
Abstract
Recent studies have proposed two subtypes of autoimmune pancreatitis (AIP): type 1 AIP (IgG4-related pancreatitis) as the pancreatic manifestation of IgG4-related disease (IgG4-RD) and type 2 related with a granulocytic epithelial lesion. Among the involved organs in type 1 AIP, association of pancreatic and biliary lesions is extremely high. On genetic backgrounds, innate immunity and acquired immunity, Th2-dominant immune status, regulatory T cells (Treg) or B cells (Breg), and complement activation via a classical pathway may be involved in the development of type 1 AIP. Although the role of IgG4 remains unclear in type 1 AIP, IgG4 may contribute to the clearance of immune complexes or termination of the inflammatory process by preventing the formation of large immune complexes with blocking Fc-mediated effector functions of IgG1. Production of IgG4 in patients with type 1 AIP is upregulated by IL-10 secreted from inhibitory costimulatory molecule (ICOS)-positive inducible Tregs and CD19+CD24hiCD27hi Bregs and B-cell-activating factor belonging to the tumor necrosis factor family (BAFF) from monocytes and/or basophils with toll-like receptors (TLRs)/nucleotide-binding oligomerization domain-like receptors (NODRs) stimulation. Fibrosis may be induced by TGF-beta secreted from ICOS-negative inducible Tregs. Further studies are necessary to clarify the pathogenic mechanism of AIP.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Systemic Lupus Erythematosus
- Autoimmune Pancreatitis
- Tubulointerstitial Nephritis
- Salivary Gland Lesion
- Pancreatic Manifestation
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Recent studies suggested the existence of two subtypes of autoimmune pancreatitis (AIP) [1–5]: type 1 AIP (IgG4-related pancreatitis as the pancreatic manifestation of IgG4-related disease (IgG4-RD)) and type 2 AIP (idiopathic duct-centric chronic pancreatitis or AIP with a granulocytic epithelial lesion). The characteristic features of type 1 AIP are swelling of the pancreas, increased serum IgG4 levels, abundant infiltration of IgG4+ plasmacytes with storiform fibrosis, obliterative phlebitis, and other organ involvements (OOIs) such as sclerosing cholangitis, sclerosing sialadenitis, and retroperitoneal fibrosis [4, 6]. The OOIs appear synchronously or metachronously with the pancreatic lesion(s), share the same pathological conditions, and show favorable response to steroid therapy [7]. The lesions are usually detected by imaging and blood tests (CT, MRI, gallium scintigraphy, FDG-PET, and IgG4); however, these should be confirmed by histological findings. The OOIs sometimes mimic, or are misdiagnosed as, primary lesions of the corresponding organs: lachrymal and salivary gland lesions for Sjögren’s syndrome, respiratory lesions for sarcoidosis, and sclerosing cholangitis for primary sclerosing cholangitis (PSC) [7, 8].
On the other hand, type 2 AIP, extremely rarely observed in Japan, is not associated with either serum IgG4 elevation or with OOIs seen in LPSP. Type 2 AIP has swelling of the pancreas, but none or very few IgG4-positive plasma cells and clinical features show a distinctly different profile associated with no biomarkers such as serum IgG4, IgG, and autoantibodies, or OOIs except for inflammatory bowel disease [1, 4]. Different from type 1 AIP, very few studies of pathogenic mechanisms in type 2 AIP have been reported except for deposition of C3c and IgG at the basement membrane of pancreatic ducts and acini, which suggests immune complex-mediated destruction of ducts and acini in type 2 AIP [9]. In this chapter, recent advances in the pathophysiology of type 1 AIP are mainly mentioned.
