
Abstract
Molybdenum (Mo) cofactor deficiency (MoCD) is characterized by neonatal seizures, high-pitch crying, convulsions, and abnormal EEG and MRI findings accompanied by rapidly progressing neurodegeneration. In the absence of treatment, patients usually die within the first years of life and show no neurodevelopmental improvement. The molecular cause of the disease is mainly due to the loss of sulfite oxidase activity, one out of four molybdenum cofactor-dependent enzymes. Sulfite oxidase catalyzes the terminal step in the oxidative degradation of cysteine; a loss of activity results in the accumulation of toxic sulfite, which in turn triggers the alteration of secondary-related metabolites such as S-sulfocysteine, thiosulfate, taurine, hypotaurine, and cystine. Xanthine oxidoreductase catalyzes the catabolism of purines from hypoxanthine to xanthine and further to uric acid, which is reduced in patients while xanthine and to a lesser extent hypoxanthine accumulate. The molybdenum cofactor (Moco) is synthesized by a three-step biosynthetic pathway, which involves gene products of the MOCS1, MOCS2, MOCS3, and GEPH loci. Depending on the mutation, type A, B, and C deficiencies are known. While MoCD types A and B are clinically indistinguishable, MoCD type C has a more severe neurological presentation due to the loss of synaptic inhibition, which is dependent on GEPHYRIN function. Dietary restriction (low cysteine and methionine) has been reported in some case, however, disease improvement was marginal. A first causative therapy has been established for MoCD type A patients and is based on the treatment with cyclic pyranopterin monophosphate, the first intermediate in the molybdenum cofactor pathway. Given the high neurotoxicity of sulfite and its related compounds, early diagnosis has been shown to be the key determinant in the treatment outcome. Patients that were treated shortly after birth and have not been exposed to extensive anticonvulsive therapy showed best clinical and neurodevelopmental outcome.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara SummaryMolybdenum (Mo) cofactor deficiency (MoCD) is characterized by neonatal seizures, high-pitch crying, convulsions, and abnormal EEG and MRI findings accompanied by rapidly progressing neurodegeneration. In the absence of treatment, patients usually die within the first years of life and show no neurodevelopmental improvement. The molecular cause of the disease is mainly due to the loss of sulfite oxidase activity, one out of four molybdenum cofactor-dependent enzymes. Sulfite oxidase catalyzes the terminal step in the oxidative degradation of cysteine; a loss of activity results in the accumulation of toxic sulfite, which in turn triggers the alteration of secondary-related metabolites such as S-sulfocysteine, thiosulfate, taurine, hypotaurine, and cystine. Xanthine oxidoreductase catalyzes the catabolism of purines from hypoxanthine to xanthine and further to uric acid, which is reduced in patients while xanthine and to a lesser extent hypoxanthine accumulate. The molybdenum cofactor (Moco) is synthesized by a three-step biosynthetic pathway, which involves gene products of the MOCS1, MOCS2, MOCS3, and GEPH loci. Depending on the mutation, type A, B, and C deficiencies are known. While MoCD types A and B are clinically indistinguishable, MoCD type C has a more severe neurological presentation due to the loss of synaptic inhibition, which is dependent on GEPHYRIN function. Dietary restriction (low cysteine and methionine) has been reported in some case, however, disease improvement was marginal. A first causative therapy has been established for MoCD type A patients and is based on the treatment with cyclic pyranopterin monophosphate, the first intermediate in the molybdenum cofactor pathway. Given the high neurotoxicity of sulfite and its related compounds, early diagnosis has been shown to be the key determinant in the treatment outcome. Patients that were treated shortly after birth and have not been exposed to extensive anticonvulsive therapy showed best clinical and neurodevelopmental outcome.
1 Introduction
In men, four molybdenum enzymes are known, each of them catalyzes either catabolic or detoxifying reactions in the body (Schwarz et al. 2009). Sulfite oxidase (SO) is localized in the intermembrane space of mitochondria and couples sulfite oxidation to the reduction of cytochrome c. Together with the recently identified mitochondrial amidoxime-reducing component (Havemeyer et al. 2006), which is believed to function in the reduction of N-hydroxylated prodrugs, they form a family of molybdenum enzyme characterized by a conserved protein-derived cysteine coordinating the molybdenum cofactor (Hille 1996). SO deficiency (see 3.6) has been reported in more than 30 cases so far and is very similar to MoCD (for details, see below). The other family of molybdenum enzymes is formed by xanthine oxidoreductase (XO) and aldehyde oxidase, catalyzing the hydroxylations of purine or other heterocyclic substrates, respectively (Schwarz et al. 2009). Their molybdenum center requires the addition of a terminal sulfido ligand, which is dependent on the Moco sulfurylase (MCSU) (Ichida et al. 2001). Consequently, a mutation in MCSU causes a deficiency of both enzymes, a disease named xanthinuria type II (see 41.9.1). While an isolated deficiency in aldehyde oxidase has not been reported in men, deficiencies in XO are well known and cause xanthinuria type I (see 41.9).
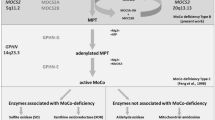
Moco deficiency (MoCD) is characterized by the simultaneous loss of all Mo-enzyme activities due to a mutational block in the biosynthesis of Moco, which can be divided into three major steps based on the two intermediates: cyclic pyranopterin monophosphate (cPMP) (Santamaria-Araujo et al. 2004), previously named precursor Z (Wuebbens and Rajagopalan 1993), and the metal-binding pterin or molybdopterin (MPT) (Johnson et al. 1984) (Scheme 1). Each of the steps involves the action of one or more proteins producing additional reaction intermediates such as pyranopterin triphosphate (Mehta et al. 2013), thio-pyranopterin phosphate (Wuebbens and Rajagopalan 2003), and adenylylated MPT (Kuper et al. 2004) (Scheme 1). Four genes have been found to encode for proteins essential for Moco biosynthesis in men. MOCS1 and MOCS2 each produce two or more proteins, being essential for steps one and two in Moco synthesis, respectively. MOCS3 is essential for the activation of MOCS2 proteins and GEPH encodes for a multifunctional protein functioning in Moco synthesis as well as glycine and GABA type A receptor clustering. A fifth gene (MCSU) is needed for the sulfuration of Moco.
