
Abstract
Human papillomaviruses (HPVs) are the causative agents of cervical and other anogenital as well as oral cancers. Approximately fifty percent of virally induced cancers in the USA are associated with HPV infections. HPVs infect stratified epithelia and link productive replication with differentiation. The viral oncoproteins, E6, E7, and E5, play important roles in regulating viral functions during the viral life cycle and also contribute to the development of cancers. p53 and Rb are two major targets of the E6 and E7 oncoproteins, but additional cellular proteins also play important roles. E5 plays an auxiliary role in contributing to the development of cancers. This review will discuss the various targets of these viral proteins and what roles they play in viral pathogenesis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Human papillomaviruses (HPVs) are small, double-stranded DNA viruses that contain circular genomes of approximately 8 kilobases (kb) in length and encode approximately eight major open reading frames (ORFs) (Howley and Lowy 2007). These viruses have a tropism for squamous epithelial tissues of the feet, hands, and anogenital tracts where they propagate via an unusual life cycle utilizing the differentiation program of the host cell (zur Hausen and de Villiers 1994). There is vast diversity among the HPVs with almost 200 different types identified to date. Even though the viruses are genetically distinct, they all infect epithelial tissues, only differing in the preferential target body location for infection (zur Hausen and de Villiers 1994). About 40 types of HPVs exhibit tropism for the genital tract, while the remaining HPVs specifically infect cutaneous tissues (Howley and Lowy 2007; Moody and Laimins 2010).
Genital HPVs can be further categorized into two main groups, high-risk (HR) and low-risk (LR) types, which is determined by the propensity of the infection to progress to malignancy. HR genital HPVs, including types 16, 18, 31, 33, 45, and 5, are frequently associated with cervical carcinomas (zur Hausen and de Villiers 1994). HPV DNA is found in over 99 % of cervical cancers, while HPV DNA exists as extrachromosomal elements, or episomes, in precancerous lesions. Although up to 50 % of HPV 16-positive and most HPV 18-positive carcinomas maintain HPV DNA as episomes (Parkin et al. 2000). While at least 10 HPV types can contribute to development of cervical cancer, three types are the major contributors: HPV 16 is found in about 50 % of cervical cancers, HPV 18 in approximately 25 %, and HPV 31 in about 10 % of cases (Fehrmann and Laimins 2003; Longworth and Laimins 2004a). Conversely, LR genital HPVs are rarely associated with malignancies and instead primarily cause benign genital warts. LR genital HPVs include types 6, 11, 42, 43, and 44, of which types 6 and 11 are responsible for about 90 % of all genital warts (Lorincz et al. 1992).
High-risk HPVs (HR-HPVs) are the causative agents of most cervical cancers accounting for up to 5 % of all human cancers (Stanley 2010). Cervical cancer is the second most common cancer in women worldwide with over 500,000 new cancer cases diagnosed each year and is the third leading cancer killer, causing nearly 300,000 deaths in women annually (Ferlay et al. 2008). The use of the Papanicolaou smear (Pap smear) has reduced the incidence of cervical cancers by over 80 % in the USA in the past 50 years (Moody and Laimins 2010). Additionally, the Federal Drug and Administration (FDA) recently approved the use of two prophylactic polyclonal vaccines targeted against the most common HPV types associated with cervical cancers and genital warts. The use of these vaccines is approved for males and females between the ages of 10 and 25. Gardasil® is the quadrivalent vaccine (HPV 6, 11, 16, 18) developed by Merck, and Cervarix® is the bivalent (HPV 16, 18) vaccine developed by GlaxoSmithKline. Both of these vaccines are effective in preventing initial HPV infection (Markowitz et al. 2007). These vaccines are recommended for individuals, who have never been sexually active since they can only prevent new infections, and not treat existing infections or used as a cancer treatment. Immunization with one of these vaccines along with annual Pap smear screening is the most effective prevention strategy.
HPV infections are sexually transmitted and rank as the most common sexually transmitted viral infections (Markowitz et al. 2007). Recent analyses indicate that approximately 20 million Americans are currently infected with HPV. Further studies by the American Social Health Association estimate that 75 % of sexually active individuals between the ages of 15 and 49 have been infected with HPV at some point in their lives. It is also estimated that more than half of infection-related cancers in females are attributed to HPV (zur Hausen 2009). Together, these findings demonstrate the major public health impact HPV infection and cervical cancer has in the USA and globally. This exemplifies the need for continued research for prevention and treatment of HPV infections and HPV-related cancers.
