced vasodilation with subsequent spikes in ICP; intubation should be instituted in the patient with hypoxia, hypoventilation, excessive sedation or loss of protective airway reflexes. Hypotension will lead to a drop in CPP with attendant ischemia and should be treated with normal saline infusion to attain euvolemia. Additional measures to decrease ICP such as elevating the head of the bed and keeping the patient’s head at midline should be commenced emergently. Hyperthermia should be treated with antipyretics or cooling measures to decrease metabolic demand. Pain should be aggressively treated to prevent spikes in blood pressure or ICP.
Patients who are stabilized with these measures should undergo emergent head imaging. A noncontrast head CT or rapid-sequence MRI can quickly identify a new mass lesion, obstructive hydrocephalus, or hemorrhage. Negative head imaging should prompt further workup. MR angiography and venography can be helpful in finding occult thrombosis. Mass lesions may warrant MRI for further evaluation prior to operative intervention. A lumbar puncture should not be performed without confirmatory negative head imaging to rule out the risk of downward herniation with CSF release. Opening pressure should ideally be measured with the patient’s legs extended or with the patient in the sitting position. Although there are not clear guidelines for normal ICP in children, values >20 mmHg are considered elevated in adults.
Patients who have progressively worsening neurologic exams or clear signs of herniation on physical examination or imaging require more aggressive interventions. Hyperosmolar therapy with mannitol or hypertonic saline can be used to decrease the total brain water content. Mannitol is started at doses of 0.25–1 g/kg of a 20 % solution and repeated every 6 h until serum osmolarity is raised to between 300 and 320 mOsm/L (Pitfield et al. 2012). Hypertonic saline has the advantage of not causing osmotic diuresis so may be preferred in the hypovolemic patient (Khanna et al. 2000). Additional treatment measures will generally be tailored to the underlying etiology.
5.4.3 Differential Diagnosis
5.4.3.1 Soft Tissue
A large meta-analysis of published data suggests that signs of increased ICP such as headache and vomiting are the most common presenting features in patients with newly diagnosed brain tumors, whether located supratentorially or in the posterior fossa (Wilne et al. 2007). In the patient with a known CNS lesion, development of new symptoms of increased ICP should raise concern for tumor progression leading to obstructive hydrocephalus. While corticosteroids are not standard therapy for increased ICP, the vasogenic edema associated with a mass lesion can be reduced with dexamethasone and should be considered in patients with primary brain lesions causing increased ICP (Andersen and Jensen 1998). Additional treatment may include surgical resection with or without placement of a ventriculoperitoneal shunt (VPS). For patients with unresectable disease, either due to multifocality or other comorbidities, radiation therapy or stereotactic radiosurgery can be considered.
5.4.3.2 Cerebrospinal Fluid
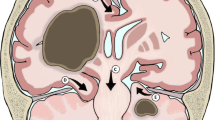
Hydrocephalus is an excess of CSF within the skull. Increased production of CSF is relatively rare, but can be seen in patients with choroid plexus papilloma (Eisenberg et al. 1974). More commonly, drainage of CSF is obstructed due to mass effect on the ventricular system. Cranial venous drainage can also be impeded by obstruction of the great vessels in superior vena cava syndrome, which is discussed in detail in Chap. 4. CNS tumors arising from the third and fourth ventricles can easily block the flow of CSF and produce symptoms of obstructive hydrocephalus (Fig. 5.3). An external ventricular drain (EVD) may help relieve the pressure from such an obstruction in the short term, allowing for attempted total resection in a more controlled fashion. Unfortunately, up to one-third of patients with posterior fossa masses will develop postoperative hydrocephalus requiring long-term shunting (Culley et al. 1994). Based on this statistic, some neurosurgeons will opt for preemptive VPS placement. A recent retrospective review of children with posterior fossa masses identified several risk factors to predict the need for a VPS: age <2 years, papilledema, severity of preoperative hydrocephalus and cerebral metastases (Riva-Cambrin et al. 2009).

Obstructive hydrocephalus. This teenage boy presented with a gradual history of headaches, ataxia and progressively worsening vomiting. T2-weighted MRI taken at the time of his presentation to medical care demonstrates a cystic mass arising in the posterior fossa. The lateral ventricles and third ventricle, seen with dark CSF on this sequence, are massively dilated in this example of obstructive hydrocephalus. Pathology after gross total resection was consistent with low-grade glioma
Malignant leptomeningeal involvement, as seen in leukemia as well as primary CNS tumors such as medulloblastoma, can occasionally be severe enough to block resorption of CSF through the arachnoid granulations. This condition can be treated by VPS placement while systemic therapy is instituted though the shunt may become clogged by extensive cellular material.
Finally, malfunction or infection of a previously placed VPS is an important diagnostic consideration. Diagnosis via VPS tapping and need for externalization or revision should be established based on neurosurgical consultation.
5.4.3.3 Hemorrhage and Thrombosis
An excess of blood in the cranial vault can be caused by hemorrhage (e.g., within the tumor or tumor bed) or by thrombosis with poor venous drainage. Thrombosis is a well-described complication of asparaginase therapy in ALL. L-asparaginase has been shown to decrease levels of antithrombin and plasminogen leading to a prothrombotic state (Leone et al. 1993). Simultaneous depletion of fibrinogen can also predispose to hemorrhage. Thrombosis in childhood ALL patients is not uncommon; one meta-analysis reported a prevalence of 3.2 %, with roughly half of the events CNS thromboembolic events, and the majority of those sinovenous thromboses (Athale and Chan 2003).
Sinovenous thrombosis should be treated with anticoagulation to prevent clot extension. Repletion of antithrombin using pooled human concentrate (Thrombate) or recombinant antithrombin (ATryn) is required to achieve therapeutic anticoagulation with either unfractionated or low molecular weight heparin. In addition, cryoprecipitate can be used to replete fibrinogen up to normal levels (i.e., >150 mg/dL), treating ongoing coagulopathy and decreasing the risk of bleeding. Fresh frozen plasma can be used in lieu of these products, but has limited amounts of antithrombin and fibrinogen, which are the most consistently decreased in studies of asparaginase therapy. Minor hemorrhages seen in association with sinovenous thrombosis may make the decision to anticoagulate more difficult, but the use of a reversible agent (unfractionated heparin), frequent monitoring of hemostatic parameters, and careful neurologic monitoring in an intensive care unit can decrease the risks associated with this strategy. Without treatment, sinovenous thrombosis can progress to infarction with worsened hemorrhagic complications.