Human MoCD is a rare autosomal recessive disorder, which mostly affects neonates and is characterized by progressive brain injury leading to early childhood death. More than 100 cases with MoCD have been reported (Johnson and Duran 2001; Reiss and Hahnewald 2011), which nearly all share a predominant deterioration of the central nervous system as main disease feature mimicking hypoxic-ischemic encephalopathy (Vijayakumar et al. 2011). Given the similarities to isolated SO deficiency, the major cause of neurodegeneration is sulfite accumulation. Besides a general cellular toxicity of sulfite, the formation of secondary metabolites, such as S-sulfocysteine (SCC), or the depletion of metabolites, such as cystine, is believed to contribute to neuronal dysfunction (Tan et al. 2005).
Mutations in patients with MoCD have been identified in three of the four Moco synthetic genes (Reiss et al. 1998a, b), and prenatal diagnosis in carrier families has been established (Reiss et al. 1999a). Today more than 64 disease-causing mutations are known in MOCS1, MOCS2, and GEPH (Reiss and Hahnewald 2011). In nearly all cases, disease-causing mutations result in a complete loss of enzyme function due to frameshifts, splice site and nonsense mutations, or missense mutations affecting highly conserved or invariant residues. Two thirds of all MoCD patients carry mutations (>40) in the Mocs1 gene (Reiss and Hahnewald 2011). Approximately one third of MoCD patients are affected in the second step of Moco synthesis; 23 disease-causing mutations have been found in both open reading frames affecting encoded by the MOCS2 gene. Interestingly, no MoCD patient with a MOCS3 mutation has been found so far, which might be due to an additional function of MOCS3 in tRNA thiolation (Chowdhury et al. 2012). In most MoCD cases, homozygous mutations are found due to the high consanguinity of heterozygous parents, while compound heterozygous cases are the minority.
Only two cases with GEPH mutations have been described so far. One patient was the last of three affected infants that all died in the neonatal period (day 12, 29, and 3, respectively), with symptoms of MoCD (Reiss et al. 2001). Genetic analysis revealed an early stop codon resulting in a total loss of GEPHYRIN expression. As a result, both functions of GEPYHRIN, Moco synthesis as well as receptor clustering, were impaired. A second case with a missense mutation in GEPH affected the catalytic site of GEPHYRIN E-domain (Reiss et al. 2011). Given that the binding site of both glycine (Schrader et al. 2004) and GABA receptors (Maric et al. 2011) is distinct from Moco synthesis (Llamas et al. 2006), an isolated MoCD can be assumed in this case, as confirmed by typical symptoms of MoCD.
The vast majority of MoCD patients present a very severe neurological phenotype. Only a handful cases have been reported with mild forms of the disease. To our knowledge, only one mild presentation was reported for MoCD type A deficiency (Arenas et al. 2009) affecting the splicing of MOCS1 exon 9 probably affecting the mitochondrial maturation and/or targeting of MOCS1AB translation products thus leading to a reduced but not completely absent cPMP-synthetic activity (G. Schwarz, 2013). All other mild cases carry either mutations in MOCS2 thus showing only partially impaired molybdopterin synthesis (Johnson et al. 2001) (G. Schwarz, 2013) or mutations in the SUOX gene (Barbot et al. 1995; Del Rizzo et al. 2013; Touati et al. 2000).
Two MoCD animal models are currently available. The gephyrin knockout mouse develops both symptoms of impaired synaptic inhibition and MoCD (Feng et al. 1998). Geph –/– neonates appeared externally normal and failed to suckle, and following mild tactile stimuli, they retained rigid with a hyperextended posture and exhibited apnea; animals usually died within 12 h after birth due to respiratory failure. Due to the high prevalence of MoCD type A, a Mocs1 knockout mouse was generated (Lee et al. 2002). Mocs1 -/- mice display a severe phenotype characterized by a retarded growth, abnormal behavior, lack of feeding, and death within the first 11 days of life (Lee et al. 2002). As observed in humans, biochemical characterization of mocs1 −/− mice revealed elevated urinary sulfite and xanthine levels, uric acid was undetectable, and Mo-enzyme activities were absent.
A substitution therapy was established in mocs1 –/– mice showing a phenotypic normalization and reconstitution of Mo-enzyme activities following repeated intrahepatic injections of purified cPMP (Schwarz et al. 2004). Improvement of treated animals was directly correlated with cPMP injections as withdrawal of cPMP caused death within 10–14 days following the final injection, and subsequent reduction in Mo-enzyme activities was observed. Treated animals showed normal behavior and were fertile.
Given the positive results in mocs1 –/– mice, a first treatment attempt was initiated in a MoCD type A patient. Treatment was started at day 36 of life with a starting dose of 80 μg/kg body weight per intravenous infusion (Veldman et al. 2010). Urinary markers of SO and XO deficiency returned within days to almost normal readings. Clinically, the patient became more alert a few days after treatment was started; convulsions and twitching disappeared as documented by an electroencephalogram showing the return of rhythmic elements and markedly reduced epileptiform discharges (Veldman et al. 2010).
