
Abstract
The word mutation was coined in 1901 by Hugo De Vries to describe “sudden, spontaneous and drastic alterations in the hereditary material of Oenothera”, the evening primrose. Mutations occur in the genome of all living organisms and vary in importance, ranging from single base-pair changes to extensive chromosomal rearrangements. They can occur either in somatic or germ cells, at all stages of development, and are transmitted to daughter cells except when they cause death or a severe selective disadvantage.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
7.1 The Importance of Mutations
The word mutation was coined in 1901 by Hugo De Vries to describe “sudden, spontaneous and drastic alterations in the hereditary material of Oenothera”, the evening primrose.Footnote 1 Mutations occur in the genome of all living organisms and vary in importance, ranging from single base-pair changes to extensive chromosomal rearrangements. They can occur either in somatic or germ cells, at all stages of development, and are transmitted to daughter cells except when they cause death or a severe selective disadvantage.
When mutations occur in somatic cells with high mitotic activity, such as cells of the bone marrow, intestinal mucosa, lung or skin, or when the mutations in question interfere with the mechanisms that regulate the cell cycle or differentiation, then the affected cells may become carcinomatous. When mutations occur in the cells contributing to the germ line they may be transmitted to the next generation and, in this case, a proportion of the offspring will be heterozygous for a new mutant allele. This category of mutations is precisely the one we will focus on in this chapter.
Germinal mutations, by definition, generate new alleles that enter the gene pool of the species and contribute to an increase in polymorphism. Most of these new alleles have no effects or effects that do not influence the fitness of the affected individuals, and for this reason they are called “neutral mutations”. A small proportion of these new mutations may result in a better adaptation of the animals to their environment.Footnote 2 Finally, some mutations have deleterious effects, frequently leading to pathological conditions. In this case, and if we consider that almost all genes in the mouse genome have an equivalent in the human genome, it is obvious that many among the new mutant alleles found in the mouse species represent potentially interesting models of human genetic diseases.
Mutations can affect all genomic regions, with a wide range of consequences at the phenotypic level. They are either dominant, semi-dominant (heterozygotes have a less severe phenotype than homozygous mutants), co-dominant (both alleles are equally expressed) or recessive. Detailed study of the phenotype of these new mutant alleles is part of the process of genome annotation, and is of great importance for the characterization of gene function(s).
The occurrence of spontaneous mutations in mammalian genomes results from errors occurring either during meiosis or in the process of DNA replication which are not mended by the cellular (DNA) repair mechanisms. These repair mechanisms are very sophisticated, with specific enzymes constantly checking the integrity of cellular DNA during and after replication, but the system is sometimes defective or saturated and fails. Taking this into account, one understands that there is no way to prevent mutations from occurring, and that the spontaneous mutation rate is a basic parameter that each species must cope with. In addition, many agents such as radiation, some chemicals, and some viruses and transposons can increase the rate of mutations well above the spontaneous rate. Some of these agents, as we will discuss in this chapter, have been used over the last fifty years for performing experimental mutagenesis.
Experimental mutagenesis can be “phenotype-driven”, when unknown genes are identified based on the phenotypic changes associated with at least one of the mutant alleles. In this case, the structure of the gene affected by the mutation is elucidated afterwards, by positional cloning, depending on the potential interest of the mutant allele. Experimental mutagenesis can also be “genotype- or gene-driven”, whereby mutations are massively induced and then sought only in pre-selected genes or DNA regions of unknown function, for example for the purpose of genome annotation. As we will see, experimental mutagenesis is relatively simple to achieve, but its efficiency depends upon the mutagenic treatment as well as on the protocols used for the characterization of the mutant phenotypes.
In this chapter, we will describe in some detail the different types of mutations that can affect a mammalian genome and their consequences. We will then discuss the different protocols that can be used for the induction of mutations in the mouse germline, with special emphasis on chemical mutagenesis, which is highly efficient and accordingly has become widespread.
7.2 The Different Types of Mutations
When considered at the DNA level, mutations are generally classified into two categories:
-
chromosomal mutations, which are detectable by the observation of morphological changes at the karyotype level, and
-
point mutations, when no alteration in chromosome integrity is detectable.
This classification into chromosomal mutations and point or gene mutations dates back to a time when the microscope was the only tool available to visualize changes in the hereditary material. Since then, the notion of point mutation has changed and now covers a group of structurally defined changes occurring in the DNA. We will describe these changes, from the simplest to the more complex, and in so doing we will realize that the classification mentioned above, in fact, is not really stringent. However, it is convenient from a didactic point of view and thus we will adopt it.Footnote 3
The geneticist H.J. Muller,Footnote 4 who did pioneering research on experimental mutagenesis in Drosophila flies using X-rays, proposed a classification of the mutations into five categories based on the effect of the genetic change on gene activity. The first category, the amorphic mutations, consisted of those mutations that completely abolish the activity of the gene and were equivalent to null or loss of function alleles. Hypomorphic mutations were associated with reduced activity compared to the wild-type allele, while hypermorphic mutations were the opposite, with an increased activity. Neomorphic mutations were the group of mutations exhibiting a new function, and antimorphic alleles were mutations with dominant negative effects.
7.2.1 Mutations Resulting from Base-Pair Substitutions in the Coding Sequences
The information gathered from recent efforts of systematic sequencing of the genome of various mouse strains have revealed that nucleotide substitutions are the most frequent type of mutations. We will then take a simple example of this type of mutation and discuss its consequences. This example will be the DNA codon 5′-TGT-3′, which is transcribed as UGU and encodes the amino acid cysteine (Cys, or C when using the one-letter code) and, for the time being, we will focus on the nucleotide at the third position of this codon (T)Footnote 5 (Table 7.1).
The first substitution is when the thymine (T) in the DNA strand is replaced by a cytosine (C) (this substitution of a pyrimidine for another pyrimidine is called a transition). In this case, the resulting mRNA codon becomes UGC but, like UGU, it still encodes the same Cys residue. This type of mutation has no effect on the protein sequence, and for this reason, it is said to be silent or synonymous. In this case, the base substitution is detected only when it suppresses or creates a restriction site or, when comparing sequences, as a single nucleotide polymorphism (SNP).
Synonymous mutations are common findings (~23 %) in the sequence databases of the different mouse inbred strains, especially those recently derived from wild specimens (Beier 2000; Sakuraba et al. 2005; Frazer et al. 2007; Keane et al. 2011; Yang et al. 2011; Arnold et al. 2012), and this observed frequency is in keeping with theoretical computations. Indeed, if we consider that each of the 61 sense codons (64 − 3) can mutate to one of nine different codons after the substitution of one or the other of the three nucleotides, we can calculate that out of these 549 (61 × 9) possible mutations around 25 % are synonymous while most others (75 %) are not (Graur 2003). If we take a closer look at the distribution of all these mutations we may notice that the synonymous mutations are much more frequent when the substitutions occur on the third nucleobase of the codon (70 %) than when they affect one of the other two positions. This is, of course, because the code is degenerated.
Synonymous mutations occur constantly and regularly, even if at a low rate. They are also relatively stable and have virtually no impact on the phenotype. For these reasons they represent an interesting class of polymorphism for evolutionists and can be considered as a molecular clock useful, for example, for assessing the time of divergence between any two species or strains (Gilman 1972).Footnote 6
These synonymous SNPs, when considered with the other flanking SNPs of the same type on the same chromosome, can also be used for identifying the phylogenetic origin of the chromosome (or haplotype) in question. We will come back to this point when discussing the inheritance of complex or quantitative traits (Keane et al. 2011) (Chap. 10).
An interesting observation is that, in mammals, some synonymous codons are found more frequently than others, even when the codons in question encode the same amino acid. For example, the 5'-AGA-3' and 5'-AGG-3' DNA codons both encode the amino acid arginine (R), but AGA is six times more frequent than AGG in the transcripts. A similar observation can be made with the codons 5'-ACA-3' and 5'-ACG-3', which both encode the amino acid threonine (T), but ACA is five times more frequent than ACG in the transcripts. The reason(s) for such a bias in codon use is (are) not yet elucidated: they may be related to the fact that the mutation rate is not the same for the four different nucleotides (discussed later); alternatively, the bias in the codon usage may be related to the fact that the synonymous codons are not equivalent in terms of efficiency at the translational level; some of the codons have a selective advantage over the others.
Let us now assume that the third nucleotide of the same 5'-TGT-3' codon, the T, is replaced by an adenine (A)—this change is designated a transversion (i.e., the substitution of a pyrimidine for a purine). This mutation results in the incorporation of the UGA codon instead of UGU in the mRNA transcript, but this is the signal for the termination of polypeptide synthesis, or stop codon. The resulting mutations are called nonsense mutations, generating null or non-functional alleles. Analysis of the sequencing data from positional cloning (in human and mouse) of mutant alleles with a deleterious effect reveals that mutations of this type represent around 4–5 % of the overall point mutations found in the coding sequences.
The functional consequences of nonsense mutations depend on the type of protein encoded by the gene and the potential existence of other genes capable of achieving the same or similar function(s). If the protein has an important function in cellular metabolism and if the gene is present as a single copy, the mutation generally leads to cell and/or embryonic death when in the homozygous state (recessive lethal). If, however, the encoded protein is not essential or if it is expressed only in a limited number of cells—for example, only the cells that are involved in the synthesis of melanin pigment (melanocytes)—then only the hair coat and retina of the animal are affected by the mutation, resulting in albinism (the consequence of a null allele of the tyrosinase-encoding gene Tyr-Chr 7). All intermediates between these two extreme cases are possible. Typically, inactive alleles resulting from a stop codon have no phenotypic expression when heterozygous, except in the case of haplo-insufficiency or parental imprinting of the normal allele (see Chap. 6).
mRNAs with a premature stop codon are in general rapidly degraded by specific exonucleases.Footnote 7 However, in some cases where the stop codon occurs close to the 3' end of the gene (in the last exon, for example), the transcript often escapes mRNA decay and the abnormal (truncated) protein may have a dominant negative effect of variable intensity.
The reciprocal mutations, where one of the three stop codons 5'-TAA-3', 5'-TGA-3', and 5'-TAG-3' reverts to a non stop-codon, are called read-through mutations. These mutations are exceptional and only a very small number have been reported (Noveroske et al. 2000). This is understandable if we consider the relatively small target the three stop codons represent (9 bp altogether) compared to the rest of the exonic sequences.
The last substitution we must consider is when the third base of the codon 5'-TGT-3' for thymine (T) is replaced by a guanine (G); this change is another transversion. This substitution changes the mRNA codon UGU to UGG, and a different amino acid (Trp—tryptophan or W) is inserted into the polypeptide chain instead of the original cysteine. These mutations are called non-synonymous or missense, and their effects are almost unpredictable because they depend upon the site where the substitution occurred and the type of amino acid replacement. This sort of mutation is by far the most common type found in sequencing data from positional cloning of mutant alleles with a deleterious effect. In some cases, the change has extremely limited effects and only some biophysical characteristics of the protein, such as, for example, its electrical charge, are altered. In the case of altered electrical charge, the proteins are designated electrophoretic variants; they are easily identified by electrophoresis in a non-denaturing gel, but the function of the protein remains generally unchanged (see Chap. 4).
The β-chain of mouse hemoglobin (HBB, encoded by the Hbb gene on Chr 7) has been extensively studied in wild mice because it represents an interesting system for evaluating the functional divergence of duplicated genes during evolution. In these studies, it has been observed that amino acid changes in the β-globin chain are very common among the different species that are close relatives of the laboratory mice in the genus Mus, but all these “mutant” molecules (called isoforms) are perfectly functional (Runck et al. 2009).
Another example of a non-synonymous mutation is worth mentioning: the Tyr c-h or Himalayan allele at the Tyr locus in the mouse. This spontaneous mutation is common in mammals and an orthologous mutant allele also exists in the rat, the Siamese breed of cats, the rabbit, and several other mammalian species. In the mouse, the mutation was found to be the consequence of an A → G transition at nucleotide 1,259 of the Tyr gene, which results in an amino acid change at position 420 from histidine to arginine (His420Arg—a structurally important change). Because of this mutation, melanin synthesis in Tyr c-h/Tyr c-h homozygous mice becomes temperature-sensitive; the pigment is synthesized normally in the fur at around 20 °C but not at ~30 °C. As a result, the mice have a different fur color at their extremities (the tip of their nose, tail, limbs, and ear are normally pigmented because the temperature is lower at these parts of the body, while the rest of the mouse is not or weakly pigmented). The Himalayan allele, which is of ancient origin, has been relatively easy to detect and propagate because it made the mice quite eye-catching without altering their health. However, if such mutations occur in genes encoding proteins with an important role in homeostasis of the organism, the consequences, although unpredictable, might be severe.
So far we have only considered the mutations that are the consequences of substitutions occurring at the third position of the 5′-TGT-3′ DNA codon. This codon was selected as an example because it is one of the rare types that can produce the three classes of mutations (synonymous, nonsense, and missense) with a single base-pair replacement at the same position. However, as we already mentioned and because of the degeneracy of the genetic code, mutations at the first and second nucleotides of mRNA codons are generally more deleterious in terms of consequences than mutations at the third position. Using the same permutation as explained above, we can calculate, for example, that substitutions at the first or second position would generate a missense mutation in 91 and 96 % of the cases, respectively.Footnote 8 Even if this theoretical computation must be corrected, taking into account that the nucleotides are not represented in equal proportions in the mouse DNA, and accordingly that all 64 codons are not equally frequent, this percentage of non-synonymous mutations is very close to the data actually collected after positional cloning of hundreds of mutations and analysis of mouse genome sequences.Footnote 9
Although predictions concerning the possible deleterious effects associated with missense mutations are difficult and always depend on the genomic context, a number of observations that have accumulated over time provide some indications. For example, it has been observed repeatedly that non-synonymous mutations replacing an aliphatic amino acid with an aromatic one (for example TCG → TGG) have deleterious consequences in most cases. The same applies to the mutations replacing one of the two amino acids containing a sulphur (S) atom (Cys or Met) by another amino acid not containing the S atom. Most amino acid substitutions occurring in the highly conserved domains of proteins almost always have deleterious consequences. Finally, missense mutations leading to an important structural change at the C-terminus often have severe effects by hampering the correct folding of the protein, as is the case for progressive motor neuronopathy (Tbce pmn) (Fig. 7.1).