In the event of anaphylaxis to E. coli asparaginase, Erwinia asparaginase is often substituted as an alternative therapy. Giving equivalent doses, a European trial did find a reduced incidence of coagulation abnormalities with Erwinia asparaginase compared to E. coli, though the frequency of thrombotic events was not reported (Duval et al. 2002). The same study was stopped early, however, due to superior survival in the E. coli asparaginase arm, and so routine substitution of Erwinia asparaginase for concerns of thrombosis is not recommended. The frequency of thrombotic events appears to be similar in patients treated with the longer-acting PEG-asparaginase as compared to those treated with L-asparaginase (Silverman et al. 2010).
The decision to restart asparaginase following sinovenous thrombosis is a complex one; limited data suggest that asparaginase can be safely restarted (Grace et al. 2011). The recurrence risk was higher in adults, consistent with the observation that the overall risk of venous thromboembolism increases from puberty throughout adulthood (White 2003).
Prophylaxis with purified antithrombin is not recommended in pediatric oncology (Mattioli Belmonte et al. 1991). The PARKAA (Prophylactic Antithrombin Replacement in Kids with Acute Lymphoblastic Leukemia Treated with Asparaginase) study, a randomized trial comparing prophylactic antithrombin repletion to observation, found a nonstatistical trend towards fewer thromboses in the prophylaxis group but lacked power to make conclusive recommendations (Mitchell et al. 2003).
5.4.3.4 Idiopathic Intracranial Hypertension
Idiopathic intracranial hypertension (IIH), previously termed pseudotumor cerebri, is a poorly understood syndrome characterized by elevated ICP without any obvious structural cause. Several chemotherapeutic agents can predispose the pediatric oncology patient to IIH. Corticosteroids, and specifically their withdrawal, have been associated with the development of IIH in children (Neville and Wilson 1970). IIH is also a commonly reported side effect of treatment with vitamin A analogs, including all-trans retinoic acid (ATRA) in the treatment of acute promyelocytic leukemia and isotretinoin and fenretinide in the treatment of neuroblastoma (Bigby and Stern 1988; Smith et al. 1992; Children’s Oncology Group [CCG 09709] et al. 2006).
Treatment of IIH can include serial large-volume CSF drainage through lumbar punctures or the use of acetazolamide. In cases of ATRA-related IIH, ATRA is often held with additional supportive steps to help alleviate symptoms (Holmes et al. 2012). Rechallenge with ATRA can be attempted if symptoms subside. The most severe complication of IIH is visual loss due to optic nerve ischemia; patients should therefore have routine ophthalmologic follow-up. Occasionally IIH is severe enough to require surgical CSF shunting to prevent further optic nerve damage (Chern et al. 2012).
5.5 Cerebrovascular Disease in Pediatric Cancer Patients
Cerebrovascular disease is one of the most devastating disorders and can present as either cerebrovascular accident (CVA, also referred to as stroke) or cerebrovascular malformations such as aneurysms. For the pediatric oncology patient, we will focus on stroke as the manifestation of cerebrovascular disease. Stroke is a disabling consequence of childhood cancer—and childhood cancer treatment—that remains poorly understood. Stroke can occur in the perioperative setting or may be treatment related and is broadly classified as hemorrhagic or ischemic. Ischemic stroke, which represents about 55 % of pediatric CVA, is further subdivided into large and small vessel stroke.
Stroke is generally defined as the abrupt onset of focal neurologic deficits referable to a vascular distribution and lasting >24 h. In children, especially the very young, stroke can also present as AMS or seizures (Zimmer et al. 2007; Hartman et al. 2009; Beslow et al. 2010). A recent retrospective analysis reported that children <1 year of age (n = 11) commonly presented with AMS or seizures, whereas older children (n = 65) commonly presented with focal weakness in ischemic stroke (Abend et al. 2011). Neurologic deficits that last <24 h are generally classified as a transient ischemic attack (TIA). Thus, the diagnosis of stroke is made based on the patient’s clinical presentation with laboratory studies and brain imaging as diagnostic correlates. Silent strokes seen on imaging alone are referred to as infarcts.
Cerebral ischemia is caused by a reduction in blood flow to the brain. Clinical symptoms occur within seconds to minutes due to the lack of glycogen storage in neurons, leading to energy failure. If blood flow is not restored quickly, infarction or death of brain tissue occurs. A generalized reduction in blood flow due to hypotension usually produces syncope. If the blood flow is not restored quickly, infarction between the border zones of major cerebral artery distribution occurs, referred to as a “watershed infarct,” presenting with proximal arm and leg weakness. Focal ischemia is often due to thrombosis of cerebral vessels or by emboli from other sources (e.g., the heart). Hemorrhagic stroke causes symptoms due to mass effect on surrounding brain structures and by direct toxic effect of blood. Pediatric stroke research has mainly focused on ischemic stroke, and therefore our knowledge of pediatric hemorrhagic stroke remains limited (Zimmer et al. 2007). In cases where bleeding occurs within a brain lesion (such as a primary brain tumor), pediatric oncologists often refer to this as intratumoral bleeding rather than hemorrhagic stroke. Here, for consistency, intratumoral hemorrhage is included within the category of hemorrhagic stroke.
5.5.1 Presentation of Stroke in Pediatric Patients
The clinical presentation of a CVA depends on the territory of brain involved. Table 5.3 lists key clinical findings based on the vascular territory, mainly attributable to ischemic stroke. In young children focal neurological deficits can be more subtle and harder to elicit.