2 Nomenclature
No. | Disorder | Alternative name | Abbreviation | Gene symbol | Chromosomal localization | Affected protein | OMIM no. | Subtype |
|---|---|---|---|---|---|---|---|---|
12.1 | Molybdenum cofactor deficiency A | MoCo deficiency, complementation group A | MoCD type A | MOCS1 | 6p21.3 | MOCS1A, MOCS1AB | 603707 | All forms |
12.2 | Molybdenum cofactor deficiency B | MoCo deficiency, complementation group B | MoCD type B | MOCS2 | 603708 | MOCS2A, MOCS2B | 603708 | All forms |
12.3 | Molybdenum cofactor deficiency C | MoCo deficiency, complementation group C | MoCD type C | GEPH | 14q23.3 | GEPHYRIN | 603930 | All forms |
3 Metabolic Pathway
Moco Biosynthesis
Proteins encoded by the MOCS1 gene catalyze the first step in Moco biosynthesis (Fig. 12.1), the conversion of GTP into cPMP. MOCS1 was first reported to produce a bicistronic transcript encoding for two open reading frames, MOCS1A and MOCS1B (Reiss et al. 1998b); however, later studies demonstrated that this transcript only encodes for functional MOCS1A, which catalyzes the ring opening reaction of GTP (Hänzelmann et al. 2002). MOCS1A is believed to use the same kind of chemistry as shown for its bacterial orthologue, MoaA, an S-adenosylmethionine (SAM)-dependent radical enzyme containing two [4Fe-4S] clusters (Hänzelmann and Schindelin 2004). Alternative splicing of MOCS1 was found to produce two other types of transcripts with a single open reading frame encoding for MOCS1AB fusion proteins (Gray and Nicholls 2000). These proteins only harbor MOCS1B activity due to the truncation of functionally important C-terminal residues within the C-terminus of MOCS1A (Hänzelmann et al. 2002). MOCS1A is believed to initiate the transformation of 5′-GTP by abstracting the 3′ proton from the ribose resulting in the formation of pyranopterin triphosphate (Mehta et al. 2013), which is subsequently converted into cPMP by the activity of MOCS1AB.
The second step in Moco biosynthesis (Fig. 12.1) is catalyzed by MPT synthase, which converts cPMP into MPT and is encoded by the bicistronic MOCS2 gene. The two overlapping open reading frames (MOCS2A and MOCS2B) are translated by a ribosomal leaky scanning mechanism producing both subunits in an approximately equimolar ratio (Stallmeyer et al. 1999). MOCS3 encodes for the Moco sulfurase (Matthies et al. 2004), which is required for the ATP-dependent thiolation of MOCS2A.
The third and final step in Moco synthesis (Fig. 12.1) involves the adenylylation of MPT (forming MPT-AMP) and subsequent molybdate-dependent hydrolysis of MPT-AMP resulting in Moco. Both reactions are dependent on GEPHYRIN, which encodes for a multi-domain cytosolic protein composed of an N-terminal G-domain (GEPH-G, MPT-AMP synthesis), a central C-domain, and a C-terminal E-domain (GEPH-E, molybdenum insertion, Scheme 1). Besides Moco biosynthesis, GEPHYRIN functions as cytosolic membrane-associated receptor-clustering protein, being essential for the formation of inhibitory synapses (Fritschy et al. 2008). The GEPH gene is highly mosaic with 27 exons distributed over 760 kb of genomic DNA on chromosome 14q32. At least six of these exons are subject to alternative splicing producing more than ten different splice variants (Rees et al. 2003).

Moco biosynthesis, Mo enzymes, and deficiencies. Intermediates of Moco synthesis are cyclic pyranopterin monophosphate (cPMP), metal-binding pterin (MPT), and adenylylated MPT (MPT-AMP). Moco undergoes an additional sulfuration prior to its incorporation into xanthine oxidase and aldehyde oxidase. In the absence of this function, xanthinuria type II is developed (41.9.1). Proteins involved in Moco synthesis are shown. Note that the individual domains of GEPHYRIN catalyze two subsequent steps in Moco synthesis. Deficiencies in MOCS1, MOCS2, and GEPH cause MoCD type A (12.1), type B (12.2), and type C (12.3), respectively. Isolated deficiencies in SO and XO cause 3.6 and 41.9. For details, see the relevant chapters
Cysteine Catabolism, Sulfite Toxicity, and Altered Metabolites
MoCD is clinically very similar to the less prevalent isolated SO deficiency implicating sulfite toxicity as a major underlying cause for neurodegeneration in MoCD patients. SO, which is localized in the mitochondrial intermembrane space, oxidizes sulfite to sulfate; thus, no accumulation of sulfite in the cytosol or extracellular compartments is seen (Scheme 2). In MoCD and SO deficiency, sulfite accumulates first in mitochondria where it has been shown to increase reactive oxygen species (Zhang et al. 2004). Sulfite also decreases ATP synthesis in mitochondria when respiring on glutamate, while other respiratory substrates such as malate, α-ketoglutarate, and succinate did not show any sulfite vulnerability (Zhang et al. 2004). The mechanism underlying the inhibition of ATP synthesis reduction has been related to glutamate dehydrogenase inhibition by sulfite, which in the brain, due to the fact that glutamate dehydrogenase operates in the direction of oxidative deamination, will lead to a decrease in the availability of α-ketoglutarate and other tricarboxylic acids, resulting in an overall decrease in ATP synthesis. Inhibition of glutamate dehydrogenase may also affect the metabolism of other neurotransmitters, such as GABA, contributing to the accelerated injury in neuronal rather than nonneuronal tissue, as observed in MoCD.