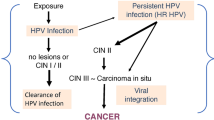
Infection with genital HPVs in most individuals can last for up to 2 years due to the ability of the virus to evade the innate immune surveillance, and thus delaying onset of an adaptive immune response. This immune evasion is due in part to HPVs not being cytolytic and the fact that viral proteins are expressed at very low levels (Bodily and Laimins 2011). In addition, HPV-induced innate immune evasion results in a delayed onset of the adaptive immune response, facilitating the persistence and productive replication of the virus. Eventually, most people are able to mount an effective cell-mediated immune response, which clears the viral infection (Stanley 2008). However, up to 20 % of women fail to clear the infection and are at high risk of developing cervical carcinoma (Bodily and Laimins 2011). Although persistent HR-HPV infection is the single most important contributing factor to the development of cervical cancer, other risk factors such as immunosuppression, cigarette smoking, and infection with human immunodeficiency virus (HIV) also contribute to progression to malignancy (Markowitz et al. 2007; Bodily and Laimins 2011; zur Hausen 1996).
The development of cervical carcinoma is not a rapid event as it typically occurs over a period of several decades. In the cases where initial infection is not cleared by immune surveillance, precancerous lesions can typically develop within a decade and cervical cancer within several decades. To understand how viral infection can progress to cervical cancer, it is first important to examine the unusual life cycle of the virus and genomic arrangement.
1.1 The HPV Life Cycle
The HPV life cycle is intimately associated with the differentiation program of epithelial cells (Fig. 1). To understand the unusual life cycle of HPV, it is first important to understand the normal epithelial differentiation program. In normal epithelial cells, the only actively dividing cells are present in the basal layers of the stratified epithelium, which consist of transit amplifying (TA) cells and stem cells. TA cells are defined as cells that are proliferating and can terminally differentiate. In contrast, stem cells have the potential to proliferate indefinitely, but divide infrequently in order to replenish the TA cell pool. Once cells in the basal layer divide and one daughter cell migrates suprabasally and differentiates, it loses its ability to remain active in the cell cycle. The differentiating cells go through a number of events including changes in gene expression and desquamation (Bodily and Laimins 2011).

Differentiation-dependent HPV Life Cycle. The HPV life cycle is intimately associated with the differentiation program of epithelial cells. HPV infects actively dividing cells in basal layer of the epithelium via a micro-abrasion. Following entry, viral gene expression is activated and episomal HPV DNA is maintained at approximately 50–100 copies per cell. HPV oncoproteins then enable the infected cells that exit the basal layer to remain active in the cell cycle. Once the infected cells begin to differentiate, the late promoter is activated, which results in the onset of the productive phase of the viral life cycle. During this phase, viral DNA amplification occurs and viral protein expression is increased. Finally, synthesis of viral capsids and packaging occurs in the uppermost differentiated layer of the epithelium, followed by release of the progeny virions
Upon infection, the virus establishes its double-stranded DNA genome in the nuclei of infected host cells (Howley and Lowy 2007). HPV gains entry to cells in the basal layer of the epithelium that become exposed through micro-abrasions (Moody and Laimins 2010). Infection of the basal layer allows the virus to establish a persistent infection, as the basal cells are the only cells of the epithelium undergoing active replication (Moody and Laimins 2010). Since the HPV genomes are only 8 kb in size, they do not encode viral polymerases or other enzymes required for viral replication. The virus must therefore rely on host cell replication machinery to facilitate viral DNA synthesis (Moody and Laimins 2010). Following entry, viral genomes are established in the nucleus as extrachromosomal plasmids, or episomes. In the infected basal cells, early viral gene expression is activated, and genome copy numbers are maintained at approximately 20–100 copies per cell (Moody and Laimins 2010). As HPV-infected basal cells divide, one of the infected daughter cells remains in the basal layer. The other daughter cell migrates away from the basal layer and begins to differentiate, resulting in the activation of the late viral promoter. This results in the onset of the productive phase of the life cycle, which includes viral DNA amplification, with copy number increasing to over 100 copies of HPV DNA per cell, and the onset of capsid gene expression. Finally, synthesis of viral capsids and packaging of viral genomes occur in the uppermost differentiated layer of the epithelium, ultimately resulting in the release of the progeny virions.
The signals that control the induction of late viral events in the life cycle are not well characterized, but studies have shown that HPV oncoproteins enable infected cells in the suprabasal layer to remain active in the cell cycle and to reenter S phase or arrest in G2/M to allow for viral amplification. This alteration of cell cycle control is essential for activation of the productive phase of the life (Moody and Laimins 2010). Furthermore, studies have indicated that viral proteins E6, E7, E1^E4, and E5 are needed for this activation, and these activities will be briefly summarized below.