Missense mutations. The severe mouse neurological syndrome called progressive motor neuronopathy is the consequence of a missense mutation (Tbce pmn-Chr 13) affecting the gene encoding the tubulin-specific chaperone E protein (TBCE). This missense mutation leads to the replacement of the very last amino acid of the protein, a tryptophan residue at position 524, by a glycine (in short: Trp524Gly). This change, which is unique to the mutant mouse and is not found in any other species, has consequences for the stability of the protein, and this probably explains the relatively late onset of the pathology (adapted from Martin et al. 2002)
Accumulation of new data of this kind contributes to the enrichment of databases, and all of these findings are important for a better understanding of the molecular mechanisms leading to genetic diseases. In this matter, it must be kept in mind that the information gathered from observations made in the mouse are universal and accordingly apply to all mammalian species. In human, around 56 % of the mutations resulting in a pathology are point mutations of the nonsense or missense types. Analysis of a large number of nucleotide substitutions associated with disorders shows that the most common substitutions are T to C, C to T, A to G, and G to A (Krawczak et al. 1998). In humans, the most common type of single nucleotide substitution is the CpG dinucleotide that mutates to TpG at a frequency which is about five times higher than mutations in all other dinucleotides (Youssoufian et al. 1988; Antonarakis et al. 1995; Krawczak et al. 1998). There is no reason to think that this frequency might be different in the mouse.
7.2.2 Base-Pair Substitutions in the Non-coding Regions
Base-pair substitutions in non-coding regions of the genome are innumerable, and the data gathered from mouse, rat, and human sequencing efforts provide many examples of such substitutions that, in most instances, have been recorded as mere SNPs with no detectable phenotypes. Exceptions are when the changes occur in splicing sites or in regulatory regions. These two kinds of mutations represent, respectively, 9.3 % and 1.9 % of the mutations associated with a pathological syndrome in humans, and it is likely that the proportion is similar in the mouse.
Mutations that interfere with the splicing process result in exon skipping or in the reciprocal defect known as intron retention. In some other instances, a cryptic splicing site is activated after a single base-pair substitution, and this results in the incorporation of a DNA segment of intronic origin into the transcript and possibly into the encoded protein (Figs. 7.2 and 7.3).

Examples of splicing defects generated by nucleotide substitutions. a Schematic representation of a normal gene. Exons are shown as grey boxes and introns as lines between exons. a′ represents the mature mRNA transcript after splicing of all introns. b A nucleotide substitution in a 3′ splicing site results in the skipping of an exon. c A nucleotide substitution leads to the activation of a cryptic splicing site and results in the incorporation of some intronic sequence into the mRNA transcript. d A nucleotide substitution deactivates the normal splicing site, while a cryptic one is used a few base-pairs downstream in the intronic sequence. e The substitution leads to the skipping of the last exon. All of these situations have been observed after positional cloning of mouse mutations

Mutations resulting in abnormal splicing. Lrp4 mdig and Lrp4 dan are two independent recessive mutations affecting the gene encoding the mouse lipoprotein receptor 4 (Lrp4-Chr 2). a Schematic representation of exons 14–17 of the Lrp4 gene indicating skipping of exon 15 in Lrp4 mdig/Lrp4 mdig mice. b RT-PCR amplifications performed on total cDNAs with specific primers (green arrows) allow the detection of an amplification product of the expected size in wild type (+/+) whereas only a faint band is observed with Lrp4 dan/Lrp4 dan cDNA. This is because a retroviral insertion in intron 2 of the Lrp4 dan allele hampers the transcription of a messenger RNA. However, the retroviral insertion does not suppress the transcription entirely since a faint band can be observed with cDNAs from homozygous Lrp4 dan/Lrp4 dan. PCR amplification with the same primers yields a product shorter than expected in homozygous Lrp4 mdig/Lrp4 mdig mice. Here again, skipping of exon 15 is probably not absolute since a faint band is still observable. c and d Genomic sequence in Lrp4 +/Lrp4 + and Lrp4 mdig/Lrp4 mdig co-isogenic mice. An A → T transversion alters the splicing donor site 3′ of exon 15 (from Simon-Chazottes et al. 2006)
All types of splicing defects that are theoretically possible have been actually identified in the mouse, altering more or less significantly the function of the encoded protein. A situation that is quite common and has severe consequences is when a 3′ splicing site (3′ss) is altered, leading to the attachment of a stretch of intronic DNA at the 3′ end of the mRNA molecule. In this case a number of amino acid residues are added to the C-terminus of the protein until, by chance, a stop codon occurs to terminate the aberrant transcription. In this case the protein is almost always abnormally folded and accordingly non-functional. Sometimes it also happens that cryptic 3′ or 5′ splice sites are activated after a single point mutation. In this case the consequences are unpredictable although, in general, severe.
Unlike for the splicing sites, mutations affecting DNA binding sites or regulatory regions are not common. This is either because these sites do not represent an important target in which mutations can occur or, alternatively, because mutations occurring at these sites have consequences that are not critical and accordingly are more or less tolerated or compensated for.
Most of the spontaneous mutations which have been found in the mouse, and which have been characterized at the molecular level after positional cloning, have been explained by the observation of a non-ambiguous structural defect. Among the few exceptions, one may cite the case of the Agtpbp1 pcd allele at the ATP/GTP binding protein 1 locus (formerly known as Purkinje cell degeneration—pcd; Chr 13). At this locus six spontaneous alleles and five chemically induced alleles have been reported, which all belong to the same complementation group (i.e., they fail to complement each other in a complementation test). For all the mutant alleles, obvious changes have been described in the coding region or splicing sites except for the original Agtpbp1 pcd allele. For this allele, Northern blot analysis failed to detect a transcript in all tissues of homozygotes except for the testis, where reduced levels were noted. In this case, the researchers suggested that the structural defect for this mutation should likely be in a regulatory region. However, as of today, the question is still open (Fernandez-Gonzalez et al. 2002).
With the rapid development of DNA sequencing techniques and the concomitant reduction in costs, it is likely that many regions of the mammalian genomes suspected of having particular importance in the regulation of gene expression will be easily compared between different strains or subspecies. In so doing, many point mutations of potential interest are likely to be discovered outside of splicing sites and regulatory regions. The discovery of a point mutation in the seed region of miRNA96, which is responsible for or associated with the semi-dominant deafness phenotype of Diminuendo mice (Mir96 Dmdo), is a good example and might be the first in a long series of such findings (Lewis et al. 2009).
7.2.3 Insertions, Deletions, and Duplications
Insertions are mutations resulting from the intercalation of a DNA sequence of variable size into the genome. The reciprocal alterations, those that are characterized by a missing sequence or portion of DNA, are called deletions. Insertions/deletions can be as small in size as a single nucleotide or, on the other hand, they can expand over several kilobases of DNA, affecting a variable number of genes on a chromosome and sometimes making their analysis difficult.
When aligning DNA sequences in the non-coding regions it is not always easy to select the appropriate designation between insertion and deletion. Sometimes it is noted that a single nucleotide makes a difference, but it is impossible to determine whether the mutation represents an insertion in one of the sequences or a deletion in the other. The situation is even more complex when this single nucleotide difference is frequent and co-localized across different strains. For these cases, geneticists have coined the word indel (from insertion/deletion), indicating their ignorance concerning the historical sequence of the molecular change and the co-existence of the two forms as alleles. In short, an indel is a gain or loss in nucleotides, at a specific site, that is polymorphic in a given species.
Microindels are indels that result in a net gain or loss of 1–50 nucleotides. Insertions, deletions, and indels are potentially innumerable since nucleotides can be either deleted or inserted almost anywhere in a DNA strand as a consequence of aberrant replication, unequal crossing-over or transposition. Interestingly, however, deletions are more commonly observed in practice than insertions in both the mouse and human genomes (17 % versus 6.4 %, respectively).
Deletions or insertions of nucleotides have consequences when they occur in an open reading frame (ORF), in the close vicinity of splicing sites or at DNA binding sites. When they occur in an ORF and have a size greater or less than three nucleotides (i.e., a number not divisible by three), they result in frameshift mutations, whose effects are similar to those of the mutations occurring in the splicing sites and are transcribed, in general, into aberrant mRNA molecules (Perez et al. 2013). When indels have a size of three or a multiple of three nucleotides, they result in the incorporation of additional amino acids into the protein chain, and their effects are difficult to predict. One such example has been described for another allele at the same Agtpbp1 locus (already mentioned above), the Agtpbp1 pcd-5J allele of spontaneous origin. Positional cloning of this mutation demonstrated that, in this case, a GAC triplet was inserted at position 775, adding an additional aspartic acid (Asp) to the protein. Northern blotting demonstrated comparable expression to that of wild-type mice, indicating normal RNA expression. However, Western blot analysis showed that the protein level is dramatically reduced (Chakrabarti et al. 2006).
Many mouse mutations of spontaneous origin, or discovered via studies of the effects of radiation on the germline, are the consequence of deletions encompassing several contiguous genes. Although common, this type of mutation is of limited interest for modeling human defects or even for annotating the mouse genome, because it is in general difficult to establish a direct link between a particular phenotypic trait and the genotypic defect. The mouse mutation oligotriche (olt-Chr 9) is an example of such a deletion. This mutation has been found to be a 234-kb deletion affecting no less than six contiguous genes: Vill, Plcd1, Dlec1, Acaa1b, and parts of Ctdspl and Slc22a14, but the gross phenotypic expression is relatively modest: some hair loss on the hind legs and male sterility due to severe sperm defects (Runkel et al. 2012).
Duplications are another type of mutation whose effects and consequences are similar to insertions. The gene encoding the leptin receptor (Lepr-Chr 4), with all its many alleles, is a good example illustrating both indels and duplications (see Fig. 7.4).

Mutations resulting from duplications and deletions. In the mouse, over 15 spontaneous mutations have been reported at the locus of the gene encoding the leptin receptor (Lepr-Chr 4). This gene normally consists of 18 exons and has multiple splice variants, comprising at least five isoforms. a Among these mutant alleles, Lepr db-Pas1 is the consequence of a partial duplication that spans the entirety of exons 4 and 5, plus 21 bp of coding exon 6 (as well as the two introns between exons 4 and 6). This produces a null allele that is unable to encode a functional receptor (from Liu et al. 1998). b Another spontaneous allele, Lepr db-Pas2, is the consequence of a 1-bp deletion producing a frameshift in exon 12, altering another domain of the protein. The mutant allele is inactive and the mouse becomes obese and diabetic
7.2.4 Triplet Expansions
Triplet expansion or trinucleotide expansion is a defect in DNA replication that is responsible for a dozen severe human diseases (i.e., Huntington disease, Friedreich ataxia, X-fragile syndrome, Kennedy syndrome, Steinert myotonic dystrophy, to mention just a few). These diseases are characterized at the DNA level by an increase in the number of tandemly repeated specific trinucleotides, for example CGG, CTG, CAA or CAG, occurring in specific genes and caused by slippage during DNA replication. Huntington disease (HD), for example, is caused by the expansion of CAG repeats in the gene encoding huntingtin (HTT). The number of CAG repeats increases with age in some patients and encodes an expanded glutamine (Gln) tract within the huntingtin protein. When the number of repeats passes a critical number (actually 36 for HTT), then the enlarged polyglutamine fragment in the protein leads to the formation of the huntingtin aggregates that are observed in the brain as well as in some other tissues, leading to severe pathologies.
Although the mechanism leading to triplet expansion is only poorly understood, geneticists have established that the number of repeats is frequently variable from tissue to tissue in the same patient, suggesting that distinct expansion processes can occur in different tissues. Human geneticists have also established strong correlations between the length of the triplet repeats and the severity of the disease.
Such spontaneous cases of trinucleotide expansions have not been reported in the mouse but mouse models of HD, displaying phenotypes relevant to the human disease, have been created by transgenesis (Menalled and Chesselet 2002). These models will aid the understanding of the fundamental mechanisms underlying unstable triplet expansion in humans, and hopefully will also provide useful targets for inhibiting disease development.
7.2.5 Mutations Resulting from the Insertion of Mobile Elements
As we discussed in Chap. 6, many mobile elements (retrotransposons, retroviruses, LINEs, SINEs, etc.) are well-known and quantitatively important components of the mouse genome. These elements move within the genome by duplication and retro-transposition and, depending on their integration site, they may have a mutagenic action. Many such mutations have been identified in the mouse. For example, the dilute (Myo5a d-Chr 9) mutation, a very ancient mutation of the mouse with several alleles, is the result of the integration of the ecotropic murine leukemia virus Emv-3 into the myosin VA (Myo5a) gene. The a (non-agouti-Chr 2) mutation is also the consequence of the insertion of a 5.5-kb virus-like element (VL30) into the first intron of the agouti gene, which interferes with the transcription process. At the same Agouti locus, we previously reported the case of the dominant mutation A vy (viable yellow), which is the consequence of the insertion of an intra-cisternal A-particle (IAP or retrotransposon) into a non-coding exon at the 5′ end of the agouti gene. Similarly, the spontaneous mutation spastic (Glrb spa-Chr 3) results from the insertion of a 7.1-kb LINE-1 element within intron 6 of the gene encoding the glycine receptor, beta subunit (Mülhardt et al. 1994). Finally, the hairless (Hr hr-Chr 14) mutation in mice was caused by the insertion of a murine leukemia virus into intron 6 that results in aberrant splicing of the Hr gene (Stoye et al. 1988). Some strategies have been designed to make use of the capacity of transposons to move in the mammalian genome, for the induction of new mutations in the mouse and mostly in the rat. We will come back to this point later in this chapter (Sect. 7.6).
