
Abstract
Fungal diseases represent an important immunological paradigm because they can result from either defective immune recognition or overreacting inflammatory responses. The ever-growing number of patients suffering from life-threatening fungal diseases as a consequence of advances in medical care has created a pressing need to clarify both the molecular and cellular bases of fungal virulence as well as the mechanisms of host and fungal adaptation underlying immune homeostasis. Understanding the dynamics of the host–fungus interaction is central to the design of novel antifungal therapies and provides the foundation for successful vaccination strategies. This chapter reviews recent advances in the knowledge of adaptive immunity to fungi, positioning them within the conceptual framework of resistance-based versus tolerance-based antifungal responses, and how these mechanisms can be exploited for improved management of severe fungal infections and diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Treg Cell
- Allergic Bronchopulmonary Aspergillosis
- Chronic Mucocutaneous Candidiasis
- Th17 Cell Response
- Chronic Mucocutaneous Candidiasis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
I. Introduction: Dynamics of the Host–Fungus Interaction
The past two decades have seen an unprecedented number of fungal diseases (Fisher et al. 2012). The fungal kingdom is characterized by enormous biodiversity, with over 70,000 known species and an estimated 1.5 million species, 150–400 of which have already been associated with human and/or animal disease. In general, pathogenic fungi (e.g., Aspergillus fumigatus, Cryptococcus neoformans, Pneumocystis jirovecii and the thermally dimorphic Histoplasma capsulatum, Paracoccidioides brasiliensis, Coccidioides immitis and posadasii, Blastomyces dermatitidis, and Sporothrix schenckii) are distinguished from commensals (e.g., Malassezia spp. and Candida albicans) by their strategies for survival and replication within a host, or for transmission from a host, that eventually lead to cellular and tissue damage (Romani 2011). Pathogenicity tactics can vary a great deal and often define unique signatures for specific fungal species, as these involve particular mechanisms for gaining access to the host, adhering to and colonizing a niche, evading immune defenses, and multiplying (Rappleye and Goldman 2008). Indeed, disease onset is often critically dependent on the ability of fungi to reversibly switch morphotypes in infection, a trait that, on the other hand, has forced the host immune system to continuously evolve its repertoire of cross-regulatory and overlapping antifungal responses at different body sites (Romani 2011). Thus, in the context of a dynamic host–fungus interaction, the strategies used by the host to limit fungal infectivity are necessarily assorted in order to cope with the multitude of fungal survival strategies; in retaliation, fungi have developed their own elaborate tactics to evade or modulate host defenses and to survive.
Given the advances in medical care witnessed in the last few decades, specifically regarding transplantation and cancer treatment, the number of immunocompromised patients has risen, resulting in an increased incidence of fungal diseases (Kontoyiannis et al. 2010; Pappas et al. 2010). For this reason, opportunistic fungi, such as Candida and Aspergillus spp. have become major concerns in clinical care of immunocompromised patients. Despite the ability of fungi to survive and persevere within the human host, the truth is that fungal diseases in immunocompetent hosts are fairly uncommon, indicating that fungi have evolved particular adaptation mechanisms that allow them to persist relatively unnoticed by the host’s immune system (Cooney and Klein 2008). This “peaceful” coexistence may digress into overt disease under conditions of immune deregulation, which may modify the environmental conditions perceived by the fungus. Fungi are very proficient at sensing their surroundings and in responding to cues that promote their survival in changing environments. One such example is the sensing of mammalian interleukin (IL)-17A, which triggers virulence by increasing fungal adhesion and filamentous growth, leading to enhanced biofilm formation and resistance to local antifungal defenses (Zelante et al. 2012). Besides the immunologically dynamic context, fungi have often to sustain extreme environmental abiotic stress conditions, the adaptation to which relies on profound metabolic changes (Grahl et al. 2011).
Thus, in the absence of a relatively inert immune response, fungal adaptation to hostile surroundings may trigger complex genomic microevolution (Odds and Jacobsen 2008; Magditch et al. 2012) and structural and metabolic adjustments that, by “awakening” host immunity, may paradoxically contribute to disease. Although such adaptation mechanisms may improve fungal fitness, they can nonetheless provide important insights into potential therapeutic targets (Richie et al. 2009).
Given that existing antifungal therapy is often toxic and ineffective, there is currently a pressing demand for the development of antifungal vaccination strategies (Cassone and Casadevall 2012; Iannitti et al. 2012). For this purpose, a clear understanding of the mechanisms of adaptive immunity is ultimately required to foster the development of vaccines or strategies aiming at modulating the host’s immune response. The permanent interaction between the host and fungi and the commensal relationship with some of them may pose significant challenges in eliciting durable protective immunity, owing to repeated exposure or sensitivity to fungal antigens. A balanced host–fungus relationship and the concomitant fine tuning of pro- and anti-inflammatory signaling to allow host survival irrespective of pathogen elimination is a prerequisite for coexistence and requires the concerted actions of both innate and adaptive immune systems (Romani 2011). Therefore, generation of antifungal immunity presents a challenge that relies on a precarious equilibrium between pathogen clearance and tissue damage restriction, while preserving the host microbiota ecology.
This chapter focuses on new findings on adaptive immunity to the major medically important fungi and emphasizes how dendritic cells (DCs), through the discrimination of fungal molecular patterns, prime responses that nurture and shape the differentiation of T cell subsets. Also discussed is the contribution of T cell subsets to resistance and tolerance mechanisms of antifungal immune protection and how these mechanisms can be exploited for effective antifungal vaccine design.
II. Resistance and Tolerance Mechanisms in Antifungal Protection
The immune system protects from infections primarily by detecting and eliminating invading pathogens through a variety of host resistance effector mechanisms (Romani 2011; Medzhitov et al. 2012). Resistance is meant to reduce pathogen burden during infection through innate and adaptive immune mechanisms, whereas tolerance mitigates the substantial cost to host fitness of resistance. Even in the absence of overt tissue damage, resistance mechanisms commonly occur at a cost to normal tissue function, thus causing immunopathology. This means that the optimal immune response is determined by the balance between efficient pathogen clearance and an acceptable level of immunopathology.
Inflammation is an essential process required for immune resistance, particularly at mucosal tissues, during the transition from the rapid innate to the slower adaptive response. However, the downside of this powerful mechanism of protection against fungi is the collateral damage to the host. These side effects may be more devastating than infection itself. Thus, the ability to tolerate a pathogen’s presence is a distinct host defense strategy that may have evolved to favor protective mechanisms without pathogen killing (Romani 2011; Medzhitov et al. 2012). A plethora of tolerance mechanisms, although not as well known as resistance mechanisms, protect the host from immune- or pathogen-induced damage (Cobbold et al. 2010; Saraiva and O’Garra 2010). Therefore, the term “tolerance” is semantically used here to refer to the multitude of anti-inflammatory mechanisms, including immunological tolerance (i.e., unresponsiveness to self-antigens). At this stage, however, whether “unwanted” immune responses against “self ” environmental antigens and commensal microorganisms occur is not clearly defined, although there is evidence that fungal sensitization contributes to auto-reactivity against self-antigens due to shared epitopes with homologous fungal allergens (Zeller et al. 2008).
T helper (Th)1 and Th17 cells, which provide antifungal resistance, and T regulatory (Treg) cells, which limit the inflammation-associated deleterious effects, crucially contribute to the activation and preservation of these two disparate antifungal mechanisms. Combined deficiency of the Th1 and Th17 pathways predisposes to fungal diseases (van de Veerdonk et al. 2011), thus emphasizing the important role played by both pathways in protection against fungi (Moraes-Vasconcelos et al. 2005; Romani 2011; Hardison and Brown 2012). Thus, the Th1/Th17 pathways and Treg cells, capable of fine-tuning protective antimicrobial immunity to lessen harmful immune pathology, are key components of the current view of immunity to fungi. The enzyme indoleamine 2,3-dioxygenase 1 (IDO1) and its downstream catabolites sustain this delicate balance by providing the host with adequate protective immune mechanisms without necessarily eliminating the pathogen or causing undesirable tissue damage (Zelante et al. 2009). As a result of their ability to induce differentiation of Treg cells and inhibit Th17 cells, IDO1 is critical to cell lineage commitment in experimental fungal infections and contributes to the overall outcome of inflammation, allergy, and Th17-driven inflammation in these infections. Under these circumstances, the Th17 pathway, by inhibiting tryptophan catabolism, may instead favor pathology and provides evidence accommodating the apparently paradoxical association of chronic inflammation with fungal disease (Romani et al. 2008b).
