
Abstract
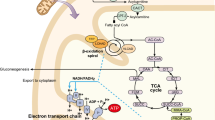
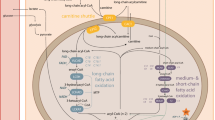
A dozen genetic defects in the fatty acid-oxidation pathway are currently known. Nearly all of these defects present in early infancy as acute, life-threatening episodes of hypoketotic, hypoglycemic coma induced by fasting [1–3]. In some of the disorders, there may also be chronic skeletal-muscle weakness or acute exercise-induced rhabdomyolysis and acute or chronic cardiomyopathy. Recognition of the fatty acid-oxidation disorders is often difficult, because patients can appear well until they undergo prolonged fasting.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Preview
Unable to display preview. Download preview PDF.
Similar content being viewed by others

References
Coates PM, Stanley CA (1992) Inherited disorders of mitochondrial fatty acid oxidation. Prog Liver Dis 10: 123–38
Hale DE, Bennett MJ (1992) Fatty acid oxidation disorders: a new class of metabolic diseases. J Pediatr 121: 1–11
Saudubray JM, Martin D, de Lonlay et al. (1999) Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis 22: 488–502
Stanley CA, DeLeeuw S, Coates PM et al. (1991) Chronic cardiomyopathy and weakness or acute coma in children with a defect in carnitine uptake. Ann Neurol 30: 709–716
Demaugre F, Bonnefont J, Mitchell G et al. (1988) Hepatic and muscular presentations of carnitine palmitoyl transferase deficiency: two distinct entities. Pediatr Res 24: 308–311
Stanley CA, Sunaryo F, Hale DE et al. (1992) Elevated plasma carnitine in the hepatic form of carnitine palmitoyltransferase-1 deficiency. J Inherit Metab Dis 15: 785–789
Vianey-Saban C, Mousson B, Bertrand C et al. (1993) Carnitine palmitoyl transferase I deficiency presenting as a Reye-like syndrome without hypoglycemia. Eur J Pediatr 152: 334–338
Falik-Borenstein ZC, Jordan SC, Saudubray JM et al. (1992) Brief report: renal tubular acidosis in carnitine palmitoyltransferase type 1 deficiency. N Engl J Med 327: 24–27
Stanley CA, Hale DE, Berry GT et al. (1992) Brief report: a deficiency of carnitine-acylcarnitine translocase in the inner mitochondrial membrane. N Engl J Med 327: 19–23
Pande SV, Brivet B, Slama A et al. (1993) Carnitine-acylcarnitine translocase deficiency with severe hypoglycemia and auriculo ventricular block. Translocase assay in permeabilized fibroblasts. J Clin Invest 91: 1247–1252
Chalmers RA, Stanley CA, English N, Wigglesworth JS (1997) Mitochondrial carnitine-acylcarnitine translocase deficiency presenting as sudden neonatal death. J Pediatr 131: 220–225
Morris AAM, Olpin SE, Brivet M et al. (1998) A patient with carnitine-acylcarnitine translocase deficiency with a mild phenotype. J Pediatr 132: 514–516
DiMauro S, DiMauro PMM (1973) Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science 182: 929–931
Demaugre F, Bonnefont JP, Colonna M et al. (1991) Infantile form of carnitine palmitoyltransferase II deficiency with hepatomuscular symptoms and sudden death. Physiopathological approach to carnitine palmitoyltransferase II deficiencies. J Clin Invest 87: 859–864
Taroni F, Verderio E, Garavaglia B et al. (1992) Biochemical and molecular studies of carnitine palmitoyltransferase II deficiency with hepatocardiomyopathic presentation. Prog Clin Biol Res 375: 521–531
Taroni F, Verderio E, Dworzak F et al. (1993) Identification of a common mutation in the carnitine palmitoyltransferase II gene in familial recurrent myoglobinuria patients. Nat Genet 4: 314–320
Hale DE, Batshaw ML, Coates PM et al. (1985) Long-chain acyl coenzyme A dehydrogenase deficiency: an inherited cause of nonketotic hypoglycemia. Pediatr Res 19: 666–671