7.2.6 Mutations Due to Non-homologous Recombination or Non-homologous End Joining
Non-homologous DNA recombination or non-homologous end joining (NHEJ) occur in mammalian genomes when double-strand DNA breaks are imprecisely repaired, leading to loss (or duplication) of a segment of nucleotides. Spontaneous mutations that are the consequence of NHEJ have been reported in humans (for example, a β-thalassemia leading to hemoglobin Lepore syndrome). To date, no such mutations have been reported in the mouse, although they may theoretically occur spontaneously. However, the NHEJ DNA repair mechanism, along with homologous recombination, is the molecular basis of new genome editing technologies with engineered nucleases (this point will be discussed in Chap. 8).
7.2.7 Copy Number Variations
As already discussed in Chap. 6, structural changes that result in copy number variations (CNVs) in a specific chromosomal region are common in all genomes. In the mouse, approximately 100 genomic regions across the 19 autosomes have been shown to harbor CNVs, ranging in size from 20 kb to 2 Mb, with more than 90 % sequence conservation. These CNVs may be considered to be mutations of a new class: the “multi-duplications”. They certainly affect gene expression by altering the transcript dosage and, accordingly, the phenotypic variability in genetic diseases by affecting the penetrance of the trait (Cutler and Kassner 2008). CNVs probably play an important role in quantitative genetics.
7.3 Spontaneous Mutation Rates
Spontaneous mutation rates are difficult to assess accurately in any mammalian species for a number of reasons. First, it is clear that only a fraction of the mutations are detectable at the phenotypic level, and this fraction fluctuates from one locus to the next. For example, dominant alleles resulting in lethality in utero or shortly after birth and those impairing the reproductive capacities of animals are often not even identified as heritable traits. Another major difficulty in the detection of mutations is that some of them frequently exhibit wide variations in expressivity or have a very subtle phenotypic expression and accordingly are underevaluated. Recessive mutations are easier to detect because they are in general observed recurrently, especially when they occur in an inbred strain but, even in this case, many mutations have not been identified simply because they have a late onset or because they are expressed only in some particular conditions. For example, most inbred mouse strains are susceptible to experimental infections with flaviviruses (yellow fever or dengue, for example) while a few others are resistant. This susceptibility is caused by a recessive mutation (Oas1b locus-Chr 5) and was discovered incidentally during an experiment, but the mice of both strains (resistant and susceptible) look perfectly “normal” for all other characteristics and, for this reason, the mutation remained undetected for many years. Similarly, some strains are susceptible to the antiparasitic drug ivermectin while most others are resistant but, here again, the mutation is cryptic, conditional, and can be detected only when the drug is administered. In conclusion, one can say that the identification of a spontaneous mutant phenotype depends upon the quality and accuracy of the phenotyping and, as a consequence of this, one must bear in mind that mutation rates are, in general, underestimated unless they are computed at a specific locus.
The first estimation concerning the mutation rate towards a recessive allele was published in 1966 by two scientists at The Jackson Laboratory (Schlager and Dickie 1966, 1967). Their estimations were established from the observation of 1,349,725 interstrain F1 progeny at five specific coat-color loci (non-agouti, a; brown, formerly b and now Tyrp1 b; albino, formerly c and now Tyr c; dilute, formerly d and now Myo5a d; and leaden, formerly ln and now Mlph ln).Footnote 10 The authors reported 12 mutations from the wild-type allele towards a recessive allele (forward mutations) and calculated an average mutation rate of 8.9 × 10−6 per locus per gamete (with 4.6–15.5 × 10−6 as 95 % confidence limits).
Another estimation based on similar crosses, although between different strains, was published a few years later by (Russell and Russell 1996). A total of 1,485,036 F1 progeny were scored at seven loci (the same five as above except leaden Mlph ln, plus pink-eyed dilution Oca2 p, piebald Ednrb s, and short ear Bmp5 se) and the authors calculated a rate of 6.6 × 10−6 mutations per locus per generation.Footnote 11 In addition to the “complete” mutations, the same authors also found several “mosaic” mutations at five loci, which led them to calculate a corrected mutation rate of 11 × 10−6 per locus per generation (Fig. 7.5).

Assessing the mutation rate at specific loci. Mice of the PT stock are homozygous for seven recessive mutant alleles involved in the determinism of coat color. When crossed with mice of the C57BL/6 inbred strain (which are non-agouti a/a and homozygous for the wild type allele at the other six loci), all F1 are expected to have a non-agouti (a/a = solid black) coat color phenotype. Phenodeviants, with a coat color different from the expected one (boxed), are potentially heterozygous for a new recessive allele at one of the six loci of the PT stock, and their status must be characterized by additional crosses. This historical PT stock, developed at Oak Ridge by W. Russell and colleagues, has been extensively used for assessing the mutagenic activity of radiation or chemical compounds. Another similar stock, the HT stock, with different alleles has been developed at MRC Harwell
These mutation rates, calculated independently, are relatively close to each other and definitely represent a good estimation for the loci described above. However, this rate (~10 × 10−6 mutation per locus per generation) is certainly not representative of the “average” mouse locus because the same scientists at The Jackson Laboratory reported a total of only 28 recessive mutations at 26 different loci from a total of 83,368,463 mice examined, yielding an overall spontaneous recessive mutation rate of 6.7 × 10−7 per locus per gamete (95 % confidence limits: 5.1–8.7 × 10−7). This rate, which is only 1/13th of the rate calculated for the forward mutations at the five/seven specific coat-color loci, is probably a better estimate of the overall spontaneous mutation rate towards a recessive allele in the mouse. This was confirmed by scientists working at Harwell using an independent tester stock, the so-called HT stock, homozygous for six recessive alleles with only one recessive allele (non-agouti a) in common with the PT stock.
Schlager and Dickie also recorded the number of mutations towards a dominant allele. They collected this information by observing breeding colonies during a 3-year period (36 mutations were collected from a total of 67,161,745 mice), yielding an estimated spontaneous mutation rate of 0.54 × 10−6 per locus per gamete, with 95 % confidence limits of 0.38–0.74 × 10−6 (Schlager and Dickie 1967).
A careful analysis of the mutations (both recessive and dominant) collected by the scientists at The Jackson Laboratory indicated that there are great differences in the mutation rates at the different loci. As we already mentioned, this is certainly a consequence of the fact that many mutant alleles escape detection either because of their unobtrusive (or very severe!) phenotype or late onset phenotype. This may also be explained by differences in the size of the different loci at the DNA level or the splitting of the coding regions into many exons, offering a wider target to the mutagenic events. However, these two explanations are clearly not sufficient to explain some of the observed differences, and it is now well established that some genes have an unexpectedly higher mutation rate than average. This is the case, for example, with the gene encoding the Kit receptor tyrosine kinase (Kit-Chr 5), in which 18 spontaneous mutant alleles were recorded in a population of mice analyzed by Schlager and Dickie during their survey.Footnote 12 This is also the case with a locus on chromosome 4, where no less than seven independent mutations were found in a single experiment (Kiernan et al. 2002). Other examples are the non-agouti locus (a-Chr 2) with 58 spontaneous alleles, and the dilute locus (Myo5a-Chr 9) with 53 alleles. Regardless of the loci and observed variations in the mutation rates, these rates remain very low. This explains why mammalian geneticists, like other geneticists, have invested in the development of strategies to increase the rates of mutation.
7.4 Mutagenesis in the Mouse
Over the last century, mice have been extensively used by geneticists as “living test tubes” for assessing the genetic hazards associated with the domestic use of nuclear energy. Mice have also been used by toxicologists for assessing the mutagenic activity of potentially hazardous chemical compounds in the human environment (drugs, food additives, pollutants, pesticides, etc.), and hundreds of mutations of all types have been produced as “by-products” of these activities. These mutations, in addition to the spontaneous mutations that were previously collected at low frequency in breeding facilities, have been instrumental for the development of mouse genetic maps because, at that time, they were the only available genetic markers. They also provided geneticists with many potentially interesting models of human diseases. However, experimental mutagenesis sensu stricto, which means the treatment of animals with known mutagenic agents to purposefully increase the mutation rate, is only recent.
7.4.1 Gametogenesis and Experimental Mutagenesis
Experimental mutagenesis consists of exposing progenitors of either sex to a mutagenic agent, with the aim of increasing the occurrence of novel mutations in the progenies of the treated animals. For practical reasons, only male progenitors are exposed to the mutagens because spermatogenesis is a continuous process, starting at puberty and lasting several months or even years. In females, on the contrary, gametogenesis is a cyclic process and the number of cells that are potential targets for mutagenesis is much reduced in adult mice (Fig. 7.6).

Histological appearance of seminiferous epithelium. Sections from an adult mouse testis indicating several stages of the spermatogenetic process. SC = Sertoli cell, Sg = spermatogonia, ZS = zygotene spermatocytes, PS = pachytene spermatocytes, Me = meiotically dividing spermatocytes, step 2 RSp = step 2 round spermatids, step 11 Esp = step 11 elongating spermatids, step 15 Esp = step 14 elongating spermatids, stage II, stage XI, and stage XII = tubules in different stages of the spermatogenic cycle (Figure courtesy of Dr. Dianne Creasy, Huntingdon Life Sciences, East Millstone, NJ, USA)
Spermatogonia are the stem cells of the male germline. When they divide they produce two daughter cells: a spermatogonium type A0, which stays in the pool of stem cells, and a spermatogonium type A1 that undergoes several mitotic rounds, producing A2, A3, A4, and Intermediate types, and finally type B spermatogonia. The type B spermatogonia divide and form pre-leptotene spermatocytes, which are almost identical to type B spermatogonia in appearance, but they become much larger as they duplicate their chromosomes to form tetraploid cells and proceed through meiotic prophase (zygotene, pachytene, diplotene, and diakinesis). The first meiotic division produces two short-lived diploid secondary spermatocytes, which rapidly divide again (second meiotic division) to produce four round haploid spermatids. These round spermatids then undergo a complex morphological transformation into spermatozoa, developing condensed heads covered by an acrosome and attached to a motile tail, which are then shed into the tubular lumen (spermiation). In theory, a single A1 spermatogonium would give rise to 256 sperm cells in 5 weeks, but there is some attrition of cells during spermatogenesis so that the actual number of sperm is smaller than the theoretical maximum. A few of these mature sperm cells will fertilize ova, and most others are eliminated while a new cycle of spermatogenesis follows. The duration of the spermatogenetic cycle is much shorter in the mouse than in most other species; spermatogonia become mature spermatids that are released into the lumen in only 5 weeks (Russell et al. 1990). By comparison, the spermatogenic cycle is 8 weeks in the rat and 10 weeks in humans. It then takes another 1–2 weeks for the released sperm to reach the tail of the epididymis, where they are stored prior to ejaculation.
Mutagenic agents (physical or chemical) exert their effects as soon as they are in contact with the genetic material of the treated mice and this effect terminates, in general, immediately or shortly after treatment ends. The cells that have been mutagenized repair most of the damage resulting from the treatment, but, depending on the severity of this damage, some cells may recover and pass genetic alterations to their daughter cells while others die and are eliminated. The success of a mutagenic treatment is reflected in the percentage of cells that survive and carry a mutation, and the higher this percentage the better. As we will discuss, this depends upon the mutagenic treatment, the type of cells exposed to the mutagen, the dose and duration of the treatment, and the dose rate and the possible splitting of the dose.
Because spermatogenesis is a continuous and precisely timed process, we can calculate the precise stage of development of a specific germ cell, at the time of exposure to a mutagen, depending on the time elapsed between the treatment and the fertile mating. For example, if male mice are exposed to a mutagen and mated 3–4 weeks later, the embryos that result from the mating will have originated from germ cells that were mature spermatids (post-meiotic stage) or spermatozoa entering the epididymis at the time of treatment. In contrast, if the mating takes place more than 7 weeks after the treatment, the embryos result from cells that were exposed as spermatogonia. When the stem cells of spermatogenesis (i.e., the spermatogonia A0) are successfully mutagenized, the male becomes a permanent provider of mutations. On the other hand, when the targeted cells are post-meiotic (spermatids or spermatozoa), the mutagenesis is transient.
An important point to mention is that a very efficient selection process operates during gametogenesis to eliminate the mutations that may have occurred either spontaneously or after the mutagenic treatment. This process is much more efficient during the early (diploid) phases of gametogenesis, where the cells divide and have an active metabolism with efficient DNA repair mechanisms, than during the haploid phase, when the cells differentiate but no longer undergo mitosis. In the same way, meiosis occurring at the spermatocyte stage is an efficient filter to eliminate the chromosomal rearrangements that interfere with the normal distribution of chromosomes in the daughter cells. Reciprocal translocations or inversions, for example, are strongly counter-selected when they occur in spermatogonia, whereas many of them are transmitted to the offspring when induced in early spermatids.
When males receive a mutagenic treatment, the number of affected stem cells depends on the dose. If the dose is elevated, most spermatogonia are killed and the male becomes permanently sterile. Conversely, if the dose is too low, the lethal effect is limited but the mutation rate is low and the experiment might not be successful. Selecting the best dose is very important and may require preliminary experiments.
7.4.2 The Induction of Mutations by Radiation
Hermann Muller (1927) was among the first to report that X-rays can cause mutations and chromosomal damage in Drosophila flies. However, most of the knowledge geneticists have gathered concerning the mutagenic effects of radiations in the mouse results from research conducted at MRC Harwell in England and at Oak Ridge National Laboratory in the United States. An excellent review of these fundamental studies, which may still be useful, can be accessed online in the book “Biology of the Laboratory Mouse” in a chapter by Green and Roderick (1966).
