
Abstract
Blumeria graminis causes powdery mildew disease on several important small grain cereals and several grasses. These ascomycete fungi, like all members of the Erysiphales (Leotiomycetes), are obligate biotrophs. They have a distinct life-cycle characterized by a mainly epiphytic growth habit on the epidermis of leaves and stems. The fungi penetrate the epidermal cells only to form haustoria surrounded by a host membrane. The Blumerias occur as distinct formae speciales that only infect one host species. The polycyclic asexual phase is most commonly observed; here the airborne conidia, which form the eponymous “powder”, are responsible for the dispersal and infection. The rarer sexual cycle usually occurs once a year, at the end of the host growing season. The size of the Blumeria genomes, over 120 Mb, is much larger than those of related ascomycetes. This is due to an extraordinary expansion of the retrotransposons evident as repetitive DNA. At the same time, the protein coding gene complement is reduced to about 6500: gene families are smaller, there are fewer paralogs; some metabolic pathways are missing altogether, notably those responsible for nitrate and sulfate metabolism. In the face of this large scale reduction in gene number, we observe a massive expansion of genes encoding small secreted proteins that are candidate effectors of pathogenicity. Understanding the role played by these candidate effectors, which constitute over 8 % of the total genes, is a challenge for research in the coming years.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
7.1 Introduction
7.1.1 Agricultural Relevance
Powdery mildews are regarded as some of the most common plant diseases. They are caused by fungi that infect a wide variety of hosts including food crops (cereals, fruit, and vegetables) and ornamentals. The disease is easily recognized because of the abundant white/off white conidia on the surface of infected leaves, stems, and flowers (Fig. 7.1a). The airborne conidia form a dry “powder”—hence their name (Glawe 2008). Their importance in agriculture is due to their ubiquity and the impact they have on productivity. Although they do not kill their host, or produce toxic metabolites, they reduce yields and the quality of produce to levels that render the crops economically unviable. In extreme cases, rapid epidemics can result in total crop loss.

Life cycle of the barley powdery mildew fungus, Blumeria graminis f. sp. hordei. a Heavily infected barley displaying masses of asexual spores that are produced on all leaf surfaces and appear as an off-white powder. The dry powder is easily dispersed by air. b Asexual spore (conidium) 30 min after germination on a solid surface (glass in this case); bar: 20 µm. The primary germ tube (arrow) is a structure with a determinate growth. c On a leaf, the secondary germ tube develops a slightly swollen and hooked appressorium (visible here 8 h after inoculation, arrow). The appressorium is separated from the secondary germ tube by a septum. The fungal structures are stained here with a lectin (wheat germ agglutinin, WGA) coupled to a fluorescent dye (Alexa 488). d The host plant responds to the activity of both the primary tubes and appressoria by producing a papilla (arrow), visible here as a trypan-blue positive apposition below the fungal structures, 24 h after inoculation. e Two days after inoculation, some of the attempted infections will succeed and a haustorium will develop inside the plant’s epidermal cell (arrow). This feeding structure takes up nutrients from the plant and enables the fungal hyphae to start expanding on the outer surface of the leaves. Note the absence of a clear papilla in correspondence of the successful penetration, whereas a papilla is still visible below the primary germ tube. f An isolated haustorium (stained with WGA-Alexa 488). The perihaustorial membrane (arrow) is a plant-derived membrane continuous with, but distinct from the plasmalemma. g Three days after inoculation, the epiphytic hyphae are visible on the leaf surface (low magnification scanning electron micrograph). Secondary appressoria (arrows) enable the fungus to penetrate further cells and develop more haustoria. h Epidermal cells that subtend a mature colony may accommodate more than one haustorium. Note that the plant cells remain alive at this time. It is assumed that the host immune system is very effectively suppressed at this stage. i Four days after inoculation, foot cells (arrow) develop and grow at about right angles to the surface hyphae. These foot cells are the base of the conidiophores that produce chains of conidia acropetally
The powdery mildews of cereals including wheat, barley, oats, and rye are caused by Blumeria graminis. All of these are agriculturally relevant, but the global importance of wheat and barley and their susceptibility to mildews are the reasons why B. graminis is regarded as one of the top fungal diseases of plants (Dean et al. 2012), and is why this fungus was the first powdery mildew whose genome was fully sequenced and annotated. B. graminis is currently the reference and model for research in the biology of the powdery mildews fungi that infect other hosts such as grapevine, cucurbits, strawberries, roses, and plantain.
In temperate regions with relatively high rainfall, powdery mildews are endemic and ubiquitous. In wheat and barley, protection from this disease is achieved by the combined use of fungicides and resistant host varieties. Genetic resistance in wheat and barley can be determined by “classical”, dominant, resistance (R) genes that conform to typical gene-for-gene interactions. Typically, deployment of varieties carrying such R genes is accompanied by rapid evolution of new virulent powdery mildew strains, which breaks down the conferred protection (Wolfe and McDermott 1994). One notable exception is resistance controlled by the mlo genes in barley (Jørgensen 1992). Mlo genes are different; resistance is determined by recessive alleles and, importantly, so far has turned out to be essentially durable in the field. It has been proposed that developing resistance based on mlo homologs in other plants may be a useful strategy for crop improvement and prevention against additional diseases (Acevedo-Garcia et al. 2014).
7.1.2 Taxonomic Position
Powdery mildew fungi are Ascomycetes and all belong to the order Erysiphales of the class Leotiomycetes. As such, they are closely allied to Botrytis and Sclerotinia . This affinity is amply confirmed by the similarities between sequences of the protein coding genes. Surprisingly, the obligate biotrophic life cycles and infection strategies of the Erysiphales are in some ways diametrically opposite of those of the necrotrophs Botrytis and Sclerotinia. Comparative genomics between groups that are at the extremes of the trophic spectrum, but are taxonomically proximal, reveal fundamental insights into the genetic basis for the various life strategies (Spanu 2012).
The powdery mildews are thought to have originated over a 100 million years (Myr) ago, in the Cretaceous, about the same time as the expansion and the explosive diversification of flowering plants. Within this group, the genus Blumeria diverged as a monophyletic line around 60 Myr ago (Takamatsu 2004). More recent diversification and evolution of B. graminis into host-specialized formae speciales is likely to have occurred over the last 10 Myr, coincidentally with the diversification and expansion of the host cereal plants (Oberhaensli et al. 2011). Data from genome sequencing contributes toward understanding these evolutionary events. This is particularly so with regard to the most recent evolution of virulence associated with agriculture in prehistoric times (Wicker et al. 2013; Hacquard et al. 2013).