Hemorrhagic stroke in children, especially the very young, often presents with nonspecific signs and symptoms such as AMS or seizures; headache is often reported in older children. One study assessed the clinical presentation of 51 children ≥6 years of age and found that 73 % presented with headache, 57 % with AMS, 39 % with focal neurological signs, 33 % with nausea/vomiting, and 16 % with seizures or other symptoms such as dysphasia and abnormal gait (Lo 2011).
5.5.2 Differential Diagnosis of Stroke
In the child with acute onset of new neurologic symptoms, the initial differential diagnosis is broad and includes limited focal movement secondary to pain, Todd’s paralysis after seizure, complex migraines, intoxication presenting as AMS or ataxia, medication side effects, and peripheral nerve injuries, among others. Despite increasing stroke awareness in the adult population, the diagnosis of stroke in children continues to be significantly delayed (McGlennan and Ganesan 2008; Rafay et al. 2009; Srinivasan et al. 2009). A recent report of children suffering from arterial ischemic stroke documented a median time >24 h between clinical onset and imaging confirmation.
5.5.3 Etiology of Stroke in Pediatric Cancer Patients
The underlying etiology of stroke in pediatric cancer patients is complex and poorly studied. In about 30 % of children presenting with an acute ischemic stroke, no cause could be elucidated despite extensive investigations (Roach 2000). No long-term, prospective study is available to better define the risk and underlying mechanisms of stroke in pediatric cancer patients. Current studies lack rigorous imaging correlates of stroke symptoms and are often based on self-reports from patients or their caregivers.
Stroke can occur during neurosurgery in pediatric intracranial tumors with close proximity of tumor and intracranial vessels. Further, several chemotherapeutic agents have been associated with increased risk of stroke such as mentioned with asparaginase and notably with the monoclonal antibody bevacizumab, which is directed against vascular endothelial growth factor receptor (VEGFR). In adult patients bevacizumab has been associated with a small but significant increase in arterial thrombotic events as well as risk for intracerebral hemorrhage (Taugourdeau-Raymond et al. 2012). The incidence of bevacizumab-associated cerebral hemorrhage in children is unknown.
Previous studies have shown that children treated for CNS tumors, leukemia, Hodgkin lymphoma, and other cancers carry a significantly increased stroke risk (Fig. 5.4), with cranial radiation therapy (CRT) a particularly strong risk factor (Bowers et al. 2005, 2006; Haddy et al. 2011). How CRT increases stroke risk in cancer survivors is not well understood. Few risk factors for stroke and arteriopathy in children treated with CRT have been described and include optic pathway tumors associated with NF-1, younger age and higher radiation dose to the circle of Willis (Grill et al. 1999). Current literature has focused on arteriopathies that develop within a relative short time following CRT (Bitzer and Topka 1995; Laitt et al. 1995; Omura et al. 1997; Grill et al. 1999; Fouladi et al. 2000). Moyamoya, a specific type of cerebral arteriopathy characterized by progressive stenosis of the terminal internal carotid arteries, has been shown more prevalent in children who undergo CRT for ALL and brain tumors as compared to children who do not receive CRT (Kikuchi et al. 2007; Ullrich et al. 2007). Others have shown that children treated with CRT are at increased risk for lacunar strokes, thought secondary to small vessel vasculopathy (Fouladi et al. 2000).

Ischemic stroke. This 19-year-old young woman, whose MRI is shown in this figure, presented with acute dysarthria and right-handed sensory changes. The diffusion-weighted images shown here demonstrate bilateral strokes with restricted diffusion, which appear bright in this sequence. The patient had a history of Hodgkin lymphoma diagnosed 4 years prior which had relapsed and subsequently treated with an autologous and then allogeneic hematopoietic stem cell transplant. Although the patient did have irradiation as part of her therapy, no arteriopathy could be detected on magnetic resonance angiography to explain the origin of her stroke
There is an additional body of literature that raises concern for long-term effects of radiation therapy by accelerating atherosclerosis, the most common etiology of stroke in adults (Dorresteijn et al. 2005). In pediatric and adult cancer patients, neck irradiation increased cervical carotid artery wall thickness, a marker for atherosclerosis (O’Leary et al. 1999; Hollander et al. 2002; Dorresteijn et al. 2005; Meeske et al. 2009). In a retrospective cohort study of 367 adult patients with head and neck tumors, neck irradiation increased risk of carotid artery-related strokes by almost tenfold compared to normal adults, and that risk was enhanced by hypertension (Dorresteijn et al. 2002). In animal models, irradiation of hypercholesterolemic mice and rabbits leads to accelerated atherosclerotic plaque formation (Vos et al. 1983). Hence, it is plausible that CRT leads to accelerated intracranial atherosclerosis and thereby increases long-term stroke risk as childhood cancer survivors become young adults.
5.5.4 Management of Acute Stroke
In the child with acute neurologic deficits, the initial evaluation should include a detailed neurologic examination to localize the site of stroke as well as careful review of the prior history and current medications. Brain imaging studies should be performed at the time of presentation if the onset of symptoms is acute. The type of imaging used will depend on availability and on the age of the child. MRI is preferred over CT given the radiation exposure associated with CT as well as the differences in time delay and resolution. CT will reliably identify hemorrhage as the cause of stroke but may fail to show ischemic stroke if the images are obtained within the first hours of presentation, if the stroke is small, and especially if located within the posterior fossa, due to bone artifact. MRI will detect hemorrhage and small strokes even in the early hours after onset of symptoms. MR angiography can identify stenosis of large intracranial vessels as well as arterial dissection. Transcranial Doppler (TCD) is another method to noninvasively assess and follow narrowing of intracranial vessels. Additional imaging with conventional angiography might be indicated depending on severity. Children with stroke should be treated by a specialized team including a pediatric stroke specialist, a pediatric neurosurgeon, an interventional radiologist and a rehabilitation team to ensure the best possible outcome.