In short, we can say that all types of radiation are mutagenic, provided they have sufficient energy to come into contact with the genetic material. Cosmic radiation, a mixture of photons and high-energy protons originating from outer space, constantly showers on all living organisms and is probably responsible of many “spontaneous” mutations. In contrast, UV radiation, consisting of photons with a wavelength between 100 and 400 nm, is mutagenic (and carcinogenic!) only for the cells of the epidermis. Their energy is insufficient to reach the gonads, and accordingly their impact on the genetic material of mammalian species is virtually nil.
Countless experiments have been performed to understand the mutagenic effects of electromagnetic (X- and γ-rays) and corpuscular (protons and β-particles) radiation. These types of radiation are mutagenic because they have a direct effect on the chromosomes and DNA strands; they produce breakages or deletions that are more or less efficiently repaired, depending on the extent of the damage and the efficiency of the repair mechanisms. They are also mutagenic because they produce ionization as they dissipate their energy into living matter, producing a very large number of hydroxyl and hydroperoxyl free radicals that are highly reactive and diffusible elements. From the experiments conducted by health physicists between 1950 and 1970, it was concluded that the mutagenic activity and the type of mutations produced by exposure to radiation depend on a physical parameter known as linear energy transfer (LET), and, of course, on the dose distributed, the duration of exposure, and whether the dose is fractionated. Heavy particles like protons or α-particles have a very high LET and dissipate their energy over a short distance while passing through living matter. Accordingly, they exhibit high mutagenic activity and produce extensive chromosomal breakage. On the other hand, photons such as X- and γ-rays have a much lower LET and are much less mutagenic, producing mostly point mutations or small-sized deletions. For X- and γ-rays, the rate of induced mutations varies linearly with the dose from 0 to 7 grays (abbreviated Gy).Footnote 13 Beyond 7 Gy repair mechanisms are saturated, and many cells are affected by several mutations and die.
When a dose of X- or γ-rays is distributed over a short period of time (at high dose-rate), the mutagenic effect of the radiation is more intense compared to the same dose distributed over a longer period of time. Similarly, a single dose of radiation is more damaging to the genetic material than the same dose split into several sessions. This is a consequence of the fact that the DNA repair mechanisms are saturated when the dose is delivered over a short period of time. Males, whose germ cells are constantly in mitotic activity (from puberty until death), are more susceptible to the mutagenic effects of radiation than females, whose germ cells are resting at the time of birth.
All germ cells are sensitive to radiation, but haploid cells (post-meiotic stages, with n chromosomes) are more sensitive than spermatogonia (2n) because the DNA repair mechanisms are almost inactive in these highly differentiated cells. As we already mentioned, mutations induced in spermatogonia may be transmitted to the offspring throughout the life of the mutagenized animal, whereas mutations affecting the post-meiotic haploid cells are transmitted only during the short lifespan of these cells (3 weeks), provided that they fertilize an oocyte.
In mice, the mutation rate after exposure of spermatogonia to 10 Gy of X-rays at 0.9 Gy/min, split into two doses of 5 Gy distributed 24 h apart, was reported to be ~ 500 × 10−6 per locus per gamete, compared to the spontaneous rate of ~10 × 10−6 as mentioned above (Russell 1962, 1963). This mutation rate seems to be the highest possible for X-and γ-rays (~50× the spontaneous rate). This increase in mutation rate due to the splitting of the dose suggests that the first irradiation imposes some sort of synchronization and enhances mutability of the cells, while the second dose yields more mutations than otherwise expected. One could, in theory, obtain a higher frequency of point mutations by using neutrons. However, most mutations produced by this type of radiation are deletions that are frequently incompatible with the survival of heterozygotes. Deletions are also difficult to analyze in the molecular context, in particular when they encompass more than one gene, as is often the case.
7.4.3 The Induction of Mutations by Chemicals
Studies of the mutagenic activities of chemicals were initiated by C. Auerbach (Auerbach and Robson 1946; Auerbach 1962), who first reported that 1,1′-thiobis[2-chloroethane], a chemical warfare agent known as “mustard gas” and used during World War I, could cause mutations in Drosophila flies. Since these initial studies, toxicologists have identified a large number of chemicals with mutagenic activity. Identification of such molecules has been rationalized by the introduction of laboratory tests using bacteria (for example, the Ames test developed in the 1970s; Ames et al. 1973). More recently, transgenic mice with several copies of bacterial genes integrated into their genome have been developed as tools for mutation assays; for example, the lacI model, commercially available as the Stratagene Big Blue® mouse, and the lacZ model, available as the Muta™Mouse (Wahnschaffe et al. 2005a, b). These tests, which are very sensitive, relatively inexpensive, and simple to use, allowed the establishment of a very long (and ever-increasing) list of substances with demonstrated mutagenic activity in mammals.
Chemical mutagens have been classified into four categories based on the type of interaction they have with DNA (Vogel and Rohrborn 1970). The first category includes molecules known as base analogs. Molecules of this category (6-amino-purine, for example) are mistakenly used by bacteria during DNA synthesis, leading to the production of transitions or transversions after replication. However, these substances have not been found to be mutagenic in mammals, probably because the metabolic pathways leading to the synthesis of nucleotides are not exactly the same in mammals and in bacteria.
Intercalating agents represent another important group of mutagens. Examples include acridine orange, ethidium bromide, and proflavine. These molecules insert into the DNA helix and bind covalently to the bases of the two strands, leading to deletions occurring during the next round of replication. As with the base analogs, these substances have little effect on pre-meiotic germ cells of mammals and have not been used frequently for the purpose of experimental mutagenesis. Some of these agents, however, are active on post-meiotic germ cells and induce translocations and deletions at a low rate.
The third class of mutagens includes the deaminating agents that are best represented by nitrous acid and sodium bisulfite. Because deamination of guanine (G) or adenine (A) occurs spontaneously in most eukaryotic cells, it has been suggested that deaminating agents might be mutagenic by increasing the basic, natural level of deamination. This, however, has never been clearly demonstrated in mammals, and these agents are not currently used for mutagenesis.
Alkylating agents are, by far, the most potent mutagens in mammals (mustard gas belongs to this category). Molecules of this type transfer alkyl radicals (methyl, CH3, or ethyl, C2H5) onto DNA bases, particularly on adenine but also on guanine. If this alkylation is not repaired promptly by the DNA repair mechanisms, transitions or transversions ensue during the next step of replication.
Alkylating agents are mutagenic in the mouse, but most of them are only active on post-meiotic germ cells (type-2 spermatocytes or spermatids). Among these substances, we must mention the anticarcinogenic drugs TEPA and Thiotepa™, ethyl methane sulfonate (EMS), methyl methane sulfonate (MMS), triethylenemelamine (TEM), procarbazine, and chlorambucil, all of which have been used during the last thirty years as chemical mutagens in the mouse.
In 1979, William Russell from Oak Ridge National Laboratory reported that a simple alkylating agent, N-ethyl-N-nitroso-urea (ENU), has considerable mutagenic power, and even more remarkably, that this substance is active on both pre- and post-meiotic germ cells (Russell et al. 1979). These observations had a major impact on genetic research and must be considered as an important milestone in the history of mouse genetics (Fig. 7.7).

Alkylating agents with mutagenic activity. a Methyl-methane-sulfonate (MMS). b Ethyl-methane-sulfonate (EMS). c N-Ethyl-N-nitrosourea (ENU). d N, N′, N″-Triethylenethiophosphoramide (ThioTEPATM). All four molecules are potent mutagens but ENU is, by far, the most potent and the only one active on pre-meiotic germ cells
ENU is generally sold in the form of a light yellow powder in dark glass bottles, sealed with a rubber stopper. This packaging makes the chemical relatively easy to handle safely. The molecule is light-, heat-, and pH-sensitive and does not dissolve easily in water, but adding a few drops of ethanol avoids this drawback. The mutagenic activity of ENU results from its capacity to transfer an ethyl group to oxygen or nitrogen radicals in the DNA molecule, inducing mis-pairing and ultimately leading to base-pair substitutions or deletions (Van Zeeland et al. 1989; Vogel and Natarajan 1995). In fact, the mutagenic activity of ENU results from two mechanisms acting in opposite directions: the alkylation of the DNA molecule resulting in the creation of adducts on the one hand, and the efficiency of the enzymatic DNA repair mechanisms on the other. In spermatogonia, the ENU-alkylated nitrogen atoms are efficiently repaired, while ENU-alkylated oxygen atoms are repaired with a much lower efficiency.
Many ENU-induced germline mutations have been studied at the molecular level after positional cloning and it has been found that, in the great majority of cases, adenine (A) is the main target of ENU activity with the primary genetic alteration being either AT to TA transversions or AT to GC transitions (Justice et al. 1999).
The mutagenic activity of ENU has been evaluated using several tests (Russell et al. 1979; Favor 1986; Lewis 1991; Lewis et al. 1991, 1992; Favor 1994; Ashby et al. 1997; Schmezer and Eckert 1999). In his initial paper, (Russell et al. 1979) found 35 confirmed mutations at the seven specific loci mentioned above (those homozygous in the PT stock) among 7,584 offspring in the treated group (one injection of 250 mg/kg of body weight), compared to 28 mutations among 531,500 mice in the control group. This indicated a mutation rate 90 times higher than the spontaneous rate and five times higher than for 6 Gy of γ-rays.
Plotting the mutation rates calculated with the same “multiple loci” assay to the doses of ENU injected in male mice, (Favor et al. 1990) observed that the mutation rate for ENU increased roughly linearly with dose, from the threshold dose of ~34 mg/kg of body weight up to 300 mg/kg, a dose that seems to be the highest tolerable by an adult mouse. If the dose remains low, say less than 30 mg/kg of body weight, the mutation rates are not significantly different from the rate of spontaneous mutations in the same assay. Favor’s calculations can be summarized in the following two formulae:
MR × 10−5 = (1.2 ± 0.3) for D < 33.9 mg/kg
MR × 10−5 = (1.2 ± 0.3) + (0.4 ± 0.05) × (D – (33.9 ± 5.0)) for D ≥ 33.9 mg/kg
where MR = mutation rate and D = dose in mg/kg of body weight.
The threshold effect observed by Favor and colleagues is probably explained by the fact that, when the number of alkylated sites remains low, the repair mechanisms can cope, but when it becomes high or very high, these mechanisms become saturated and mis-pairing increases in proportion to the dose of mutagen.
W. Russell and colleagues reported a few years after their initial publication that three or four injections of 100 mg/kg of body weight, each delivered at weekly intervals, enhanced the mutation rates by a factor 1.8 and 2.2, respectively, compared with a single dose of 250 mg/kg of body weight, while allowing greater survival and fertility of the treated mice (Russell et al. 1982a, b; Hitotsumachi et al. 1985). With such a treatment, the maximum mutation rate of 125–152 × 10−5 per locus could be obtained that roughly corresponds to 150 times the spontaneous mutation rate. It is probably difficult, if not impossible, to increase this mutation rate further because the risk of inducing dominant lethal damage would then be maximized (Fig. 7.8).

A dose–response analysis of ethylnitrosourea (ENU)-induced recessive locus-specific mutations in treated spermatogonia. Predicted locus-specific mutation rates (MR × 10−5) following ENU treatment of spermatogonia in the mouse. This diagram represents the dose–effect linear relationships for the mutagen, between ~34 mg/kg of body weight (the threshold dose under which there is no detectable effect) and 300 mg/kg of body weight, which seems to be the highest dose tolerated by the mouse. This linear model was computed based on extensive data from Neuherberg (Germany) and Oak Ridge (USA) (adapted from Favor et al. 1990)
This linear dose relationship for induced mutation rates at these seven specific loci demonstrates the extraordinary power of ENU as a mutagen, but cannot adequately predict the absolute rate of induced mutation at an “average” locus in the mouse genome. Lewis and co-workers, for example, calculated the number of electrophoretic variants induced at 32 loci after treatment with increasing doses of ENU (from 0 to 250 mg/kg of body weight) in DBA/2 and C57BL/6 male mice (Lewis 1991). In these experiments, the mutation rates again appeared to increase linearly with dose but were on average 2.6 times lower than for the “multiple loci” test performed by Russell and colleagues. This latter observation, which has been reported by many other scientists with different tests, indicates that the sensitivity of a locus to the mutagenic activity of ENU probably depends on a variety of parameters such as its “molecular” size, the gene structure (density in A-T, number of introns, etc.), and presumably several other unknown parameters. It is likely that some regions of DNA are more susceptible than others to the mutagenic activity of ENU, validating the idea that hot spots of mutagenesis exist in the mouse genome (Kiernan et al. 2002; Arnold et al. 2012).
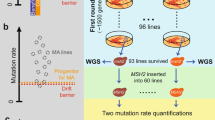
The mutation frequency, established by Russell and co-workers for seven specific loci, was later refined by Bode (1984) in another experimental context. Bode considered that, from an optimally mutagenized male, one can expect to obtain, on average, one mutation at a given locus per 1,500 of its gametes. It must, however, be kept in mind that a given male can produce only a limited number of mutations, and this number is dependent on the number of targets that have been hit by the mutagen. From his experimental data, Bode concluded that this number is close to 500 with a dose of 250 mg/kg of mouse body weight. This important observation means that, when the ultimate goal of an experiment is to produce a great variety of mutations, as is often the case, it is strongly recommended to inject a quite large batch of males rather than to breed many offspring from only a few males.