7.1.3 Life Cycle
Like all powdery mildews, B. graminis is an obligate biotroph. This means that it requires a living host to grow, develop, and complete a life cycle. The life-cycle is simple when compared to some other fungal pathogens.
The most commonly observed cycles are asexual, which represent the principle driver of epidemics. They begin when an ascospore or a conidiospore (conidium) lands on an appropriate host. Germination is rapid and, unusually for plant pathogenic fungi, happens in the absence of liquid water. A primary germ tube emerges from the conidium a few minutes after touching a solid surface (Fig. 7.1b). On a plant, growth of the primary germ tube stops and is limited to a few µm in length. It is thought that the main role of this structure is surface sensing (Carver et al. 1995; Yamaoka et al. 2006). Shortly after the primary germ tube stops elongating, a second germ tube emerges. On a host plant, the secondary germ tube differentiates an elongated, slightly swollen, and often hooked appressorium , which is clearly visible 4–8 h after germination (Fig. 7.1c). Many studies have investigated the role played by surface molecules in determining development in B. graminis (reviewed in Both and Spanu 2004). In the absence of a plant or appropriate artificial signals, the conidia produces abortive and unusual developmental programs. For example, on glass many short germ tubes similar to primary germ tubes are formed; on water agar, the germ tubes continue to grow into untypically long hyphae, which eventually stop growing, possibly because of the inability to process nutrients. In the presence of a plant, the appressorium develops a penetration peg. This peg, utilizes a combination of turgor pressure and plant cell-wall degrading enzymes to facilitate penetration through the cell wall of the epidermis (Pryce-Jones et al. 1999). Commonly, the plant produces a papilla (Fig. 7.1d) and further penetration is restricted. Even on fully compatible host plants, only a small percentage of the penetrations are successful and lead to an established infection.
Once the hypha penetrates through the cell wall, the fungus differentiates a haustorium (Fig. 7.1e). The haustorium is surrounded by a membrane (Fig. 7.1f, arrow) that is continuous with the plasmamembrane, but has distinct properties (Hückelhoven and Panstruga 2011). The haustorium is the only structure likely to take up nutrients in any significant way. It is clear that there are processes that actively suppress host defense at this stage, and it is not unusual to observe multiple penetrations of a single host epidermis cell (Fig. 7.1h).
After a functional haustorium is established, the fungus continues to develop hyphae on the epidermal surface. These hyphae are usually visible 2 days after inoculation and develop small secondary appressoria (Fig. 7.1g, arrow) from which further penetration pegs and haustoria are formed. About 3–5 days from inoculation, “foot cells” develop on the epiphytic structures, from which the asexual conidiophores grow perpendicularly to the surface of the epidermis (Fig. 7.1i). Asexual conidia are produced acropetally on the conidiophores. At this stage, the colonies become visible to the naked eye, then expand and start producing abundant conidia. The conidia are dispersed by air currents and no liquid water is necessary. The asexual cycles are therefore short and may be repeated many times throughout the season of the host’s growth, and thus, can support epidemics that spread extremely fast. The capacity to produce masses of conidia to drive swift, airborne epidemics cannot be overstated: one calculation estimated that up to 1019 conidia were produced per month during one such epidemic in Europe (Wolfe and McDermott 1994).
The sexual cycle usually occurs at the end of the host’s life, when the leaves senesce and dry. Sexual compatibility in B. graminis is determined by one of two alleles at a relatively simple mating-type locus (Brewer et al. 2011). When two individuals of opposite mating types grow in proximity to one another, hyphae fuse and a chasmothecium (fruiting body) develops. Karyogamy, meiosis and ascogenesis take place inside these structures. Sexual recombination takes place at this stage. The chasmothecia can act as long-lived resting structures, capable of surviving for long periods in inclement conditions (overwintering or “oversummering”). In mild, humid weather, they break open and the ascospores are liberated, disperse, and can go on to infect a new host.
In some environments, it is possible for B. graminis to persist in conditions when the main crop is not grown, by infecting volunteer plants that act as “green bridges” (Liu et al. 2012). In these conditions, the sexual cycle is not necessary for survival and propagation of infection. It is probable that in the agricultural settings of temperate regions (such as Europe) where hosts are grown both in spring and winter, most of the propagation of powdery mildews is actually asexual. This is now confirmed by the first comparative genomic re-sequencing studies (see below) (Wicker et al. 2013; Hacquard et al. 2013).
7.1.4 Host Range
Some powdery mildew fungi are polyphagous generalists and are capable of infecting a wide variety of dicotyledonous hosts (Jones et al. 2001). Unlike these, B. graminis displays very narrow host range: it infects only some Pooideae, a sub family of the Poaceae (the true grasses). Moreover, within the B. graminis species, eight formae speciales, which only infect one host species, have been observed (Hiura 1978). For example B. graminis f. sp. hordei only infects barley, whereas B. graminis f. sp. tritici grows exclusively on wheat. These narrow host specificities are genetically determined and, although it has been possible to cross some of these in the laboratory, the progeny do not appear to be very viable and these events do not occur commonly in the wild (Walker et al. 2011). Genetic and cytological analyses of the events that follow infection indicate that adapted formae speciales induce a form of short range susceptibility (Olesen et al. 2003) in the host cells and those in the immediate vicinity (Olesen et al. 2003).
7.1.5 Genetic Tractability and Functional Genomics
The fact that it is possible to obtain crosses between individuals with appropriate mating types , has enabled some significant application of classical genetics. This, however, is very laborious and requires long-term experiments given the constraints imposed by obligate biotrophy and the fact that the sexual cycle is much longer than the asexual one (months, as opposed to days). These analyses have been employed with some degree of success to create genetic maps (Pedersen et al. 2002a) and to identify avirulence genes in B. graminis f. sp. hordei (Skamnioti et al. 2008).
At present, powdery mildews are not readily transformable. In spite of some reports of successful expression of GUS reporter genes in B. graminis (Christiansen et al. 1995; Chaure et al. 2000), these findings could not be reproduced reliably enough to be used effectively in practice. It is not clear exactly what the limiting factors that cause these difficulties are. The frequency of transformation is low and, although expression of heterologous reporter genes such as GFP is possible (James K Brown and Alejandro Perez-Garcia, personal communication), the transgenes appear to be relatively unstable and they are lost after many rounds of subculture. These difficulties are clearly a stumbling block and have imposed some limits on the application of functional genomic techniques to powdery mildews.