5.5.4.1 Ischemic Stroke
After an ischemic stroke is diagnosed, the patient should be monitored and treated in a pediatric intensive care unit. Treatment is mainly supportive. Similar to hemorrhagic stroke, the first steps are to stabilize the child and assess the airway, breathing and circulation. Permissive hypertension is indicated to assure collateral flow to the area of ischemia and minimize injury. Fever is detrimental and children should be treated with antipyretics to achieve normothermia. Glucose levels should be carefully monitored and normalized as needed. Intravenous recombinant tissue plasminogen activator (tPA) is used in the acute setting in patients ≥18 years of age within the first 4.5 h after onset of symptoms if no contraindications to tPA are present. There is limited understanding of the safety and efficacy of tPA in pediatric patients, and therefore it is not yet the standard of care in this patient population. The Thrombolysis in Pediatric Stroke Study (TIPS) is a multicenter trial with the goal to assess the safety and efficacy of tPA in children with ischemic stroke; results from this study are pending (Amlie-Lefond et al. 2009). To assess the underlying etiology of ischemic stroke, children often have vascular imaging performed, particularly MR angiography. The necessity of assessment of an underlying prothrombotic state in pediatric stroke is unclear, and testing has not been shown useful in improving clinical outcomes (Raffini 2008). For all pediatric stroke cases, the reported prevalence of prothrombotic conditions ranges from 20–50 % (Mackay and Monagle 2008). Assessment of the most common thrombophilias including antithrombin deficiency, protein C or protein S deficiency, hyperhomocysteinemia, prothrombin 20210 gene mutation, elevated FVIII, factor V Leiden mutation, elevated lipoprotein(a) and antiphospholipid antibodies can be considered (Raffini 2008; Roach et al. 2008). Pregnancy should be excluded in teenage girls presenting with stroke and discontinuation of estrogen-containing contraceptives should be considered (Roach et al. 2008).
Prior studies have shown that the risk of stroke recurrence in children with an underlying arteriopathy is high and therefore these children should be prophylaxed with either antiplatelet agents or anticoagulation (Fullerton et al. 2007). However, systematic studies of secondary stroke prevention in children with arteriopathies, including children with irradiation-induced vasculopathies, are not available in the literature. A special writing group of the American Heart Association Council and the Council on Cardiovascular Disease in the Young has issued consensus guidelines for management of infants and children with stroke (Roach et al 2008). These guidelines outline specific treatment recommendations for children with an ischemic stroke based on the underlying condition including Moyamoya syndrome which can be associated with CRT in pediatric cancer patients.
5.5.4.2 Hemorrhagic Stroke
Hematomas can expand over several hours from initial presentation and monitoring in the pediatric intensive care unit is indicated. The initial management of the child with hemorrhagic stroke should include assessment of the airway, breathing and circulation. Blood pressure management is directed towards normal blood pressure values for age. Children are at risk to develop hydrocephalus and placement of an EVD may be required. A Camino® bolt can be used to monitor ICP if no hydrocephalus is present. Awake patients can be monitored with serial examinations, whereas children with depressed level of consciousness will benefit from invasive ICP monitoring to maintain adequate CPP. The use of recombinant factor VII (rFVIIa) to halt hemorrhage in pediatric patients is still under investigation and currently not considered standard therapy (McQuilten et al. 2012). Pediatric neurosurgery should be consulted immediately for cerebellar hemorrhage, as evacuation is often indicated. Patients with thrombocytopenia should be treated with platelet transfusions to maintain a platelet count ≥100 × 109/L. The role of routine seizure prophylaxis in the absence of seizures is unclear. Adult patients with hemorrhagic strokes who were treated with prophylactic AEDs had worse outcome than those who were just monitored (Messe et al. 2009). Currently there are no data available for children. A special writing group of the American Heart Association Council and the Council on Cardiovascular Disease in the Young has issued consensus guidelines for management of infants and children with stroke; potential relevant recommendations in pediatric nontraumatic hemorrhagic stroke include utilization of cerebral angiography if MR angiography is nondiagnostic, adequate factor replacement in children with coagulopathy, and optimization of respiratory effort, control of hypertension, control of seizures, and management of ICP to help stabilize the patient (Roach et al. 2008).
5.5.5 Outcome of Acute Stroke
Currently there are no reported data that address the outcome of pediatric cancer patients after stroke. However, stroke is an important cause of disability in the general pediatric population. After stroke, children suffer from motor and sensory deficits, cognitive decline and epilepsy. Outcome research in pediatric stroke has been hampered by the lack of validated and standardized outcome measures (Engelmann and Jordan 2012). Recently the Pediatric Stroke Outcome Measure (PSOM) was published with a reported inter-rater variability of 0.93 (95 % CI 0.76–0.98) (Kitchen et al. 2012). Development and systematic use of such validated assessments will be important to fully understand the impact of stroke on pediatric patient as well as assess efficacy of current treatment modalities.
5.6 Summary
The diversity of neurologic complications in pediatric oncology, and the frequency with which they occur, demand a careful awareness on the part of physicians caring for these patients. The approaches described above highlight the unique effects of cancer and cancer treatment on the nervous system as well as the lack of evidence basis for much of the current standards of care.