The mutagenic activity of ENU has been assessed directly at the DNA level in several laboratories, by performing a careful characterization of the number and type of nucleotide substitutions induced, then by matching the nature of these substitutions to the phenotype of the affected mice—if any (Justice et al. 1999; Noveroske et al. 2000; Concepcion et al. 2004; Quwailid et al. 2004; Keays et al. 2006; Takahasi et al. 2007; Arnold et al. 2012).Footnote 14 The results of these analyses indicate that ENU induces mutations at a frequency of one for every 0.7–1.9 Mbp of genomic DNA, depending upon the strain and dose. Analysis of the mutations confirms that AT-to-TA transversions occur in about 44 % of the cases while AT-to-GC transitions occur in about 38 % of the cases. When they fall within the coding regions these substitutions cause missense mutations (64 %), splicing defects (26 %) or nonsense mutations (10 %). Another interesting observation, which is a direct consequence of the observed AT-to-TA and AT-to-GC bias mentioned above, is that some amino acid changes are more likely to occur after ENU treatment than others. As such, it must be kept in mind that ENU mutagenesis does not merely increases the spontaneous mutation rate but its action is biased towards certain amino acid changes.
ENU has also been used as a mutagen in the rat and has proved efficient. In this species, however, the dose must be reduced to 90 mg/kg of body weight (Mashimo et al. 2010) and, here again, splitting of the doses has proved more efficient than a single dose.
ENU is a powerful, easy-to-use, inexpensive, and remarkably efficient mutagen. Its effectiveness varies with the strain of mouse treated, and this is why it is absolutely essential to calibrate the experimental parameters as precisely as possible before embarking on a new mutagenesis project. Non-optimal use of the mutagen (i.e., using a dose that is either too high or too low) will inevitably lead to a waste of both time and animal lives.
7.5 Protocols of Experimental Mutagenesis
When a male mouse is treated with a mutagen, for example by performing a single injection of 250 mg ENU per kilogram of body weight, it stays fertile for a few days after the treatment and then becomes sterile for a period spanning 10–18 weeks (Oakberg and Crosthwait 1983). This sterility period is a consequence of spermatogonial cell killing and it is, in large part, strain- and dose-dependent. BTBR, BALB/c, C3H/He, C57BL/6, and DBA/2 strains have been used for many years, in particular for the large ENU mutagenesis programs conducted in Germany, England, and the USA (Hrabe de Angelis et al. 2000; Nolan et al. 2000; Arnold et al. 2012). These strains appeared to be relatively resistant to ENU, although a relatively higher percentage of C57BL/6 males did not recover fertility after the ENU treatment (Lewis et al. 1991, 1992). Strain FVB, which has several advantages over the other strains for the production of embryos for transgenesis, appeared quite susceptible to ENU, and, accordingly, is not a good choice for experimental mutagenesis (Justice et al. 2000).
While information concerning the toxicity of ENU for the different strains of mice is available, information about the differences in mutation rates is scarce. In an experiment aimed at the production of electrophoretic mutant proteins, Lewis and colleagues (Lewis et al. 1991) made use of C57BL/6 and DBA/2 males, mated to DBA/2 and C57BL/6 females respectively, and did not observe any statistically significant differences in mutation rate between the two strains. Considering the many experiments that have been performed with the classical laboratory inbred strains and the mutagen ENU, one would conclude that, if inter-strain differences in mutation rate were important, this would have been noticed, but this is not the case.
After the sterility period, the spermatogonia that survive ENU treatment progressively repopulate the testis, the sperm concentration rises progressively and the males regain fertility and produce spermatozoa derived from the several different clones of mutagenized spermatogonia. In the sperm population (and later in the embryos), all types of mutations are present but, while dominant mutations can be observed directly in the F1 (or G1) progeny, recessive mutations must be homozygous to express a phenotype. This requires two more generations and the establishment of so-called individual or micro-pedigrees.
The production of mutations in laboratory rodents can be achieved either genome-wide (i.e., at any locus), or in more or less precisely targeted regions, depending on the aim of the experiment and the protocol used. These protocols do not depend upon the mutagen and can apply to radiation as well as to chemicals. We will review the most commonly used mutagenesis strategies.
7.5.1 Phenotype-Driven, Genome-Wide Mutagenesis
A phenotype-driven, genome-wide mutagenesis program consists of four successive steps. First, males (G0) are treated with the mutagen and then mated with females of the same strain, or of another inbred strain, once they have recovered from the sterility period.Footnote 15 Unusual phenotypes (often called phenodeviants) that could result from dominant mutations are then looked for by careful examination of the offspring of this first cross (G1 population).
In the second step, G1 males, which are all potential carriers of recessive mutations at a number of unknown loci, are gathered for the establishment of individual micro-pedigrees. For this, each G1 male is mated to a few females, either of the same or from a different strain, and a sample of six G2 females, offspring of this cross, is selected and crossed (backcrossed in this case) to their father to produce a G3 population. This G3 generation is then carefully examined for the detection of possible recessive mutations. The rigorous and systematic examination of the G3 progeny is part of the phenotyping process and requires much care. Indeed, the higher the number of parameters screened, the higher the number of mutations detected (Fig. 7.9).

Phenotype-driven genome-wide mutagenesis. Phenotype-driven mutagenesis consists of four successive steps. In the first step, males are treated with the powerful mutagen ENU (see text for doses) and mated with 2–3 females after recovery from a 10 to 13-week sterility period (this the G0 generation). The entire G1 progeny is then carefully scrutinized, looking for possible dominant mutations (arrow). In the second step, males of the G1 generation (which are potential heterozygous carriers of recessive mutations of all kinds) are selected for the establishment of micro-pedigrees. First, they are mated with females of either the same or a different strain, and 4–6 female offspring (G2) are backcrossed to their G1 father. Finally, the progenies of the G1 male × G2 female offspring (G3) are subjected to careful phenotypic examination (for example, in a “Mouse Clinic”). Micro-pedigrees producing mutant phenotypes are then isolated for in-depth analysis. The number of G2 females and their G3 offspring are established after statistical computation to optimize the possibility of detection of new phenotypes
Because their deleterious effects are compensated for by the presence of a normal allele in heterozygotes, the recessive mutations induced in the G0 males recur in G3 of the same micro-pedigree and, accordingly, they are easier to detect and preserve than the dominant mutations, which, in most instances, appears only once in the G1 population.
In these micro-pedigrees, when six heterozygous (+/mut?) G2 females are backcrossed to the individual G1 males and a minimum of ten G3 offspring are phenotyped per G2 female, the probability of not detecting, just by chance, a recessive mutation with a visible phenotype that would have been heterozygous in the +/mut? G1 males is less than 2 % at the 95 % confidence level.
Bode et al. (1988), followed by McDonald et al. (1994), were among the first to use a whole-genome, phenotype-driven ENU mutagenesis program to produce relevant animal models of phenylketonuria (PKU-OMIM 261640). G0 males were treated with ENU, the G1 male offspring were mated to females of the same strain to produce the G2 progeny, and finally the G1 males and their G2 female offspring were intercrossed to produce the G3 progeny. Blood samples from G1, G2, and G3 mice were analyzed by using the popular Guthrie test, a biochemical test that was used some years ago for detecting elevated levels of phenylalanine in the blood of human newborns.Footnote 16 In these experiments, three independent mutant alleles were identified in the G3 populations (hph1, hph2, and Pah hph5). In addition, it is interesting to note that, using such a phenotype-driven genome-wide strategy, the biochemical pathways at work in the catabolism of the amino acid phenylalanine were literally “dissected” out, with one mutation identified at each biochemical step. This was done in exactly the same manner in which the bacterial geneticists of the early days disentangled the metabolic pathways in bacteria (McDonald 1995).
Nowadays, after much progress in genotyping and phenotyping, several projects have been undertaken by which the G1 and G3 progenies of ENU-mutagenized males have been systematically and extensively phenotyped using a number of criteria by a team of specialists in so-called “mouse clinics”. Many interesting mutations have been discovered in these projects that would probably not have been noticed in other laboratories (Hoebe and Beutler 2005; Massironi et al. 2006; Arnold et al. 2012). Among the many interesting mutations identified are Clock, which modifies the circadian rhythm of affected mice (Wilsbacher et al. 2000), and Ticam1 Lps2, which results in impaired defense mechanisms against viral and bacterial diseases (Beutler et al. 2007). In a European project comprising six different laboratories and focusing on deafness syndromes, no less than thirteen new independent genes involved in inner ear differentiation and pathology were identified by ENU mutagenesis (Quint and Steel 2003).
The genome-wide production of recessive mutations is a tedious enterprise that requires both intensive animal care and large breeding programs. The advantage of this approach is that no a priori assumptions are made about the genes involved in any pathway. Phenotype-driven mutagenesis is thus an effective method for the identification of novel genes. Numerous projects are now in progress in several laboratories worldwide, where groups of novel mutations, once identified, are roughly phenotyped, mapped to a chromosome, and finally made available to the scientific community for further study. There is no doubt that genome annotation will benefit from all these programs, even if a significant amount of work remains to be achieved after a gene is identified in the form of a mutant allele.
7.5.2 The Induction of New Mutant Alleles at Specific Loci
As we remarked above, the main advantage of the genome-wide, phenotype-driven mutagenesis approach is that most of the mutations collected are new alleles appearing at loci where no mutations had ever been isolated before. However, in some cases, it may be desired to induce new alleles at a given locus, for example to explore the possibility that the severity of a given phenotype might be allele-dependent.
The induction of new alleles at a specific locus is well illustrated by the so-called “multiple loci test”, which was used for the assessment of spontaneous mutation rates, and which we introduced earlier in this chapter. Using this test several new alleles (in particular at the Tyr-albino locus) have been induced after treatment with mutagens in the gametes of wild-type male partners, and were then observed directly in the F1 progeny of these males after mating with females homozygous for a set of recessive viable alleles (Rinchik and Carpenter 1999). A similar strategy can be applied to any situation where the production of a series of new alleles at a given locus might be informative, and is ideal when at least one viable recessive allele is available.
Bode (1984) used ENU to produce new alleles at the Brachyury (T), quaking (qk) and tufted (tf) loci by mutagenizing +++/+++ (wild-type) male mice and crossing them to females with the genetic constitution T qk tf/++ tf. In the F1 progeny of these mice the researcher found three tufted phenotypes [tf], one quaking [qk], and one with a short tail (t-interacting or t int) out of 5,172 offspring. In similar experiments, Justice and Bode (1986) and Bode et al. (1988) produced several new alleles at the same three loci, with some of the new alleles at the quaking locus exhibiting interesting and unexpected properties (Justice and Bode 1990; Cox et al. 1999).
Chapman and colleagues performed a similar experiment (Chapman et al. 1989) with the aim of understanding why mice affected by the X-linked Dmd mdx mutation, homologous to the human mutation producing Duchenne muscular dystrophy, were not clinically affected. Chapman hypothesized that this striking phenotypic difference might be because the original Dmd mdx mutation hits a domain of the gene encoding dystrophin that is not functionally essential, and supposed that alleles affecting one of the other four domains of the protein might have more severe effects. To test this hypothesis he created four new alleles at the Dmd locus (Dmd cv2, Dmd cv3, Dmd cv4, and Dmd cv5) by ENU mutagenesis. These new mutant alleles were detected by checking for an increase in the creatine phosphokinase (CPK) plasmatic levels in the female progeny of ENU treated +/Y males crossed with Dmd mdx/Dmd mdx homozygous females.Footnote 17 The result of these experiments was that all five alleles, the four ENU-induced and the original one, had a very similar phenotype with no obvious muscular pathology, although the four mutations were found to affect totally different domains. Later, it was demonstrated that mice homozygous for the original Dmd mdx allele and one of the ENU-induced series (Dmd cv5) had a weaker effect than the other three alleles on the electro-retinogram (ERG) phenotype of the mutant mice (Figs. 7.10 and 7.11).

Targeted chemical mutagenesis. Male mice are mutagenized and then mated to females homozygous for a recessive allele (m) at a specific locus. The G1 offspring of this type of cross are expected to be all wild type. Any deviation from this phenotype must be considered a possible new mutant allele at the m locus, especially if some similarities exist between the new phenotype and the phenotype of the female (m). For example, this strategy allowed the generation of an allelic series at the dystrophin gene (Dmd) (Chapman et al. 1989)

Targeted chemical mutagenesis. A male homozygous for a polymorphic protein is treated and then crossed with a female homozygous for another electrophoretic variant, and the G1 progeny are analyzed with the same technique. Mice nos. 1 and 5, as expected, are heterozygous for the two parental forms. Mouse no. 2 is heterozygous for an inactive allele (dotted line) inherited from its (mutagenized) father. Mouse no. 3 is heterozygous for the maternal form and a new functional electrophoretic variant derived from its father
This observation indicated that the position of the mutation in the dystrophin-encoding gene, although it had no effect on the muscular phenotype, nonetheless had some direct consequences on the ERG phenotype (Pillers et al. 1999). This contributed to the fine annotation of the different domains of the Dmd gene, but did not explain the phenotypic differences between the human pathology and the mouse model.
A variation of the above-mentioned strategy is to analyze the electrophoretic pattern of enzymatic proteins in an interstrain F1 hybrid where one parent (usually the male) has been mutagenized. Such an “electrophoretic multiple loci test” has been successfully used to identify new mutations at loci encoding for enzymatic proteins (Johnson and Lewis 1981; Marshall et al. 1983; Lewis et al. 1991, 1992).
ENU mutagenesis has also been used to induce mutant alleles in the genes encoding the β-chain of hemoglobin (Peters et al. 1986) as well as to produce several null or functionally different alleles (Charles and Pretsch 1987; Pretsch et al. 1994).
The production of new alleles is also interesting in that it allows the production of slightly different animal models. An excellent example of this situation is provided by the existing animal models of human citrullinemia type I (Perez et al. 2010), where it was demonstrated that some alleles, because they hit a different domain of the protein, appeared to be much better animal models of the human syndrome of citrullinemia (OMIM 215700).