Immunogenic Backgrounds
Based on immunogenic backgrounds, abnormal conditions of immune responses may be involved in the development of AIP [8]. However, immunogenic factors have been in a few series, mainly from Asian countries, and then not conclusive. As type 2 AIP is rare in Asia, these data are deemed to reflect immunogenic factors in type 1 AIP. Susceptibility to AIP may be associated with the class II antigen of the major histocompatibility complex (MHC), polymorphism of nuclear factor (NF)-kB, and Fc-receptor-like (FCRL) 3 genes expressed on B cells [10, 11]. Two studies of HLA association with AIP have been reported from the Japanese [10] and Korean group [11]. In the Japanese patients with AIP, HLA haplotype DRB1*0405-DQB1*0401 (class II) and ABCF1 proximal to C3-2-11, telomeric of HLA-E (class I) are susceptibility to AIP, but not in the Korean patients. However, substitution of aspartic acid to nonaspartic acid at DQβ1 may be a predictive factor for relapse of AIP in Korean patients [11]. FCRL3 polymorphisms are linked to various autoimmune diseases, such as rheumatoid arthritis, autoimmune thyroid disease, and systemic lupus erythematosus (SLE) in the Japanese population [12, 13]. However, Fc-receptor-like 3 gene polymorphisms is not correlated with the DRB1*0405-DQB1*0401 haplotype, suggesting that while both are related to AIP susceptibility in the Japanese population, they are part of distinct underlying mechanisms of disease development [12, 13].
Innate immunity is important in the development of acquired immunity or autoimmune diseases. Although polymorphisms in toll-like receptor (TLR)-4 gene have been linked with several autoimmune and allergic diseases, it seems not to play an important role in AIP [14]. On the other hand, an inhibitory molecule, cytotoxic T lymphocyte antigen-4 (CTLA-4; CD152) expressed on the activated memory T cells and CD4 + CD25+ regulatory T cells (Tregs) was independently reported as a susceptibility factor for AIP in the Taiwanese [15] and Japanese population [16]. CTLA-4 acts as a negative regulator of T-cell responses by competing with the CD28 molecule for engagement with the B7 molecules CD80 and CD86 on antigen-presenting cells [17]. Umemura T et al. [16] reported that the 3′ untranslated region of CTLA-4 + 6230 SNP plays a pivotal role for both susceptibility (+6230G/G genotype) to and protection (haplotype of the +6230A allele) from AIP, while exon 1 + 49 SNP is not associated with AIP in the Japanese patients. They also found that +49A/A or +6230A/A genotypes may be associated with the recurrence of the disease, which is observed in Graves’ disease, type 1 diabetes, and clearance of hepatitis B virus [16]. On the other hand, Chan MC et al. [15] have reported that CTLA-4 SNPs have shown significantly higher frequencies of the +49G allele in patients with AIP than in controls, but not with other subtypes of chronic pancreatitis. TNF-alpha promoter 863A was significantly associated with higher risk of AIP. Racial and geographical differences may be associated with SNPs of the different locus of CTLA-4 [15]. Soluble isoform of CTLA4 (sCTLA4) is reported to be elevated in patients with autoimmune diseases, such as autoimmune thyroid disease, SLE, and myasthenia gravis [17]. Therefore, the sCLTA4 molecule may have a dual role of maintaining self-tolerance and enhancing immune responses by blocking the interaction of CD80 on antigen-presenting cells and CTLA4 on T cells.
Innate Immunity
Recently, abnormal innate immunity has been demonstrated in type 1 AIP. Activation of NOD-2 and TLR ligands on monocytes or basophils from patients with type 1 AIP enhances IgG4 production by B cells through signaling pathways mediated by B-cell-activating factor family (BAFF) and IL-13 and thereby induce systemic IgG4 responses [18, 19]. Although specific microorganisms still remain unclear, several candidates have been reported. Molecular mimicry among microbes and target antigens may be a possible mechanism to break down immune tolerance. The hypothesis is based on the concept that infectious agents share one or more epitopes with self-components or infectious agents cause bystander activation of immune cells with autoaggressive potential [20–23]. Guarneri and colleagues showed significant homology between human CA-II and alpha-CA of H. pylori, a fundamental enzyme for bacterial survival and proliferation in the stomach [21]. Moreover, the homologous segments contain the binding motif of DRB1*0405, which confers a risk for AIP development [21]. The plasminogen-binding protein (PBP) peptide newly identified in European patients with AIP shows homology with an amino acid sequence of PBP of H. pylori and with ubiquitin-protein ligase E3 component n-recognin 2 (UBR2), an enzyme highly expressed in acinar cells of the pancreas [20], while European patients with AIP did not necessarily show LPSP as the typical histopathology in IgG4-related diseases. These findings suggest that gastric H. pylori infection might trigger AIP in genetically predisposed subjects. Although no previous data of viral factors in human IgG4-RD, animal models suggested possible roles of the innate immune system in the development of immune-mediated cholangiopancreatitis and sialadenitis through activation of TLR3/TLR4 by infiltrating pathogen-associated molecular patterns (PAMP) and/or antigen(s) derived from virus or commensal bacteria in addition to virulent bacteria [24–26].