Alternative approaches to manipulate gene expression for the functional analysis of genes in B. graminis have been more successful. The first well-established method is host-induced gene silencing (HIGS) (Douchkov et al. 2005; Dong et al. 2006; Nowara and Schweizer 2007; Nowara et al. 2010). In HIGS, fungal genes are targeted by the expression in the host of an inverted repeat RNA (separated by an intron) that interferes with mRNA function (RNAi). Typically, leaves are bombarded with particles coated with plasmid DNA that drive the production of RNAi . The production of fungal RNAi in the epidermal cells results in the down-regulation of target RNA in the fungus. Although the precise mechanisms for RNA transfer for host to pathogen are not clear, HIGS can be applied to various other plant-microbe interactions as well (Nunes and Dean 2012). A derivative of HIGS has even been used to modulate parasite gene expression in the plant-plant parasite systems (Bandaranayake and Yoder 2013). HIGS has now been applied with success to validate, experimentally, candidate effector genes first identified in studies of the proteome (Zhang et al. 2012b; Pliego et al. 2013).
Further, approaches to investigate the functionality of powdery mildew genes include virus induced gene silencing (VIGS) , virus gene over-expression (VOX), and the delivery of effectors by appropriate bacteria such as Xanthomonas spp. (Wise et al., in preparation).
It has been noted that all these methodologies rely on transient expression or delivery of genes and proteins in infected plants and that this may result in artifacts that are difficult to control. It may therefore be desirable to complement and confirm these studies using stable transgenic plants (Spanu and Panstruga 2012). Achieving stable transformation of the powdery mildew fungi in a reliable, reproducible manner at sufficient efficiency and frequency is still a highly desirable goal.
7.2 The Genome
Sequencing the genome of an obligate biotrophic pathogen posed various challenges. Some of these challenges were predictable, others were not; some but not all were mastered. The first evident difficulty was obtaining sufficient DNA for sequencing and free of DNA from non-B. graminis sources. The quality and quantity of DNA required depended on the method eventually used for sequencing. For example, the first stages of the original B. graminis f. sp. hordei sequencing project relied on dideoxynucleotide (Sanger) sequencing, and therefore on the preparation of appropriate libraries in fosmid vectors. With the advent of “next generation” sequencing platforms (454 Pyrosequencing, Illumina, SOLiD), a reduce quantity and quality was adequate. In the case of B. graminis f. sp. tritici, a BAC tiling approach was used, which required relatively large DNA fragments as the starting material. Fortunately, sufficient material of adequate size could be obtained from conidia collected from infected leaves. For B. graminis, this material is relatively abundant, can be isolated in relatively pure form, with little or no DNA from host cells, or other microbes. Care is needed to remove unknown contaminating substances, which co-purify with DNA (and RNA) and inhibit the activity of many enzymes such as Taq polymerase, reverse transcriptase, restriction enzymes used in the down-stream molecular biological processes.
The next challenge, which was not predicted, was related to the genome size. At the start of the project, it was not known exactly how large the B. graminis genomes were. Assumptions were made, based on the values obtained for related ascomycetes, that envisaged values of 30–40 Mb; these very optimistic predictions assumed that, being an obligate pathogen, the B. graminis genome was likely to be smaller because of probable gene loss associated with obligate parasitism. The genomes are now known to be in excess of 120 Mb. Fortunately, the advent of next generation sequence platforms delivered very high coverage at relatively low cost.
The third hurdle was the exceptionally high proportion of repetitive DNA present in all powdery mildew genomes analyzed to date. Although it was known that B. graminis genomes contained repetitive DNA that originated from retrotransposons (Pedersen et al. 2002b), the scale of this and the difficulties posed to assembly were underestimated. As discussed later, this problem has not been solved yet.
In spite of the challenges, a number of strains of B. graminis from both formae speciales. hordei and tritici have now been sequenced, partially assembled and deeply annotated thanks to the joint efforts of the international community researching powdery mildews (Spanu et al. 2010; Hacquard et al. 2013; Wicker et al. 2013). The salient findings from these first projects are summarized here.
7.2.1 Genome Structure
It is quite remarkable that we are still uncertain about the overall structure of powdery mildews genomes. There are few published cytogenetic studies. Based on a combination of gel electrophoresis and microscopy of metaphase chromosomes, B. graminis f. sp. hordei was estimated to have at least seven chromosomes (Borbye et al. 1992), although this may be an underestimate. Genetic analyses have detected a number of linkage groups: the most detailed published map to date identifies 34 linkage groups (Pedersen et al. 2002a). Sequencing the genomes of B. graminis has not helped in this matter, because of the difficulties in assembling highly repetitive-rich sequences. The original sequence of B. graminis f. sp. hordei (Spanu et al. 2010) is highly fragmented into 6898 supercontigs. More recent attempts at assembling B. graminis f. sp. tritici genome have been facilitated by the use of tiled BAC libraries in addition to conventional “shot gun” sequencing (Wicker et al. 2013); however, even in this case there are still 250 BAC contigs representing 82 % of the genome. Ten sequences resembling conserved telomere ends (tandem repeats of TTAGGG motifs) were identified suggesting a minimum number of five chromosomes. However, the authors cautioned that this is likely an underestimate due to the difficulty in cloning telomeric end sequences. This issue currently remains unsolved and awaits improvements in the strategies for sequencing and assembling repetitive DNA.
Two recent comparative resequencing studies of various strains of both the B. graminis f. sp. hordei (Hacquard et al. 2013) and tritici (Wicker et al. 2013) genomes found that these genomes are made up of “mosaics” of monomorphic and polymorphic sequences. These findings support the view that extant populations of cereal mildews are the result of mainly clonal (asexual) propagation interspersed by rare sexual cycles that allow outbreeding. This is an important finding, because it underscores the likely importance of (retro)transposition and expansion of repetitive DNA in the generation of variation in these mildews.
7.2.2 Repetitive DNA
All the powdery mildew fungal genomes analyzed so far are exceptionally rich in repetitive DNA. Estimates vary depending on the methods used to calculate this and on the actual size of the genomes; in the wheat-infecting B. graminis f. sp. tritici, more than 90 % of genome is made of repetitive DNA (Wicker et al. 2013). In fact, powdery mildews are considered the genomes with the most repetitive DNA, to date.