Sulfite is a strong reductant and will therefore reduce disulfide bridges, primarily in membrane and extracellular proteins thus affecting their folding, stability, and function. Probably first, sulfite reacts with cystine leading to the formation of the secondary metabolite S-sulfocysteine (SSC, 12.2) (Rupar et al. 1996). SSC is very abundant in MoCD patients, and its excretion in urine is detectable shortly after birth and increases with age (Veldman et al. 2010), which supports the view that sulfite is cleared during maternal gestation (Belaidi et al. 2012; Reiss et al. 2005). SSC is structurally similar to glutamate, is able to bind to NMDA receptors, and therefore postulated as main agent responsible for seizure development and subsequent brain damage in MoCD (Gorman and Griffiths 1994; Olney et al. 1975). In fact, early studies in rats demonstrated that subcutaneous administration of SSC induces the same type of brain damage that glutamate and other excitatory amino acids are known to cause (Olney et al. 1975). In contrast to the neurotransmitter glutamate, whose release in the extracellular compartment is highly controlled by vesicular fusion and cellular reuptake, SSC is continuously produced by sulfite accumulation. In the absence of a specific transporter, SSC is assumed to persist in the extracellular compartment and/or inhibits the uptake of glutamate (Dunlop et al. 1991) thus potentiating its excitotoxic effect leading to neuronal death. Besides SSC, taurine is also elevated in MoCD (Belaidi and Schwarz 2013) and known to be neuroprotective by playing an important role in glutamate and GABA signaling (El Idrissi and Trenkner 1999); however, this positive effect seems to be erased by SSC toxicity.
SSC formation goes hand in hand with cystine depletion (Barbot et al. 1995; Johnson and Duran 2001). Cystine is the major transport form of cysteine in plasma (Fig. 12.2). In the brain, cystine is taken up into glial cells, where it is reduced and incorporated into glutathione (GSH), the major antioxidant in neuronal tissue and most abundant low molecular weight thiol in animal cells (0.5–10 mM) (Wu et al. 2004). An increase in cysteine supply via oral or intravenous administration enhances GSH synthesis and prevents GSH deficiency in humans (Townsend et al. 2003). Thus, cysteine is generally considered to be the limiting amino acid for GSH synthesis in humans, rats, and pigs (Chung et al. 1990; Jahoor et al. 1999), and its depletion will have a major impact on cell viability. Despite the dramatically reduced levels of plasma cystine in MoCD, no information is available regarding GSH concentrations in affected patients, which together with SSC, cystine, and glutamate levels in cerebrospinal fluid may further contribute to the understanding of the underlying neurodegeneration in MoCD.
Besides oxidative cysteine catabolism, which usually contributes to 80–90 % of sulfur elimination, there is also a nonoxidative degradation, which involves the contribution of one of the three enzymes, cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), and 3-mercaptopyruvate sulfurtransferase (MSPT, Fig. 12.2). All three enzymes have been shown to be involved in cysteine-dependent production of hydrogen sulfide (Kamoun 2004), which was considered to be toxic as reported in several poisoning cases (Nam et al. 2004), while other studies suggest a functional role as neural messenger (Baranano et al. 2001). Hydrogen sulfide is further oxidized to thiosulfate in mitochondria by three sequential enzymatic reactions (Fig. 12.2) (Hildebrandt and Grieshaber 2008). First, the mitochondrial membrane flavoprotein-quinone oxidoreductase (SQR) converts sulfide to a protein-bound persulfide and transfers two electrons to the ubiquinone pool (Kabil and Banerjee 2010). Next, the persulfide is handed over to a sulfur dioxygenase, which converts the persulfide molecule to sulfite using molecular oxygen. Finally, a sulfur transferase adds a second persulfide molecule from SQR to sulfite yielding the final product thiosulfate (Hildebrandt and Grieshaber 2008). The relative contribution of the nonoxidative pathway in cysteine catabolism is usually low and insensitive to cysteine dietary intake (Bagley and Stipanuk 1994), while nonoxidative cysteine catabolism is increased in case of MoCD or SO deficiency as observed by high levels of thiosulfate excretion. A similar disbalance can be seen if CDO activity is lost as observed in the CDO -/- mice (Ueki et al. 2011), showing to some degree similar symptoms to another animal model, Ethe1 −/− mice, that accumulate both sulfide and thiosulfate (Tiranti et al. 2009).
MoCD patients have been reported to present homocysteinemia (Graf et al. 1998; Sass et al. 2003) suggesting a general reduction in methionine as well as S-adenosylmethionine levels. The molecular cause of these upstream alterations in MoCD are not understood, but might reflect a general “leak” of cysteine due to SSC accumulation, which subsequently alters the methionine-cysteine balance. Furthermore, recent studies demonstrated a sulfite-dependent reduction of pyridoxal 5′-phosphate (PLP) in cerebrospinal fluid (CSF) (Footitt et al. 2011). Sulfite is one of the nucleophiles known to be able to react with PLP. As a result of reduced PLP levels, the excretion of other metabolites such as α-aminoadipic semialdehyde (Mills et al. 2012).