Possible Roles of IgG4 in Type 1 AIP
Although the association of IgE-mediated allergy and IgG4 antibodies is well known, IgG4 characteristics are still poorly understood. IgG4 is involved in an immune process referring to as “Fab-arm exchange,” which is a swapping of a heavy chain and attached light chain (half-molecule) with a heavy-light chain pair from another molecule; this usually results in asymmetric antibodies with two different antigen-combining sites (Fig. 3.1a) [27]. While these modified antibodies are heterobivalent, they behave as monovalent antibodies. Another aspect of IgG4 is that it mimics IgG rheumatoid factor (RF) activity by interacting with IgG, namely, Fc-mediated aggregation (Fig. 3.1b) [28]. IgG4 seems to be associated with a pathogenic effect in a few situations. In pemphigus, recognition of skin autoantigens (desmogleins) by IgG4 is at the origin of the disease process [29]. A most recent study of structural determinants of human IgG4-Fc using crystallization suggested that Fc-Fc interactions are compatible with intact IgG4 molecules and may provide a model for the formation of aggregates of IgG4 that can cause disease pathology in the absence of antigen [30].

Characteristic forms of IgG4. (a) Schematic representation of the generation of bispecific IgG4 antibodies by the exchange of half-molecules (“Fab-arm exchange”) (With permission from the publisher of van der Neut Kolfschoten et al. [27]). IgG4 Fab-arm exchange occurs by the exchange of a heavy chain-light chain pair (half-molecule) of one IgG4 molecule with that of another IgG4 molecule. The IgG4 molecule may thereby acquire two distinct Fab arms and become bispecific. The Fc structure remains essentially unchanged apart from potential changes due to differences in glycosylation or allotype. Fab-arm exchange is proposed to be stochastic and dynamic. (b) On the left: IgG4 Fc interacts with Ig Fc. On the right: IgM RF recognizes IgG in a “classical” Fab-Fc recognition (With permission from Kawa et al. [28])
Another recent data on regulating IgG4 showed that IgG4-related diseases may reflect an excessive production of anti-inflammatory cytokines such as IL-10 that triggers an overwhelming expansion of IgG4-producing plasma cells [31–35]. Increased peripheral inducible memory Tregs are positively correlated with serum levels of IgG4 [33]. In addition, prominent infiltration of Tregs upregulated IL-10 in livers of patients with IgG4-SC [31]. These findings suggest that IgG4 do not act as a pathogenic factor nor as an anti-inflammatory factor in IgG4-RD. Further studies are necessary to clarify the precise role of IgG4 in IgG4-RD.
The Complement System
Patients with active stage of AIP occasionally show decreased complement (C3, C4) with elevated circulating immune complex as well as serum levels of IgG4 [36]. Both tubulointerstitial nephritis (TIN) and membranous glomerulonephritis are often observed in IgG4-related kidney disease (IgG4-KD), and deposition of immune complex (IgG and C3) was observed on the tubular basement membrane as well as the glomerular basement membrane [37, 38]. As the complements cannot connect at the Fc portion of IgG4, the classical pathway of complement activation through IgG1 may be involved in the development of AIP rather than mannose-binding lectin or alternative pathways through IgG4 [39]. Moreover, IgG4 bound to other isotype such as IgG1, IgG2, and IgG3 with an Fc-Fc interaction immune complex in patients with AIP [28], and then IgG4 may contribute to the clearance of immune complexes or termination of the inflammatory process by preventing the formation of large immune complexes with blocking Fc-mediated effector functions of IgG1.