Repetitive DNA is almost entirely derived from Class I (retro)transposons (Wicker et al. 2013; Spanu et al. 2010). The majority of these transposons are Long Interspersed Nuclear Elements (LINEs), followed by Long Terminal Repeat (LTR) retrotransposons. There are smaller numbers of Short Interspersed Nuclear Elements (SINEs) and only very few DNA based (Class II) transposons (Spanu et al. 2010; Parlange et al. 2011). The extraordinary wealth of repeats therefore appears to be largely due to retrotransposon activity. There is currently no indication of heightened retrotransposition activity per se in the mildew genomes. However, it is notable that Repeat Induced Point mutation (RIP) is absent in the mildews. RIP is one of the mechanisms that is known to be involved in controlling the proliferation of repetitive DNA in filamentous ascomycetes. None of the powdery mildew genomes analyzed so far appear to have any genes that encode key enzymes that promote RIP (Spanu et al. 2010); moreover, a systematic analysis of all repeats showed that there is very little evidence that RIP has ever happened except in very few instances within some unclassified, and possibly very old, repeat sequences (Joelle Amselem, in preparation). It is therefore possible that loss of RIP led to the accumulation of the repetitive DNA that is the product of transposon activity.
We have found that the repetitive DNA in the B. graminis genomes is homogeneously distributed throughout the genome, which does not have distinct regions with significantly different proportions of repetitive DNA/protein coding genes. This is in contrast with the Leptosphaeria maculans genome, where there are clear isochores of repetitive DNA interspersed with isochores of gene-rich DNA (Parlange et al. 2009). It also differs notably from the peculiar situation in Fusarium oxysporum, where repeat-rich “lineage specific” chromosomes are found (Ma et al. 2010).
A detailed comparative analysis of a few selected, but representative, loci in the B. graminis f. sp. hordei and B. graminis f. sp. tritici highlighted some noteworthy features (Oberhaensli et al. 2011). The protein coding genes are highly conserved and syntenic between the two formae specialis. In contrast, the repetitive rich interspersed regions (Fig. 7.2) are extremely diversified. The picture that emerges is that of “two-speed genomes”, with DNA that is relatively stable (protein coding genes) embedded into areas that are much more dynamic (the retrotransposons) and subject to relatively fast reshuffling, deletions, and additions.

Details of the synteny in two genomic loci in the B. graminis f. sp. tritici and B. graminis f. sp. hordei genomes. The protein coding genes (gray) are numbered and their direction of transcription is given by the arrows. Sequence conservation is shown by the gray connectors and the levels of sequence similarity indicated by the intensity of the shading. The retrotransposons are shown as white blocks. From these images, it is clear how the protein coding genes are highly conserved, whereas the vast majority of the transposons differ. These events result in genomes that appear to be made up of portions of DNA that evolves at two different speeds: the high-speed retro-elements, which form much of the repetitive DNA, and protein coding genes evolving at a lower speed (Courtesy of Simone Oberhaensli, modified from Oberhaensli, Parlange et al. (2011)
In other plant pathogens, there are many cases in which repeat- and transponson-rich regions are associated with effectors , i.e., genes responsible for modulating virulence through interactions with the host immunity and metabolism. This is true in the oomycetes (Raffaele and Kamoun 2012; Raffaele et al. 2010; Haas et al. 2009) and in fungi such as Fusarium (Ma et al. 2010) and Leptosphaeria (Parlange et al. 2009). In B. graminis f. sp. hordei, candidate effector genes appear to be commonly linked to certain retrotranspons (Pedersen et al. 2012). Direct association between a specific transposable element and a set of effector gene has been recently found in F. oxysporum where this association was used successfully to identify new effector genes (Schmidt et al. 2013). In all these examples, it has been suggested that the physical proximity of effector genes and transposable elements influences effector gene plasticity. The dynamic nature of the transposons enhances evolution of effectors. This, in turn, increases the ability of the pathogens to evolve in response to the evolution of host immunity. In the powdery mildews, these observations may help to explain the terms of the trade-off inherent with increasing genome size. That is, the cost of large genomes full of active retrotransponsons is paid with the “evolutionary currency” of more swiftly adaptable effectors (Spanu 2012). And the cost of large genomes with many active retrotransposons includes gene loss—as we see in the next section.
7.2.3 Protein-Coding Genes
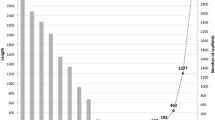
The genome of B. graminis f. sp. hordei was annotated by a combination of automated prediction and manual curation (Spanu et al. 2010). The latter was the result of a community-based activity, which produced a highly curated set of protein coding genes, supported by multiple lines of evidence. At the time of writing this chapter, 6470 and 6540 genes are identified in the barley powdery mildew (http://www.blugen.org/) and wheat powdery mildew genomes (Wicker et al. 2013), respectively. It is clear that the overall number of genes in these fungi is about half of those found in closely related ascomycetes, and more akin to the numbers in the hemiascomycete yeasts. In fact, if the respective genome sizes of related ascomycetes are taken into account, the overall protein-coding gene density of both sequenced B. graminis formae specialis is very much lower than the average of both eu- and hemiascomycetes (Fig. 7.3).

Density of the protein coding genes in the genomes of selected ascomycete fungi. On the left, a tree illustrates the relative taxonomic positions of the species (note the length of the tree branches have no significance in relation to taxonomic distance). The histogram on the left illustrates the gene density of the sequenced genomes. The average gene densities of the hemiascomycete genomes (black) and euascomycete genomes (green) are shown here. The gene density of the sequenced cereal powdery mildews (red) and of the truffle fungus, are clear outliers. This is the result both of their larger genomes and lower numbers of gene, as described in the text
Detailed analysis of the missing genes noted that this reduction can be attributed to: (1) the near absence of paralogs; (2) the reduction in the size of particular gene families; (3) the absence of genes encoding enzymes in some primary and secondary metabolic pathways. For example, the complement of polysaccharide degrading enzymes is drastically reduced in comparison to other ascomycetes (O’Connell et al. 2012), and there are only two genes encoding polyketide synthases and non-ribosomal peptide synthases (Spanu et al. 2010). A systematic analysis identified 99 conserved “ascomycete core genes ” missing in B. graminis. Interestingly, where tested, these genes were present and fully expressed during the biotrophic phase of the true hemibiotroph, Colletotrichum higginsianum: this led to the suggestion that the genes are not missing because they are detrimental for biotrophy (Spanu 2012).