References
Abend NS, Beslow LA, Smith SE et al (2011) Seizures as a presenting symptom of acute arterial ischemic stroke in childhood. J Pediatr 159:479–483
Acquaviva A, Marconcini S, Municchi G et al (2003) Non-Hodgkin lymphoma in a child presenting with acute paraplegia: a case report. Pediatr Hematol Oncol 20:245–251
Adrogue HJ, Madias NE (2000) Hyponatremia. N Engl J Med 342:1581–1589
Amlie-Lefond C, Chan AK, Kirton A et al (2009) Thrombolysis in acute childhood stroke: design and challenges of the thrombolysis in pediatric stroke clinical trial. Neuroepidemiology 32:279–286
Andersen C, Jensen FT (1998) Differences in blood-tumour-barrier leakage of human intracranial tumours: quantitative monitoring of vasogenic oedema and its response to glucocorticoid treatment. Acta Neurochir (Wien) 140:919–924
Angelini P, Plantaz D, De Bernardi B et al (2011) Late sequelae of symptomatic epidural compression in children with localized neuroblastoma. Pediatr Blood Cancer 57:473–480
Athale UH, Chan AK (2003) Thrombosis in children with acute lymphoblastic leukemia: part I. Epidemiology of thrombosis in children with acute lymphoblastic leukemia. Thromb Res 111:125–131
Baker WJ, Royer GL Jr, Weiss RB (1991) Cytarabine and neurologic toxicity. J Clin Oncol 9:679–693
Benesch M, Weber-Mzell D, Gerber NU et al (2010) Ependymoma of the spinal cord in children and adolescents: a retrospective series from the HIT database. J Neurosurg Pediatr 6:137–144
Bertsch H, Rudoler S, Needle MN et al (1998) Emergent/urgent therapeutic irradiation in pediatric oncology: patterns of presentation, treatment, and outcome. Med Pediatr Oncol 30:101–105
Beslow LA, Licht DJ, Smith SE et al (2010) Predictors of outcome in childhood intracerebral hemorrhage: a prospective consecutive cohort study. Stroke 41:313–318
Bigby M, Stern RS (1988) Adverse reactions to isotretinoin. A report from the Adverse Drug Reaction Reporting System. J Am Acad Dermatol 18:543–552
Bitzer M, Topka H (1995) Progressive cerebral occlusive disease after radiation therapy. Stroke 26:131–136
Bowers DC, McNeil DE, Liu Y et al (2005) Stroke as a late treatment effect of Hodgkin’s Disease: a report from the Childhood Cancer Survivor Study. J Clin Oncol 23:6508–6515
Bowers DC, Liu Y, Leisenring W et al (2006) Late-occurring stroke among long-term survivors of childhood leukemia and brain tumors: a report from the Childhood Cancer Survivor Study. J Clin Oncol 24:5277–5282
Chern JJ, Tubbs RS, Gordon AS et al (2012) Management of pediatric patients with pseudotumor cerebri. Childs Nerv Syst 28:575–578
Children’s Oncology Group (CCG 09709), Villablanca JG, Krailo MD et al (2006) Phase I trial of oral fenretinide in children with high-risk solid tumors: a report from the Children’s Oncology Group (CCG 09709). J Clin Oncol 24:3423–3430
Culley DJ, Berger MS, Shaw D et al (1994) An analysis of factors determining the need for ventriculoperitoneal shunts after posterior fossa tumor surgery in children. Neurosurgery 34:402407; discussion 407–408
Daley MF, Partington MD, Kadan-Lottick N et al (2003) Primary epidural burkitt lymphoma in a child: case presentation and literature review. Pediatr Hematol Oncol 20:333–338
David KA, Picus J (2005) Evaluating risk factors for the development of ifosfamide encephalopathy. Am J Clin Oncol 28:277–280
De Bernardi B, Pianca C, Pistamiglio P et al (2001) Neuroblastoma with symptomatic spinal cord compression at diagnosis: treatment and results with 76 cases. J Clin Oncol 19:183–190
de Laat P, Te Winkel ML, Devos AS et al (2011) Posterior reversible encephalopathy syndrome in childhood cancer. Ann Oncol 22:472–478
Dorresteijn LD, Kappelle AC, Boogerd W et al (2002) Increased risk of ischemic stroke after radiotherapy on the neck in patients younger than 60 years. J Clin Oncol 20:282–288
Dorresteijn LD, Kappelle AC, Scholz NM et al (2005) Increased carotid wall thickening after radiotherapy on the neck. Eur J Cancer 41:1026–1030
Drachtman RA, Cole PD, Golden CB et al (2002) Dextromethorphan is effective in the treatment of subacute methotrexate neurotoxicity. Pediatr Hematol Oncol 19:319–327
Dufourg MN, Landman-Parker J, Auclerc MF et al (2007) Age and high-dose methotrexate are associated to clinical acute encephalopathy in FRALLE 93 trial for acute lymphoblastic leukemia in children. Leukemia 21:238–247
Dunton SF, Nitschke R, Spruce WE et al (1986) Progressive ascending paralysis following administration of intrathecal and intravenous cytosine arabinoside. A Pediatric Oncology Group study. Cancer 57:1083–1088
Duval M, Suciu S, Ferster A et al (2002) Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of Cancer-Children’s Leukemia Group phase 3 trial. Blood 99:2734–2739
Eisenberg HM, McComb JG, Lorenzo AV (1974) Cerebrospinal fluid overproduction and hydrocephalus associated with choroid plexus papilloma. J Neurosurg 40:381–385
Engelmann KA, Jordan LC (2012) Outcome measures used in pediatric stroke studies: a systematic review. Arch Neurol 69:23–27
Fouladi M, Langston J, Mulhern R et al (2000) Silent lacunar lesions detected by magnetic resonance imaging of children with brain tumors: a late sequela of therapy. J Clin Oncol 18:824–831
Fullerton HJ, Wu YW, Sidney S et al (2007) Risk of recurrent childhood arterial ischemic stroke in a population-based cohort: the importance of cerebrovascular imaging. Pediatrics 119:495–501
George R, Jeba J, Ramkumar G et al (2008) Interventions for the treatment of metastatic extradural spinal cord compression in adults. Cochrane Database Syst Rev (4):CD006716
Glantz MJ, LaFollette S, Jaeckle KA et al (1999) Randomized trial of a slow-release versus a standard formulation of cytarabine for the intrathecal treatment of lymphomatous meningitis. J Clin Oncol 17:3110–3116