Autoantibodies and Candidate of Target Antigens
Patients with type 1 AIP generally show several autoantibodies in addition to increased IgG and IgG4 [8]. Although some patients have nonspecific antibodies such as antinuclear antibody (ANA), there is scarcely association of IgG4-RD and well-known autoimmune diseases such as Sjögren’s syndrome and SLE. From the view of IgG4 function, a big mystery whether IgG4-related disease is autoimmune or allergic disease is addressed. However, occasional coexistence of OOIs leads us the concept that there may be common target antigens in the involved organs such as the pancreas, salivary gland, biliary tract, lung, renal tubules, and so on. Although the disease-specific antibodies have not been identified at this moment, several disease-related antibodies such as anti-lactoferrin (LF) [40, 41], anti-carbonic anhydrase (CA)-II [40–43], anti-CA-IV [44], anti-pancreatic secretory trypsin inhibitor (PSTI) [45], anti-amylase-alpha [46], anti-HSP-10 [47], and anti-plasminogen-binding protein (PBP) peptide autoantibodies [20] have been reported. Although the patients show increased serum levels of IgG4, the major subclass of these autoantibody is not necessarily IgG4, but often IgG1 [45]. CA-II, CA-IV, LF, and PSTI are distributed in the ductal cells of several exocrine organs, including the pancreas, salivary gland, biliary duct, lung, and renal tubules [40, 41]. Although all peptides have not been studied, immunization with CA-II or LF-induced systemic lesions such as pancreatitis, sialadenitis, cholangitis, and interstitial nephritis in the mice models similar to human IgG4-related diseases [48, 49]. The high prevalence of these antibodies suggests that these may be the candidates for the target antigens in type 1 AIP. Among the involved organs in IgG4-RD, recent studies suggest an extremely high association of pancreatic and biliary lesions [50]. As both peribiliary glands in the biliary tract and pancreatic duct glands associated with pancreatic ducts in human are intermingled with small amounts of pancreatic exocrine acini [51], and the biliary tree-derived stem cells constitute a pancreatic organogenesis in mice [52], Nakanuma et al. have proposed a new concept of the “biliary diseases with pancreatic counterparts” [53], in which targets of type 1 AIP and IgG4-SC may be periductal glands around the bile and pancreatic ducts.
Diabetes mellitus is complicated with 43 ~ 68 % of the patients with AIP, but autoantibodies against glutamic acid decarboxylase, beta-cell, or tyrosine phosphatase-like protein associated type 1A DM are rarely observed [54]. These findings suggest that islet cells may not be targeted in the development of DM associated with AIP.
Role of B Cells
In addition to steroid and immune modulators, the B-cell depletion by rituximab, which reduces only IgG4, but not IgG1, IgG2, or IgG3, is useful in the therapeutic strategy in IgG4-RD [55, 56]. A recent study showed expansion of IgG4+ B-cell receptor (BCR) clones in blood and tissue of patients with active IgG4-cholangiopathy, and disappearance by corticosteroid treatment [57]. A recent study showed that the increased CD19+CD24highCD38high Bregs may suppress the disease activity of type 1 AIP, whereas the decreased CD19+CD24highCD27+ Bregs might be involved in the development of type 1 AIP [58]. These findings suggest that specific B-cell responses may have a pivotal role in the pathogenesis of IgG4-RD such as type 1 AIP and IgG4-SC.
Th1 and Th2 Immune Balance
The effector cells in IgG4-related diseases have been poorly understood. Presence of autoantibodies; infiltration of CD4+/CD8 + T cells, plasmacytes, and B cells; and expression of HLA-DR antigens in the pancreas [41] suggest that acquired immunity may be involved in the development of AIP. The CD4 + T cells differentiate from naïve T cells (Th0) to Th1, Th2, Th17, and regulatory T cells (Treg). IL-12 induces Thl cells, which produce IL-2, tumor necrosis factor (TNF)-alpha, and IFN-gamma; mediate cellular immunity, macrophage activation, and cytotoxicity; and help in B-cell production of opsonizing and complement-fixing antibodies [54]. IL-4 induces Th2 cells which produce IL-4, IL-5, IL-6, and IL-10 and promote humoral and allergic responses [59]. TGF-beta, IL-6, IL-21, and IL-23 induce Th17 cells, which secret IL-17 and may be involved in the inflammation in mice. In some patients with AIP, Th1 cells but not Th17 cells are predominant over Th2-type cells in the periphery. In the livers of IgG4-SC patients, however, a Th2-type immune reaction [60, 61] is induced in addition to a Th1 response [60, 62]. The discrepancy may be explained by the shift of Th2 cells from the periphery to local tissues or different disease stages. The mouse model with depletion of Tregs by neonatal thymectomy (nTx) [62] and the WBN/Kob model (Sakaguchi et al. 2002) support the hypothesis that Th1 cells mainly act as effectors in the initial early stage. Th2 cytokines may be involved in the progression of the disease process, especially the maturation and proliferation of local B cells and plasmacytes.