Cysteine catabolism and S-containing metabolites that are altered in MoCD and SO deficiency. Transsulfuration pathway, oxidative and nonoxidate cysteine catabolism are summarized with the involved enzymes and metabolites. Changes in MoCD are highlighted in blue with corresponding arrows indicating an increase or decrease in concentration in comparison to control individuals. Enzyme abbreviations are as follows: MAT methionine-S-adenosyl transferase, MT methyltransferase, SAHH S-adenosylhomocysteine hydrolase, BHMT betaine-homocysteine methyltransferase, MT methionine synthase, CBS cystathionine β-synthase, CSE cystathionine γ-lyase (cystathionase), GCS γ-glutamylcysteine synthetase, GS glutathione synthetase, CDO cysteine dioxygenase, CSD cysteinesulfinate decarboxylase, AAT aspartate aminotransferase, SO sulfite oxidase, MPST 3-mercaptopyruvate sulfurtransferase, SQR quinone oxidoreductase, SDO sulfur dioxygenase, and ST sulfur transferase
4 Signs and Symptoms
The first human patient with MoCD was described in 1978 (Duran et al. 1978) and was presented in his neonatal period with initial feeding difficulties, therapy-resistant seizures, high-pitch crying followed by severe neurological abnormalities, lens dislocation of the eyes, and major dysmorphic features of the head. At the time of identification, the chemical nature of Moco was not known, neither its biosynthesis. Based on the identified alterations in the biomarkers of two Mo-dependent enzymes, XO and SO, a defect in either molybdenum metabolism or transport has been proposed (Duran et al. 1978). Since then, more than 100 cases with MoCD have been reported (Johnson and Duran 2001; Reiss and Hahnewald 2011), which nearly all share a predominant deterioration of the central nervous system as main disease feature mimicking hypoxic-ischemic encephalopathy (Vijayakumar et al. 2011). In general, first symptoms are observed within the first days of life, which are initially presented by feeding difficulties accompanied by intractable seizures with a predominant opisthotonus and an exaggerated startle reaction (Reiss and Hahnewald 2011). Disease progression is accompanied by psychomotor retardation due to progressive cerebral atrophy and ventricular dilatation, which are typical in brain MRI of patients. In addition, major radiological features of the disease include global cerebral edema, cystic encephalomalacia, cortical and white matter atrophy, focal or bilateral changes within the globi pallidi and subthalamic regions, and dysgenesis of the corpus callosum and ventriculomegaly (Arslanoglu et al. 2001; Bayram et al. 2013; Vijayakumar et al. 2011). Patients that survive the neonatal period show essentially no neuronal development, are unable to make coordinated movements, require tube feeding and show no signs of communication with their environment, and usually die within their first years of life (Johnson and Duran 2001).
5 Reference and Pathological Values
See also disorder 41.9
6 Diagnosis/Diagnostic Flowchart

Flowchart to diagnose MoCD
7 Specimen Collection
Specimen collection as early as possible in those cases in which a treatment attempt with cPMP (currently as experimental drug) is undertaken, specimen to be collected prior to the first dose of cPMP.
Test | Sample requirement |
|---|---|
Sulfite dipstick | Fresh urine, bedside dipstick test |
S-sulfocysteine in urine | Urine, frozen 1 ml |
S-sulfocysteine in plasma | EDTA blood, 1 ml |
Uric acid in urine | Urine |
Uric acid in plasma | EDTA blood, 1 ml |
Xanthine in urine | Urine |
Xanthine in plasma | EDTA blood, 1 ml |
8 Prenatal Diagnosis Table for All Disorders and Sample Requirement
Disorder | Material | Timing/trimester |
|---|---|---|
All MoCD types | Chorionic villus cells | I |
Cultured amniocytes | II | |
Amniotic fluid (might be positive for SSC, unclear diagnostic accuracy) | III |
9 DNA Testing Table for All Disorders and Sample Requirement
In MoCD patients, mutations have been found in MOCS1, MOCS2, and GEPH; no mutations were found in the MOCS3 gene. Given the highest frequency of MOCS1 mutations, analysis of this gene is suggested before testing for the MOCS2 gene. Only if no mutation has been found in either of the two MOCS genes, sequence analysis of GEPH is suggested. Primers and protocols have been published previously by Reiss et al. (2001, 1998b, 1999b).
10 Treatment Summary
Up to today, there is no approved specific therapy for MoCD available, and the prognosis remains poor with most affected patients dying in infancy and early childhood. Some few long-term survivors with a delayed onset of symptoms have been described (Johnson and Duran 2001).
Treatment in all age groups is mainly focused on seizure control, which is notoriously difficult, and other symptomatic and supportive approaches. In the early neonatal period, prolonged apnea, probably a result of status epilepticus, can make mechanical ventilation necessary, and some few patients might need mild ionotropic support for low blood pressure. Nasogastric feeding tubes or percutaneous gastrostomy tubes for feeding are almost always needed. Later in life, botulinum toxin injections can become indicated in the treatment of spastic quadriplegia.
Some reports on sulfur-restricted diet document marginal to moderate improvement in seizure control. Pyridoxine supplementation has been described to improve seizure control in selected patients (Boles et al. 1993; Del Rizzo et al. 2013; Touati et al. 2000).
Recently, replacement therapy with cyclic pyranopterin monophosphate (cPMP) has been described in patients with MoCD type A since this subtype of MoCD features a mutational block in the Moco biosynthesis upstream of cPMP (Veldman et al. 2010). Preliminary data indicate that if this therapy is initiated within the first week of life, significant improvement in neurodevelopmental outcome is possible (Hitzert et al. 2012). cPMP is currently available in Compasionate Use programs only, but clinical trials are expected to commence in the very near future. Reference to www.clinicaltrials.gov is recommended for most recent updates on this development.
MoCD is characterized by the enormous neurotoxicity of sulfite and related metabolites. This and secondary neuronal injury by continuous seizures and status epilepticus together with high-dose anticonvulsive therapy is believed to be major cause of rapidly progressing neurodegeneration. As a result, very early diagnosis and start of treatment is required to preserve significant neurocognitive function.
With cPMP emerging as a potential causative therapy at least for a subset of patients, sulfite removal by hemofiltration has been tried as a bridge to therapy; results of the efficacy and safety of this intervention are pending (J. Aschner et al. 2012, Vanderbuilt University, personal communication). Also, in patients in whom prenatal diagnosis is available, early delivery to prevent the risk of in utero brain damage by elevated sulfite levels in late pregnancy might be considered in individual cases.