III. Fungi and Inflammation: Evolving Concepts
As in autoimmunity and chronic inflammation, an imbalance between pro- and anti-inflammatory signals may prevent successful host–fungal interaction, thus leading to infection and disease (Romani and Puccetti 2007). Indeed, despite the occurrence of severe fungal infections in immunocompromised patients, clinical evidence indicates that fungal disease also occurs in the setting of a heightened inflammatory response, in which immunity occurs at the expense of host damage and pathogen eradication (Perfect 2012). Although inflammation is an essential component of the protective response, fungi have evolved ways to exploit and subvert it, thereby affecting their ability to persist in the host and pathogenicity (Romani and Puccetti 2008). A hyperinflammatory response does, in fact, enhance the virulence of some fungi. This is well illustrated by the commensal lifestyle of Malassezia spp. in normal skin, possibly due to the downregulation of inflammation via tumor growth factor-β1 (TGF-β1) and IL-10 (Ashbee 2006). In contrast, in atopic dermatitis and psoriasis, the skin barrier enhances release of allergens and molecules involved in hyperproliferation, cell migration, and disease exacerbation. Additional fungal diseases are also important examples of such dichotomy. For example, in chronic mucocutaneous candidiasis (CMC), C. albicans yeasts persist in recurring lesions of the skin, nails, and mucous membranes (Lilic 2002). Although CMC has occasionally been associated with autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (a condition of dysfunctional T cell activity), evidence has highlighted the contribution of deregulated inflammation and immune responses to disease pathogenesis (Liu et al. 2011; Puel et al. 2011; van de Veerdonk et al. 2011). As already mentioned, fungal sensing of IL-17A is a newly described mechanism by which host inflammation may favor fungal infectivity and promote the transition from fungal commensalism to infection (Zelante et al. 2012). Thus, commensals or ubiquitous fungi have evolved a contingency-based system during co-evolution to guarantee their persistence in an inflammatory host environment.
The main implication of these findings is that, at least in specific clinical settings, it is a heightened inflammatory response that probably compromises a patient’s ability to eradicate infection and not an “intrinsic” susceptibility to infection that determines a state of chronic or intractable diseases (Romani and Puccetti 2007). The conceptual principle highlighting a truly bipolar nature of the inflammatory process in infection is best exemplified by the occurrence of severe fungal infections in patients with chronic granulomatous disease (Romani et al. 2008a) or with immune reconstitution syndrome, an entity characterized by localized and systemic inflammatory reactions and worsening disease in opportunistic and non-opportunistic infections that are associated with immunological recovery (Gupta and Singh 2011; Perfect 2012). Additionally, a high incidence of fungal infections and sensitization to Aspergillus spp. has been described in the hyper-IgE syndrome, in which increased levels of pro-inflammatory gene transcripts have been found (Antachopoulos et al. 2007; Holland et al. 2007). These observations suggest that an inflammatory loop hampering a patient’s capacity to counter infection seems to be at work, at least in specific clinical settings. The manipulation of this loop may offer strategies to control or prevent exacerbations of these diseases.
IV. Activation of Antifungal Immunity
A. Dendritic Cells
Immunity to fungi is a dynamic interplay between every arm of the immune system. Innate immune mechanisms are used by the host to respond to a range of fungal pathogens in an acute and conserved fashion (as discussed in chapter “Receptor-Ligand Interactions in Fungal Infections”). The induction of innate immunity shapes the development of the adaptive immune response. As sentinels of the immune system, DCs are responsible for sampling antigenic material in the environment, shaping T cell responses through secretion of cytokines, and priming T cells via antigen presentation (Steinman 2012). Priming of T cells by DCs is mediated by pathogen-associated antigens on major histocompatibility complex class I (MHC-I) or MHC class II (MHC-II) molecules for priming of CD8+ or CD4+ T cells, respectively. After priming of naïve T cells, the response is generally described as Th1, Th2, Th17, or Treg (described in detail in a later section B) based on the pattern of cytokine production. Thus, the ability to control the fate of the immune response makes DCs both central to balancing antifungal immunity and a prime target for vaccination strategies.
For years, DC biologists have oscillated between two apparently opposing concepts: functional specialization of DC subsets (division of labor) and plasticity (multitasking). More recently, a third hypothesis is gathering support: crosstalk between functionally distinct DC subsets. This reveals a previously unappreciated hierarchy of organization within the DC system, and provides a conceptual framework for understanding how cooperation between functionally distinct, yet plastic, DC subsets can shape adaptive immunity and immunological memory (Pulendran et al. 2008).
1. The Role of DCs in Antifungal Immunity
DCs are uniquely proficient at decoding the fungus-associated antigens and translating them into qualitatively different adaptive T cell immune responses (Romani et al. 2002; Romani 2011; Roy and Klein 2012; Wuthrich et al. 2012). DCs have the exceptional competence to initiate distinct adaptive antifungal immune responses as a result of cooperation between subsets (Romani and Puccetti 2006; Romani et al. 2008a) and activation of distinct intracellular signaling pathways (Bonifazi et al. 2009, 2010). The functional plasticity of DCs is mostly defined by the discriminative recognition of different fungal species and morphotypes by the full range of pattern recognition receptors (PRRs). As a matter of fact, whole−genome transcriptional analysis of fungus-pulsed DCs revealed the presence of a specific transcriptional program governing fungal recognition (Rizzetto and Cavalieri 2010). Among DC subsets, plasmacytoid (p)DCs have a prominent role in fungal infections. Not only are pDCs rapidly recruited and activated in response to fungi at mucosal sites, such as the lung (Bonifazi et al. 2010; Ramirez-Ortiz et al. 2011; Carvalho et al. 2012; De Luca et al. 2012) and gut (De Luca et al. 2007; Bonifazi et al. 2009), but infusion of pDCs in bone marrow-transplanted mice has been found to trigger Th1/Treg cell priming, eventually leading to fungal growth restriction, limited inflammatory pathology and, interestingly, transplantation tolerance (Romani et al. 2006). More recently, Toll-like receptor (TLR) 3-dependent recognition of fungal RNA by CD8+ DCs induced potent cytotoxic CD8+ T cell responses to A. fumigatus, both in mice and humans (Carvalho et al. 2012). TLR3 deficiency renders mice highly susceptible to aspergillosis, and a TLR3 mutation affecting the ability of CD8+ DCs to cross-present antigens renders hematopoietic stem cell-transplanted patients more susceptible to invasive aspergillosis.
The activation of distinct signaling pathways in DCs translates recognition of fungi into distinct inflammatory and adaptive immune responses (Bonifazi et al. 2009, 2010). The screening of signaling pathways in DCs through a systems biology approach was exploited for the development of therapeutics to attenuate inflammation in experimental fungal infections and diseases. In vivo targeting inflammatory [phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR)] or anti-inflammatory [signal transducer and activator of transcription (STAT3)/IDO1] DC pathways by intranasally delivered small interfering RNA (siRNA) modified resistance and tolerance to infection. Thus, the screening of signaling pathways in DCs through a systems biology approach may be exploited for the development of siRNA therapeutics to attenuate inflammation in respiratory fungal infections and diseases (Bonifazi et al. 2010).
DCs are now being exploited to improve vaccine efficacy (Steinman 2008). The potential use of tolerogenic pDCs as negative cellular vaccines to induce experimental transplantation tolerance has been suggested (Turnquist and Thomson 2008).
Over recent years, experimental models have shown that it is possible to exploit the mechanisms that normally maintain immune homeostasis and tolerance to self-antigens to induce tolerance to allo-antigens (Waldmann and Cobbold 2004; Martinic and von Herrath 2006). Like natural tolerance, transplantation tolerance is achieved through control of T cell reactivity by central and peripheral mechanisms of tolerance.
Fungus-pulsed DCs or RNA-transfected DCs acted as potent fungal vaccines in experimental hematopoietic stem cell transplantation (Bozza et al. 2003), a model in which autologous reconstitution of host stem cells is greatly reduced due to the benefit of a long-term, donor-type chimerism in more than 95% of the mice and low incidence of graft rejection. Protection was associated with myeloid and T cell recovery, the activation of CD4 + Th1 lymphocytes, and the concomitant IL-10-driven Treg cells. Thus, tolerogenic DCs proved to be pivotal in the generation of some form of dominant regulation that ultimately controlled inflammation, pathogen immunity, and tolerance in transplant recipients eventually leading to prevention of graft-versus-host reaction and reduction of aspergillosis incidence rates. These results, along with the finding that fungus-pulsed DCs could reverse T cell anergy of patients with fungal diseases, further supports the utility of targeting DCs for antifungal vaccination strategies (Bozza et al. 2004).