Regulatory T Cells
From naïve Th0 cells, TGF-beta can induce CD4+CD25+ regulatory T cells (Tregs), which have potent inhibitory function via the transcription factor Foxp3 to CD4+ T-cell-mediated immune responses such as Th1, Th2, and Th17 [61]. This suppressive function is mediated by transforming growth factor β (TGF-β) and IL-10, and/or cell-to-cell contact via ligation of CTLA-4. Foxp3 is a member of the forkhead/winged-helix family of transcriptional regulators and functions as the master regulator in the development and function of CD4+CD25+ regulatory T cells (Tregs) classified as naturally occurring CD4+CD25+ Tregs (nTregs) originating in the thymus and adaptive Tregs (aTregs) induced in the periphery by different antigens [63]. As Tregs expressing Foxp3 are critical in the transfer of immune tolerance, deficient Tregs induce various autoimmune diseases in animal studies [64]. In type 1 AIP, circulatory naïve (CD45RA+) Tregs are significantly decreased in the peripheral blood, whereas memory (CD45RA-) Tregs are significantly increased [33]. In addition, prominent infiltration of Tregs with upregulation of IL-10 is observed in the liver of type 1 AIP and IgG4-SC patients [31, 32]. Moreover, IL-10 and TGF-beta are secreted from ICOS+ and ICOS- inducible aTregs, respectively. These findings suggest that increased memory Tregs in the periphery and local tissues may be an inhibitory immune response against inflammation, although decreased naïve Tregs may be pathogenic.
Hypothesis for the Pathogenesis of Type 1 AIP
The neonatally thymectomized (nTx)-BALB/c mice models showed that immunization with CA-II or LF can induce pancreatitis, cholangitis, and sialadenitis similar to human IgG4-RD [62]. These findings suggest that depletion of naïve Tregs may induce macrophage/T-cell activation and further proinflammatory reactions may occur during the early stage of the disease as direct cytotoxicity effects through Fas ligand expression. WBN/Kob rat models with congenital decreased peripheral Tregs spontaneously develop sclerotic cholangitis, sialadenitis, thyroiditis, and tubulointerstitial nephritis [65]. These animal models suggest that CD4+/CD8+ T cells play major roles in the development of primary lesions similarly to human IgG4-related diseases; however, the counterpart of IgG4 in mice IgG subclasses has not been identified.
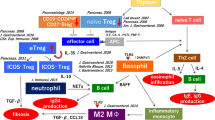
Based on these findings, we proposed the pathogenesis of type 1 AIP [8] (Fig. 3.2). The basic concept is the biphasic mechanism of “induction” and “progression.” An initial response to unknown disease-specific antigens including self-antigens (LF, CA-II, CA-IV, and PSTI) or microorganisms (bacteria or virus) might be induced by decreased naïve Tregs and/or CD19+CD24hiCD38hi Bregs followed by a Th1-type immune response with the release of proinflammatory cytokines (IFN-γ, IL-1beta, IL-2, TNF-α). In progression, Th2-type immune responses producing IgG, IgG4, and autoantibodies may be involved in pathophysiology. Production of IgG4 may be upregulated by increased IL-10 from ICOS+ inducible aTregs and CD19+CD24hiCD27hi Bregs and BAFF from monocytes and/or basophils. Fibrosis may be induced by TGF-beta secreted from ICOS-inducible aTregs.

Hypothesis for the pathogenesis of type 1 AIP as an IgG4-related disease (With permission from the publisher of Okazaki et al. [8]). In the central tolerance, naïve and natural regulatory T cells (Tregs) derived from the thymus suppress autoreactive CD4 or CD8 cells in the normal state. The basic concept is the biphasic mechanism of “induction” and “progression.” An initial response to unknown disease-specific antigens including self-antigens (LF, CA-II, CA-IV, and PSTI) or microorganisms (bacteria or virus) might be induced by decreased naïve Tregs and/or CD19+CD24hiCD38hi Bregs followed by a Th1-type immune response with the release of proinflammatory cytokines (IFN- γ, IL-1beta, IL-2, TNF-α). In progression, Th2-type immune responses producing IgG, IgG4, and autoantibodies may be involved in pathophysiology. Production of IgG4 may be upregulated by increased IL-10 from ICOS+ inducible aTregs and CD19+CD24hiCD27hi Bregs and BAFF from monocytes and/or basophils. Fibrosis may be induced by TGF-beta secreted from ICOS- inducible aTregs. iTreg inducible memory regulatory T cell, TE effector T cell, nTreg naturally occurring naive regulatory T cell, BAFF B-cell-activating factor family, Breg regulatory B cell, ICOS inhibitory costimulatory molecule
Conclusion
Recent advances support the concept of AIP and subtypes, type 1 and type 2 AIP. Type 1 AIP is a pancreatic manifestation of IgG4-RD. Although the pathogenic mechanism remains unclear, innate and acquired immunity, Tregs, and B cells may be involved in the development of type 1 AIP. Further studies are necessary to clarify the pathogenesis including genetic backgrounds, disease-specific antigens, and the role of IgG4.