The condition set above—that at least one recessive and viable mutant allele for the locus of interest is available to allow the production of other mutant alleles—is not an absolute prerequisite, and alternative strategies are possible. Let us suppose, for example, that other alleles are desired at the Mut locus, which to date has only been characterized by the unviable (or sterile) mutation mut 1. In this case, several F1 (or G1) males, potentially heterozygous for many new ENU-induced mutations (among which is a potentially new mut 2 allele?) are produced and then crossed to +/mut 1 females. If, by chance, a mouse with an abnormal [mut] phenotype is detected in the progeny of one of these females, this suggests that a new mut 2 allele at the Mut locus has very likely been induced by the treatment. The new allele can then be recovered from the G1 progeny.
7.5.3 The Induction of Mutations in Specific Regions of the Genome
Many strategies have been used to induce and identify the mutations in a specific chromosomal region. Here, we describe three of these strategies that may be of interest in the future: the first makes use of deletions, the second uses consomic or congenic strains, and the last strategy requires a set of overlapping inversions.
Using deletions to detect recessive mutations can only be applied to regions where haploidy is compatible with life. The basic principle is that, when a mutation is induced in the chromosomal segment in front of a deletion, a new phenotype (often lethal) is observed when the chromosome carrying the induced mutation and the deleted chromosome are associated in the same genome. In these conditions, the breeding protocol requires more than one generation, since the induced mutation must be kept in the heterozygous state while it is revealed by the deletion. The deletion strategy has been used many times (Justice et al. 1997; Rinchik and Carpenter 1999) and has been included in modern mutagenesis programs (Nolan et al. 2000) to identify potential models of human diseases (Fig. 7.12).

Using deletions to detect recessive mutations in a specific region. A male mouse is mutagenized and then mated to females heterozygous for a recessive marker a, and for a viable deletion (Δ) (its phenotype is [a]). Most offspring of this cross have a wild-type phenotype, except when a recessive viable mutation m is induced by the mutagen in the chromosome segment encompassed by the deletion. In this case, a new phenotype is observed (m). In the cases where the induced mutation is lethal, the progenies are reduced in size and the mice heterozygous for a are also heterozygous for m, the new mutation (except for a few recombinants)
The use of consomic strains is an interesting strategy to safely collect the mutations induced in a particular chromosome. Consomic strains (see Chap. 9) are strains in which an entire chromosome has been backcrossed from a donor strain into a different recipient or background strain. Such strains are completely identical for all chromosome pairs but one. These are not common, but at least one set exists (Nadeau et al. 2000), and this is sufficient for the strategy to be applicable. The strategy, presented in Fig. 7.13, is an interesting approach to studying the mutations that have a weak effect or that require sophisticated tests for their detection, because it is possible to establish a co-isogenic strain where the newly induced mutations are safely stored before being studied. This is a great advantage when populations (not only individuals) are to be compared at the phenotypic level; for example, histocompatibility, susceptibility to infectious diseases, and QTL analysis. The same co-isogenic strain that is homozygous for the targeted chromosome can be used several times in successive rounds of mutagenesis experiments, resulting in the progressive accumulation of several new alleles in the targeted chromosome (Fig. 7.13).

Accumulating mutations in a specific chromosome. Male mice are mutagenized and then mated to female mice consomic for a specific (targeted) chromosome (solid black). F1 male offspring of this cross are mutagenized again and crossed to female mice of the same strain, consomic for the same chromosome. The same cycle of mutagenesis—cross with a consomic partner is perpetuated a number of times, and in so doing mutations accumulate only on the targeted chromosome at each generation. Finally a few female offspring of this series of backcross are selected by microsatellite genotyping and backcrossed to their consomic father. This allows re-establishment of a fully co-isogenic strain with many independent mutations accumulated in the same chromosome pair. These mice are then carefully phenotyped. If one of the induced mutations is lethal, the experiment cannot be completed, but the induced mutations can be kept and studied in another context, for example after outcrossing. This strategy requires very little work and a very limited number of animals to be used. This can easily be coupled with a gene-driven mutagenesis experiment, reducing the time spent on genotyping
The use of a set of overlapping inversions is similar in principle to the use of consomic strains and is reminiscent of the balancer chromosome developed in the past by Muller and colleagues for the collection of X-ray induced mutations in Drosophila melanogaster. An example of this strategy was the use of a genetically engineered inversion in chromosome 11, which has been described in detail (Zheng et al. 1999; Kile et al. 2003).
Many other strategies have been used to generate and keep mutations in specific areas of the mouse genome that cannot be described in detail here. We will just briefly mention that Shedlovsky et al. (1986, 1988), using a specially designed strategy, were able to induce and study a dozen new lethal alleles within a region spanning two centiMorgans on each side of the T/t region on mouse chromosome 17.
7.5.4 A Gene-Driven Strategy for the Production of Mutations at Specific Loci
With the expansion of advanced techniques for the structural analysis of DNA, approaches have been developed that are based on the direct, in vitro detection of DNA alterations, either at specific loci or in specific regions of the genome. These techniques, when applied to the offspring of mutagenized males, allow the production of new mutations in specific regions, ultimately into a preselected (or targeted) gene.
The strategy generally consists of four steps. First, adult males of an inbred strain are treated with an appropriate dose of mutagen (ENU in most instances) and then mated with females of the same inbred strain for the production of a large G1 population.Footnote 18 In the second step, sperm samples are collected from adult G1 offspring of this initial cross and stored deep-frozen for performing future in vitro fertilization. Simultaneously, DNA samples from the same G1 males are prepared, cross-referenced with the sperm samples, and stored (Fig. 7.14).

Genotype-driven mutagenesis. Male mice are treated with ENU and mated to females (preferably of the same inbred strain) once they have recovered from the sterile period (G0). A large number of G1 males, which all are heterozygous carriers of a great number of independent point mutations (mostly base-pair changes), are then bred. Sperm samples from each G1 mouse are collected and preserved deep-frozen, while DNA samples from the same mice are processed and stored with the same reference. Identification of the mutations generated by the ENU treatment in a specific target (a gene or any other specific sequence) is carried out by molecular techniques to identify DNA mismatches, or directly by sequencing. Once the base-pair changes are identified and considered potentially interesting (stop codons, missense, etc.), the corresponding sperm cells are thawed and heterozygous mice are produced by in vitro fertilization with oocytes of the same background strain. A major advantage of this method is that it produces all types of point mutations, not only knockouts. A drawback is the difficulty of and time required for identifying the mutations in the targeted region. With the rapid expansion of new sequencing techniques, the identification step should be somewhat easier
The third step consists of the analysis of the DNA sequence of all G1 mice, looking for any structural changes that may have occurred in a selected and well-delimited region of the genome. This can be achieved by using a sensitive, high-throughput, physical technique, detecting all single nucleotide mismatches after pooling of the DNA samples. This can also be achieved by direct sequencing or SNP genotyping.
When a mutation is found and registered as potentially interesting (i.e., excluding synonymous base-pair changes but retaining nonsense or missense mutations with predicted severe effects), the fourth and last step is performed: the sample of sperm cells corresponding to the potentially interesting mutant mouse is thawed, oocytes of the same strain are fertilized in vitro and implanted in pseudo-pregnant mothers, and, once born, the potentially heterozygous offspring are bred and crossed in order to produce homozygous offspring whose phenotype is then observed. In this micro-pedigree, the molecular characterization of the offspring is fundamental.
This gene-driven protocol allows the production of all types of mutations (and not only knockouts) in all regions of the genome (coding and non-coding). A drawback is the difficulty and time required for identifying the mutations in the targeted regions. However, with the rapid expansion of modern sequencing techniques, the identification step should be somewhat simplified and shortened in the near future.
The gene-driven or targeted mutagenesis approach has several advantages. It is fast and relatively inexpensive compared to other gene-driven strategies (for example, the engineering of knockouts in ES cells—see Chap. 8). Once identified in a batch of frozen sperm cells, a mutation can be retrieved and made available as heterozygous adult mice in 4–5 months’ time. Another interesting point is that a repository comprising a very large number of (non-characterized) mutant alleles can be established by progressively accumulating and storing samples of deep-frozen sperm cells from ENU-treated mice. As we already mentioned, and as observed by direct sequencing of samples prepared from ENU-treated mice, one expects ~0.7–1.9 nucleotide change(s) to be induced per Mbp of mouse DNA after the injection of a single dose of 250 mg/kg. If we consider that the mouse genome consists of 2.7 × 109 bp, one can then expect between ~2,000 and 5,000 de novo substitutions in each G1 progeny from an ENU-treated male mouse. If these nucleotide changes are randomly distributed, one can then expect between ~30 and 75 of the latter to be in the coding DNA or the splicing sites, of which ~25–60 will generate a missense, a nonsense or a splicing defect (77 %).
In addition to these theoretical considerations (but based on actual sequencing data!), one can also calculate that a repository with frozen sperm samples from 20,000 individual G1 animals will be a resource with the potential presence of six independent mutations at any gene of the mouse genome (at the 5 % risk level).
The identification of specific gene alterations can be achieved using pooled DNA samples and run concurrently in several different laboratories to increase the efficiency and ultimately lower the cost of mutagenesis. The final advantage is that, in a species such as mouse where sperm cells can be frozen for long periods and thawed for fertilization, there is no time limit for the identification of mutations. Several laboratories have already published interesting results in this manner (Coghill et al. 2002; Augustin et al. 2005; Michaud et al. 2005; Gondo 2008; Gondo et al. 2010), demonstrating that this gene-driven strategy for the induction of mutations in the mouse might be very promising. This is even truer if we consider that the technique in question can also be applied to the annotation of DNA sequences that are highly conserved across different species; for example, those that are transcribed into non-coding RNAs or not transcribed at all, and whose function is still under scrutiny.
7.6 Other Techniques for the Production of Mutations in the Mouse
In addition to those described earlier in this chapter, a few other strategies have been proposed in the past for the induction of novel mutant alleles in the mouse genome. Most of these techniques have not proved to be significantly more advantageous than the techniques currently in use (ENU mutagenesis in particular) and, for this reason, they have been abandoned. However, exceptions must be made for two strategies that have demonstrated some real advantages. The first consists of treating embryonic stem cells (ES cells) with chemical mutagens (ENU or EMS): this approach will be discussed in the next chapter. The second strategy consists of using transposable elements as insertional mutagens in the mouse, just as the P elements were used in Drosophila melanogaster, i.e. with the assumption that, when by chance the random insertion of a transposon occurs into a gene, it generally hinders the transcription of a normal mRNA at or near the insertion site and causes a loss-of-function mutation. This technique is known as transposon-based insertional mutagenesis or TIM. We will describe it briefly.
As discussed in Chap. 5, transposable elements (TEs or transposons) are short DNA sequences that move (transpose) within the genome of a great variety of organisms, including bacteria, plants, insects, and vertebrates, by using a cut-and-paste mechanism (i.e., with no RNA intermediate). This mechanism of transposition requires a specific structure of the transposon, with inverted repeats at both ends, and a specific enzyme (a transposase or transposonase), which is synthesized either by the TE itself (in the case of autonomous transposons) or “in trans” by an independent gene (in the case of non-autonomous transposons). Transposons are very active in the genome of plants and bacteria, as well as in some other species, and play an important role in evolution.Footnote 19 In mammalian genomes, on the other hand, transposons are inactive and the transposase-encoding genes are degenerated and no longer functional.
Starting from these observations, geneticists had the clever idea to “synthesize” a transposon by genetic engineering using an active transposase in the context of a mammalian genome. To do this, they selected the sequence of a transposon of the Tc1/mariner family active in fish (salmon) and, taking into account some phylogenetic data, they could “resurrect” a functional transposon system that they judiciously named Sleeping Beauty (SB10) in memory of its historical origins. SB10 was confirmed active in the mouse and rat genomes, inducing mutations by transposition as expected (Ivics et al. 1997).
In experiments making use of the SB10 transposon system, two transgenic strains are prepared independently, one carrying the transposon proper (sometimes modified to carry a marker cassette that helps track the animal carriers of a novel mutant allele) and the other expressing the indispensable transposase. When desired, the two strains are crossed to generate F1s in which transposition can occur. In the mouse, the frequency of SB transposition was estimated to be in the range of 0.2–2.0 events per spermatid (Copeland and Jenkins 2010). Although the rate of production of transposon knockout mutations (TKOs) is less than the rate of mutations resulting from ENU treatment, the TKOs are, in most instances, easier to identify and to clone. By outcrossing the animals carrying the TKO mutations of interest, one can separate the transgene-encoding transposase from the other components of the SB system (the mutator element) and transposition immediately stops.
To illustrate the use of transposons as mutagens and the great versatility of this strategy, we recommend a set of interesting publications (Carlson et al. 2003; Lu et al. 2007; Takeda et al. 2007, 2008; Largaespada 2009; Ivics et al. 2011; Furushima et al. 2012). Finally, a review paper by Copeland and Jenkins (2010) is a beautiful illustration of the contribution of the SB10 system to the analysis of the determinism of cancer and the discovery of cancer genes.
In the mouse, and as we will explain in the next chapter, the transposon Sleeping Beauty as well as another one called piggyBac have been used extensively both for the transfection and the production of mutations in ES cell lines in vitro.
If transposon-based insertional mutagenesis has some obvious advantages for the production of mutations, it is also interesting for the transfer of genes with stable expression in mouse ES cells. Finally, it may also have applications enabling the persistent expression of therapeutic genes in patients.
7.7 Conclusions
Spontaneous mutations, which are generally identified through the observation of an abnormal phenotype, present several advantages. The first and probably the most important is that they are produced at virtually no cost and are in general freely available. Another advantage is that they have, in general, an obvious phenotype given that they are identified based on observation. Also, spontaneous mutations represent a great variety of molecular events, such as deletions, insertions, and point mutations, generating not only loss-of-function alleles but also hypomorphic and hypermorphic alleles. The problem is that not all mutant genes have an obvious phenotype or, conversely, the phenotype of some mutant alleles is sometimes so severe that affected offspring die in utero.