2. Metabolic Regulation of DC Plasticity in Response to Fungi
A wealth of evidence indicates that acquisition of an immunogenic or tolerogenic phenotype is a trait of a specific subset or lineage of DCs, but that it is an environmentally acquired feature. In this regard, the tryptophan metabolic pathway pivotally contributes to DC regulation, such that tolerance and Treg induction can be mediated by IDO1-expressing DCs (Orabona et al. 2004) (see below). In response to fungi, IDO1 expression was found to confer tolerogenic properties to DCs (Zelante et al. 2009) such that C. albicans-pulsed, IDO1-expressing gut DCs ameliorated experimental colitis (Bonifazi et al. 2009). By subverting the morphotype-specific program of activation of DCs, environmental factors and fungi themselves qualitatively affect DC functioning and Th/Treg cell selection in vivo, ultimately impacting on fungal virulence. Thus, the current view accommodates the concept of virulence as an important component of fungal fitness within the plasticity of immune responses orchestrated by DCs. Indeed, impaired DC maturation and function have been associated with disease in patients with CMC (Ryan et al. 2008).
B. T Cell Responses in Fungal Infection
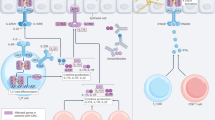
Studies on nonmammalian hosts have provided means to examine the molecular elements of fungal virulence and host innate immunity (Fuchs and Mylonakis 2006; Mylonakis et al. 2007; Peleg et al. 2008). In higher organisms, however, innate sensing mechanisms are capable of distinguishing different fungal morphotypes and are hard-wired to activate distinct adaptive immune responses with protective and nonprotective functions against the different fungal species. It has been suggested that a memory-based immune mechanism may have evolved in vertebrates to accommodate colonization by symbiotic microbes while retaining the capacity to oppose their infectivity (McFall-Ngai 2007). This suggests that the adaptive immune system has co-evolved with ubiquitous or commensal fungi, with a price to be paid for this permissiveness. Stimulation of antigen-presenting macrophages, DCs, and, more recently, epithelial cells (ECs) leads to activation and recruitment of lymphocytes and the development of Th cell−specific antifungal responses. There is extensive plasticity in T cell responses to fungi (Fig. 6.1). The heterogeneity of the CD4+ and CD8+ T cell repertoire may account for the multiplicity and redundancy of effector mechanisms through which T lymphocytes participate in the control of fungal infections. Once committed, T cells express effector functions largely, but not exclusively, through release of cytokines, most notably interferon (IFN)-γ, tumor necrosis factor (TNF)-α and IL-17/IL-22, which are instrumental in mobilizing and activating antifungal effectors, thus providing prompt and effective control of infectivity once the fungus has established itself in tissues or spread to internal organs (Romani 2011).

T cell activation in fungal infection. Dendritic cells (DCs) are at the crossroads of antifungal immunity and tolerance because both inflammatory and tolerogenic DCs may arise from their common immature progenitors. Following fungal recognition by specific pattern recognition receptors, inflammatory DCs may trigger the expression of subset-associated transcription factors and differentiation of naïve CD4+ T cells into T helper 1 (Th1), Th2, or Th17 cells, which may induce cytokine secretion and activation of specific antifungal immunity programs. On the other hand, plasmacytoid DCs may activate CD8+ cytotoxic lymphocytes (CTLs) that, by displaying direct fungicidal activity, also contribute to antifungal immunity. In addition, indoleamine 2,3-dioxygenase 1 (IDO1)-competent tolerogenic DCs produce kynurenines (Kyn) and cytokines that contribute to the expansion of interleukin (IL)-10-secreting T regulatory (Treg) cells, crucial for the maintenance of tolerance to the fungus. MR mannose receptor, IFN interferon, TLR Toll-like receptor, T-bet T-box expressed in T cells, Gata3 GATA-binding protein 3, Rorc retinoic acid receptor (RAR)-related orphan receptor C
1. Th1 Cells
Generation of a dominant Th1 response driven by IL-12 is essentially required for the expression of protective immunity to fungi and is compulsory for the design of effective antifungal vaccines (Spellberg et al. 2008). Through the release of the signature cytokine IFN-γ and by helping the production of opsonizing antibodies, induction of Th1 cells is instrumental for the optimal activation of phagocytes at sites of infection. Of interest, Th1 cell-based cross-protection for different fungi has been recently demonstrated to be achievable using a single immunogenic epitope from the A. fumigatus cell wall, thus providing attractive opportunities for immunotherapeutic strategies (Stuehler et al. 2011). The failure to deliver activating signals to effector phagocytes may predispose patients to overwhelming infections, limit the therapeutic efficacy of antifungals and antibodies, and favor persistency and/or commensalism (Romani 2011).
As noted above, Th cell skewing is determined by the way that DCs respond to the combination of fungal-derived TLR and C-type lectin receptor (CLR) signaling. Interestingly, dectin-1-mediated signals can alter the Th1 profile of CD4+ T cell responses to fungal infection by decreasing the production of IL-12 and IFN-γ in innate cells and consequent expression of T-box expressed in T cells (T-bet) in A. fumigatus-specific CD4+ T cells, thus enabling Th17 differentiation (Rivera et al. 2011).
Further supporting the pivotal role of Th1 cell activation, immunological studies on patients with polar forms of paracoccidioidomycosis demonstrate an association between Th1-biased reactivity and asymptomatic and mild forms of infection, as opposed to the correlation of Th2 cell responses with severe disease and poor prognosis. Thus, the finding that estradiol favors Th1-type immunity may explain why paracoccidioidomycosis is much more frequent in men than in women (Pinzan et al. 2010).
2. Th2 Cells
Progressive fungal infections, especially those acquired through the respiratory tract, are eventually associated with a shift in dominance from Th1 to Th2. IL-4 and IL-13 provide the most potent proximal signals for commitment of naïve T cells to the Th2 cell lineage. Despite often occurring coincidentally with vigorous Th1 responses, Th2 reactivity results in a net effect of poor control of the fungal burden because it induces alternatively activated macrophages that are permissive for intracellular fungal growth and may be impervious to signaling by IFN-γ (Voelz et al. 2009). In addition, Th2 reactivity alters pulmonary physiology such that airway resistance increases, thus compounding the severity of infection and contributing to fungus-associated allergic responses (Jain et al. 2009). Accordingly, attenuating Th2 responses to A. fumigatus in cystic fibrosis patients with allergic bronchopulmonary aspergillosis (Kreindler et al. 2010) or limiting IL-4 production in experimental models of histoplasmosis (Szymczak and Deepe 2009) restored antifungal resistance.
For the vast majority of fungi, Th2 responses are associated with pathogenic allergic responses. The contradiction is pneumocystosis, in which a Th2 cell-dependent humoral immunity affords some degree of protection (Rapaka et al. 2010). In addition, alternatively activated macrophages driven by IL-13 and amplified by IL-33 displayed enhanced fungicidal activity regardless of the antibody-dependent effect, a finding that further supports a contribution of Th2 cells to fungal clearance (Bhatia et al. 2011).
3. Th17 Cells
Over the past several years, the Th1/Th2 dichotomy has been replaced by the belief that the “fates” for developing CD4+ T cells and associated cytokines are more flexible than formerly anticipated (Zhou et al. 2009). Th17 cells are a separate lineage of effector Th cells contributing to immune pathogenesis previously attributed to the Th1 lineage (Kaufmann and Kuchroo 2009). Th17 cells have an important function in the host response against extracellular pathogens, but they are also associated with the pathogenesis of many autoimmune and allergic disorders. It is now well accepted that Th17 cell activation occurs in fungal infections (Hernandez-Santos and Gaffen 2012), mainly through the spleen tyrosine kinase/caspase recruitment domain-containing protein 9 (Syk/CARD9), myeloid differentiation primary response protein 88 (MyD88), and mannose receptor (MR), signaling pathways in DCs and macrophages leading to the production of unique cytokines such as IL-17, IL-17F, and IL-22. This signaling is inhibited by Raf-1 kinase and TIR domain–containing adapter–inducing interferon-beta (TRIF)/type I IFN pathways, indicating that the molecular pathways defining activation or inhibition of Th17 cells are present downstream of both CLRs and TLRs. Indeed, the central role of Th17 cells in antifungal immunity is supported by studies reporting Mendelian susceptibility to fungal infections of individuals with inborn errors of dectin-1, CARD9, STAT1, STAT3 and, specifically, IL-17 immunity (Ferwerda et al. 2009; Glocker et al. 2009; Puel et al. 2011; van de Veerdonk et al. 2011) (Table 6.1). In experimental fungal infections, however, IL-17-dependent immunity has been reported to be either essential (Huang et al. 2004; Saijo et al. 2007; Conti et al. 2009; Wuthrich et al. 2011) or not (Lin et al. 2009; De Luca et al. 2010b; Hardison et al. 2010). This suggests that the protective or detrimental effects of this pathway may depend on the stage and site of infection, probably influenced by environmental stimuli that induce cells to produce cytokines of the IL-17 family, including IL-22 (see below). In this regard, recent evidence has demonstrated that Th17 cells can be segregated into pathogenic or nonpathogenic cells, depending on whether they produce granulocyte macrophage colony stimulating factor (GM-CSF) or IL-10, respectively (Codarri et al. 2011; El-Behi et al. 2011). These Th17 phenotypes are still to be described in the context of fungal diseases (Wuthrich et al. 2012).