Glantz MJ, Cole BF, Forsyth PA et al (2000) Practice parameter: anticonvulsant prophylaxis in patients with newly diagnosed brain tumors. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 54:1886–1893
Goldsby RE, Liu Q, Nathan PC et al (2010) Late-occurring neurologic sequelae in adult survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. J Clin Oncol 28:324–331
Grace RF, Dahlberg SE, Neuberg D et al (2011) The frequency and management of asparaginase-related thrombosis in paediatric and adult patients with acute lymphoblastic leukaemia treated on Dana-Farber Cancer Institute consortium protocols. Br J Haematol 152:452–459
Greenwood MJ, Dodds AJ, Garricik R et al (2003) Posterior leukoencephalopathy in association with the tumour lysis syndrome in acute lymphoblastic leukaemia–a case with clinicopathological correlation. Leuk Lymphoma 44:719–721
Grill J, Couanet D, Cappelli C et al (1999) Radiation-induced cerebral vasculopathy in children with neurofibromatosis and optic pathway glioma. Ann Neurol 45:393–396
Gupta V, Srivastava A, Bhatia B (2009) Hodgkin disease with spinal cord compression. J Pediatr Hematol Oncol 31:771–773
Guyatt G, Gutterman D, Baumann MH et al (2006) Grading strength of recommendations and quality of evidence in clinical guidelines: report from an American College of Chest Physicians task force. Chest 129:174–181
Haddy N, Mousannif A, Tukenova M et al (2011) Relationship between the brain radiation dose for the treatment of childhood cancer and the risk of long-term cerebrovascular mortality. Brain 134:1362–1367
Hamadani M, Awan F (2006) Role of thiamine in managing ifosfamide-induced encephalopathy. J Oncol Pharm Pract 12:237–239
Hardesty DA, Sanborn MR, Parker WE et al (2011) Perioperative seizure incidence and risk factors in 223 pediatric brain tumor patients without prior seizures. J Neurosurg Pediatr 7:609–615
Hartman AL, Lunney KM, Serena JE (2009) Pediatric stroke: do clinical factors predict delays in presentation? J Pediatr 154:727–732
Hayes FA, Thompson EI, Hvizdala E et al (1984) Chemotherapy as an alternative to laminectomy and radiation in the management of epidural tumor. J Pediatr 104:221–224
Herzig RH, Hines JD, Herzig GP et al (1987) Cerebellar toxicity with high-dose cytosine arabinoside. J Clin Oncol 5:927–932
Hinchey J, Chaves C, Appignani B et al (1996) A reversible posterior leukoencephalopathy syndrome. N Engl J Med 334:494–500
Hoffman KE, Yock TI (2009) Radiation therapy for pediatric central nervous system tumors. J Child Neurol 24:1387–1396
Hollander M, Bots ML, Del Sol AI et al (2002) Carotid plaques increase the risk of stroke and subtypes of cerebral infarction in asymptomatic elderly: the Rotterdam study. Circulation 105:2872–2877
Holmes D, Vishnu P, Dorer RK et al (2012) All-trans retinoic acid-induced pseudotumor cerebri during induction therapy for acute promyelocytic leukemia: a case report and literature review. Case Rep Oncol Med 2012:313057
Huang EA, Feldman BJ, Schwartz ID et al (2006) Oral urea for the treatment of chronic syndrome of inappropriate antidiuresis in children. J Pediatr 148:128–131
Huisman TA (2009) Pediatric tumors of the spine. Cancer Imaging 9:S45–S48
Inaba H, Khan RB, Laningham FH et al (2008) Clinical and radiological characteristics of methotrexate-induced acute encephalopathy in pediatric patients with cancer. Ann Oncol 19:178–184
Isome K, Matsubara K, Taki T et al (2011) Spinal cord compression by epidural involvement over 21 vertebral levels in acute lymphoblastic leukemia. J Pediatr Hematol Oncol 33:153–157
Kaal EC, Vecht CJ (2004) The management of brain edema in brain tumors. Curr Opin Oncol 16:593–600
Kenz S, Haas CS, Werth SC et al (2011) High sensitivity to tolvaptan in paraneoplastic syndrome of inappropriate ADH secretion (SIADH). Ann Oncol 22:2696
Khanna S, Davis D, Peterson B et al (2000) Use of hypertonic saline in the treatment of severe refractory posttraumatic intracranial hypertension in pediatric traumatic brain injury. Crit Care Med 28:1144–1151
Khasraw M, Holodny A, Goldlust SA et al (2012) Intracranial hemorrhage in patients with cancer treated with bevacizumab: the Memorial Sloan-Kettering experience. Ann Oncol 23:458–463
Kikuchi A, Maeda M, Hanada R et al (2007) Moyamoya syndrome following childhood acute lymphoblastic leukemia. Pediatr Blood Cancer 48:268–272
Kirkpatrick JP, van der Kogel AJ, Schultheiss TE (2010) Radiation dose-volume effects in the spinal cord. Int J Radiat Oncol Biol Phys 76:S42–S49
Kitchen L, Westmacott R, Friefeld S et al (2012) The pediatric stroke outcome measure: a validation and reliability study. Stroke 43:1602–1608
Klein SL, Sanford RA, Muhlbauer MS (1991) Pediatric spinal epidural metastases. J Neurosurg 74:70–75
Kupfer A, Aeschlimann C, Wermuth B et al (1994) Prophylaxis and reversal of ifosfamide encephalopathy with methylene-blue. Lancet 343:763–764
Laitt RD, Chambers EJ, Goddard PR et al (1995) Magnetic resonance imaging and magnetic resonance angiography in long term survivors of acute lymphoblastic leukemia treated with cranial irradiation. Cancer 76:1846–1852
Land VJ, Shuster JJ, Crist WM et al (1994) Comparison of two schedules of intermediate-dose methotrexate and cytarabine consolidation therapy for childhood B-precursor cell acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Clin Oncol 12:1939–1945
Leone G, Gugliotta L, Mazzucconi MG et al (1993) Evidence of a hypercoagulable state in patients with acute lymphoblastic leukemia treated with low dose of E. coli L-asparaginase: a GIMEMA study. Thromb Haemost 69:12–15