When mutant allelic forms of a gene are not readily available, the only possible approach for gene annotation is to generate de novo mutations. Thus, the discovery of the extraordinary virtues of ENU as a mutagen can certainly be regarded as a milestone in the history of mouse genetics. With this substance at our disposition, it is now possible to produce and store a great number of new mutant alleles for each protein-coding gene, and all these mutations are a valuable tool for genome annotation. Another advantage is that it is now also possible to induce mutations in those regions of the genome that are highly conserved but whose function is not yet elucidated. The only drawback that should be considered is that mutagens act randomly, forcing us to make a sometimes lengthy and costly selection among the collected mutations. In this regard, and as we will discuss in the next chapter, the widespread availability of a variety of genetic engineering technologies, including new genome editing tools, has opened the field to the creation of subtle modifications in the mouse genome at will. Even though the identification of genes accountable for single-gene phenotypes is very important, in particular in the context of gene annotation, most of the pathologies that affect human patients are not “monogenic” but are influenced by multiple genes with additive or synergistic effects. As such, our present challenge is to advance the genetic analysis of complex traits.
Notes
- 1.
Hugo de Vries also used the word “sport” to define the same sort of sudden genetic changes.
- 2.
Resistance to the rodenticide warfarin is a good example of the mutations that occurred in wild populations, generating a selective advantage.
- 3.
Chromosomal rearrangements such as translocations of all types, transpositions, deletions, duplications, inversions, etc. have been discussed in Chap. 3 (Cytogenetics) and 5 (The Mouse Genome).
- 4.
Hermann J. Muller was awarded the Nobel Prize in Physiology or Medicine for the “discovery that mutations can be induced by X-rays”.
- 5.
Here we write the codon sequences as they are read on the sense (5′ to 3′) strand of DNA. In these conditions, they read the same as the RNA codons (with the exception that T is replaced by U in mRNAs). However, it must be kept in mind that the mRNA transcripts are synthesized using the antisense strand of DNA (3′ to 5′) as a template.
- 6.
The average spontaneous mutation rate at the DNA level has been estimated to be 2.2 × 10−9 per nucleotide per year in the human species (Kumar and Subramanian 2002).
- 7.
This is referred as nonsense-mediated mRNA decay.
- 8.
Four substitutions of the first nucleobase result in a synonymous codon (lysine or arginine codons). No substitution of the second nucleobase leads to a synonymous codon.
- 9.
In mouse nuclear DNA, the G + C content is 41.70 %, indicating that codons making use of these two nucleotides are under-represented in this species.
- 10.
Computations of the mutation rates were made on several interstrain F1 hybrids expected to be all heterozygous for one or several of the recessive coat color alleles and the corresponding wild-type allele. In such an F1 population, the mice with a non-wild-type phenotype are potential carriers of a new mutant allele. This was confirmed by setting up separate crosses.
- 11.
These observations were made on the F1 progeny of a cross between a tester stock, known as PT stock, homozygous for seven fully penetrant recessive alleles, and mice homozygous for the wild-type alleles at the same seven loci.
- 12.
On a total of 36 dominant mutations.
- 13.
Since 1970, the gray (Gy) has replaced the rad as a unit of absorbed radiation in terms of energy per unit of mass. One gray corresponds to one joule of energy absorbed per kilogram of living matter. One Gy is equal to 100 rads.
- 14.
The publication by Arnold et al. (2012) is a rich source of information calculated on a very large sample.
- 15.
The choice of the strain must be considered with care depending on the future use of the mutant potentially discovered. If mutations are induced, it will definitely be important to identify the background strain in which the mutation occurred.
- 16.
The Guthrie test (a bacterial assay) was routinely used for the neonatal diagnostic of phenylketonuria. It is now replaced either by an immunoassay or by a tandem mass spectrometry assay that measures the amino acid proportions.
- 17.
An increase in the plasma level of creatine phosphokinase (CPK) in these F1 mice reveals some damage to the muscular tissue, and is often an indication of the likely occurrence of a new mdx allele.
- 18.
In this type of experiment it is necessary to exclusively use mice of an inbred strain to enable the non-ambiguous characterization of the mutations potentially induced in the progeny by the mutagen.
- 19.
The transposons were discovered and studied in maize by Nobel laureate B. McClintock, precisely because of their mutagenic activity.
References
Ames BN, Lee FD, Durston WE (1973) An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc Ntl Acad Sc USA 70:782–786
Antonarakis SE, Kazazian HH, Gitschier J, Hutter P, de Moerloose P, Morris MA (1995) Molecular etiology of factor VIII deficiency in hemophilia A. Adv Exp Med Biol 386:19–34
Arnold CN, Barnes MJ, Berger M, Blasius AL, Brandl K, Croker B, Crozat K, Du X, Eidenschenk C, Georgel P, Hoebe K, Huang H, Jiang Z, Krebs P, La Vine D, Li X, Lyon S, Moresco EM, Murray AR, Popkin DL, Rutschmann S, Siggs OM, Smart NG, Sun L, Tabeta K, Webster V, Tomisato W, Won S, Xia Y, Xiao N, Beutler B (2012) ENU-induced phenovariance in mice: inferences from 587 mutations. BMC Res 5:577
Ashby J, Gorelick NJ, Shelby MD (1997) Mutation assays in male germ cells from transgenic mice: overview of study and conclusions. Mutat Res 388:111–122
Auerbach C (1962) Mutation: an introduction to research on mutagenesis. Part I: methods. Oliver and Boyd, Edinburgh
Auerbach C, Robson JM (1946) Chemical production of mutations. Nature 157:202
Augustin M, Sedlmeier R, Peters T, Huffstadt U, Kochmann E, Simon D, Schöniger M, Garke-Mayerthaler S, Laufs J, Mayhaus M, Franke S, Klose M, Graupner A, Kurzmann M, Zinser C, Wolf A, Voelkel M, Kellner M, Kilian M, Seelig S, Koppius A, Teubner A, Korthaus D, Nehls M, Wattler S (2005) Efficient and fast targeted production of murine models based on ENU mutagenesis. Mamm Genome 16:405–413
Beier D (2000) Sequence-based analysis of mutagenized mice. Mamm Genome 11:594–597
Beutler B, Du X, Xia Y (2007) Precis on forward genetics in mice. Nat Immunol 8:659–664
Bode VC (1984) Ethylnitrosourea mutagenesis and the isolation of mutant alleles for specific genes located in the T region of mouse chromosome 17. Genetics 108:457–470
Bode VC, McDonald JD, Guénet JL, Simon D (1988) hph-1: a mouse mutant with hereditary hyperphenylalaninemia induced by ethylnitrosourea mutagenesis. Genetics 118:299–305
Carlson CM, Dupuy AJ, Fritz S, Roberg-Perez KJ, Fletcher CF, Largaespada DA (2003) Transposon mutagenesis of the mouse germline. Genetics 165:243–256
Chakrabarti L, Neal JT, Miles M, Martinez RA, Smith AC, Sopher BL, La Spada AR (2006) The Purkinje cell degeneration 5 J mutation is a single amino acid insertion that destabilizes Nna1 protein. Mamm Genome 17:103–110
Chapman VM, Miller DR, Armstrong D, Caskey CT (1989) Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc Natl Acd Sc USA 86:1292–1296
Charles DJ, Pretsch W (1987) Linear dose-response relationship of erythrocyte enzyme-activity mutations in offspring of ethylnitrosourea-treated mice. Mutat Res 176:81–91
Coghill EL, Hugill A, Parkinson N, Davison C, Glenister P, Clements S, Hunter J, Cox RD, Brown SD (2002) A gene-driven approach to the identification of ENU mutants in the mouse. Nat Genet 30:255–256
Concepcion D, Seburn KL, Wen G, Frankel WN, Hamilton BA (2004) Mutation rate and predicted phenotypic target sizes in ethylnitrosourea-treated mice. Genetics 168:953–959
Copeland NG, Jenkins NA (2010) Harnessing transposons for cancer genes discovery. Nat Rev Cancer 10:696–706
Cox RD, Hugill A, Shedlovsky A, Noveroske JK, Best S, Justice MJ, Lehrach H, Dove WF (1999) Contrasting effects of ENU induced embryonic lethal mutations of the quaking gene. Genomics 57:333–341
Cutler G, Kassner PD (2008) Copy number variation in the mouse genome: implications for the mouse as a model organism for human disease. Cytogenet Genome Res 123:297–306
Favor J (1986) The frequency of dominant cataract and recessive specific-locus mutations in mice derived from 80 or 160 mg ethylnitrosourea per kg body weight treated spermatogonia. Mutat Res 162:69–80
Favor J (1994) Specific-locus mutations tests in germ cells of the mouse: an assessment of the screening procedures and the mutational events detected. In: Mattison DR, Olsham AF (eds) Male-mediated developmental toxicity. Plenum Press, New York, pp 23–36
Favor J, Sund M, Neuhauser-Klaus A, Ehling UH (1990) A dose-response analysis of ethylnitrosourea-induced recessive specific-locus mutations in treated spermatogonia of the mouse. Mutat Res 231:47–54
Fernandez-Gonzalez A, La Spada AR, Treadaway J, Higdon JC, Harris BS, Sidman RL, Morgan JI, Zuo J (2002) Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science 295:1904–1906
Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, Pethiyagoda CL, Stuve LL, Johnson FM, Daly MJ, Wade CM, Cox DR (2007) A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature 448:1050–1053
Furushima K, Jang CW, Chen DW, Xiao N, Overbeek PA, Behringer RR (2012) Insertional mutagenesis by a hybrid piggyBac and sleeping beauty transposon in the rat. Genetics 192:1235–1248
Gilman JG (1972) Hemoglobin beta chain structural variation in mice: evolutionary and functional implications. Science 178:873–874
Gondo Y (2008) Trends in large-scale mouse mutagenesis: from genetics to functional genomics. Nat Rev Genet 9:803–810
Gondo Y, Fukumura R, Murata T, Makino S (2010) ENU-based gene-driven mutagenesis in the mouse: a next-generation gene-targeting system. Exp Anim 59:537–548
Graur D (2003) Single-base mutation—in nature encyclopedia of the Human Genome Macmillan Publishers Ltd
Green EL, Roderick TH (1966) Radiation genetics. In: Green EL (ed) Biology of the laboratory mouse. Dover Publications, New York, pp 165–185
Hitotsumachi S, Carpenter DA, Russell WL (1985) Dose-repetition increases the mutagenic effectiveness of N-ethyl-N-nitrosourea in mouse spermatogonia. Proc Ntl Acd Sc USA 82:6619–6621
Hoebe K, Beutler B (2005) Unraveling innate immunity using large scale N-ethyl-N-nitrosourea mutagenesis. Tissue Antigens 65:395–401
Hrabe de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, Marschall S, Heffner S, Pargent W, Wuensch K, Jung M, Reis A, Richter T, Alessandrini F, Jakob T, Fuchs E, Kolb H, Kremmer E, Schaeble K, Rollinski B, Roscher A et al (2000) Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet 25:444–447
Ivics Z, Hackett PB, Plasterk RH, Izsvák Z (1997) Molecular reconstruction of sleeping beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91:501–510
Ivics Z, Izsvák Z, Chapman KM, Hamra FK (2011) Sleeping beauty transposon mutagenesis of the rat genome in spermatogonial stem cells. Methods 53:356–365
Johnson FM, Lewis SE (1981) Mutation-rate determinations based on electrophoretic analysis of laboratory mice. Mutat Res 82:125–135
Justice MJ, Bode VC (1986) Induction of new mutations in a mouse t-haplotype using ethylnitrosourea mutagenesis. Genet Res 47:187–192
Justice MJ, Bode VC (1990) ENU-induced allele of brachyury (Tkt1) exhibits a developmental lethal phenotype similar to the original brachyury (T) mutation. J Exp Zool 254:286–295
Justice MJ, Zheng B, Woychik RP, Bradley A (1997) Using targeted large deletions and high-efficiency N-ethyl-N-nitrosourea mutagenesis for functional analyses of the mammalian genome. Methods 13:423–436
Justice MJ, Noveroske JK, Weber JS, Zheng B, Bradley A (1999) Mouse ENU mutagenesis. Hum Mol Genet 8:1955–1963
Justice MJ, Carpenter DA, Favor J, Neuhauser-Klaus A, Hrabé de Angelis M, Soewarto D, Moser A, Cordes S, Miller D, Chapman V, Weber JS, Rinchik EM, Hunsicker PR, Russell WL, Bode VC (2000) Effects of ENU dosage on mouse strains. Mamm Genome 11:484–488
Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellåker C, Whitley H, Cleak J, Janowitz D, Hernandez-Pliego P, Edwards A, Belgard TG, Oliver PL, McIntyre RE, Bhomra A, Nicod J, Gan X, Yuan W, van der Weyden L, Steward CA, Bala S, Stalker J, Mott R, Durbin R, Jackson IJ, Czechanski A, Guerra-Assunção JA, Donahue LR, Reinholdt LG, Payseur BA, Ponting CP, Birney E, Flint J, Adams DJ (2011) Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477:289–294