An alternative explanation is that the missing genes are simply not needed for the powdery mildew lifestyle. For example, hydrophobins are ubiquitous in filamentous fungi (Wösten 2001) and are considered important in water-mediated dissemination of conidia (Whiteford and Spanu 2001) and traversing the challenging water-air barrier (Wösten and Willey 2000). Powdery mildews are unusual fungi, as they do not require liquid water for dissemination or germination of their spore/conidia, and this may explain the absence of hydrophobin genes. Genes encoding alcohol dehydrogenases are also absent from powdery mildews. Since powdery mildew fungi inhabit exclusively the aerial surfaces of plants and are consistently exposed to aerobic environments, they may have dispensed with genes that are essential for anaerobic metabolism.
7.2.4 Gene Loss Convergence in Biotrophs
When the first group of biotrophic plant pathogen genomes were sequenced and annotated, one aspect that caught our attention was the extraordinary similarity in the genes and pathways that were either lost or reduced when compared to hemibiotroph or non-pathogenic related species. For instance, similar reductions in polysaccharide degrading enzymes were also observed in the rusts (basidiomycetes, obligate biotrophic pathogens) (Duplessis et al. 2011) and downy mildew (oomycetes, obligate biotrophic pathogen) (Baxter et al. 2010); carbohydrate transporters are fewer in both B. graminis (Wicker et al. 2013) and P. graminis (basidiomycetes, obligate biotrophic pathogen) (Duplessis et al. 2011); lower numbers of genes encoding enzymes devoted to secondary metabolite synthesis was observed in B. graminis, Tuber melanosporum (ascomycete, non-obligate biotroph mutualistic symbiont) and Ustilago maydis (basidiomycete, non-obligate biotrophic pathogen) (Spanu et al. 2010).
The most striking instances of convergence are probably those related to the loss of entire metabolic pathways. The genes that encode enzymes needed for inorganic nitrate and inorganic sulfate assimilation are missing in B. graminis (Wicker et al. 2013; Spanu et al. 2010), the rusts (Duplessis et al. 2011), and the oomycete downy mildews (Baxter et al. 2010) and white rusts (Kemen et al. 2011). Both B. graminis and rusts also lack the same genes encoding enzymes involved aromatic amino acids synthesis and degradation (Fig. 7.4) (Wicker et al. 2013).

Examples of primary metabolic pathways missing in the cereal powdery mildews. a Aromatic amino acid catabolism/degradation. b Aromatic amino acid biosynthesis. c Organic sulfur metabolism, and biosynthesis of the key co-factor for sulfite reductase. The genes missing from B. graminis that encode the metabolic enzymes are shown as capital/large typeface. All the genes marked with an asterisk (*) are also missing in the rust genomes Puccinia graminis and P. triticina (modified from Wicker, Oberhaensli et al. (submitted))
The data suggest that some pathways, in particular those involved with primary metabolism , are not active in these biotrophs. The evident corollary for this is that rust and powdery mildew fungi obtain the respective nitrogen, sulfur and amino acids from their plant hosts. This raises the question of whether these microbes are obligate because they are auxotrophic for the metabolites in question. In fact, this interpretation is probably too simple; if this were true, it would be possible to culture the powdery mildew fungi in vitro simply by supplying complex media that contain aromatic aminoacids, and sources of organic nitrogen and sulfur. An alternative explanation is that obligate biotrophs may require complex signaling cues to regulate their residual metabolic pathways (Spanu 2006), and these cues are only obtainable from appropriate living host plants.
It is not clear why the same sets of genes are missing from unrelated biotrophs: clearly this is a striking example of convergent evolution . It could be that the costs of these particular metabolic steps are high enough that, given an abundant source of ready alternatives, there was an advantage to losing exactly these genes/proteins. This hypothesis remains to be tested.
7.2.5 Gene Amplification: Effector-like Proteins
Plant pathogens deploy effector proteins to manipulate host immunity and metabolism (Göhre and Robatzek 2008). The importance of genes encoding effectors and effector-like proteins in B. graminis is highlighted by the fact that, unlike most genes described in the previous sections, effector gene families are expanded, and some have expanded to a remarkable extent. For example, in B. graminis f. sp. hordei, 491 Candidate Secreted Effector Proteins (CSEPs) were identified and characterized (Pedersen et al. 2012). Overall, in barley powdery mildew the CSEPs account for over 7 % of the predicted protein coding genes. This number may actually represent an underestimate, because in that work we restricted the assignment to genes encoding proteins, which do not have evident orthologs in non-mildew fungi (as determined by absence of significant hits in BLAST searches). The CSEPs are all predicted to be secreted. In other studies, we have identified effector-like proteins by functional screening candidates that are specifically abundant in the haustoria (Pliego et al. 2013); these effectors include proteins that do have orthologs in other species; for example the BEC1019 effector is similar to proteases commonly found in other fungi. Furthermore, one of the features of many CSEPs is that they have been subject to very intense diversifying selection during the evolution of the mildews; this is evident in the very high non-synonymous/synonymous changes observed between paralogs (Pedersen et al. 2012). In B. graminis f. sp. tritici, this observation was used to identify a further 165 Candidate effector Proteins (CEPs) that are actually devoid of a conventional secretion signal, in addition to 437 CSEPs (Wicker et al. 2013).
The absence of evident sequence-determined orthologs notwithstanding, one subset of CSEP families includes proteins that are predicted to have a structural “fold” reminiscent of fungal RNAses (Pedersen et al. 2012). Most of the proteins are not likely to be enzymatically active RNAses because they lack conserved amino acid residues known to be required for hydrolase activity. Remarkably, they all appear to be descended from a single ancestral “proto-effector” gene. This gene was subjected to multiple rounds of duplication followed by very strong diversification. Only the predicted RNAse-like structural motifs are conserved overall. Two of the eight barley powdery mildew effectors verified functionally (BEC1011 and BEC1054) are part of this group (Pliego et al. 2013). Most of the CSEPs that are conserved in the powdery mildew pathogens of non-cereals are members of the RNAse-like families (Spanu et al. 2010; Tollenaere, personal communication). It therefore appears that these effectors proteins have been of central importance for the evolution of the whole Erysiphales clade.