1.2 The HPV Genome
The small, double-stranded DNA genome of all HPVs is approximately 8 kb in size. On average, HPVs encode eight major ORFs which are expressed from polycistronic mRNAs transcribed from a single DNA strand (Howley and Lowy 2007) (Fig. 2). The early proteins, E1, E2, E6, and E7, are expressed early in infection in undifferentiated cells and have drastically different functions. Sequences within the upstream regulatory region (URR) located in the non-coding region of the genome are responsible for regulation of viral transcription and replication. Expression of HPV gene products is directed from two different promoters, the early promoter and the late promoter (Moody and Laimins 2010). The early promoter, termed P97 in HPV 31, is located upstream of the E6 ORF and directs expression of early (E) gene products in undifferentiated cells. Early proteins include E1, E2, E6, E7, E1^E4, and E5. Translation of HPV messages occurs by a leaky scanning mechanism resulting in high levels of E6 and E7, but low levels of E1^E4 and E5 protein synthesis. The E1 and E2 proteins function in replication and transcription control, while E1^E4 modulates late viral functions. The late promoter, P742 in HPV 31, directs the expression of late (L) gene products and is located within the E7 ORF. Importantly, P742 is activated upon epithelial cell differentiation. Late proteins include L1 and L2 as well as E1^E4, and E5, and these are all expressed from P742 (Moody and Laimins 2010).

Linear arrangement of HPV genome. The HPV genome is represented in linear form here for simplicity. The genomes are small, circular, double-stranded DNA genomes of 8 kilobases (kb) in size. There are on average 8 open reading frames (ORFs) (E1, E2, E4, E5, E6, E7, L1, and L2) expressed from a single polycistronic transcript transcribed from a single strand of DNA. The upstream regulatory region (URR) is located in the non-coding region and contains sequences responsible for regulating viral transcription and replication. Three general groups of HPV genes that are regulated during differentiation are the virus early promoter (P 97 ), the differentiation-dependent late promoter (P 742 ), and two polyadenylation signals (PolyA). P 97 is located upstream of the E6 ORF and directs expression of early genes in undifferentiated cells. P 742 is located within the E7 ORF, is activated upon differentiation of the host cells, and directs expression of late gene products. E6 and E7 are oncogenes involved in replication competence. E1 and E2 are genes involved in viral DNA replication and regulation of viral transcription. E4 and E5 are genes involved in late functions. L1 and L2 are the capsid proteins
1.3 Oncoproteins: E5, E6, and E7
The E6 and E7 proteins are expressed upon initial HPV infection of host keratinocytes, while E5 is primarily expressed in the late phase of the life cycle. Although these proteins all contribute to promoting tumor growth in host cells, each of these proteins has distinct functions. As previously discussed, E1 and E2 are responsible for replication and regulation of viral transcription, whereas E6 and E7 proteins are largely responsible for modulating cell cycle progression. In the HR-HPVs, E6 and E7 act as oncoproteins that are necessary for the development of genital carcinomas. Conversely, no such function has been demonstrated for LR-HPV proteins. While both E6 and E7 proteins are localized to the host nucleus, the E6 proteins are also detected in the cytoplasm of HPV-infected cells (Howley and Lowy 2007; Moody and Laimins 2010). Studies have shown that expression of E6 proteins is sufficient for immortalization of human mammary epithelial cells and transformation of NIH3T3 fibroblasts; however, expression of both E6 and E7 proteins is required for efficient immortalization of human keratinocytes (Howley and Lowy 2007).
E6 proteins are approximately 150 amino acids (18 kilodaltons) in size and contain two zinc-binding domains consisting of four Cys–X–X–Cys motifs (Howley and Lowy 2007). Studies have identified various E6-mediated activities that are mediated by interactions with over a dozen different proteins. One of the well-characterized interactions is the binding of E6 proteins to the tumor suppressor protein p53, affecting p53-dependent cell cycle regulation. The p53 protein is important in regulating the G1/S and G2/M cell cycle checkpoints following DNA damage (Slee et al. 2004; Oren 2003). Studies have shown that E6 proteins form complexes with an E3 cellular ubiquitin ligase, E6-associated protein (E6AP), and p53, resulting in rapid proteasomal (26S) degradation of p53 (Scheffner et al. 1990). In a normal response to DNA damage or unscheduled induction of replication, p53 is activated via various modifications. The activation of this short-lived transcription factor results in modulation of the cell cycle and in, some cases, activation of apoptotic processes. Activated p53 forms a homotetramer that transcriptionally activates expression of cell cycle regulatory proteins, such as cyclin kinase inhibitor p21, which is responsible for inducing a G1/S arrest (Ko and Prives 1996). The activation of p53 can also induce programmed cell death (apoptosis). Cell cycle arrest allows the cell to repair the damage to the DNA prior to entry to S phase. In the event that the damage is too extensive, the cell triggers apoptosis to prevent a cell from replicating damaged DNA. In addition, p53 is activated following viral infection. Given that HPV relies on host cell machinery and S phase entry to replicate its genome, the virus has devised a mechanism to disrupt normal p53 action. The E6-mediated proteasomal degradation of p53 results in the deregulation of the cell cycle, which allows the virus to persist and replicate its genome (Moody and Laimins 2010).