Keays DA, Clark TG, Flint J (2006) Estimating the number of coding mutations in genotypic- and phenotypic-driven N-ethyl-N-nitrosourea (ENU) screens. Mamm Genome 17:230–238
Kiernan AE, Erven A, Voegeling S, Peters J, Nolan P, Hunter J, Bacon Y, Steel KP, Brown SDM, Guénet JL (2002) ENU mutagenesis reveals a highly mutable locus on mouse chromosome 4 that affects ear morphogenesis. Mamm Genome 13:142–148
Kile BT, Hentges KE, Clark AT, Nakamura H, Salinger AP, Liu B, Box N, Stockton DW, Johnson RL, Behringer RR, Bradley A, Justice MJ (2003) Functional genetic analysis of mouse chromosome 11. Nature 425:81–86
Krawczak M, Ball EV, Cooper DN (1998) Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am J Hum Genet 63:474–488
Kumar S, Subramanian S (2002) Mutation rates in mammalian genomes. Proc Natl Acad Sv USA 99:803–808
Largaespada DA (2009) Transposon mutagenesis in mice. Meth Mol Biol 530:379–390
Lewis MA, Quint E, Glazier AM, Fuchs H, De Angelis MH, Langford C, van Dongen S, Abreu-Goodger C, Piipari M, Redshaw N, Dalmay T, Moreno-Pelayo MA, Enright AJ, Steel KP (2009) An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat Genet 41:614–618
Lewis SE (1991) The biochemical specific-locus test and a new multiple-endpoint mutation detection system: considerations for genetic risk assessment. Environ Mol Mut 18:303–306
Lewis SE, Barnett LB, Sadler BM, Shelby MD (1991) ENU mutagenesis in the mouse electrophoretic specific-locus test, 1. Dose-response relationship of electrophoretically-detected mutations arising from mouse spermatogonia treated with ethylnitrosourea. Mutat Res 249:311–315
Lewis SE, Barnett LB, Shelby MD (1992) ENU mutagenesis in the mouse electrophoretic specific locus test. 2. Mutational studies of mature oocytes. Mutat Res 296:129–133
Liu SM, Leibel RL, Chua SC Jr (1998) Partial duplication in the Leprdb-Pas mutation is a result of unequal crossing over. Mamm Genome 9:780–781
Lu B, Geurts AM, Poirier C, Petit DC, Harrison W, Overbeek PA, Bishop CE (2007) Generation of rat mutants using a coat color-tagged sleeping beauty transposon system. Mamm Genome 8:338–346
Marshall RR, Raj AS, Grant FJ, Heddle JA (1983) The use of two-dimensional electrophoresis to detect mutations induced in mouse spermatogonia by ethylnitrosourea. Can J Genet Cytol 25:457–466
Martin N, Jaubert J, Gounon P, Salido E, Haase G, Szatanik M, Guénet JL (2002) A missense mutation in Tbce causes progressive motor neuronopathy in mice. Nat Genet 32:443–447
Mashimo T, Ohmori I, Ouchida M, Ohno Y, Tsurumi T, Miki T, Wakamori M, Ishihara S, Yoshida T, Takizawa A, Kato M, Hirabayashi M, Sasa M, Mori Y, Serikawa T (2010) A missense mutation of the gene encoding voltage-dependent sodium channel (Nav1.1) confers susceptibility to febrile seizures in rats. J Neurosci 30:5744–5753
Massironi SM, Reis BL, Carneiro JG, Barbosa LB, Ariza CB, Santos GC, Guénet JL, Godard AL (2006) Inducing mutations in the mouse genome with the chemical mutagen ethylnitrosourea. Braz J Med Biol Res 39:1217–1226
McDonald JD (1995) Using high-efficiency mouse germline mutagenesis to investigate complex biological phenomena: genetic diseases, behavior, and development. Proc Soc Exp Biol Med 209:303–308
McDonald JD, Trischler M, Stoorvogel W, Ullrich O (1994) The PKU mouse project: its history, potential and implications. Acta Paediatr Suppl 407:122–123
Menalled LB, Chesselet MF (2002) Mouse models of Huntington’s disease. Trends Pharmacol Sci 23:32–39
Michaud EJ, Culiat CT, Klebig ML, Barker PE, Cain KT, Carpenter DJ, Easter LL, Foster CM, Gardner AW, Guo ZY, Houser KJ, Hughes LA, Kerley MK, Liu Z, Olszewski RE, Pinn I, Shaw GD, Shinpock SG, Wymore AM, Rinchik EM, Johnson DK (2005) Efficient gene-driven germ-line point mutagenesis of C57BL/6 J mice. BMC Genom 6:164
Mülhardt C, Fischer M, Gass P, Simon-Chazottes D, Guénet JL, Kuhse J, Betz H, Becker CM (1994) The spastic mouse: aberrant splicing of glycine receptor beta subunit mRNA caused by intronic insertion of L1 element. Neuron 13:1003–1015
Muller HJ (1927) Artificial transmutation of the gene. Science 66:84–87
Nadeau JH, Singer JB, Matin A, Lander ES (2000) Analysing complex genetic traits with chromosome substitution strains. Nat Genet 24:221–225
Nolan P, Peters J, Strivens M, Rogers D, Hagan J, Spurr N, Gray IC, Vizor L, Brooker D, Whitehill E, Washbourne R, Hough T, Greenaway S, Hewitt M, Liu X, McCormack S, Pickford K, Selley R, Wells C, Tymowska-Lalanne Z et al (2000) A systematic genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat Genet 25:440–443
Noveroske JK, Weber JS, Justice MJ (2000) The mutagenic action of N-ethyl-N-nitrosourea in the mouse. Mamm Genome 11:478–483
Oakberg EF, Crosthwait CD (1983) The effect of ethyl-, methyl- and hydroxyethyl-nitrosourea on the mouse testis. Mutat Res 108:337–344
Perez CJ, Jaubert J, Guénet JL, Barnhart KF, Ross-Inta CM, Quintanilla VC, Aubin I, Brandon JL, Otto NW, DiGiovanni J, Gimenez-Conti I, Giulivi C, Kusewitt DF, Conti CJ, Benavides F (2010) Two hypomorphic alleles of mouse Ass1 as a new animal model of citrullinemia type I and other hyperammonemic syndromes. Am J Pathol 177:1958–1968
Perez CJ, Dumas A, Vallières L, Guénet JL, Benavides F (2013) Several classical mouse inbred strains, including DBA/2, NOD/Lt, FVB/N, and SJL/J, carry a putative loss-of-function allele of Gpr84. J Hered 104:565–571
Peters J, Ball ST, Andrews SJ (1986) The detection of gene mutations by electrophoresis, and their analysis. Prog Clin Biol Res 209B:367–374
Pillers DA, Weleber RG, Green DG, Rash SM, Dally GY, Howard PL, Powers MR, Hood DC, Chapman VM, Ray PN, Woodward WR (1999) Effects of dystrophin isoforms on signal transduction through neural retina: genotype-phenotype analysis of Duchenne muscular dystrophy mouse mutants. Mol Genet Metab 66:100–110
Pretsch W, Favor J, Lehmacher W, Neuhauser-Klaus A (1994) Estimates of the radiation-induced mutation frequencies to recessive visible, dominant cataract and enzyme-activity alleles in germ cells of AKR, BALB/c, DBA/2 and (102xC3H)F1 mice. Mutagenesis 9:289–294
Quint E, Steel KP (2003) Use of mouse genetics for studying inner ear development. Curr Top Dev Biol 57:45–83
Quwailid MM, Hugill A, Dear N, Vizor L, Wells S, Horner E, Fuller S, Weedon J, McMath H, Woodman P, Edwards D, Campbell D, Rodger S, Carey J, Roberts A, Glenister P, Lalanne Z, Parkinson N, Coghill EL, McKeone R, Cox S, Willan J, Greenfield A, Keays D, Brady S, Spurr N, Gray I, Hunter J, Brown SDM, Cox RD (2004). A gene-driven ENU-based approach to generating an allelic series in any gene. Mamm Genome 15:585–591
Rinchik EM, Carpenter DA (1999) N-ethyl-N-nitrosourea mutagenesis of a 6- to 11-cM subregion of the Fah-Hbb interval of mouse chromosome 7: Completed testing of 4557 gametes and deletion mapping and complementation analysis of 31 mutations. Genetics 152:373–383
Runck AM, Moriyama H, Storz JF (2009) Evolution of duplicated β-globin genes and the structural basis of hemoglobin isoform differentiation in Mus. Mol Biol Evol 11:2521–2532
Runkel F, Hintze M, Griesing S, Michels M, Blanck B, Fukami K, Guénet JL, Franz T (2012) Alopecia in a viable phospholipase C delta 1 and phospholipase C delta 3 double mutant. PLoS ONE 7(6):e39203
Russell LB, Russell WL (1996) Spontaneous mutations recovered as mosaics in the mouse specific-locus test. Proc Natl Acad Sci USA 93:13072–13077
Russell LD, Ettlin RA, SinhaHikim AP, Clegg ED (1990) Histological and histopathological evaluation of the testis. Cache River Press, Clearwater
Russell WL (1962) An augmenting effect of dose fractionation on radiation-induced mutation rate in mice. Proc. National Acad. Sc. USA 48:1724–1728
Russell WL (1963) The effect of radiation dose rate and fractionation on mutation in mice. In: Sobels F (ed) Repair from genetic radiation damage, vol 4. Pergamon Press, New York, p 205–217
Russell WL, Kelly EM, Hunsicker PR, Bangham JW, Maddux SC, Phipps EL (1979) Specific locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proc Ntl Acad Sc USA 76:5818–5819
Russell WL, Hunsicker PR, Carpenter DA, Cornett CV, Guinn GM (1982a) Effect of dose fractionation on the ethylnitrosourea induction of specific-locus mutations in mouse spermatogonia. Proc Ntal acad Sc USA 79:3592–3593
Russell WL, Hunsicker PR, Raymer GD, Steele MH, Stelzner KF, Thompson HM (1982b) Dose—response curve for ethylnitrosourea-induced specific-locus mutations in mouse spermatogonia. Proc Natl Acad Sc USA 79:3589–3591
Sakuraba Y, Sezutsu H, Takahasi KR, Tsuchihashi K, Ichikawa R, Fujimoto N, Kaneko S, Nakai Y, Uchiyama M, Goda N, Motoi R, Ikeda A, Karashima Y, Inoue M, Kaneda H, Masuya H, Minowa O, Noguchi H, Toyoda A, Sakaki Y, Wakana S, Noda T, Shiroishi T, Gondo Y (2005) Molecular characterization of ENU mouse mutagenesis and archives. Biochem Biophys Res Commun 336:609–616
Schlager G, Dickie MM (1966) Spontaneous mutation rates at five coat-color loci in mice. Science 151:205–206
Schlager G, Dickie MM (1967) Spontaneous mutation and mutation rates in the house mouse. Genetics 57:319–330
Schmezer P, Eckert C (1999) Induction of mutations in transgenic animal models: BigBlue and Muta Mouse. Int Agency Res Cancer-Res Publ 146:367–394
Shedlovsky A, Guénet JL, Johnson LL, Dove WF (1986) Induction of recessive lethal mutations in the T/t-H-2 region of the mouse genome by a point mutagen. Genet Res 47:135–142
Shedlovsky A, King TR, Dove WF (1988) Saturation germline mutagenesis of the murine t region including a lethal allele at the quaking locus. Proc Ntl Acad Sc USA 85:180–184
Simon-Chazottes D, Tutois S, Kuehn M, Evans M, Bourgade F, Cook S, Davisson MT, Guénet JL (2006) Mutations in the gene encoding the low-density lipoprotein receptor LRP4 cause abnormal limb development in the mouse. Genomics 87:673–677
Stoye JP, Fenner S, Greenoak GE, Moran C, Coffin JM (1988) Role of endogenous retroviruses as mutagens: the hairless mutation of mice. Cell 54:383–391
Takahasi KR, Sakuraba Y, Gondo Y (2007) Mutational pattern and frequency of induced nucleotide changes in mouse ENU mutagenesis. BMC Mol Biol 8:52
Takeda J, Keng VW, Horie K (2007) Germline mutagenesis mediated by Sleeping Beauty transposon system in mice. Genome Biol 8(Suppl 1):S14
Takeda J, Izsvák Z, Ivics Z (2008) Insertional mutagenesis of the mouse germline with sleeping beauty transposition. Meth Mol Biol 435:109–125
Van Zeeland AA, Mohn GR, Mullenders LH, Natarajan AT, Nivard M, Simons JW, Venema J, Vogel EW, Vrieling H, Zdzienicka MZ et al (1989) Relationship between DNA-adduct formation, DNA repair, mutation frequency and mutation spectra. Annali dell’Instituto superiore di sanita (Ann 1st Super Sanita) Istituto Superiore di Sanita (ISDIS) 2003 25:223–228
Vogel EW, Natarajan AT (1995) DNA damage and repair in somatic and germ cells in vivo. Mutat Res 330:183–208
Vogel F, Rohrborn G (1970) Chemical mutagenesis in mammals and man. Springer, New York, p 519
Wahnschaffe U, Bitsch A, Kielhorn J, Mangelsdorf I (2005a) Mutagenicity testing with transgenic mice. Part I: Comparison with the mouse bone marrow micronucleus test. J Carcinog 4:3
Wahnschaffe U, Bitsch A, Kielhorn J, Mangelsdorf I (2005b) Mutagenicity testing with transgenic mice. Part II: comparison with the mouse spot test. J Carcinog 4:4
Wilsbacher LD, Sangoram AM, Antoch MP, Takahashi JS (2000) The mouse clock locus: sequence and comparative analysis of 204 kb from mouse chromosome 5. Genome Res 10:1928–1940
Yang H, Wang JR, Didion JP, Buus RJ, Bell TA, Welsh CE, Bonhomme F, Yu AH, Nachman NW, Pialek J et al (2011) Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet 43:648–655
Youssoufian H, Antonarakis SE, Bell W, Griffin AM, Kazazian HH Jr (1988) Nonsense and missense mutations in hemophilia A: estimate of the relative mutation rate at CG dinucleotides. Am J Hum Genet 42:718–25
Zheng B, Sage M, Cai WW, Thompson DM, Tavsanli BC, Cheah YC, Bradley A (1999) Engineering a mouse balancer chromosome. Nat Genet 22:375–378
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Guénet, JL., Benavides, F., Panthier, JJ., Montagutelli, X. (2015). Mutations and Experimental Mutagenesis. In: Genetics of the Mouse. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-44287-6_7
Download citation
DOI: https://doi.org/10.1007/978-3-662-44287-6_7
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44286-9
Online ISBN: 978-3-662-44287-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)