Emergency Treatment Table for All Disorders and Medication Requirements
In the event, cPMP therapy will be an option, hemofiltration may be considered as emergency treatment option. During hemofiltration, careful monitoring of anticonvulsant plasma levels is required.
Standard Treatment Table for All Disorders and Medication Requirements
Anticonvulsive therapy should be performed according to local standards and supportive ICU treatment.
Experimental Treatment Table for All Disorders and Medication Requirements
Currently, experimental cPMP therapy is the only available causative treatment for MoCD type A patients. Patients that meet the criteria summarized in Table Requirements for cPMP treatment can be considered for treatment.
Following treatment initiation additional analyses are required to justify continuation.
References
Arenas M, Fairbanks LD, Vijayakumar K, Carr L, Escuredo E, Marinaki AM (2009) An unusual genetic variant in the MOCS1 gene leads to complete missplicing of an alternatively spliced exon in a patient with molybdenum cofactor deficiency. J Inherit Metab Dis 32:560–569
Arslanoglu S, Yalaz M, Goksen D, Coker M, Tutuncuoglu S, Akisu M, Darcan S, Kultursay N, Ciris M, Demirtas E (2001) Molybdenum cofactor deficiency associated with Dandy-Walker complex. Brain Dev 23:815–818
Bagley PJ, Stipanuk MH (1994) The activities of rat hepatic cysteine dioxygenase and cysteinesulfinate decarboxylase are regulated in a reciprocal manner in response to dietary casein level. J Nutr 124:2410–2421
Bamforth FJ, Johnson JL, Davidson AGF, Wong LTK, Lockitsch G, Applegrath DA (1990) Biochemical investigation of a child with molybdenum deficiency. Clin Biochem 23:537–542
Baranano DE, Ferris CD, Snyder SH (2001) Atypical neural messengers. Trends Neurosci 24:99–106
Barbot C, Martins E, Vilarinho L, Dorche C, Cardoso ML (1995) A mild form of infantile isolated sulphite oxidase deficiency. Neuropediatrics 26:322–324
Bayram E, Topcu Y, Karakaya P, Yis U, Cakmakci H, Ichida K, Kurul SH (2013) Molybdenum cofactor deficiency: review of 12 cases (MoCD and review). Eur J Paediatr Neurol 17:1–6
Belaidi AA, Schwarz G (2013) Molybdenum cofactor deficiency: metabolic link between taurine and S-sulfocysteine. Adv Exp Med Biol 776:13–19
Belaidi AA, Arjune S, Santamaria-Araujo JA, Sass JO, Schwarz G (2012) Molybdenum cofactor deficiency: a new HPLC method for fast quantification of s-sulfocysteine in urine and serum. JIMD Rep 5:35–43
Boles RG, Ment LR, Meyn MS, Horwich AL, Kratz LE, Rinaldo P (1993) Short-term response to dietary therapy in molybdenum cofactor deficiency. Ann Neurol 34:742–744
Chowdhury MM, Dosche C, Lohmannsroben HG, Leimkuhler S (2012) Dual role of the molybdenum cofactor biosynthesis protein MOCS3 in tRNA thiolation and molybdenum cofactor biosynthesis in humans. J Biol Chem 287:17297–17307
Chung TK, Funk MA, Baker DH (1990) L-2-oxothiazolidine-4-carboxylate as a cysteine precursor: efficacy for growth and hepatic glutathione synthesis in chicks and rats. J Nutr 120:158–165
Del Rizzo M, Burlina AP, Sass JO, Beermann F, Zanco C, Cazzorla C, Bordugo A, Giordano L, Manara R, Burlina AB (2013) Metabolic stroke in a late-onset form of isolated sulfite oxidase deficiency. Mol Genet Metab 108:263–266
Dunlop J, Fear A, Griffiths R (1991) Glutamate uptake into synaptic vesicles – inhibition by sulphur amino acids. Neuroreport 2:377–379
Duran M, Beemer FA, van de Heiden C, Korteland J, de Bree PK, Brink M, Wadman SK, Lombeck I (1978) Combined deficiency of xanthine oxidase and sulphite oxidase: a defect of molybdenum metabolism or transport? J Inherit Metab Dis 1:175–178
El Idrissi A, Trenkner E (1999) Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J Neurosci 19:9459–9468
Feng G, Tintrup H, Kirsch J, Nichol MC, Kuhse J, Betz H, Sanes JR (1998) Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity [see comments]. Science 282:1321–1324
Footitt EJ, Heales SJ, Mills PB, Allen GF, Oppenheim M, Clayton PT (2011) Pyridoxal 5′-phosphate in cerebrospinal fluid; factors affecting concentration. J Inherit Metab Dis 34:529–538
Fritschy JM, Harvey RJ, Schwarz G (2008) Gephyrin: where do we stand, where do we go? Trends Neurosci 31:257–264
Gorman A, Griffiths R (1994) Sulphur-containing excitatory amino acid-stimulated inositol phosphate formation in primary cultures of cerebellar granule cells is mediated predominantly by N-methyl-D-aspartate receptors. Neuroscience 59:299–308
Graf WD, Oleinik OE, Jack RM, Weiss AH, Johnson JL (1998) A homocysteinemia in molybdenum cofactor deficiency. Neurology 51:860–862
Gray TA, Nicholls RD (2000) Diverse splicing mechanisms fuse the evolutionarily conserved bicistronic MOCS1A and MOCS1B open reading frames. RNA 6:928–936
Hänzelmann P, Schindelin H (2004) Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. Proc Natl Acad Sci U S A 101:12870–12875