Vianey-Saban C, Divry P, Brivet M et al. (1998) Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: clinical characteristics and diagnostic considerations in 30 patients. Clin Chim Acta 269: 43–62
Mao LF, Chu C, Luo MJ et al. (1995) Mitochondrial beta-oxidation of 2-methyl fatty acids in rat liver. Arch Biochem Biophys 321: 221–228
Wanders RJ, Denis S, Ruiter JP, Ijlst L, Dacremont G (1998) 2,6-Dimethylheptanoyl-CoA is a specific substrate for long-chain acyl-CoA dehydrogenase (LCAD): evidence for a major role of LCAD in branched-chain fatty acid oxidation. Biochim Biophys Acta 1393: 35–40
Stanley CA, Hale DE, Coates PM et al. (1983) Medium-chain acyl-CoA dehydrogenase deficiency in children with nonketotic hypoglycemia and low carnitine levels. Pediatr Res 17: 877–884
Wilson CJ, Champion MP, Collins JE, Clayton PT, Leonard JV (1999) Outcome of medium chain acyl-CoA dehydrogenase deficiency after diagnosis. Arch Dis Child 80: 459–462
Yokota I, Coates PM, Hale DE, Rinaldo P, Tanaka K (1992) The molecular basis of medium chain acyl-CoA dehydrogenase deficiency: survey and evolution of 985A-G transition, and identification of five rare types of mutation within the medium chain acyl-CoA dehydrogenase gene. Prog Clin Biol Res 375: 425–440
Yokota I, Coates PM, Hale DE, Rinaldo P, Tanaka K (1991) Molecular survey of a prevalent mutation, 985A-to-G transition, and identification of five infrequent mutations in the medium-chain Acyl-CoA dehydrogenase (MCAD) gene in 55 patients with MCAD deficiency. Am J Hum Genet 49: 1280–1291
Bhala A, Willi SM, Rinaldo P et al. (1995) Clinical and biochemical characterization of short-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr 126: 910–915
Coates PM, Hale DE, Finocchiaro G, Tanaka K, Winter SC (1988) Genetic deficiency of short-chain acyl-coenzyme A dehydrogenase in cultured fibroblasts from a patient with muscle carnitine deficiency and severe skeletal muscle weakness. J Clin Invest 81x71–175
Gregersen N, Winter VS, Corydon MJ et al. (1998) Identification of four new mutations in the short-chain acyl-CoA dehydrogenase (SCAD) gene in two patients: one of the variant alleles, 51C -s T, is present at an unexpectedly high frequency in the general population, as was the case for 625G -s A, together conferring susceptibility to ethylmalonic aciduria. Hum Mol Genet 7: 619–627
Poll-The BT, Bonnefont JP, Ogier H et al. (1988) Familial hypoketotic hypoglycaemia associated with peripheral neuropathy, pigmentary retinopathy and C6–C14 hydroxydicarboxylic aciduria. A new defect in fatty acid oxidation? J Inherit Metab Dis 2: 183–185
Tyni T, Pihko H (1999) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Review article. Acta Paediatr 88: 237–245
Dionisi-Vici C, Burlina AB, Bertini E et al. (1991) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical and therapeutic considerations. J Pediatr 118: 744–746
Rocchiccioli F, Wanders RJ, Aubourg P et al. (1990) Deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase: a cause of lethal myopathy and cardiomyopathy in early childhood. Pediatr Res 28: 657–662
Jackson S, Kier RS, Bartlett K et al. (1992) Combined defect of long-chain 3-hydroxyacyl-CoA dehydrogenase, 2-enoyl-CoA hydratase and 3-oxoacyl-CoA thiolase. Prog Clin Biol Res 375: 327–337
Treem WR, Rinaldo P, Hale DE et al. (1994) Acute fatty liver of pregnancy and long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Hepatology 19: 339–345
Treem WR, Shoup ME, Hale DE et al. (1996) Acute fatty liver of pregnancy, hemolysis, elevated liver enzymes, and low platelets syndrome, and long chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency [see comments]. Am J Gastroenterol 91: 2293–2300