References
Chari ST, Kloeppel G, Zhang L, et al. Histopathologic and clinical subtypes of autoimmune pancreatitis: the Honolulu consensus document. Pancreas. 2010;39:549–54.
Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. New Engl J Med. 2001;344:732–8.
Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas–an autonomous pancreatic disease? Am J Dig Dis. 1961;6:688–98.
Shimosegawa T, Chari ST, Frulloni L, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the international association of pancreatology. Pancreas. 2011;40(3):352–8.
Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40:1561–8.
Kawaguchi K, Koike M, Tsuruta K, Fujita N, et al. Lymphoplasmacytic sclerosing pancreatitis with cholangitis: a variant of primary sclerosing cholangitis extensively involving pancreas. Hum Pathol. 1991;22:387–95.
Umehara H, Okazaki K, Masaki Y, et al. A novel clinical entity, IgG4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22:1–14
Okazaki K, Uchida K, Koyabu M, et al. Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease. J Gastroenterol. 2011;46:277–88.
Klöppel G, Detlefsen S, Chari ST, et al. Autoimmune pancreatitis: the clinicopathological characteristics of the subtype with granulocytic epithelial lesions. J Gastroenterol. 2010;45:787–93.
Kawa S, Ota M, Yoshizawa K, et al. HLA DRB10405-DQB10401 haplotype is associated with autoimmune pancreatitis in the Japanese population. Gastroenterology. 2002;122(5):1264–9.
Park do H, Kim MH, Oh HB, et al. Substitution of aspartic acid at position 57 of the DQbeta1 affects relapse of autoimmune pancreatitis. Gastroenterology. 2008;134(2):440–6.
Kochi Y, Yamada R, Suzuki A, et al. A functional variant in FCRL3, encoding Fc receptor-like 3, is associated with rheumatoid arthritis and several autoimmunities. Nat Genet. 2005;37(5):478–85.
Umemura T, Ota M, Hamano H, Katsuyama Y, Kiyosawa K, Kawa S. Genetic association of Fc receptor-like 3 polymorphisms with autoimmune pancreatitis in Japanese patients. Gut. 2006;55(9):1367–8.
Umemura T, Katsuyama Y, Hamano H, et al. Association analysis of Toll-like receptor 4 polymorphisms with autoimmune pancreatitis. Hum Immunol. 2009;70(9):742–6.
Chang MC, Chang YT, Tien YW, et al. T-cell regulatory gene CTLA-4 polymorphism/haplotype association with autoimmune pancreatitis. Clin Chem. 2007;53(9):1700–5.
Umemura T, Ota M, Hamano H, et al. Association of autoimmune pancreatitis with cytotoxic T-lymphocyte antigen 4 gene polymorphisms in Japanese patients. Am J Gastroenterol. 2008;103(3):588–94.
Ueda H, Howson JM, Esposito L, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423(6939):506–11.
Watanabe T, Yamashita K, Fujikawa S, et al. Involvement of activation of toll-like receptors and nucleotide-binding oligomerization domain-like receptors in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis Rheum. 2012;64:914–24.
Watanabe T, Yamashita K, Sakurai T, Kudo M, Shiokawa M, Uza N, et al. Toll-like receptor activation in basophils contributes to the development of IgG4 related disease. J Gastroenterol. 2013;48:247–53.
Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med. 2009;361:2135–42.
Guarneri F, Guarneri C, Benvenga S. Helicobacter pylori and autoimmune pancreatitis: role of carbonic anhydrase via molecular mimicry? J Cell Mol Med. 2005;9(3):741–4.
Kountouras J, Zavos C, Chatzopoulos D. A concept on the role of Helicobacter pylori infection in autoimmune pancreatitis. J Cell Mol Med. 2005;9(1):196–207.