Hänzelmann P, Schwarz G, Mendel RR (2002) Functionality of alternative splice forms of the first enzymes involved in human molybdenum cofactor biosynthesis. J Biol Chem 277:18303–18312
Havemeyer A, Bittner F, Wollers S, Mendel R, Kunze T, Clement B (2006) Identification of the missing component in the mitochondrial benzamidoxime prodrug-converting system as a novel molybdenum enzyme. J Biol Chem 281:34796–34802
Hildebrandt TM, Grieshaber MK (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J 275:3352–3361
Hille R (1996) The mononuclear molybdenum enzymes. Chem Rev 96:2757–2816
Hitzert MM, Bos AF, Bergman KA, Veldman A, Schwarz G, Santamaria-Araujo JA, Heiner-Fokkema R, Sival DA, Lunsing RJ, Arjune S et al (2012) Favorable outcome in a newborn with molybdenum cofactor type A deficiency treated with cPMP. Pediatrics 130:e1005–e1010
Ichida K, Matsumura T, Sakuma R, Hosoya T, Nishino T (2001) Mutation of human molybdenum cofactor sulfurase gene is responsible for classical xanthinuria type II. Biochem Biophys Res Commun 282:1194–1200
Jahoor F, Jackson A, Gazzard B, Philips G, Sharpstone D, Frazer ME, Heird W (1999) Erythrocyte glutathione deficiency in symptom-free HIV infection is associated with decreased synthesis rate. Am J Physiol 276:E205–E211
Johnson JL, Duran M (2001) Molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. In: Scriver C, Beaudet A, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 3163–3177
Johnson JL, Hainline BE, Rajagopalan KV, Arison BH (1984) The pterin component of the molybdenum cofactor. Structural characterization of two fluorescent derivatives. J Biol Chem 259:5414–5422
Johnson JL, Coyne KE, Rajagopalan KV, Van Hove JL, Mackay M, Pitt J, Boneh A (2001) Molybdopterin synthase mutations in a mild case of molybdenum cofactor deficiency. Am J Med Genet 104:169–173
Kabil O, Banerjee R (2010) Redox biochemistry of hydrogen sulfide. J Biol Chem 285:21903–21907
Kamoun P (2004) Endogenous production of hydrogen sulfide in mammals. Amino Acids 26:243–254
Kuper J, Llamas A, Hecht HJ, Mendel RR, Schwarz G (2004) Structure of molybdopterin-bound Cnx1G domain links molybdenum and copper metabolism. Nature 430:803–806
Lee H-J, Adham IM, Schwarz G, Kneussel M, Sass J-O, Engel W, Reiss J (2002) Molybdenum cofactor-deficient mice resemble the phenotype of human patients. Hum Mol Genet 11:3309–3317
Llamas A, Otte T, Multhaup G, Mendel RR, Schwarz G (2006) The mechanism of nucleotide-assisted molybdenum insertion into molybdopterin. A novel route toward metal cofactor assembly. J Biol Chem 281:18343–18350
Maric HM, Mukherjee J, Tretter V, Moss SJ, Schindelin H (2011) Gephyrin-mediated gamma-aminobutyric acid type a and glycine receptor clustering relies on a common binding site. J Biol Chem 286:42105–42114
Matthies A, Rajagopalan KV, Mendel RR, Leimkuhler S (2004) Evidence for the physiological role of a rhodanese-like protein for the biosynthesis of the molybdenum cofactor in humans. Proc Natl Acad Sci U S A 101:5946–5951
Mehta AP, Hanes JW, Abdelwahed SH, Hilmey DG, Hanzelmann P, Begley TP (2013) Catalysis of a new ribose carbon-insertion reaction by the molybdenum cofactor biosyn-thetic enzyme MoaA. Biochemistry 52(7):1134–1136
Mills PB, Footitt EJ, Ceyhan S, Waters PJ, Jakobs C, Clayton PT, Struys EA (2012) Urinary AASA excretion is elevated in patients with molybdenum cofactor deficiency and isolated sulphite oxidase deficiency. J Inherit Metab Dis 35:1031–1036
Nam B, Kim H, Choi Y, Lee H, Hong ES, Park JK, Lee KM, Kim Y (2004) Neurologic sequela of hydrogen sulfide poisoning. Ind Health 42:83–87
Olney JW, Misra CH, de Gubareff T (1975) Cysteine-S-sulfate: brain damaging metabolite in sulfite oxidase deficiency. J Neuropathol Exp Neurol 34:167–177
Rees MI, Harvey K, Ward H, White JH, Evans LI, Duguid IC, Hsu CC, Coleman SL, Miller J, Baer K et al (2003) Isoform heterogeneity of the human gephyrin gene (GPHN), binding domains to the glycine receptor and mutation analysis in hyperekplexia. J Biol Chem 278:24688–24696
Reiss J, Hahnewald R (2011) Molybdenum cofactor deficiency: mutations in GPHN, MOCS1, and MOCS2. Hum Mutat 32:10–18
Reiss J, Christensen E, Kurlemann G, Zabot M-T, Dorche C (1998a) Genomic structure and mutational spectrum of the bicistronic MOCS1 gene defective in molybdenum cofactor deficiency type A. Hum Genet 103:639–644
Reiss J, Cohen N, Dorche C, Mandel H, Mendel RR, Stallmeyer B, Zabot MT, Dierks T (1998b) Mutations in a polycistronic nuclear gene associated with molybdenum cofactor deficiency. Nat Genet 20:51–53
Reiss J, Christensen E, Dorche C (1999a) Molybdenum cofactor deficiency: first prenatal genetic analysis. Prenat Diagn 19:386–388
Reiss J, Dorche C, Stallmeyer B, Mendel RR, Cohen N, Zabot MT (1999b) Human molybdopterin synthase gene: genomic structure and mutations in molybdenum cofactor deficiency type B. Am J Hum Genet 64:706–711