The sheer number and diversity of CSEP and CEP genes, their specific expression in haustoria , and their very existence in the face of widespread gene loss elsewhere, underscores their centrality in the powdery mildews genome. Effector diversity is driven by coevolution with plant hosts that are themselves under pressure to detect effectors and use them as alarm triggers (Takken and Rep 2010). If this is true, though, the question remains why so many are actually maintained in the powdery mildew genomes, rather than simply jettisoned when the plant evolves to recognizing them. An illuminating insight into this conundrum is given by an exhaustive and in depth transcriptome-wide analysis of gene expression in B. graminis f. sp. hordei during early stages of infection (Hacquard et al. 2013). This study shows that there are specific groups of CSEP genes that are activated at specific stages of the early infection. Intriguingly, the effectors genes activated at the earliest stages, are also those that appear to have been under the strongest diversifying selection; in various cases, CSEPs belonging to the same families are expressed specifically at different times. This suggests that CSEPs expressed early in infection are devoted to challenging the plant’s immune system, and are under pressure to diversify by coevolutionary pressure. The CSEPs expressed later, in mature haustoria, may play roles in the maintenance of biotrophy (Hacquard et al. 2013).
7.3 Perspectives
The most pressing challenge posed by the B. graminis genomes is that of their fragmentation: assembly of the sequences has clearly failed to deliver anything that even approaches a truly finished status where all, or practically all, of the DNA in the genome is accounted for. This is important for a number of reasons.
We have seen here that, like other biotrophic plant pathogens, the powdery mildew fungi appear to encode fewer protein coding genes than related fungi. Some of these losses may be the result of convergent evolution and simply reflect functions that are no longer necessary in life restricted to a living plant. Other losses may be important in understanding fundamental principles underlying biotrophy or obligate biotrophy. The robustness of these conclusions in this area will be limited until we are certain beyond reasonable doubt that all the genome sequences are accounted for.
Without an accurate, detailed, and complete assembly, the map of the large scale genome structure is uncertain. The finished assembly of other fungal genomes has had significant impact on our understanding of the evolution of those plant pathogens. For example, in Fusarium it allowed the discovery of the effector-rich lineage specific chromosomes that play key roles in disease and host range (Ma et al. 2010).
The repetitive DNA in the powdery mildew genomes is particularly affected by partial assembly. It may be argued that this just represents the subpopulation of retrotransposons, transposons, and the genomic “trash” associated with their activity. However, the many observations that effector genes are often associated with repetitive DNA (Sacristán et al. 2009; Ridout et al. 2006; Schmidt et al. 2013), and that effectors are likely to be key to understanding important aspects of pathogen biology, means that resolving the structure of these parts of the genome accurately, at high resolution, could yield unexpected and invaluable insights.
The difficulties faced in the assembly of genomes with large proportions of long stretches of repetitive DNA sequence will be overcome by new developments in sequencing technology, or by combinations of different techniques. For example, single molecule ultra-long reads exceeding 10 kb have been reported (Niedringhaus et al. 2011). These methodologies may enable the spanning of the repetitive regions unequivocally between non-repetitive elements; at present, their principal drawback is the relative inaccuracy due to elevated error rates. Therefore, it may be useful to couple these long reads with very deep sequencing with current short read techniques (e.g., Illumina). An additional strategy could be the application of optical mapping (Zhang et al. 2012a) of the B. graminis genomes. In principle, the single molecule restriction analysis has the potential of resolving the true structure of very long repetitive DNA. In combination with the sequencing techniques described above, current difficulties may be overcome (Lin et al. 2012). Finally, we observed that the B. graminis f. sp. hordei genome sequence was mostly congruent with the existing genetic maps (Pedersen et al. 2002a). At present, the density of molecular/physical/sequence markers on these maps is too low to improve the genome assemblies. The creation of high density genetic maps based on sequence markers may provide a useful orthogonal cross-check to corroborate the assemblies obtained with sequencing.
Population genomics data holds many promises to our understanding powdery mildews, particularly when the tools available for direct genetic manipulation are restricted. Arguably, “Nature has carried out lots of experiments out there”: recent advances in understanding the evolution of pathogens in plant disease demonstrate the power of these approaches (Stukenbrock and Bataillon 2012). In my view, similar studies in powdery mildews promise to explain much with regard to the evolutionary dynamics of mildews in relation to their hosts in both agriculture and the natural environment. Ultimately, this understanding will contribute toward mitigating the impact of powdery mildews on our food and other crops resources.