Th17 cells are present in the fungus-specific T cell memory repertoire in humans (Acosta-Rodriguez et al. 2007; Bozza et al. 2009) and mediate some (Wuthrich et al. 2011), but not all (De Luca et al. 2012), vaccine-induced protection in mice. Interestingly, as in mice (De Luca et al. 2012), human host defense against A. fumigatus relies on Th1 rather than Th17 cell responses (Chai et al. 2010), and CMC patients (with or without autosomal dominant hyper-IgE syndrome) have defective Th17 and Th1 cell responses (Ryan et al. 2008; van de Veerdonk et al. 2011). This could be explained by the notion that Th17 cells, although found early during the initiation of an immune response, are involved in a broad range of both Th1- and Th2-type responses.
Indeed, a role for Th17 cells in supporting Th1 cell responses has been shown in experimental mucosal candidiasis (Conti et al. 2009; De Luca et al. 2010b). In addition, in experimental aspergillosis, enhanced Th2 cell responses and fungal allergy are observed in the absence of IL-17A receptor signaling (our unpublished observations). Thus, these findings point to an important regulatory function of the Th17 cell pathway in promoting Th1-type and restraining Th2-type immunity.
It is intriguing that Th17 cell responses are dampened by C. albicans (Cheng et al. 2010) and that failure to do so eventually results in chronic inflammation and failure to resolve the infection (Zelante et al. 2007; Loures et al. 2009). In this regard, it is fascinating that fungi are able to sense mammalian IL-17 in their surrounding environment and turn on molecular programs that result in enhanced virulence and survival aptitude (Zelante et al. 2012).
The mechanisms that link inflammation to chronic infection may lie in an inability to control inflammation following IL-17A-dependent neutrophil mobilization, thus preventing optimal protection and favoring fungal persistence. Thus, the Th17 cell pathway could be involved in the immunopathogenesis of chronic fungal disease, in which persistent fungal antigens may promote immune deregulation, as demonstrated in patients with autoimmune polyendocrine syndrome type 1 and in the absence of autoimmune regulator (AIRE), in which excessive Th17-type responses to fungi have been observed (Ahlgren et al. 2011).
As noted above, Th17 cells can concomitantly synthesize IL-22, a member of the IL-10 family of cytokines, which has been shown to play a more important role than IL-17 in host defense in the lung and gut (Zenewicz and Flavell 2008). Our recent findings suggest that the IL-23/IL-22/defensins pathway is crucially involved in the control of fungal growth at mucosal and nonmucosal sites in both candidiasis and aspergillosis, particularly in conditions of Th1 deficiency. Interestingly, memory IL-22+ CD4+ cells specific for C. albicans are present in humans (Liu et al. 2009) and are defective in patients with CMC (Eyerich et al. 2008). Thus, further tweaking the Th17 model, Th17 cells may exert their protective role in fungal infections through IL-22. Indeed, IL-22 has recently been demonstrated to be required for the control of C. albicans growth at mucosal sites in the absence of Th1 and Th17 cells (De Luca et al. 2010b). Specifically, IL-22 produced by NKp46+ innate lymphoid cells expressing the aryl hydrocarbon receptor directly targeted intestinal ECs and induced STAT3 phosphorylation and release of S100 calcium binding protein A8 (S100A8) and S100A9, peptides known to have antifungal activity and anti-inflammatory effects. Thus, in the relative absence of protective Th1/Treg, IL-22 + Th17 cells may fulfill the role of a protective response that exploits primitive effector defense mechanisms of antifungal resistance, as demonstrated also for experimental bacterial diseases (Aujla et al. 2008). Consistent with this role for IL-22, patients with autosomal dominant hyper-IgE syndrome, owing to dominant-negative mutations of STAT3, have a defective Th17 cell response to C. albicans (Milner et al. 2008). Accordingly, C. albicans-specific IL-22+ CD4+ memory T cells are present in healthy individuals (Liu et al. 2009) but are lacking in CMC patients (Eyerich et al. 2010), an observation pointing to IL-22 production in the mucosa as a primitive mechanism of resistance against fungi under conditions of limited inflammation. Of interest, dectin-1-mediated production of IL-22 in the lung has also been demonstrated to contribute to early innate immune resistance to A. fumigatus (Gessner et al. 2012), although it paradoxically promoted lung inflammation and immunopathology during persistent fungal exposure in an allergy model (Lilly et al. 2012). These seemingly discrepant findings further add to the complexity of IL-22 function in antifungal mucosal immunity and point to the existence of regulatory events leading to its production that depend on the stage and site of infection.
4. Treg Cells
During infection, the immune response must eliminate the fungus while limiting infection-associated costs to host fitness and restoring a homeostatic environment. Treg cells, by means of their anti-inflammatory activity, play a central role in this process. In experimental fungal infections, inflammatory immunity and immune tolerance in the respiratory or gastrointestinal mucosa have been shown to be controlled by the coordinated activation of different Treg cell subsets. Because Treg cell responses may handicap the efficacy of protective immunity, Treg cell activity decreases host tissue damage but may conversely promote fungal persistence (Romani and Puccetti 2006) and, eventually, immunosuppression (Ferreira et al. 2010). Some cells with this function, such as CD4+ Foxp3+ natural Tregs (nTregs), exist regardless of the presence of infectious stimuli, whereas others may be induced as a consequence of infection (iTregs) or in conditions of impaired co-stimulatory signaling and in the presence of deactivating cytokines and drugs. This scenario is crucially exemplified in experimental aspergillosis, in which inflammation was controlled at an early stage by nTregs suppressing neutrophils whereas, later tolerogenic iTregs inhibited Th2 cells and prevented fungal allergy (Montagnoli et al. 2006).
As already discussed, a reciprocal relationship has been described between the development of Foxp3+ Treg and effector Th17 cells, so that naïve T cell activation in the presence of innate stimuli redirects iTreg generation to Th17 generation. In this regard, CD4+ CD25+ Foxp3+ Treg cells have been recently found to promote IL-17 upregulation and contribute to suppression of mucosal candidiasis in vivo (Pandiyan et al. 2011). Thus, by controlling the quality and magnitude of effector innate and adaptive responses, the spectrum of Treg cell skills may go from protective tolerance and immune homeostasis to dominant effector activities. Furthermore, this suggests that this degree of interaction between fungi and the host immune system determines whether a fungus is perceived as commensal or pathogen, and that this definition may evolve constantly.
5. Indoleamine 2,3-Dioxygenase 1 is a Critical Regulator of Tolerance to Fungi
IDO1 is an IFN-γ-inducible intracellular enzyme that catalyzes the catabolism of tryptophan (Puccetti and Grohmann 2007; Mellor and Munn 2008). The effects of IDO1 activity are tryptophan deficiency, excess tryptophan breakdown products (kynurenines), and consumption of reactive oxygen species. IDO1 and kynurenines serve many roles in fungal infections. A number of studies have established that the proper control of infection and associated inflammatory reactions require IDO1 induction and consequent production of tryptophan metabolites with immunoregulatory activities, contributing to the maintenance of the Treg/Th17 balance (Romani et al. 2008b). As already mentioned, IDO1-expressing DCs are regarded as regulatory DCs specialized in antigen-specific deletional tolerance or induction of CD4+ CD25+ Treg cells. These findings disclose a mutual interaction between DCs and Treg cells for the preservation of immunological tolerance. Indeed, IDO1 blockade greatly exacerbated experimental fungal infections and the associated inflammatory pathology, and swept away resistance to re-infection, as a result of deregulated innate and adaptive immune responses caused by the impaired activation and functioning of suppressor CD4+ CD25+ Treg cells producing IL-10 (Romani and Puccetti 2006). IDO1 expression is paradoxically upregulated in patients with allergy or autoimmune inflammation, a finding pointing to the occurrence of a homeostatic mechanism to halt ongoing inflammation (Grohmann et al. 2007). During experimental fungal allergy, modulation of tryptophan catabolism via the glucocorticoid-induced tumor necrosis factor receptor (GITR) and its ligand, GITRL, inhibited Th2 cell responses and allergy and induced the expression of Foxp3+ Treg cells through IDO1-dependent mechanisms (Grohmann et al. 2007), a finding pointing to the potential relevance of IDO1 in the anti-inflammatory action of corticosteroids. As already mentioned, a reciprocal antagonistic relationship exists between IDO1 and the Th17 pathway, with IDO1 restraining Th17 responses and IL-17A inhibiting IDO1 (Zelante et al. 2007).