An additional HR E6 activity is its ability to interact with p300/CBP, a p53 co-activator (Patel et al. 1999; Zimmermann et al. 1999). The p300/CBP/E6 interaction prevents acetylation of p53, which down-regulates p53 activity, therefore blocking cell cycle arrest. The binding of p300/CBP occurs independently of E6-mediated degradation of p53 (Patel et al. 1999; Zimmermann et al. 1999). Interestingly, studies indicate that immortalization competency is not exclusively linked to p53-dependent mechanisms since E6 mutants incapable of degrading p53 are still able to immortalize cells; similarly, E6 mutants with normal degradation activity fail to immortalize cells (Kiyono et al. 1997).
One p53-independent function of E6 is the activation of telomerase by HR E6 proteins (Klingelhutz et al. 1996). Telomerase is an enzyme with four subunits that replicates telomeric DNA at the ends of chromosomes by adding hexamer repeats. Expression of its catalytic subunit, human telomerase reverse transcriptase (hTERT), plays an essential role in regulation of telomerase activity (Liu 1999). Over successive cell divisions, the telomeres become critically shortened and dysfunctional, leading to a limited cell proliferative lifespan because of the induction of senescence and irreversible cell growth arrest. In contrast, in cancers, hTERT expression is typically reactivated resulting in reconstitution of telomerase activity (Liu 1999). Studies have revealed that E6 increases expression of endogenous hTERT levels through transcriptional activation of the hTERT promoter through the action of NFX1-123, Myc and Sp-1 (Kyo et al. 2000; Howie et al. 2009; Gewin and Galloway 2001; Katzenellenbogen et al. 2009). While this activity is not the only function necessary for efficient immortalization of the host cell, it is a crucial function of E6.
Another interaction important for the ability of E6 proteins to immortalize cells is its association with several PDZ domain–containing proteins. PDZ domains are approximately 90 amino acids in size and are binding domains for a number of proteins including post-synaptic density protein (PSD-95), Drosophila disk large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1). These domains are often found in proteins responsible for cell–cell adhesion as well as cell signaling and typically localized in areas of cell–cell contact. Studies have shown that PDZ proteins MUPP-1, hDLG, hScribble, and MAGI-1, 2, 3 bind to the extreme C-terminus of HR E6 proteins resulting in degradation of the PDZ protein (Lee et al. 1997, 2000). While PDZ protein interactions with E6 proteins may contribute to malignant progression, complete characterization of the mechanisms involved is still unclear.
HR E6 proteins have also been found to interact with various other cellular factors including paxillin, the putative calcium-binding protein E6-BP, and the interferon regulatory factor IRF-3 (Patel et al. 1999; Ronco et al. 1998). Several studies have identified many cellular binding partners for LR E6 proteins such as MCM7, Bak, zyxin, and GPS2 (Kuhne and Banks 1998; Kukimoto et al. 1998; Thomas and Banks 1999). It is clear from the characterization of numerous E6-binding partners and activities, that this viral protein is essential in the viral life cycle since knockout of E6 in genomes results in loss of ability to maintain episomes (Thomas et al. 1999).
The second oncoprotein, E7, is approximately 100 amino acids in size and is able to form dimers via its C-terminus. HR E7 proteins are comprised of three conserved regions: CR1 present at the N-terminus; CR2 containing an LXCXE motif that binds the retinoblastoma protein (Rb); and CR3, which contains two zinc finger-like motifs (Dyson et al. 1992). The CR1 and CR2 domains have sequence homology to the conserved regions CR1 and CR2 in adenovirus E1A. E7 is able to transform NIH3T3 fibroblasts by itself and with increased efficiency upon co-expression of E6. In contrast to E6, E7 oncoproteins are able to immortalize human keratinocytes when expressed alone, albeit, at a very low frequency (Howley and Lowy 2007; Munger et al. 1989; Riley et al. 2003).