References
Acevedo-Garcia J, Kusch S. Panstruga R (2014) Magical mystery tour: MLO proteins in plant immunity and beyond. New Phytologist. doi:10.1111/nph.12889
Bandaranayake PCG, Yoder JI (2013) Trans-specific gene silencing of acetyl-CoA carboxylase in a root-parasitic Plant. Mol Plant Microbe Interact 26(5):575–584. doi:10.1094/mpmi-12-12-0297-r
Baxter L, Tripathy S, Ishaque N, Boot N, Cabral A, Kemen E, Tyler BM (2010) Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science 330(6010):1549–1551. doi:10.1126/science.1195203
Borbye L, Linde-Laursen I, Christiansen SK, Giese H (1992) The chromosome complement of Erysiphe graminis f.sp. hordei analysed by light microscopy and field inversion gel electrophoresis. Mycol Res 96(2):97–102. doi:10.1016/S0953-7562(09)80922-2
Both M, Spanu P (2004) Blumeria graminis f. sp. hordei, an obligate pathogen of barley. In: Talbot N (ed) Plant pathogen interactions, vol 11. Blackwell publishing, Oxford, pp 202–218
Brewer MT, Cadle-Davidson L, Cortesi P, Spanu PD, Milgroom MG (2011) Identification and structure of the mating-type locus and development of PCR-based markers for mating type in powdery mildew fungi. Fungal Genet Biol 48:704–713. doi:10.1016/j.fgb.2011.04.004
Carver TLW, Thomas BJ, Ingersonmorris SM (1995) The surface of Erysiphe graminis and the production of extracellular material at the fungus—host interface during germling and colony development. Can J Bot-Rev Can Bot 73(2):272–287
Chaure P, Gurr SJ, Spanu P (2000) Stable transformation of Erysiphe graminis, an obligate biotrophic pathogen of barley. Nat Biotechnol 18(2):205–207
Christiansen SK, Knudsen S, Giese H (1995) Biolistic transformation of the obligate plant pathogenic fungus, Erysiphe graminis f.sp. hordei. Curr Genet 29(1):100–102
Dean R, Van Kan J, Pretorius Z, Hammond-Kosack K, di Pietro A, Spanu P, Foster G (2012) The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol 13(4):414–430. doi:10.1111/J.1364-3703.2011.00783.X
Dong WB, Nowara D, Schweizer P (2006) Protein polyubiquitination plays a role in basal host resistance of barley. Plant Cell 18(11):3321–3331
Douchkov D, Nowara D, Zierold U, Schweizer P (2005) A high-throughput gene-silencing system for the functional assessment of defense-related genes in barley epidermal cells. Mol Plant Microbe Interact 18(8):755–761
Duplessis S, Cuomo CA, Lin YC, Aerts A, Tisserant E, Veneault-Fourrey C, Martin F (2011) Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc Natl Acad Sci USA 108(22):9166–9171. doi:10.1073/pnas.1019315108
Glawe DA (2008) The powdery mildews: a review of the world’s most familiar (yet poorly known) plant pathogens. Annu Rev Phytopathol 46(1):27–51. doi:doi:10.1146/annurev.phyto.46.081407.104740
Göhre V, Robatzek S (2008) Breaking the barriers: microbial effector molecules subvert plant immunity. Annu Rev Phytopathol 46(1):189–215. doi:10.1146/annurev.phyto.46.120407.110050
Haas B, Kamoun S, Zody M, Jiang R, Handsaker R, Cano L (2009) Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461(7262):393–398
Hacquard S, Kracher B, Maekawa T, Vernaldi S, Schulze Lefert P, Ver Looren van Themaat E. (2013) Mosaic genome structure of the barley powdery mildew pathogen and conservation of transcriptional programs in divergent hosts. Proc Nat Acad Sci 110 (34):13965–13970. doi:10.1073/pnas.1306807110
Hiura U (1978) Genetic basis of special forms in Erysiphe graminis
Hückelhoven R, Panstruga R (2011) Cell biology of the plant-powdery mildew interaction. Curr Opin Plant Biol 11(14):1–9
Jones H, Whipps JM, Gurr SJ (2001) The tomato powdery mildew fungus Oidium neolycopersici. Mol Plant Pathol 2(6):303–309. doi:10.1046/j.1464-6722.2001.00084.x
Jørgensen JH (1992) Discovery, characterization and exploitation of Mlo powdery mildew resistance in barley. In: Johnson R, Jellis GJ (eds) Breeding for disease resistance, vol 1. Springer, Netherlands. 141–152
Kemen E, Gardiner A, Schultz-Larsen T, Kemen AC, Balmuth AL, Robert-Seilaniantz A, Jones JDG (2011) Gene gain and loss during evolution of obligate parasitism in the white rust pathogen of Arabidopsis thaliana. PLoS Biology 9(7):e1001094
Lin H, Goldstein S, Mendelowitz L, Zhou S, Wetzel J, Schwartz D, Pop M (2012) AGORA: assembly guided by optical restriction alignment. BMC Bioinf 13(1):189
Liu N, Gong G, Zhang M, Zhou Y, Chen Z, Yang J, Liu K (2012) Over-summering of wheat powdery mildew in Sichuan Province, China. Crop Prot 34(0):112–118. doi:10.1016/j.cropro.2011.12.011
Ma L-J, van der Does HC, Borkovich KA, Coleman JJ, Daboussi M-J, Di Pietro A, Rep M (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464(7287):367–373. doi:10.1038/nature08850
Niedringhaus TP, Milanova D, Kerby MB, Snyder MP, Barron AE (2011) Landscape of next-generation sequencing technologies. Anal Chem 83(12):4327–4341. doi:10.1021/ac2010857
Nowara D, Gay A, Lacomme C, Shaw J, Ridout C, Douchkov D, Schweizer P (2010) HIGS: Host-induced gene silencing in the obligate biotrophic fungal pathogen Blumeria graminis. Plant Cell 22(9):3130–3141. doi:10.1105/tpc.110.077040
Nowara D, Schweizer P (2007) Host-induced gene silencing in Blumeria graminis. In: XIII International congress on molecular plant- microbe interactions. Sorrento, Italy
Nunes CC, Dean RA (2012) Host-induced gene silencing: a tool for understanding fungal host interaction and for developing novel disease control strategies. Mol Plant Pathol 13(5):519–529. doi:10.1111/j.1364-3703.2011.00766.x
O’Connell RJ, Thon MR, Hacquard S, Amyotte SG, Kleemann J, Torres MF, Vaillancourt LJ (2012) Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat Genet 44(9):1060–1065. doi:10.1038/ng.2372
Oberhaensli S, Parlange F, Buchmann JP, Jenny FH, Abbott JC, Burgis TA, Wicker T (2011) Comparative sequence analysis of wheat and barley powdery mildew fungi reveals gene colinearity, dates divergence and indicates host-pathogen co-evolution. Fungal Genet Biol 48(3):327–334. doi:10.1016/j.fgb.2010.10.003
Olesen KL, Carver TLW, Lyngkjaer MF (2003) Fungal suppression of resistance against inappropriate Blumeria graminis formae speciales in barley, oat and wheat. Physiol Mol Plant Pathol 62(1):37–50. doi:10.1016/s0885-5765(03)00005-5