Recent evidence indicates that the non-hematopoietic compartment also contributes to tolerance to fungi via IDO1 (Cunha et al. 2010; de Luca et al. 2010a). ECs are key players in tolerance to respiratory pathogens via an IFN-γ–IDO1 axis culminating in the inhibition of Th17 cell responses (Desvignes and Ernst 2009; de Luca et al. 2010a). IDO1 overexpression in airway ECs was found to restrain CD4+ T cell activation to A. fumigatus, an activity that was nevertheless dispensable in the presence of IDO1-expressing tolerogenic DCs. However, IDO1 induction in ECs could compensate for the lack of IDO1 on hematopoietic cells (Paveglio et al. 2011). The expression of IDO1 on ECs occurred through the TLR3/TRIF-dependent pathway, a finding consistent with the abundant expression of TLR3 both intracellularly and on the cell surface of ECs. The failure to activate IDO1 probably accounted for the lack of tolerance to the fungus observed in experimental stem cell transplantation in conditions in which either the recipient or the donor (or even more when both) were TRIF- or TLR3-deficient (de Luca et al. 2010a).
Overall, these data shed light on pathways of immune resistance and tolerance to the fungus that probably take place in a hematopoietic stem cell transplantation setting. It appears that protective tolerance to the fungus is achieved through a TLR3/TRIF-dependent pathway activating Th1/Treg cells via IDO1 expressed on both the hematopoietic and non-hematopoietic compartments. In contrast, the MyD88 pathway provides antifungal resistance, i.e., the ability to restrict fungal growth through defensins and, probably, other effector mechanisms (de Luca et al. 2010a). However, the ability of mice to clear the fungus in the relative absence of the MyD88 pathway (Bretz et al. 2008) clearly indicates redundancies and hierarchy in antifungal mechanisms of resistance. Ultimately, the finding that both C. albicans (De Luca et al. 2007) and A. fumigatus (de Luca et al. 2010a), two major human fungal pathogens, exploit the TRIF/IDO1-dependent pathway at the interface with the mammalian hosts indicates that the exploitation of tolerance mechanisms is an advantageous option.
V. Immune Memory and Antifungal Vaccines
A successful vaccination relies on the elicitation of pathogen-specific immune memory that mediates long-term protection from infection or disease. Given the plethora of fungal ligands present at the cell surface, as well as those that become available to immune sensing upon processing of the fungus by phagocytic cells, it is clear that vaccine-induced protection to attenuated fungal strains occurs through distinct PRRs and downstream signaling adapters (Wuthrich et al. 2011; De Luca et al. 2012). For instance, Th17-induced acquisition of vaccine immunity to live attenuated strains of B. dermatitidis, H. capsulatum , and C. posadasii was found to require MyD88 signaling (Wuthrich et al. 2011 ), whereas Th1-induced protection to A. fumigatus relied on TRIF (De Luca et al. 2012 ). Of interest, vaccination with purified A. fumigatus antigens was found to be dependent on the MyD88 pathway in the presence of the appropriate adjuvant (Carvalho et al. 2012; De Luca et al. 2012), a finding pointing to the crucial role of adjuvants in promoting T cell differentiation along specific effector pathways. Thus, fungal innate sensing is one critical step in mounting immune responses, eventually defining appropriate effector responses to maximize protection (Levitz and Golenbock 2012).
Although CD4+ Th1 cells have been historically considered the cornerstone of cell-mediated defense against intracellular fungi (Cassone and Casadevall 2012; Iannitti et al. 2012), CD8+ T cells have also earned a place in this category (Cutler et al. 2007). Indeed, in a mouse model of vaccination against blastomycosis, both the numbers and function of protective antifungal memory CD8+ T cells were maintained, even in the absence of CD4+ T cell help (Nanjappa et al. 2012b). Under these circumstances, a distinct lineage of CD8+ T cells able to produce IL-17 (Tc17 cells) has been found to be nonredundant for vaccine immunity to fungal infection by mediating protection in a neutrophil-dependent manner (Nanjappa et al. 2012a).
The persistence of immunological memory and how it relates to vaccination strategies is a question of central importance. Memory T cells are derived from normal T cells that have learned how to overcome a pathogen by “remembering” the strategy used to defeat previous infections (Sallusto et al. 2010). In addition to central memory T cells present in secondary lymphoid organs, which scrutinize the presence of remote pathogens via DCs, effector memory T cells reside in peripheral non-lymphoid tissues such as the skin and mucosa. The latter are heterogeneous in terms of homing receptor expression and effector function and comprise the Th1, Th2, Th17, and Treg cells and cytotoxic T lymphocytes. Memory CD8+ cytotoxic T cells are also induced in fungal infections (Nanjappa et al. 2012b) and exhibit a pleiotropic activity by mediating protection via production of IFN-γ and cytolytic activity against fungus-laden cells or the fungus itself (Carvalho et al. 2012; De Luca et al. 2012). As such, CD8+ T cells, especially if long-lasting, are regarded as ideal candidates for expansion at mucosal surfaces by vaccination strategies.
The nature of the antifungal vaccine, the route of antigen delivery, and the mode of antigen routing and presentation are important for determining the success of a fungal vaccine. Indeed, recent evidence has highlighted striking differences in antigen presentation pathways in DCs leading to the activation of CD4+ or CD8+ T cells (De Luca et al. 2012). Memory CD4+ T cells were activated by purified antigens from A. fumigatus that were routed to the endosome/lysosome-dependent MHC-II presentation pathway via MyD88 with the involvement of distinct upstream PRRs. En route to lysosomes, purified antigens were also targeted to the mildly acidic stable early endosomal compartment where uptake by MR led to presentation on MHC-I molecules and Th1 polarization. Consistently, mannosylation has been reported to significantly enhance antifungal CD8+ T cell priming (Luong et al. 2007). In contrast to soluble antigens, phagocytosed cells or particulate antigens activated CD8-dependent memory through a pathway relying on TLR3/TRIF signaling (De Luca et al. 2012). Similar to the situation for C. neoformans (Wozniak and Levitz 2008), phagocytosed cells were routed to the late endosome/lysosome compartment and also to the rat sarcoma (RAS)-related protein (Rab)14+ compartment, which is known to limit routing of antigens from early endosomes to the acidic lysosomal environment, thus limiting antigen degradation and favoring cross-presentation (Saveanu et al. 2009). The escape of fungal cells from the endosomal/lysosomal compartment to the Rab14+ compartment occurs through TLR3-dependent autophagy. Accordingly, CD8+ T cell memory to conidia was abrogated when autophagy was defective (e.g., in the absence of TLR3) or under conditions of defective endosomal alkalinization (e.g., in NADPH deficiency) (Savina et al. 2006) (Fig. 6.2). In these conditions, long-lasting antifungal protection and disease control was successfully achieved upon vaccination with purified fungal antigens that activate CD4+ T cells. Thus, CD8+ T cells can provide antifungal memory in CD4+ T cell deficiency and vice versa. These data highlight how understanding memory at a basic level, including information obtained from suitable animal models, may be exploited to personalize vaccination strategies against fungal diseases. Refinement of these approaches could lead to antifungal vaccines and adjuvants tailored to the different target populations, administered either alone or in combination with immunomodulators targeting antigen trafficking and presentation pathways. In this regard, promoting autophagy restores defective CD8+ T cell memory (De Luca et al. 2012), a finding that broadens the role of autophagy in adaptive immunity to include response to vaccines.

En route to antifungal vaccines. Antifungal vaccines undergo specific antigen presentation pathways leading to the activation of CD4+ and CD8+ T cells. Shown are results from co-localization studies using live conidia of A. fumigatus or protective and nonprotective soluble antigens (Ag). Memory, major histocompatibility complex (MHC) class II-restricted CD4+ T cells were activated by live conidia and purified Ags that were routed to the endosome/lysosome-dependent MHC class II presentation pathway. En route to lysosomes, purified fungal Ags also targeted to the mildly acidic stable early endosomal compartment and to MHC class II+ organelles. In contrast, phagocytosed cells activated MHC class I-restricted CD8+ T cells upon routing to the Rab14+ compartment, known to favor cross-presentation, and to MHC class I+ organelles. Of interest, the escape of fungal cells from endosomal/lysosomal degradation to the Rab14+ compartment occurred through activation of autophagy. MR mannose receptor, LAMP1 lysosomal-associated membrane protein 1, LC3 microtubule-associated protein 1A/1B-light chain 3, Rab14 rat sarcoma (RAS)–related protein (Rab) 14
VI. Final Remarks
The control of inflammation leading to tolerance, the molecular bases of immune regulation and deregulation, and the way in which commensal but opportunistic fungal pathogens can switch from a “friendly” affinity with the host to a pathological relationship by evading or subverting host inflammation, are challenging issues in the field of medical mycology and infection-related immunological disorders. A related question is how and whether the fungal microbiota contributes to the regulation of inflammation in health and disease. By the use of multidisciplinary approaches based on whole-genome immunogenetics, cutting-edge “omics” techniques, advanced bioinformatics, and systems biology applied to immune profiling, it will be possible to challenge existing paradigms in the fields of fungal immunopathology, thereby leading to the discovery of “commensal signatures” for the fungal biota and the development of therapeutic approaches for mucosal and systemic fungal diseases.