Kountouras J, Zavos C, Gavalas E, Tzilves D. Challenge in the pathogenesis of autoimmune pancreatitis: potential role of helicobacter pylori infection via molecular mimicry. Gastroenterology. 2007;133(1):368–9.
Haruta I, Yanagisawa N, Kawamura S, et al. A mouse model of autoimmune pancreatitis with salivary gland involvement triggered by innate immunity via persistent exposure to avirulent bacteria. Lab Invest. 2010;90:1757–69.
Nishio A, Asada M, Uchida K, et al. The role of innate immunity in the pathogenesis of experimental autoimmune pancreatitis in mice. Pancreas. 2011;40:95–102.
Yamashina M, Nishio A, Nakayama S, Okazaki T, Uchida K, Fukui T, et al. Comparative study on experimental autoimmune pancreatitis and its extrapancreatic involvement in mice. Pancreas. 2012;41:1255–62.
van der Neut Kolfschoten M, Schuurman J, Losen M, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317(5844):1554–7.
Kawa S, Kitahara K, Hamano H, et al. A novel immunoglobulin-immunoglobulin interaction in autoimmunity. PLoS One. 2008;3(2):e1637.
Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010–7.
Davies AM, Rispens T, Ooijevaar-de Heer P, et al. Structural determinants of unique properties of human IgG4-Fc. J Mol Biol. 2014;426:630–44.
Koyabu M, Uchida K, Miyoshi H, et al. Analysis of regulatory T cells and IgG4-positive plasma cells among patients of IgG4-related sclerosing cholangitis and autoimmune liver diseases. J Gastroenterol. 2010;45:732–41.
Kusuda T, Uchida K, Miyoshi H, et al. Involvement of inducible costimulator- and interleukin 10-positive regulatory T cells in the development of IgG4-related autoimmune pancreatitis. Pancreas. 2011;40:1120–30.
Miyoshi H, Uchida K, Taniguchi T, et al. Circulating naive and CD4 + CD25high regulatory T cells in patients with autoimmune pancreatitis. Pancreas. 2008;36:133–40.
Tanaka A, Moriyama M, Nakashima H, et al. Th2 and regulatory immune reactions contribute to IgG4 production and the initiation of Mikulicz disease. Arthritis Rheum. 2012;64:254–63.
Zen Y, Fujii T, Harada K, et al. Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology. 2007;45:1538–46.
Cornell LD, Chicano SL, Deshpande V, et al. Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol. 2007;31(10):1586–97.
Kawano M, Mizushima I, Yamaguchi Y, et al. Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: detailed analysis of 20 Japanese cases. Int J Rheumatol. 2012;2012:609795.
Takeda S, Haratake J, Kasai T, Takaeda C, Takazakura E. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant. 2004;19:474–6.
Muraki T, Hamano H, Ochi Y, et al. Autoimmune pancreatitis and complement activation system. Pancreas. 2006;32:16–21.
Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology. 2000;118:573–81.
Uchida K, Okazaki K, Konishi Y, et al. Clinical analysis of autoimmune-related pancreatitis. Am J Gastroenterol. 2000;95:2788–94.
Aparisi L, Farre A, Gomez-Cambronero L, et al. Antibodies to carbonic anhydrase and IgG4 levels in idiopathic chronic pancreatitis: relevance for diagnosis of autoimmune pancreatitis. Gut. 2005;54:703–9.
Nishi H, Tojo A, Onozato ML, et al. Anti-carbonic anhydrase II antibody in autoimmune pancreatitis and tubulointerstitial nephritis. Nephrol Dial Transplant. 2007;22:1273–5.
Nishimori I, Miyaji E, Morimoto K, et al. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut. 2005;54:274–81.
Asada M, Nishio A, Uchida K, et al. Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas. 2006;33:20–6.
Endo T, Takizawa S, Tanaka S, et al. Amylase alpha-2A autoantibodies: novel marker of autoimmune pancreatitis and fulminant type 1 diabetes. Diabetes. 2009;58:732–7.
Takizawa S, Endo T, Wanjia X, Tanaka S, Takahashi M, Kobayashi T. HSP 10 is a new autoantigen in both autoimmune pancreatitis and fulminant type 1 diabetes. Biochem Biophys Res Commun. 2009;386:192–6.