Reiss J, Gross-Hardt S, Christensen E, Schmidt P, Mendel RR, Schwarz G (2001) A mutation in the gene for the neurotransmitter receptor-clustering protein gephyrin causes a novel form of molybdenum cofactor deficiency. Am J Hum Genet 68:208–213
Reiss J, Bonin M, Schwegler H, Sass JO, Garattini E, Wagner S, Lee HJ, Engel W, Riess O, Schwarz G (2005) The pathogenesis of molybdenum cofactor deficiency, its delay by maternal clearance, and its expression pattern in microarray analysis. Mol Genet Metab 85:12–20
Reiss J, Lenz U, Aquaviva-Bourdain C, Joriot-Chekaf S, Mention-Mulliez K, Holder-Espinasse M (2011) A GPHN point mutation leading to molybdenum cofactor deficiency. Clin Genet 80:598–599
Reynolds AP, Harkness RA (1991) Urinary thiosulphate/creatinine concentration ratio in hospitalized children. J Inherit Metab Dis 14:938–939
Rupar CA, Gillett J, Gordon BA, Ramsay DA, Johnson JL, Garrett RM, Rajagopalan KV, Jung JH, Bacheyie GS, Sellers AR (1996) Isolated sulfite oxidase deficiency. Neuropediatrics 27:299–304
Santamaria-Araujo JA, Fischer B, Otte T, Nimtz M, Mendel RR, Wray V, Schwarz G (2004) The tetrahydropyranopterin structure of the sulfur-free and metal-free molybdenum cofactor precursor. J Biol Chem 279:15994–15999
Sass JO, Kishikawa M, Puttinger R, Reiss J, Erwa W, Shimizu A, Sperl W (2003) Hypohomocysteinaemia and highly increased proportion of S-sulfonated plasma transthyretin in molybdenum cofactor deficiency. J Inherit Metab Dis 26:80–82
Schrader N, Kim EY, Winking J, Paulukat J, Schindelin H, Schwarz G (2004) Biochemical characterization of the high affinity binding between the glycine receptor and gephyrin. J Biol Chem 279:18733–18741
Schwarz G, Santamaria-Araujo JA, Wolf S, Lee HJ, Adham IM, Grone HJ, Schwegler H, Sass JO, Otte T, Hanzelmann P et al (2004) Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli. Hum Mol Genet 13:1249–1255
Schwarz G, Mendel RR, Ribbe MW (2009) Molybdenum cofactors, enzymes and pathways. Nature 460:839–847
Stallmeyer B, Drugeon G, Reiss J, Haenni AL, Mendel RR (1999) Human molybdopterin synthase gene: identification of a bicistronic transcript with overlapping reading frames. Am J Hum Genet 64:698–705
Tan WH, Eichler FS, Hoda S, Lee MS, Baris H, Hanley CA, Grant PE, Krishnamoorthy KS, Shih VE (2005) Isolated sulfite oxidase deficiency: a case report with a novel mutation and review of the literature. Pediatrics 116:757–766
Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, Levitt MD, Prelle A, Fagiolari G, Rimoldi M et al (2009) Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med 15:200–205
Touati G, Rusthoven E, Depondt E, Dorche C, Duran M, Heron B, Rabier D, Russo M, Saudubray JM (2000) Dietary therapy in two patients with a mild form of sulphite oxidase deficiency. Evidence for clinical and biological improvement. J Inherit Metab Dis 23:45–53
Townsend DM, Tew KD, Tapiero H (2003) The importance of glutathione in human disease. Biomed Pharmacother 57:145–155
Ueki I, Roman HB, Valli A, Fieselmann K, Lam J, Peters R, Hirschberger LL, Stipanuk MH (2011) Knockout of the cysteine dioxygenase gene results in severe impairment in taurine synthesis and increased catabolism of cysteine to hydrogen sulfide. Am J Physiol Endocrinol Metab 301(4):E668–E684
van Gennip AH, Stroomer A, Plandsoen WG, Abeling NG (1991) The effect of molybdenum cofactor deficiency on the purine pattern of cerebrospinal fluid. J Inherit Metab Dis 14:364–366
Veldman A, Santamaria-Araujo JA, Sollazzo S, Pitt J, Gianello R, Yaplito-Lee J, Wong F, Ramsden CA, Reiss J, Cook I et al (2010) Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics 125:e1249–e1254
Vijayakumar K, Gunny R, Grunewald S, Carr L, Chong KW, DeVile C, Robinson R, McSweeney N, Prabhakar P (2011) Clinical neuroimaging features and outcome in molybdenum cofactor deficiency. Pediatr Neurol 45:246–252
Wu G, Fang YZ, Yang S, Lupton JR, Turner ND (2004) Glutathione metabolism and its implications for health. J Nutr 134:489–492
Wuebbens MM, Rajagopalan KV (1993) Structural characterization of a molybdopterin precursor. J Biol Chem 268:13493–13498
Wuebbens MM, Rajagopalan KV (2003) Mechanistic and mutational studies of Escherichia coli molybdopterin synthase clarify the final step of molybdopterin biosynthesis. J Biol Chem 278:14523–14532
Zhang X, Vincent AS, Halliwell B, Wong KP (2004) A mechanism of sulfite neurotoxicity: direct inhibition of glutamate dehydrogenase. J Biol Chem 279:43035–43045
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Schwarz, G., Veldman, A. (2014). Molybdenum Cofactor Disorders. In: Blau, N., Duran, M., Gibson, K., Dionisi Vici, C. (eds) Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-40337-8_12
Download citation
DOI: https://doi.org/10.1007/978-3-642-40337-8_12
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-40336-1
Online ISBN: 978-3-642-40337-8
eBook Packages: MedicineMedicine (R0)