References
Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G (2007) Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8:639–646
Ahlgren KM, Moretti S, Lundgren BA, Karlsson I, Ahlin E, Norling A, Hallgren A, Perheentupa J, Gustafsson J, Rorsman F, Crewther PE, Ronnelid J, Bensing S, Scott HS, Kampe O, Romani L, Lobell A (2011) Increased IL-17A secretion in response to Candida albicans in autoimmune polyendocrine syndrome type 1 and its animal model. Eur J Immunol 41:235–245
Antachopoulos C, Walsh TJ, Roilides E (2007) Fungal infections in primary immunodeficiencies. Eur J Pediatr 166:1099–1117
Ashbee HR (2006) Recent developments in the immunology and biology of Malassezia species. FEMS Immunol Med Microbiol 47:14–23
Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, Mcallister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK (2008) IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 14:275–281
Bhatia S, Fei M, Yarlagadda M, Qi Z, Akira S, Saijo S, Iwakura Y, Van Rooijen N, Gibson GA, St Croix CM, Ray A, Ray P (2011) Rapid host defense against Aspergillus fumigatus involves alveolar macrophages with a predominance of alternatively activated phenotype. PLoS One 6:e15943
Bonifazi P, Zelante T, D’angelo C, De Luca A, Moretti S, Bozza S, Perruccio K, Iannitti RG, Giovannini G, Volpi C, Fallarino F, Puccetti P, Romani L (2009) Balancing inflammation and tolerance in vivo through dendritic cells by the commensal Candida albicans. Mucosal Immunol 2:362–374
Bonifazi P, D’angelo C, Zagarella S, Zelante T, Bozza S, De Luca A, Giovannini G, Moretti S, Iannitti RG, Fallarino F, Carvalho A, Cunha C, Bistoni F, Romani L (2010) Intranasally delivered siRNA targeting PI3K/Akt/mTOR inflammatory pathways protects from aspergillosis. Mucosal Immunol 3:193–205
Bozza S, Perruccio K, Montagnoli C, Gaziano R, Bellocchio S, Burchielli E, Nkwanyuo G, Pitzurra L, Velardi A, Romani L (2003) A dendritic cell vaccine against invasive aspergillosis in allogeneic hematopoietic transplantation. Blood 102:3807–3814
Bozza S, Montagnoli C, Gaziano R, Rossi G, Nkwanyuo G, Bellocchio S, Romani L (2004) Dendritic cell-based vaccination against opportunistic fungi. Vaccine 22:857–864
Bozza S, Clavaud C, Giovannini G, Fontaine T, Beauvais A, Sarfati J, D’angelo C, Perruccio K, Bonifazi P, Zagarella S, Moretti S, Bistoni F, Latge JP, Romani L (2009) Immune sensing of Aspergillus fumigatus proteins, glycolipids, and polysaccharides and the impact on Th immunity and vaccination. J Immunol 183(4):2407–2414
Bretz C, Gersuk G, Knoblaugh S, Chaudhary N, Randolph-Habecker J, Hackman RC, Staab J, Marr KA (2008) MyD88 signaling contributes to early pulmonary responses to Aspergillus fumigatus. Infect Immun 76:952–958
Carvalho A, De Luca A, Bozza S, Cunha C, D’angelo C, Moretti S, Perruccio K, Iannitti RG, Fallarino F, Pierini A, Latge JP, Velardi A, Aversa F, Romani L (2012) TLR3 essentially promotes protective class I-restricted memory CD8(+) T-cell responses to Aspergillus fumigatus in hematopoietic transplanted patients. Blood 119:967–977
Cassone A, Casadevall A (2012) Recent progress in vaccines against fungal diseases. Curr Opin Microbiol 15:427–433
Chai LY, Van De Veerdonk F, Marijnissen RJ, Cheng SC, Khoo AL, Hectors M, Lagrou K, Vonk AG, Maertens J, Joosten LA, Kullberg BJ, Netea MG (2010) Anti-Aspergillus human host defence relies on type 1 T helper (Th1), rather than type 17 T helper (Th17), cellular immunity. Immunology 130:46–54
Cheng SC, Van De Veerdonk F, Smeekens S, Joosten LA, Van Der Meer JW, Kullberg BJ, Netea MG (2010) Candida albicans dampens host defense by downregulating IL-17 production. J Immunol 185:2450–2457
Cobbold SP, Adams E, Nolan KF, Regateiro FS, Waldmann H (2010) Connecting the mechanisms of T-cell regulation: dendritic cells as the missing link. Immunol Rev 236:203–218
Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B (2011) RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12:560–567
Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, Filler SG, Masso-Welch P, Edgerton M, Gaffen SL (2009) Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206:299–311
Cooney NM, Klein BS (2008) Fungal adaptation to the mammalian host: it is a new world, after all. Curr Opin Microbiol 11:511–516
Cunha C, Di Ianni M, Bozza S, Giovannini G, Zagarella S, Zelante T, D’angelo C, Pierini A, Pitzurra L, Falzetti F, Carotti A, Perruccio K, Latge JP, Rodrigues F, Velardi A, Aversa F, Romani L, Carvalho A (2010) Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 116:5394–5402
Cutler JE, Deepe GS Jr, Klein BS (2007) Advances in combating fungal diseases: vaccines on the threshold. Nat Rev Microbiol 5:13–28
De Luca A, Montagnoli C, Zelante T, Bonifazi P, Bozza S, Moretti S, D’angelo C, Vacca C, Boon L, Bistoni F, Puccetti P, Fallarino F, Romani L (2007) Functional yet balanced reactivity to Candida albicans requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. J Immunol 179:5999–6008
De Luca A, Bozza S, Zelante T, Zagarella S, D’angelo C, Perruccio K, Vacca C, Carvalho A, Cunha C, Aversa F, Romani L (2010a) Non-hematopoietic cells contribute to protective tolerance to Aspergillus fumigatus via a TRIF pathway converging on IDO. Cell Mol Immunol 7:459–470
De Luca A, Zelante T, D’angelo C, Zagarella S, Fallarino F, Spreca A, Iannitti RG, Bonifazi P, Renauld JC, Bistoni F, Puccetti P, Romani L (2010b) IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol 3:361–373
De Luca A, Iannitti RG, Bozza S, Beau R, Casagrande A, D’angelo C, Moretti S, Cunha C, Giovannini G, Massi-Benedetti C, Carvalho A, Boon L, Latge JP, Romani L (2012) CD4(+) T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J Clin Invest 122:1816–1831
Desvignes L, Ernst JD (2009) Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 31:974–985
El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A (2011) The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol 12:568–575
Eyerich K, Foerster S, Rombold S, Seidl HP, Behrendt H, Hofmann H, Ring J, Traidl-Hoffmann C (2008) Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. J Invest Dermatol 128:2640–2645
Eyerich K, Eyerich S, Hiller J, Behrendt H, Traidl-Hoffmann C (2010) Chronic mucocutaneous candidiasis, from bench to bedside. Eur J Dermatol 20:260–265
Ferreira MC, De Oliveira RT, Da Silva RM, Blotta MH, Mamoni RL (2010) Involvement of regulatory T cells in the immunosuppression characteristic of patients with paracoccidioidomycosis. Infect Immun 78:4392–4401
Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, Van Spriel AB, Venselaar H, Elbers CC, Johnson MD, Cambi A, Huysamen C, Jacobs L, Jansen T, Verheijen K, Masthoff L, Morre SA, Vriend G, Williams DL, Perfect JR, Joosten LA, Wijmenga C, Van Der Meer JW, Adema GJ, Kullberg BJ, Brown GD, Netea MG (2009) Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med 361:1760–1767
Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, Mccraw SL, Gurr SJ (2012) Emerging fungal threats to animal, plant and ecosystem health. Nature 484:186–194
Fuchs BB, Mylonakis E (2006) Using non-mammalian hosts to study fungal virulence and host defense. Curr Opin Microbiol 9:346–351
Gessner MA, Werner JL, Lilly LM, Nelson MP, Metz AE, Dunaway CW, Chan YR, Ouyang W, Brown GD, Weaver CT, Steele C (2012) Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infect Immun 80:410–417
Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, Tahami F, Jamal S, Manguiat A, Rezaei N, Amirzargar AA, Plebani A, Hannesschlager N, Gross O, Ruland J, Grimbacher B (2009) A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 361:1727–1735
Grahl N, Puttikamonkul S, Macdonald JM, Gamcsik MP, Ngo LY, Hohl TM, Cramer RA (2011) In vivo hypoxia and a fungal alcohol dehydrogenase influence the pathogenesis of invasive pulmonary aspergillosis. PLoS Pathog 7:e1002145