Tein I, De VDC, Hale DE et al. (1991) Short-chain L-3hydroxyacyl-CoA dehydrogenase deficiency in muscle: a new cause for recurrent myoglobinuria and encephalopathy. Ann Neurol 30: 415–419
Bennett MJ, Weinberger MJ, Kobori JA, Rinaldo P, Burlina AB (1996) Mitochondrial short-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: a new defect of fatty acid oxidation. Pediatr Res 39: 185–188
Kamigo T, Indo Y, Souri M et al. (1997) Medium chain 3ketoacyl-coenzyme A thilase deficiency: a new disorder of mitochondrial fatty acid beta-oxidation. Pediatr Res 42: 569–576
Roe C, Millington D, Norwood D et al. (1990) 2,4-Dienoylcoenzyme A reductase deficiency: a possible new disorder of fatty acid oxidation. J Clin Invest 85: 1703–1707
Freeman FE, Goodman SI (1985) Deficiency of electron transfer flavoprotein or electron transfer flavoprotein:ubiquinone oxidoreductase in glutaric acidemia type II fibroblasts. Proc Natl Acad Sci USA 82: 4517–4520
Thompson GN, Hsu BY, Pitt JJ, Treacy E, Stanley CA (1997) Fasting hypoketotic coma in a child with deficiency of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase. N Engl J Med 337: 1203–1207
Gibson K, Breuer J, Nyhan W (1988) 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: review of 18 reported cases. Eur J Pediatr 148: 180–186
Stanley CA (1995) Carnitine disorders. Adv Pediatr 42: 209–242
Millington DS, Terada N, Chace DH et al. (1992) The role of tandem mass spectrometry in the diagnosis of fatty acid oxidation disorders. In: Coates PM, Tanaka K (eds) New developments in fatty acid oxidation. Progress in clinical and biochemical research. New York, John Wiley-Liss, pp 339–354
Ziadeh R, Hoffman EP, Finegold DM et al. (1995) Medium chain acyl-CoA dehydrogenase deficiency in Pennsylvania: neonatal screening shows high incidence and unexpected mutation frequencies. Pediatr Res 37: 675–678
Stanley CA, Berry GT, Bennett MJ et al. (1993) Renal handling of carnitine in secondary carnitine deficiency disorders. Pediatr Res 34: 89–97
Rinaldo P, O’Shea JJ, Coates PM et al. (1988) Medium-chain acyl-CoA dehydrogenase deficiency. Diagnosis by stable-isotope dilution measurement of urinary n-hexanoylglycine and 3-phenylpropionylglycine [published erratum appears in N Engl J Med 1989, 320 (18):1227]. N Engl J Med 319: 1308–1313
Runsby G, Seakins J, Leonard J (1886) A simple screening test for medium chain acyl-CoA dehydrogenase deficiency. Lancet 2: 467
Parini R, Garavaglia B, Saudubray J et al. (1991) Clinical diagnosis of long-chain acyl-coenzyme A dehydrogenase deficiency: use of stress and fat-loading tests. J Pediatr 119: 77–80
Treem WR, Witzleben CA, Piccoli DA et al. (1986) Medium-chain and long-chain acyl CoA dehydrogenase deficiency: clinical, pathologic and ultrastructural differentiation from Reye’s syndrome. Hepatology 6: 1270–1278
Lieu YK, Hsu BY, Price WA, Corkey BE, Stanley CA (1997) Carnitine effects on coenzyme A profiles in rat liver with hypoglycin inhibition of multiple dehydrogenases. Am J Physiol 272: E359–366
Odaib AA, Schneider BL, Bennett MJ et al. (1998) A defect in the transport of long-chain fatty acids associated with acute liver failure. N Engl J Med 339: 1752–1757
Rizzo WB (1998) Inherited disorders of fatty alcohol metabolism. Minireview. Mol Genet Metab 65: 63–73
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2000 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Stanley, C.A. (2000). Disorders of Fatty Acid Oxidation. In: Fernandes, J., Saudubray, JM., Van den Berghe, G. (eds) Inborn Metabolic Diseases. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-04285-4_11
Download citation
DOI: https://doi.org/10.1007/978-3-662-04285-4_11
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-04287-8
Online ISBN: 978-3-662-04285-4
eBook Packages: Springer Book Archive