A central activity of E7 proteins is their association with the Rb family of proteins (Dyson et al. 1989). Rb, p107, and p130 are the members of this family, and their expression occurs throughout the cell cycle, regulating cell cycle progression. In order to understand how E7 can circumvent Rb-regulated cell cycle progression, it is first important to appreciate how Rb regulates the cell cycle in normal circumstances. Unphosphorylated Rb proteins and the E2F/DP1 transcription factors form complexes to repress transcription of genes involved in S phase progression (DNA synthesis) or apoptosis. E2F transcription factors regulate the transcription of proteins required for normal cellular DNA synthesis. The transition from G1 to S phase is triggered when cyclin kinase complexes phosphorylate the Rb proteins causing their release from the E2F complex relieving transcriptional repression of transcription of genes involved in DNA synthesis.
E7 alters the regulation of G1/S by promoting the constitutive expression of E2F-regulated genes by binding Rb and sequestering it away from forming E2F/DP1 complexes. This relieves transcriptional repression and allows genes required for DNA synthesis to be transcribed (Edmonds and Vousden 1989; Weintraub et al. 1995). Additionally, E7 is able to target Rb for ubiquitin-mediated proteasomal degradation, which again allows for E2F-regulated genes to be constitutively transcribed, promoting DNA synthesis (Howley and Lowy 2007; Moody and Laimins 2010). Rb proteins are also responsible for controlling cell cycle exit during epithelial differentiation; hence, E7-mediated abrogation of Rb function maintains cell cycle activity. This activity is necessary for productive viral replication to occur in the differentiated epithelial cells (Thomas et al. 1999). The importance of the Rb-E7 is also essential for the virus’ ability to maintain the genome as episomes in undifferentiated cells (Longworth and Laimins 2004b).
Another way in which E7 proteins can affect cell cycle progression is via its ability to associate with cyclins and cyclin-dependent kinases (cdk) inhibitors. For example, the association of E7 with cyclins A and E as well as cdk inhibitors p21 and p27 has been characterized in various studies (Davies et al. 1993; Funk et al. 1997; Jones et al. 1997; Ruesch and Laimins 1998; Tommasino et al. 1993; Zerfass-Thome et al. 1996). These cellular proteins affect the phosphorylation status of Rb proteins, and in doing so facilitate cell cycle progression. Specifically, E7 proteins can bind cyclins A, E, p21, and p27 resulting in an increase in cyclin A and E levels while causing a decrease in p21 and p27 levels (Jones et al. 1997; Ruesch and Laimins 1998; Tommasino et al. 1993). The net effect of these interactions is to drive progression of the cell cycle, which is necessary for facilitating the HPV life cycle in differentiating epithelia.
Alteration of Rb phosphorylation status is not the only means of affecting E2F-responsive promoters as they can also be repressed by action of histone deacetylases (HDACs). Targeting of class I HDACs is another mechanism in which E7 affects cell cycle regulation (Longworth and Laimins 2004a, b; Brehm et al. 1998). HDACs are ubiquitously expressed transcriptional co-repressors that remove acetyl groups from lysine-rich N-terminal tails of histone proteins in the nucleosome. Additionally, HDACs can directly deacetylated E2F-responsive factors resulting in the loss of their function. There are three classes of HDACs, which are classified according to sequence homology and localization of the factors in the cell. Class I HDACs are localized to the nucleus and include HDACs 1, 2, 3, and 8. Class I HDACs require binding to cofactor proteins that either modify their activity or localize them to site of action. HR E7 proteins indirectly associate with class I HDACs through direct binding to the auxiliary protein, Mi2β, via sequences in the zinc finger regions at the C-terminus (Brehm et al. 1998). This HDAC/E7 interaction specifically results in increased levels of E2F-responsive transcription in differentiating cells, which allows cells to maintain cell cycle activity (Longworth and Laimins 2004b). The binding of E7 to HDACs is also important in facilitating the viral life cycle as HPV 31 genomes with mutations abrogating the E7/HDAC interaction have slower growth, are defective in maintaining genomes as episomes, and have a limited life span (Longworth and Laimins 2004b).
Many cancers exhibit increased genomic instability, and similar effects are seen in HPV-induced cancers. HR E7 expression is responsible for inducing genomic instability in these cancers. In biopsies isolated from HPV-positive cancers, high levels of aneuploidy are observed suggesting that changes in chromosome numbers may promote the progression from low-grade lesions to malignancy. During normal cell division, centrosomes coordinate equal segregation of chromosomes to the daughter cells. Expression of E7 in human keratinocytes results in an increase in the number of cells harboring abnormal centrosome numbers, indicating E7 is a major inducer of chromosome missegregation (Duensing et al. 2000). Furthermore, E7 was found to induce chromosomal abnormalities in cells deficient of p130, Rb, and p107, indicating that this action is independent of E7’s ability to bind and/or degrade Rb (Duensing and Munger 2003).