Lim YJ, Park EK, Koh HC et al (2010) Syndrome of inappropriate secretion of antidiuretic hormone as a leading cause of hyponatremia in children who underwent chemotherapy or stem cell transplantation. Pediatr Blood Cancer 54:734–737
Lo WD (2011) Childhood hemorrhagic stroke: an important but understudied problem. J Child Neurol 26:1174–1185
Loblaw DA, Perry J, Chambers A, Laperriere NJ (2005) Systematic review of the diagnosis and management of malignant extradural spinal cord compression: the Cancer Care Ontario Practice Guidelines Initiative’s Neuro-Oncology Disease Site Group. J Clin Oncol 23:2028–2037
Lucchini G, Grioni D, Colombini A et al (2008) Encephalopathy syndrome in children with hemato-oncological disorders is not always posterior and reversible. Pediatr Blood Cancer 51:629–633
Mackay MT, Monagle P (2008) Perinatal and early childhood stroke and thrombophilia. Pathology 40:116–123
Mahoney DH Jr, Shuster JJ, Nitschke R et al (1998) Acute neurotoxicity in children with B-precursor acute lymphoid leukemia: an association with intermediate-dose intravenous methotrexate and intrathecal triple therapy–a Pediatric Oncology Group study. J Clin Oncol 16:1712–1722
Mantadakis E, Katragkou A, Papadaki E et al (2008) Spinal cord compression in an adolescent with relapsed B-precursor acute lymphoblastic leukemia and mental neuropathy. Int J Hematol 88:294–298
Mattioli Belmonte M, Gugliotta L, Delvos U et al (1991) A regimen for antithrombin III substitution in patients with acute lymphoblastic leukemia under treatment with L-asparaginase. Haematologica 76:209–214
McGlennan C, Ganesan V (2008) Delays in investigation and management of acute arterial ischaemic stroke in children. Dev Med Child Neurol 50:537–540
McQuilten ZK, Barnes C, Zatta A et al (2012) Off-label use of recombinant factor VIIa in pediatric patients. Pediatrics 129:e1533–e1540
Medina LS, Pinter JD, Zurakowski D et al (1997) Children with headache: clinical predictors of surgical space-occupying lesions and the role of neuroimaging. Radiology 202:819–824
Meeske KA, Siegel SE, Gilsanz V et al (2009) Premature carotid artery disease in pediatric cancer survivors treated with neck irradiation. Pediatr Blood Cancer 53:615–621
Messe SR, Sansing LH, Cucchiara BL et al (2009) Prophylactic antiepileptic drug use is associated with poor outcome following ICH. Neurocrit Care 11:38–44
Mitchell L, Andrew M, Hanna K et al (2003) Trend to efficacy and safety using antithrombin concentrate in prevention of thrombosis in children receiving l-asparaginase for acute lymphoblastic leukemia. Results of the PAARKA study. Thromb Haemost 90:235–244
Moritz ML, Ayus JC (2010) New aspects in the pathogenesis, prevention, and treatment of hyponatremic encephalopathy in children. Pediatr Nephrol 25:1225–1238
Morris EB, Laningham FH, Sandlund JT et al (2007) Posterior reversible encephalopathy syndrome in children with cancer. Pediatr Blood Cancer 48:152–159
Neville BG, Wilson J (1970) Benign intracranial hypertension following corticosteroid withdrawal in childhood. Br Med J 3:554–556
Norman JK, Parke JT, Wilson DA et al (2007) Reversible posterior leukoencephalopathy syndrome in children undergoing induction therapy for acute lymphoblastic leukemia. Pediatr Blood Cancer 49:198–203
O’Leary DH, Polak JF, Kronmal RA et al (1999) Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N Engl J Med 340:14–22
Ogiwara H, Nordli DR, DiPatri AJ et al (2010) Pediatric epileptogenic gangliogliomas: seizure outcome and surgical results. J Neurosurg Pediatr 5:271–276
Olcay L, Aribas BK, Gokce M (2009) A patient with acute myeloblastic leukemia who presented with conus medullaris syndrome and review of the literature. J Pediatr Hematol Oncol 31:440–447
Omura M, Aida N, Sekido K et al (1997) Large intracranial vessel occlusive vasculopathy after radiation therapy in children: clinical features and usefulness of magnetic resonance imaging. Int J Radiat Oncol Biol Phys 38:241–249
Papadopoulos KP, Garvin JH, Fetell M et al (1998) High-dose thiotepa and etoposide-based regimens with autologous hematopoietic support for high-risk or recurrent CNS tumors in children and adults. Bone Marrow Transplant 22:661–667
Parikh SN, Crawford AH (2003) Orthopaedic implications in the management of pediatric vertebral and spinal cord tumors: a retrospective review. Spine (Phila Pa 1976) 28:2390–2396
Patchell RA, Tibbs PA, Regine WF et al (2005) Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: a randomised trial. Lancet 366:643–648
Patel PN (2006) Methylene blue for management of ifosfamide-induced encephalopathy. Ann Pharmacother 40:299–303
Pelgrims J, De Vos F, Van den Brande J et al (2000) Methylene blue in the treatment and prevention of ifosfamide-induced encephalopathy: report of 12 cases and a review of the literature. Br J Cancer 82:291–294
Pitfield AF, Carroll AB, Kissoon N (2012) Emergency management of increased intracranial pressure. Pediatr Emerg Care 28:200–204; quiz 205–207
Pollono D, Tomarchia S, Drut R et al (2003) Spinal cord compression: a review of 70 pediatric patients. Pediatr Hematol Oncol 20:457–466
Pratt CB, Green AA, Horowitz ME et al (1986) Central nervous system toxicity following the treatment of pediatric patients with ifosfamide/mesna. J Clin Oncol 4:1253–1261
Rades D, Huttenlocher S, Dunst J et al (2010) Matched pair analysis comparing surgery followed by radiotherapy and radiotherapy alone for metastatic spinal cord compression. J Clin Oncol 28:3597–3604
Rafay MF, Pontigon AM, Chiang J et al (2009) Delay to diagnosis in acute pediatric arterial ischemic stroke. Stroke 40:58–64
Raffini L (2008) Thrombophilia in children: who to test, how, when, and why? Hematology Am Soc Hematol Educ Program 228–235