Parlange F, Daverdin G, Fudal I, Kuhn M-L, Balesdent M-H, Blaise F, Rouxel T (2009) Leptosphaeria maculans avirulence gene AvrLm4-7 confers a dual recognition specificity by the Rlm4 and Rlm7 resistance genes of oilseed rape, and circumvents Rlm4-mediated recognition through a single amino acid change. Mol Microbiol 71(4):851–863
Parlange F, Oberhaensli S, Breen J, Platzer M, Taudien S, Šimková H, Keller B (2011) A major invasion of transposable elements accounts for the large size of the Blumeria graminis f.sp. tritici genome. Funct Integr Genomics 11(4):671–677. doi:10.1007/s10142-011-0240-5
Pedersen C, Rasmussen SW, Giese H (2002a) A genetic map of Blumeria graminis based on functional genes, avirulence genes, and molecular markers. Fungal Genet Biol 35(3):235–246
Pedersen C, Wu BQ, Giese H (2002b) A Blumeria graminis f.sp. hordei BAC library - contig building and microsynteny studies. Curr Genet 42(2):103–113. doi:10.1007/s00294-002-0341-8
Pedersen C, Ver Loren van Themaat E, McGuffin LJ, Abbott JC, Burgis TA, Barton G, Spanu PD (2012) Structure and evolution of barley powdery mildew effector candidates. BMC Genomics 13:694. doi:10.1186/1471-2164-13-694
Pliego C, Nowara D, Bonciani G, Gheorghe D, Xu R, Surana P, Spanu P (2013) Host-Induced Gene Silencing based pathogen effector discovery in barley powdery mildew. Mol Plant-Microbe Interact 26(6):633–642. doi:10.1094/MPMI-01-13-0005-R
Pryce-Jones E, Carver T, Gurr SJ (1999) The roles of cellulase enzymes and mechanical force in host penetration by Erysiphe graminis f.sp. hordei. Physiol Mol Plant Pathol 55:175–182
Raffaele S, Farrer RA, Cano LM, Studholme DJ, MacLean D, Thines M, Kamoun S (2010) Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science 330(6010):1540–1543. doi:10.1126/science.1193070
Raffaele S, Kamoun S (2012) Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol 10(6):417–430. doi:10.1038/nrmicro2790
Ridout CJ, Skamnioti P, Porritt O, Sacristàn S, Jones JDG, Brown JKM (2006) Multiple avirulence paralogues in cereal powdery mildew fungi may contribute to parasite fitness and defeat of plant resistance. Plant Cell 18(9):2402–2414
Sacristán S, Vigouroux M, Pedersen C, Skamnioti P, Thordal-Christensen H, Micali C, Ridout CJ (2009) Coevolution between a family of parasite virulence effectors and a class of LINE-1 retrotransposons. PLoS ONE 4(10):e7463
Schmidt S, Houterman P, Schreiver I, Ma L, Amyotte S, Chellappan B, Rep M (2013) MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genomics 14(1):119
Skamnioti P, Pedersen C, Al-Chaaram GR, Holefors A, Thordal-Christensen H, Brown JKM, Ridout CJ (2008) Genetics of avirulence genes in Blumeria graminis f.sp hordei and physical mapping of AVR(a22) and AVR(a12). Fungal Genet Biol 45(3):243–252. doi:10.1016/j.fgb.2007.09.011
Spanu P (2012) The genomics of obligate (and non-obligate) biotrophs. Annu Rev Phytopathol 50:91–109. doi:10.1146/annurev-phyto-081211-173024
Spanu PD (2006) Why do some fungi give up their freedom and become obligate dependants on their host? New Phytol 171(3):447–450
Spanu PD, Abbott JC, Amselem J, Burgis TA, Soanes DM, Stuber K, Panstruga R et al. (2010) Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 330(6010):1543–1546. doi:10.1126/science.1194573
Spanu PD, Panstruga R (2012) Powdery mildew genomes in the crosshairs. New Phytol 195(1):20–22. doi:10.1111/j.1469-8137.2012.04173.x
Stukenbrock EH Bataillon T (2012). A population genomics perspective on the emergence and adaptation of new plant pathogens in agro-ecosystems. Plos Pathog 8(9). doi:10.1371/journal.ppat.1002893
Takamatsu S (2004) Phylogeny and evolution of the powdery mildew fungi (Erysiphales, Ascomycota) inferred from nuclear ribosomal DNA sequences. Mycoscience 45(2):147–157
Takken F, Rep M (2010) The arms race between tomato and Fusarium oxysporum. Mol Plant Pathol 11(2):309–314. doi:10.1111/j.1364-3703.2009.00605.x
Walker AS, Bouguennec A, Confais J, Morgant G, Leroux P (2011) Evidence of host-range expansion from new powdery mildew (Blumeria graminis) infections of triticale (xTriticosecale) in France. Plant Pathol 60(2):207–220. doi:10.1111/j.1365-3059.2010.02379.x
Whiteford JR, Spanu PD (2001) The hydrophobin HCf-1 of Cladosporium fulvum is required for efficient water-mediated dispersal of conidia. Fungal Genet Biol 32:159–168
Wicker T, Oberhaensli S, Parlange F, Buchmann JP, Shatalina M, Roffler S, Ben-David R, Dolezel J, Simkova H, Schulze-Lefert P, Spanu PD, Bruggmann R, Amselem J, Quesneville H, van Themaat EVL, Paape T, Shimizu KK, Keller B (2013) The wheat powdery mildew genome shows the unique evolution of an obligate biotroph. Nat Genet 45:1092–1096. doi:10.1038/ng.2704
Wolfe MS, McDermott JM (1994) Population genetics of plant pathogen interactions: The example of the Erysiphe graminis-Hordeum vulgare pathosystem. Annu Rev Phytopathol 32:89–113
Wösten HAB (2001) Hydrophobins: multipurpose proteins. Annu Rev Microbiol 55:625–646
Wösten HAB, Willey JM (2000) Surface-active proteins enable microbial aerial hyphae to grow into the air. Microbiology-Uk 146:767–773
Yamaoka N, Matsumoto I, Nishiguchi M (2006) The role of primary germ tubes (PGT) in the life cycle of Blumeria graminis: the stopping of PGT elongation is necessary for the triggering of appressorial germ tube (AGT) emergence. Physiol Mol Plant Pathol 69(4–6):153–159. doi:http://dx.doi.org/10.1016/j.pmpp.2007.04.003
Zhang Q, Chen W, Sun L, Zhao F, Huang B, Yang W, Wang J (2012a) The genome of Prunus mume. Nat Commun 3:1318. doi:10.1038/ncomms2290
Zhang W-J, Pedersen C, Kwaaitaal M, Gregersen PL, Mørch SM, Hanisch S, Thordal-Christensen H (2012b) Interaction of barley powdery mildew effector candidate CSEP0055 with the defence protein PR17c. Mol Plant Pathol 13(9):1110–1119. doi:10.1111/j.1364-3703.2012.00820.x
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Spanu, P.D. (2014). The Genomes of the Cereal Powdery Mildew Fungi, Blumeria graminis . In: Dean, R., Lichens-Park, A., Kole, C. (eds) Genomics of Plant-Associated Fungi: Monocot Pathogens. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-44053-7_7
Download citation
DOI: https://doi.org/10.1007/978-3-662-44053-7_7
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44052-0
Online ISBN: 978-3-662-44053-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)