Nishimori I, Bratanova T, Toshkov I, et al. Induction of experimental autoimmune sialoadenitis by immunization of PL/J mice with carbonic anhydrase II. J Immunol. 1995;154(9):4865–73.
Ueno Y, Ishii M, Takahashi S, Igarashi T, Toyota T, LaRusso NF. Different susceptibility of mice to immune-mediated cholangitis induced by immunization with carbonic anhydrase II. Lab Invest. 1998;78(5):629–37.
Okazaki K, Uchida K, Matsushita M, Takaoka M. How to diagnose autoimmune pancreatitis by the revised Japanese clinical criteria. J Gastroenterol. 2007;42:32–8.
Nakanuma Y. A novel approach to biliary tract pathology based on similarities to pancreatic counterparts: is the biliary tract an incomplete pancreas? Pathol Int. 2010;60:419–29.
Wang Y, Lanzoni G, Carpino G, et al. Biliary tree stem cells, precursors to pancreatic committed progenitors: evidence for possible life-long pancreatic organogenesis. Stem Cells. 2013;31:1966–79.
Nakanuma Y, Harada K, Sasaki M, et al. Proposal of a new disease concept “biliary diseases with pancreatic counterparts”. Anatomical and pathological bases. Histol Histopathol. 2014;29:1–10.
Okazaki K. Autoimmune pancreatitis: etiology, pathogenesis, clinical findings and treatment. The Japanese experience. JOP. 2005;6(1 Suppl):89–96.
Khosroshahi A, Carruthers MN, Deshpande V, et al. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore). 2012;91:57–66.
Topazian M, Witzig TE, Smyrk TC, et al. Rituximab therapy for refractory biliary strictures in immunoglobulin G4-associated cholangitis. Clin Gastroenterol Hepatol. 2008;6:364–6.
Maillette de Buy Wenniger LJ, Doorenspleet ME, Klarenbeek PL, et al. Immunoglobulin G4+ clones identified by next-generation sequencing dominate the B cell receptor repertoire in immunoglobulin G4 associated cholangitis. Hepatology. 2013;57:2390–8.
Sumimoto K, Uchida K, Kusuda T, et al. The role of CD19 + CD24highCD38high and CD19 + CD24highCD27+ regulatory B cells in patients with type 1 autoimmune pancreatitis. Pancreatology. 2014. http://dx.doi.org/10.1016/j.pan.2014.02.004.
Roitt I. Antibodies. In: Roitt I, editor. Roitt’s essential immunology. 9th ed. London: Blackwell Science; 1997. p. 43–62.
McGeachy MJ, Cua DJ. The link between IL-23 and Th17 cell-mediated immune pathologies. Semin Immunol. 2007;19(6):372–6.
Oukka M. Interplay between pathogenic Th17 and regulatory T cells. Ann Rheum Dis. 2007;66 Suppl 3:iii87–90.
Uchida K, Okazaki K, Nishi T, et al. Experimental immune-mediated pancreatitis in neonatally thymectomized mice immunized with carbonic anhydrase II and lactoferrin. Lab Invest. 2002;82:411–24.
Valencia X, Lipsky PE. CD4 + CD25 + FoxP3+ regulatory T cells in autoimmune diseases. Nat Clin Pract Rheumatol. 2007;3(11):619–26.
Sakaguchi S, Fukuma K, Kuribayashi K, et al. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161:72–87.
Sakaguchi Y, Inaba M, Tsuda M, et al. The Wistar Bonn Kobori rat, a unique animal model for autoimmune pancreatitis with extrapancreatic exocrinopathy. Clin Exp Immunol. 2008;152:1–12.
Acknowledgment
This study was partially supported by (1) Grant-in-Aid for Scientific Research (C) of the Ministry of Culture and Science of Japan (20590810, 24591020, 12008507), (2) the Research Program on Intractable Diseases, from the Ministry of Labor and Welfare of Japan, and (3) Grants-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, from CREST Japan Science, and Technology Agency.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Okazaki, K., Uchida, K. (2015). Pathophysiology of Autoimmune Pancreatitis. In: Kamisawa, T., Chung, J. (eds) Autoimmune Pancreatitis. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-55086-7_3
Download citation
DOI: https://doi.org/10.1007/978-3-642-55086-7_3
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-55085-0
Online ISBN: 978-3-642-55086-7
eBook Packages: MedicineMedicine (R0)