Reddick WE, Glass JO, Helton KJ et al (2005) Prevalence of leukoencephalopathy in children treated for acute lymphoblastic leukemia with high-dose methotrexate. AJNR Am J Neuroradiol 26:1263–1269
Relling MV, Pui CH, Sandlund JT et al (2000) Adverse effect of anticonvulsants on efficacy of chemotherapy for acute lymphoblastic leukaemia. Lancet 356:285–290
Riva-Cambrin J, Detsky AS, Lamberti-Pasculli M et al (2009) Predicting postresection hydrocephalus in pediatric patients with posterior fossa tumors. J Neurosurg Pediatr 3:378–385
Rivkees SA (2008) Differentiating appropriate antidiuretic hormone secretion, inappropriate antidiuretic hormone secretion and cerebral salt wasting: the common, uncommon, and misnamed. Curr Opin Pediatr 20:448–452
Roach ES (2000) Etiology of stroke in children. Semin Pediatr Neurol 7:244–260
Roach ES, Golomb MR, Adams R et al (2008) Management of stroke in infants and children: a scientific statement from a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke 39:2644–2691
Rollins N, Winick N, Bash R et al (2004) Acute methotrexate neurotoxicity: findings on diffusion-weighted imaging and correlation with clinical outcome. AJNR Am J Neuroradiol 25:1688–1695
Rubin EH, Andersen JW, Berg DT et al (1992) Risk factors for high-dose cytarabine neurotoxicity: an analysis of a cancer and leukemia group B trial in patients with acute myeloid leukemia. J Clin Oncol 10:948–953
Rubnitz JE, Relling MV, Harrison PL et al (1998) Transient encephalopathy following high-dose methotrexate treatment in childhood acute lymphoblastic leukemia. Leukemia 12:1176–1181
Ruggiero A, Rizzo D, Mastrangelo S et al (2010) Interactions between antiepileptic and chemotherapeutic drugs in children with brain tumors: is it time to change treatment? Pediatr Blood Cancer 54:193–198
Sandoval C, Kutscher M, Jayabose S et al (2003) Neurotoxicity of intrathecal methotrexate: MR imaging findings. AJNR Am J Neuroradiol 24:1887–1890
Schwenn MR, Blattner SR, Lynch E et al (1991) HiC-COM: a 2-month intensive chemotherapy regimen for children with stage III and IV Burkitt’s lymphoma and B-cell acute lymphoblastic leukemia. J Clin Oncol 9:133–138
Seet RC, Rabinstein AA, Lindell PE et al (2011) Cerebrovascular events after bevacizumab treatment: an early and severe complication. Neurocrit Care 15:421–427
Silverman LB, Supko JG, Stevenson KE et al (2010) Intravenous PEG-asparaginase during remission induction in children and adolescents with newly diagnosed acute lymphoblastic leukemia. Blood 115:1351–1353
Simon T, Niemann CA, Hero B et al (2012) Short- and long-term outcome of patients with symptoms of spinal cord compression by neuroblastoma. Dev Med Child Neurol 54:347–352
Singh S, Bohn D, Carlotti AP et al (2002) Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 30:2575–2579
Smith MA, Adamson PC, Balis FM et al (1992) Phase I and pharmacokinetic evaluation of all-trans-retinoic acid in pediatric patients with cancer. J Clin Oncol 10:1666–1673
Smith GA, Damon LE, Rugo HS et al (1997) High-dose cytarabine dose modification reduces the incidence of neurotoxicity in patients with renal insufficiency. J Clin Oncol 15:833–839
Sommers LM, Hawkins DS (1999) Meningitis in pediatric cancer patients: a review of forty cases from a single institution. Pediatr Infect Dis J 18:902–907
Spiegel RJ, Cooper PR, Blum RH et al (1984) Treatment of massive intrathecal methotrexate overdose by ventriculolumbar perfusion. N Engl J Med 311:386–388
Srinivasan J, Miller SP, Phan TG et al (2009) Delayed recognition of initial stroke in children: need for increased awareness. Pediatrics 124:e227–e234
Sterns RH, Nigwekar SU, Hix JK (2009) The treatment of hyponatremia. Semin Nephrol 29:282–299
Taugourdeau-Raymond S, Rouby F, Default A et al (2012) Bevacizumab-induced serious side-effects: a review of the French pharmacovigilance database. Eur J Clin Pharmacol 68:1103–1107
Ullrich NJ, Robertson R, Kinnamon DD et al (2007) Moyamoya following cranial irradiation for primary brain tumors in children. Neurology 68:932–938
Vos J, Aarnoudse MW, Dijk F et al (1983) On the cellular origin and development of atheromatous plaques. A light and electron microscopic study of combined X-ray and hypercholesterolemia-induced atheromatosis in the carotid artery of the rabbit. Virchows Arch B Cell Pathol Incl Mol Pathol 43:1–16
White RH (2003) The epidemiology of venous thromboembolism. Circulation 107:I4–I8
Widemann BC, Balis FM, Shalabi A et al (2004) Treatment of accidental intrathecal methotrexate overdose with intrathecal carboxypeptidase G2. J Natl Cancer Inst 96:1557–1559
Wilne S, Collier J, Kennedy C et al (2007) Presentation of childhood CNS tumours: a systematic review and meta-analysis. Lancet Oncol 8:685–689
Winick NJ, Bowman WP, Kamen BA et al (1992) Unexpected acute neurologic toxicity in the treatment of children with acute lymphoblastic leukemia. J Natl Cancer Inst 84:252–256
Zimmer JA, Garg BP, Williams LS et al (2007) Age-related variation in presenting signs of childhood arterial ischemic stroke. Pediatr Neurol 37:171–175
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Berlin Heidelberg
About this chapter
Cite this chapter
Sabnis, A., Finlay, J.L., Mueller, S. (2015). Neurologic Emergencies. In: Feusner, J., Hastings, C., Agrawal, A. (eds) Supportive Care in Pediatric Oncology. Pediatric Oncology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-44317-0_5
Download citation
DOI: https://doi.org/10.1007/978-3-662-44317-0_5
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44316-3
Online ISBN: 978-3-662-44317-0
eBook Packages: MedicineMedicine (R0)