The third oncoprotein, E5, is a small, hydrophobic membrane protein that is localized primarily to the endoplasmic reticulum, but is also found in the Golgi and plasma membrane (Conrad et al. 1993; Disbrow et al. 2005). The HPV E5 proteins are approximately 84 amino acids in size and are primarily expressed late in the viral life cycle. The function of E5 in the viral life cycle is less well understood as compared to other viral proteins; however, studies have begun to illuminate E5’s role in promoting cancer. In Bovine papillomavirus (BPV), E5 proteins exhibit efficient transforming ability of rodent fibroblasts when expressed alone (Petti et al. 1991). In contrast, HR-HPV E5 proteins exhibit very weak transforming ability when expressed alone. However, when HR-HPV E5 proteins are expressed in conjunction with E6 and E7 proteins, E5 proteins enhance transformation capacity of the cells (Stoppler et al. 1996; Valle and Banks 1995; Bouvard et al. 1994). The oncogenic potential of E5 proteins was most evidently demonstrated in estrogen-treated transgenic mice expressing E5 alone, which rapidly developed cervical cancer (Maufort et al. 2007). Although this is not the exclusive pathway by which HPVs promote tumorigenesis, these studies have shown that E5 proteins are important for the virus’ survival, possibly serving as a putative target for cervical cancer therapies (Valle and Banks 1995; Bouvard et al. 1994; DiMaio and Mattoon 2001).
A number of studies have identified proteins that associate with E5. One such association is the binding and subsequent alteration of the activity of the epidermal growth factor receptor (EGFR) (Straight et al. 1995). In addition, E5 is able to interact with the 16 kDa subunit of the vacuolar proton-ATPase, which alters endosomal pH, endocytic trafficking, and may contribute to the alteration of EGFR turnover (Conrad et al. 1993; Disbrow et al. 2005; Straight et al. 1993). Studies have also illustrated the importance of E5 in activating late functions in the productive phase of the viral life cycle. This was shown using stable keratinocyte cells lines containing HPV 31 genomes harboring wild-type or translational termination mutant E5 sequences (Fehrmann et al. 2003). Recently, a split-ubiquitin yeast two-hybrid system yielded identification of novel E5-binding partners, including a B cell receptor protein (BAP31), which is involved in the regulation of membrane protein transport. This interaction has been shown to be important for maintaining proliferative capacity of HPV-infected cells following differentiation (Regan and Laimins 2008).
2 Summary
Human papillomaviruses are important human pathogens that are responsible for the induction of a variety of human cancers. Despite the introduction of vaccines against HPV, they only protect against initial infections and no therapeutics, other than surgery, are available to treat existing HPV lesions. The E6 and E7 viral proteins provide important functions in the viral life cycle and also provide major contributions to progression to malignancy. Among the E6 and E7, cellular targets are p53, Rb, p300, and telomerase as well as a variety of other factors. The membrane-associated E5 protein can also contribute to malignant progression though its mechanism of action is unclear. Understanding the modes of action of HPV oncoproteins can provide important targets for therapeutics to treat HPV-associated cancers.