Grohmann U, Volpi C, Fallarino F, Bozza S, Bianchi R, Vacca C, Orabona C, Belladonna ML, Ayroldi E, Nocentini G, Boon L, Bistoni F, Fioretti MC, Romani L, Riccardi C, Puccetti P (2007) Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med 13:579–586
Gupta AO, Singh N (2011) Immune reconstitution syndrome and fungal infections. Curr Opin Infect Dis 24:527–533
Hardison SE, Brown GD (2012) C-type lectin receptors orchestrate antifungal immunity. Nat Immunol 13:817–822
Hardison SE, Wozniak KL, Kolls JK, Wormley FL Jr (2010) Interleukin-17 is not required for classical macrophage activation in a pulmonary mouse model of Cryptococcus neoformans infection. Infect Immun 78:5341–5351
Hernandez-Santos N, Gaffen SL (2012) Th17 cells in immunity to Candida albicans. Cell Host Microbe 11:425–435
Holland SM, Deleo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B (2007) STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357:1608–1619
Huang W, Na L, Fidel PL, Schwarzenberger P (2004) Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190:624–631
Iannitti RG, Carvalho A, Romani L (2012) From memory to antifungal vaccine design. Trends Immunol 33:467–474
Jain AV, Zhang Y, Fields WB, Mcnamara DA, Choe MY, Chen GH, Erb-Downward J, Osterholzer JJ, Toews GB, Huffnagle GB, Olszewski MA (2009) Th2 but not Th1 immune bias results in altered lung functions in a murine model of pulmonary Cryptococcus neoformans infection. Infect Immun 77:5389–5399
Kaufmann SH, Kuchroo VK (2009) Th17 cells. Microbes Infect 11:579–583
Kontoyiannis DP, Marr KA, Park BJ, Alexander BD, Anaissie EJ, Walsh TJ, Ito J, Andes DR, Baddley JW, Brown JM, Brumble LM, Freifeld AG, Hadley S, Herwaldt LA, Kauffman CA, Knapp K, Lyon GM, Morrison VA, Papanicolaou G, Patterson TF, Perl TM, Schuster MG, Walker R, Wannemuehler KA, Wingard JR, Chiller TM, Pappas PG (2010) Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001-2006: overview of the Transplant-Associated Infection Surveillance Network (TRANSNET) Database. Clin Infect Dis 50:1091–1100
Kreindler JL, Steele C, Nguyen N, Chan YR, Pilewski JM, Alcorn JF, Vyas YM, Aujla SJ, Finelli P, Blanchard M, Zeigler SF, Logar A, Hartigan E, Kurs-Lasky M, Rockette H, Ray A, Kolls JK (2010) Vitamin D3 attenuates Th2 responses to Aspergillus fumigatus mounted by CD4+ T cells from cystic fibrosis patients with allergic bronchopulmonary aspergillosis. J Clin Invest 120:3242–3254
Levitz SM, Golenbock DT (2012) Beyond empiricism: informing vaccine development through innate immunity research. Cell 148:1284–1292
Lilic D (2002) New perspectives on the immunology of chronic mucocutaneous candidiasis. Curr Opin Infect Dis 15:143–147
Lilly LM, Gessner MA, Dunaway CW, Metz AE, Schwiebert L, Weaver CT, Brown GD, Steele C (2012) The beta-glucan receptor dectin-1 promotes lung immunopathology during fungal allergy via IL-22. J Immunol 189:3653–3660
Lin L, Ibrahim AS, Xu X, Farber JM, Avanesian V, Baquir B, Fu Y, French SW, Edwards JE Jr, Spellberg B (2009) Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog 5:e1000703
Liu Y, Yang B, Zhou M, Li L, Zhou H, Zhang J, Chen H, Wu C (2009) Memory IL-22-producing CD4+ T cells specific for Candida albicans are present in humans. Eur J Immunol 39:1472–1479
Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulacsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RM, Blancas-Galicia L, Beas IM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel PR, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bue M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, Al-Muhsen S, Reichenbach J, Kobayashi M, Rosales FE, Lozano CT, Kilic SS, Oleastro M, Etzioni A, Traidl-Hoffmann C, Renner ED, Abel L, Picard C, Marodi L, Boisson-Dupuis S, Puel A, Casanova JL (2011) Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208:1635–1648
Loures FV, Pina A, Felonato M, Calich VL (2009) TLR2 is a negative regulator of Th17 cells and tissue pathology in a pulmonary model of fungal infection. J Immunol 183:1279–1290
Luong M, Lam JS, Chen J, Levitz SM (2007) Effects of fungal N- and O-linked mannosylation on the immunogenicity of model vaccines. Vaccine 25:4340–4344
Magditch DA, Liu TB, Xue C, Idnurm A (2012) DNA mutations mediate microevolution between host-adapted forms of the pathogenic fungus Cryptococcus neoformans. PLoS Pathog 8:e1002936
Martinic MM, Von Herrath MG (2006) Control of graft-versus-host disease by regulatory T cells: which level of antigen specificity? Eur J Immunol 36:2299–2303
Mcfall-Ngai M (2007) Adaptive immunity: care for the community. Nature 445:153
Medzhitov R, Schneider DS, Soares MP (2012) Disease tolerance as a defense strategy. Science 335:936–941
Mellor AL, Munn DH (2008) Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol 8:74–80
Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’shea J, Holland SM, Paul WE, Douek DC (2008) Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452:773–776
Montagnoli C, Fallarino F, Gaziano R, Bozza S, Bellocchio S, Zelante T, Kurup WP, Pitzurra L, Puccetti P, Romani L (2006) Immunity and tolerance to Aspergillus involve functionally distinct regulatory T cells and tryptophan catabolism. J Immunol 176:1712–1723
Moraes-Vasconcelos D, Grumach AS, Yamaguti A, Andrade ME, Fieschi C, De Beaucoudrey L, Casanova JL, Duarte AJ (2005) Paracoccidioides brasiliensis disseminated disease in a patient with inherited deficiency in the beta1 subunit of the interleukin (IL)-12/IL-23 receptor. Clin Infect Dis 41:e31–e37
Mylonakis E, Casadevall A, Ausubel FM (2007) Exploiting amoeboid and non-vertebrate animal model systems to study the virulence of human pathogenic fungi. PLoS Pathog 3:e101
Nanjappa SG, Heninger E, Wuthrich M, Gasper DJ, Klein BS (2012a) Tc17 cells mediate vaccine immunity against lethal fungal pneumonia in immune deficient hosts lacking CD4+ T cells. PLoS Pathog 8:e1002771
Nanjappa SG, Heninger E, Wuthrich M, Sullivan T, Klein B (2012b) Protective antifungal memory CD8(+) T cells are maintained in the absence of CD4(+) T cell help and cognate antigen in mice. J Clin Invest 122:987–999
Odds FC, Jacobsen MD (2008) Multilocus sequence typing of pathogenic Candida species. Eukaryot Cell 7:1075–1084
Orabona C, Grohmann U, Belladonna ML, Fallarino F, Vacca C, Bianchi R, Bozza S, Volpi C, Salomon BL, Fioretti MC, Romani L, Puccetti P (2004) CD28 induces immunostimulatory signals in dendritic cells via CD80 and CD86. Nat Immunol 5:1134–1142
Pandiyan P, Conti HR, Zheng L, Peterson AC, Mathern DR, Hernandez-Santos N, Edgerton M, Gaffen SL, Lenardo MJ (2011) CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity 34:422–434
Pappas PG, Alexander BD, Andes DR, Hadley S, Kauffman CA, Freifeld A, Anaissie EJ, Brumble LM, Herwaldt L, Ito J, Kontoyiannis DP, Lyon GM, Marr KA, Morrison VA, Park BJ, Patterson TF, Perl TM, Oster RA, Schuster MG, Walker R, Walsh TJ, Wannemuehler KA, Chiller TM (2010) Invasive fungal infections among organ transplant recipients: results of the Transplant-Associated Infection Surveillance Network (TRANSNET). Clin Infect Dis 50:1101–1111
Paveglio SA, Allard J, Foster Hodgkins SR, Ather JL, Bevelander M, Campbell JM, Whittaker Leclair LA, Mccarthy SM, Van Der Vliet A, Suratt BT, Boyson JE, Uematsu S, Akira S, Poynter ME (2011) Airway epithelial indoleamine 2,3-dioxygenase inhibits CD4+ T cells during Aspergillus fumigatus antigen exposure. Am J Respir Cell Mol Biol 44:11–23
Peleg AY, Tampakakis E, Fuchs BB, Eliopoulos GM, Moellering RC Jr, Mylonakis E (2008) Prokaryote-eukaryote interactions identified by using caenorhabditis elegans. Proc Natl Acad Sci USA 105:14585–14590
Perfect JR (2012) The impact of the host on fungal infections. Am J Med 125:S39–S51
Pinzan CF, Ruas LP, Casabona-Fortunato AS, Carvalho FC, Roque-Barreira MC (2010) Immunological basis for the gender differences in murine Paracoccidioides brasiliensis infection. PLoS One 5:e10757
Puccetti P, Grohmann U (2007) IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat Rev Immunol 7:817–823
Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova JL (2011) Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332:65–68
Pulendran B, Tang H, Denning TL (2008) Division of labor, plasticity, and crosstalk between dendritic cell subsets. Curr Opin Immunol 20:61–67
Ramirez-Ortiz ZG, Lee CK, Wang JP, Boon L, Specht CA, Levitz SM (2011) A nonredundant role for plasmacytoid dendritic cells in host defense against the human fungal pathogen Aspergillus fumigatus. Cell Host Microbe 9:415–424
Rapaka RR, Ricks DM, Alcorn JF, Chen K, Khader SA, Zheng M, Plevy S, Bengten E, Kolls JK (2010) Conserved natural IgM antibodies mediate innate and adaptive immunity against the opportunistic fungus Pneumocystis murina. J Exp Med 207:2907–2919
Rappleye CA, Goldman WE (2008) Fungal stealth technology. Trends Immunol 29:18–24
Richie DL, Hartl L, Aimanianda V, Winters MS, Fuller KK, Miley MD, White S, Mccarthy JW, Latge JP, Feldmesser M, Rhodes JC, Askew DS (2009) A role for the unfolded protein response (UPR) in virulence and antifungal susceptibility in Aspergillus fumigatus. PLoS Pathog 5:e1000258
Rivera A, Hohl TM, Collins N, Leiner I, Gallegos A, Saijo S, Coward JW, Iwakura Y, Pamer EG (2011) Dectin-1 diversifies Aspergillus fumigatus-specific T cell responses by inhibiting T helper type 1 CD4 T cell differentiation. J Exp Med 208:369–381
Rizzetto L, Cavalieri D (2010) A systems biology approach to the mutual interaction between yeast and the immune system. Immunobiology 215:762–769
Romani L (2011) Immunity to fungal infections. Nat Rev Immunol 11:275–288
Romani L, Puccetti P (2006) Protective tolerance to fungi: the role of IL-10 and tryptophan catabolism. Trends Microbiol 14:183–189
Romani L, Puccetti P (2007) Controlling pathogenic inflammation to fungi. Expert Rev Anti Infect Ther 5:1007–1017
Romani L, Puccetti P (2008) Immune regulation and tolerance to fungi in the lungs and skin. Chem Immunol Allergy 94:124–137
Romani L, Bistoni F, Puccetti P (2002) Fungi, dendritic cells and receptors: a host perspective of fungal virulence. Trends Microbiol 10:508–514
Romani L, Bistoni F, Perruccio K, Montagnoli C, Gaziano R, Bozza S, Bonifazi P, Bistoni G, Rasi G, Velardi A, Fallarino F, Garaci E, Puccetti P (2006) Thymosin alpha1 activates dendritic cell tryptophan catabolism and establishes a regulatory environment for balance of inflammation and tolerance. Blood 108:2265–2274
Romani L, Fallarino F, De Luca A, Montagnoli C, D’angelo C, Zelante T, Vacca C, Bistoni F, Fioretti MC, Grohmann U, Segal BH, Puccetti P (2008a) Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 451:211–215
Romani L, Zelante T, De Luca A, Fallarino F, Puccetti P (2008b) IL-17 and therapeutic kynurenines in pathogenic inflammation to fungi. J Immunol 180:5157–5162
Roy RM, Klein BS (2012) Dendritic cells in antifungal immunity and vaccine design. Cell Host Microbe 11:436–446
Ryan KR, Hong M, Arkwright PD, Gennery AR, Costigan C, Dominguez M, Denning D, Mcconnell V, Cant AJ, Abinun M, Spickett GP, Lilic D (2008) Impaired dendritic cell maturation and cytokine production in patients with chronic mucocutanous candidiasis with or without APECED. Clin Exp Immunol 154:406–414
Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N, Kinjo T, Nakamura K, Kawakami K, Iwakura Y (2007) Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol 8:39–46
Sallusto F, Lanzavecchia A, Araki K, Ahmed R (2010) From vaccines to memory and back. Immunity 33:451–463
Saraiva M, O’garra A (2010) The regulation of IL-10 production by immune cells. Nat Rev Immunol 10:170–181
Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, Greer F, Davoust J, Kratzer R, Keller SR, Niedermann G, Van Endert P (2009) IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science 325:213–217
Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, Amigorena S (2006) NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126:205–218
Spellberg B, Ibrahim AS, Lin L, Avanesian V, Fu Y, Lipke P, Otoo H, Ho T, Edwards JE Jr (2008) Antibody titer threshold predicts anti-candidal vaccine efficacy even though the mechanism of protection is induction of cell-mediated immunity. J Infect Dis 197:967–971
Steinman RM (2008) Dendritic cells and vaccines. Proc (Bayl Univ Med Cent) 21(1):3–8. PMID 18209746
Steinman RM (2012) Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 30:1–22
Stuehler C, Khanna N, Bozza S, Zelante T, Moretti S, Kruhm M, Lurati S, Conrad B, Worschech E, Stevanovic S, Krappmann S, Einsele H, Latge JP, Loeffler J, Romani L, Topp MS (2011) Cross-protective TH1 immunity against Aspergillus fumigatus and Candida albicans. Blood 117:5881–5891
Szymczak WA, Deepe GS Jr (2009) The CCL7-CCL2-CCR2 axis regulates IL-4 production in lungs and fungal immunity. J Immunol 183:1964–1974
Turnquist HR, Thomson AW (2008) Taming the lions: manipulating dendritic cells for use as negative cellular vaccines in organ transplantation. Curr Opin Organ Transplant 13:350–357
Van De Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-Van Der Graaf CA, Kullberg BJ, Van Der Meer JW, Lilic D, Veltman JA, Netea MG (2011) STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 365:54–61
Voelz K, Lammas DA, May RC (2009) Cytokine signaling regulates the outcome of intracellular macrophage parasitism by Cryptococcus neoformans. Infect Immun 77:3450–3457
Waldmann H, Cobbold S (2004) Exploiting tolerance processes in transplantation. Science 305:209–212
Wozniak KL, Levitz SM (2008) Cryptococcus neoformans enters the endolysosomal pathway of dendritic cells and is killed by lysosomal components. Infect Immun 76:4764–4771
Wuthrich M, Gern B, Hung CY, Ersland K, Rocco N, Pick-Jacobs J, Galles K, Filutowicz H, Warner T, Evans M, Cole G, Klein B (2011) Vaccine-induced protection against 3 systemic mycoses endemic to North America requires Th17 cells in mice. J Clin Invest 121:554–568
Wuthrich M, Deepe GS Jr, Klein B (2012) Adaptive immunity to fungi. Annu Rev Immunol 30:115–148
Zelante T, De Luca A, Bonifazi P, Montagnoli C, Bozza S, Moretti S, Belladonna ML, Vacca C, Conte C, Mosci P, Bistoni F, Puccetti P, Kastelein RA, Kopf M, Romani L (2007) IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol 37:2695–2706
Zelante T, Fallarino F, Bistoni F, Puccetti P, Romani L (2009) Indoleamine 2,3-dioxygenase in infection: the paradox of an evasive strategy that benefits the host. Microbes Infect 11:133–141
Zelante T, Iannitti RG, De Luca A, Arroyo J, Blanco N, Servillo G, Sanglard D, Reichard U, Palmer GE, Latge JP, Puccetti P, Romani L (2012) Sensing of mammalian IL-17A regulates fungal adaptation and virulence. Nat Commun 3:683
Zeller S, Glaser AG, Vilhelmsson M, Rhyner C, Crameri R (2008) Immunoglobulin-E-mediated reactivity to self antigens: a controversial issue. Int Arch Allergy Immunol 145:87–93
Zenewicz LA, Flavell RA (2008) IL-22 and inflammation: leukin’ through a glass onion. Eur J Immunol 38:3265–3268
Zhou L, Chong MM, Littman DR (2009) Plasticity of CD4+ T cell lineage differentiation. Immunity 30:646–655
Acknowledgments
We thank Cristina Massi-Benedetti for editorial assistance. The authors’ work was supported by the ERC Advanced Grant FUNMETA (ERC-2011-AdG-293714). Cristina Cunha was supported by the Fundação para a Ciência e Tecnologia, Portugal (contract SFRH/BD/65962/2009). We sincerely apologize to all those colleagues whose important work is not cited because of space considerations.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Cunha, C., Aversa, F., Romani, L., Carvalho, A. (2014). 6 T Cell Responses in Fungal Infections. In: Kurzai, O. (eds) Human Fungal Pathogens. The Mycota, vol 12. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-39432-4_6
Download citation
DOI: https://doi.org/10.1007/978-3-642-39432-4_6
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-39431-7
Online ISBN: 978-3-642-39432-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)