References
Bodily J, Laimins LA (2011) Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol 19(1):33–39. doi:10.1016/j.tim.2010.10.002
Bouvard V, Storey A, Pim D, Banks L (1994a) Characterization of the human papillomavirus E2 protein: evidence of trans-activation and trans-repression in cervical keratinocytes. EMBO J 13(22):5451–5459
Bouvard V, Matlashewski G, Gu ZM, Storey A, Banks L (1994b) The human papillomavirus type 16 E5 gene cooperates with the E7 gene to stimulate proliferation of primary cells and increases viral gene expression. Virology 203(1):73–80
Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391(6667):597–601. doi:10.1038/35404
Conrad M, Bubb VJ, Schlegel R (1993) The human papillomavirus type 6 and 16 E5 proteins are membrane-associated proteins which associate with the 16-kilodalton pore-forming protein. J Virol 67(10):6170–6178
Davies R, Hicks R, Crook T, Morris J, Vousden K (1993) Human papillomavirus type 16 E7 associates with a histone H1 kinase and with p107 through sequences necessary for transformation. J Virol 67(5):2521–2528
DiMaio D, Mattoon D (2001) Mechanisms of cell transformation by papillomavirus E5 proteins. Oncogene 20(54):7866–7873. doi:10.1038/sj.onc.1204915
Disbrow GL, Hanover JA, Schlegel R (2005) Endoplasmic reticulum-localized human papillomavirus type 16 E5 protein alters endosomal pH but not trans-Golgi pH. J Virol 79(9):5839–5846. doi:10.1128/JVI.79.9.5839-5846.2005
Duensing S, Munger K (2003) Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J Virol 77(22):12331–12335
Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, Crum CP, Munger K (2000) The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proce Natl Acad Sci USA 97(18):10002–10007. doi:10.1073/pnas.170093297
Dyson N, Howley PM, Munger K, Harlow E (1989) The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243(4893):934–937 (New York)
Dyson N, Guida P, Munger K, Harlow E (1992) Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J Virol 66(12):6893–6902
Edmonds C, Vousden KH (1989) A point mutational analysis of human papillomavirus type 16 E7 protein. J Virol 63(6):2650–2656
Fehrmann F, Laimins LA (2003) Human papillomaviruses: targeting differentiating epithelial cells for malignant transformation. Oncogene 22(33):5201–5207. doi:10.1038/sj.onc.1206554
Fehrmann F, Klumpp DJ, Laimins LA (2003) Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol 77(5):2819–2831
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127(12):2893–2917. doi:10.1002/ijc.25516
Funk JO, Waga S, Harry JB, Espling E, Stillman B, Galloway DA (1997) Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev 11(16):2090–2100
Gewin L, Galloway DA (2001) E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-myc. J Virol 75(15):7198–7201. doi:10.1128/JVI.75.15.7198-7201.2001
Howie HL, Katzenellenbogen RA, Galloway DA (2009) Papillomavirus E6 proteins. Virology 384(2):324–334. doi:10.1016/j.virol.2008.11.017
Howley PM, Lowy DR (2007) Papillomaviruses. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia
Jones DL, Alani RM, Munger K (1997) The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev 11(16):2101–2111
Katzenellenbogen RA, Vliet-Gregg P, Xu M, Galloway DA (2009) NFX1-123 increases hTERT expression and telomerase activity posttranscriptionally in human papillomavirus type 16 E6 keratinocytes. J Virol 83(13):6446–6456. doi:10.1128/JVI.02556-08
Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M (1997) Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci USA 94(21):11612–11616
Klingelhutz AJ, Foster SA, McDougall JK (1996) Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 380(6569):79–82. doi:10.1038/380079a0
Ko LJ, Prives C (1996) p53: puzzle and paradigm. Genes Dev 10(9):1054–1072
Kuhne C, Banks L (1998) E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J Biol Chem 273(51):34302–34309
Kukimoto I, Aihara S, Yoshiike K, Kanda T (1998) Human papillomavirus oncoprotein E6 binds to the C-terminal region of human minichromosome maintenance 7 protein. Biochem Biophys Res Commun 249(1):258–262. doi:10.1006/bbrc.1998.9066
Kyo S, Takakura M, Taira T, Kanaya T, Itoh H, Yutsudo M, Ariga H, Inoue M (2000) Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acid Res 28(3):669–677
Lee SS, Weiss RS, Javier RT (1997) Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci USA 94(13):6670–6675
Lee SS, Glaunsinger B, Mantovani F, Banks L, Javier RT (2000) Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J Virol 74(20):9680–9693
Liu JP (1999) Studies of the molecular mechanisms in the regulation of telomerase activity. FASEB J 13(15):2091–2104
Longworth MS, Laimins LA (2004a) Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev 68(2):362–372. doi:10.1128/MMBR.68.2.362-372.2004
Longworth MS, Laimins LA (2004b) The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol 78(7):3533–3541
Lorincz AT, Reid R, Jenson AB, Greenberg MD, Lancaster W, Kurman RJ (1992) Human papillomavirus infection of the cervix: relative risk associations of 15 common anogenital types. Obstet Gynecol 79(3):328–337
Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chesson H, Unger ER (2007) Quadrivalent human papillomavirus vaccine: recommendations of the advisory committee on immunization practices (ACIP). MMWR Recomm Rep 56(RR-2):1–24 (Morbidity and mortality weekly report Recommendations and reports/Centers for Disease Control)
Maufort JP, Williams SM, Pitot HC, Lambert PF (2007) Human papillomavirus 16 E5 oncogene contributes to two stages of skin carcinogenesis. Cancer Res 67(13):6106–6112. doi:10.1158/0008-5472.CAN-07-0921
Moody CA, Laimins LA (2010) Human papillomavirus oncoproteins: pathways to transformation. Nat Rev 10(8):550–560. doi:10.1038/nrc2886
Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R (1989) The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol 63(10):4417–4421
Oren M (2003) Decision making by p53: life, death and cancer. Cell Death Differ 10(4):431–442. doi:10.1038/sj.cdd.4401183
Parkin DM, Bray F, Ferlay J, Pisani P (2000) Estimating the world cancer burden: Globocan. Int J Cancer 94(2):153–156
Patel D, Huang SM, Baglia LA, McCance DJ (1999) The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J 18(18):5061–5072. doi:10.1093/emboj/18.18.5061
Petti L, Nilson LA, DiMaio D (1991) Activation of the platelet-derived growth factor receptor by the bovine papillomavirus E5 transforming protein. EMBO J 10(4):845–855
Regan JA, Laimins LA (2008) Bap31 is a novel target of the human papillomavirus E5 protein. J Virol 82(20):10042–10051. doi:10.1128/JVI.01240-08
Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM (2003) Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res 63(16):4862–4871
Ronco LV, Karpova AY, Vidal M, Howley PM (1998) Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev 12(13):2061–2072
Ruesch MN, Laimins LA (1998) Human papillomavirus oncoproteins alter differentiation-dependent cell cycle exit on suspension in semisolid medium. Virology 250(1):19–29. doi:10.1006/viro.1998.9359
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM (1990) The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63(6):1129–1136. doi:0092-8674(90)90409-8
Slee EA, O’Connor DJ, Lu X (2004) To die or not to die: how does p53 decide? Oncogene 23(16):2809–2818. doi:10.1038/sj.onc.1207516
Stanley M (2008) Immunobiology of HPV and HPV vaccines. Gynecol Oncol 109(2):S15–S21. doi:10.1016/j.ygyno.2008.02.003
Stanley M (2010) HPV: immune response to infection and vaccination. Infect Agent Cancer 5:19. doi:10.1186/1750-9378-5-19
Stoppler MC, Straight SW, Tsao G, Schlegel R, McCance DJ (1996) The E5 gene of HPV-16 enhances keratinocyte immortalization by full-length DNA. Virology 223(1):251–254. doi:10.1006/viro.1996.0475
Straight SW, Hinkle PM, Jewers RJ, McCance DJ (1993) The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J Virol 67(8):4521–4532
Straight SW, Herman B, McCance DJ (1995) The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J Virol 69(5):3185–3192
Thomas M, Banks L (1999) Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J Gen Virol 80(Pt 6):1513–1517
Thomas JT, Hubert WG, Ruesch MN, Laimins LA (1999) Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc Natl Acad Sci USA 96(15):8449–8454
Tommasino M, Adamczewski JP, Carlotti F, Barth CF, Manetti R, Contorni M, Cavalieri F, Hunt T, Crawford L (1993) HPV16 E7 protein associates with the protein kinase p33CDK2 and cyclin A. Oncogene 8(1):195–202
Valle GF, Banks L (1995) The human papillomavirus (HPV)-6 and HPV-16 E5 proteins co-operate with HPV-16 E7 in the transformation of primary rodent cells. J Gen Virol 76(Pt 5):1239–1245
Weintraub SJ, Chow KN, Luo RX, Zhang SH, He S, Dean DC (1995) Mechanism of active transcriptional repression by the retinoblastoma protein. Nature 375(6534):812–815. doi:10.1038/375812a0
Zerfass-Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz JW, Jansen-Durr P (1996) Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 13(11):2323–2330
Zimmermann H, Degenkolbe R, Bernard HU, O’Connor MJ (1999) The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J Virol 73(8):6209–6219
zur Hausen H (1996) Papillomavirus infections–a major cause of human cancers. Biochim Biophys Acta 1288(2):F55–F78
zur Hausen H (2009) Papillomaviruses in the causation of human cancers: a brief historical account. Virology 384(2):260–265. doi:10.1016/j.virol.2008.11.046
zur Hausen H, de Villiers EM (1994) Human papillomaviruses. Annu Rev Microbiol 48:427–447. doi:10.1146/annurev.mi.48.100194.002235
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Mighty, K.K., Laimins, L.A. (2014). The Role of Human Papillomaviruses in Oncogenesis. In: Chang, M., Jeang, KT. (eds) Viruses and Human Cancer. Recent Results in Cancer Research, vol 193. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-38965-8_8
Download citation
DOI: https://doi.org/10.1007/978-3-642-38965-8_8
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-38964-1
Online ISBN: 978-3-642-38965-8
eBook Packages: MedicineMedicine (R0)