
Abstract
Epigenetic control, which includes DNA methylation and histone modifications, leads to chromatin remodeling and regulated gene expression. Remodeling of chromatin constitutes a critical interface of transducing signals, such as light or nutrient availability, and how these are interpreted by the cell to generate permissive or silenced states for transcription. CLOCK-BMAL1-mediated activation of clock-controlled genes (CCGs) is coupled to circadian changes in histone modification at their promoters. Several chromatin modifiers, such as the deacetylases SIRT1 and HDAC3 or methyltransferase MLL1, have been shown to be recruited to the promoters of the CCGs in a circadian manner. Interestingly, the central element of the core clock machinery, the transcription factor CLOCK, also possesses histone acetyltransferase activity. Rhythmic expression of the CCGs is abolished in the absence of these chromatin modifiers. Here we will discuss the evidence demonstrating that chromatin remodeling is at the crossroads of circadian rhythms and regulation of metabolism and cellular proliferation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Circadian rhythms occur with a periodicity of about 24 h and regulate a wide array of metabolic and physiologic functions. Accumulating epidemiological and genetic evidence indicates that disruption of circadian rhythms can be directly linked to many pathological conditions, including sleep disorders, depression, metabolic syndrome, and cancer. Intriguingly, a number of molecular gears constituting the clock machinery have been found to establish functional interplays with regulators of cellular metabolism and cell cycle.
The Earth’s rotation around its axis leads to day–night cycles, which affects the physiology of most living organisms. Circadian (from the Latin circa diem meaning “about a day”) clocks are intrinsic, time-tracking systems that enable organisms to anticipate environmental changes (such as food availability and predatory pressure) and allow them to adapt their behavior and physiology to the appropriate time of day (Schibler and Sassone-Corsi 2002). Feeding behavior, sleep–wake cycles, hormonal levels, and body temperature are just a few examples of physiological circadian rhythms, with light being the principal zeitgeber (“time giver”). Other zeitgebers, such as feeding time and temperature, are discussed in accompanying chapters in this book (Brown and Azzi 2013; Buhr and Takahashi 2013).
The three integral parts of circadian clocks are the following: an input pathway that includes detectors to receive environmental cues (or zeitgebers) and transmits them to the central oscillator; a central oscillator that keeps circadian time and generates rhythm; and output pathways through which the rhythms are manifested via control of various metabolic, physiological, and behavioral processes. Distinguishing characteristics of circadian clocks include that they are entrainable (synchronizable by external cues), self-sustained (oscillations can persist even in the absence of zeitgebers), and temperature compensated (moderate variations in ambient temperature does not affect the period of circadian oscillation) (Merrow et al. 2005).
Circadian clocks are present in almost all of the tissues in mammals. The master or “central” clock is located in the hypothalamic suprachiasmatic nucleus (SCN), which contains 10–15,000 neurons (Slat et al. 2013). Peripheral clocks are present in almost all other mammalian tissues such as liver, heart, lung, and kidney, where they maintain circadian rhythms and regulate tissue-specific gene expression (Brown and Azzi 2013). These peripheral clocks are synchronized by the central clock to ensure temporally coordinated physiology. The synchronization mechanisms implicate various humoral signals, including circulating entraining factors such as glucocorticoids. The SCN clock can function autonomously, without any external input, but can be set by environmental cues such as light. The molecular machinery that regulates these circadian rhythms comprises of a set of genes, known as “clock” genes, whose products interact to generate and maintain rhythms (Buhr and Takahashi 2013).
A conserved feature among many organisms is the regulation of the circadian clock by a negative feedback loop (Sahar and Sassone-Corsi 2009). Positive regulators induce the transcription of clock-controlled genes (CCGs), some of which encode proteins that feedback on their own expression by repressing the activity of positive regulators. CLOCK and BMAL1 are the positive regulators of the mammalian clock machinery which regulate the expression of the negative regulators: cryptochrome (CRY1 and CRY2) and period (PER1, PER2, PER3) families. CLOCK and BMAL1 are transcription factors that heterodimerize through the PAS domain and induce the expression of clock-controlled genes by binding to their promoters at E-boxes [CACGTG]. Once a critical concentration of the PER and CRY proteins is accumulated, these proteins translocate into the nucleus and form a complex to inhibit CLOCK-BMAL1-mediated transcription, thereby closing the negative feedback loop. In order to start a new transcriptional cycle, the CLOCK-BMAL1 complex needs to be derepressed through the proteolytic degradation of PER and CRY. Core clock genes (such as Clock, Bmal1, Period, Cryptochrome) are necessary for generation of circadian rhythms, whereas CCGs (such as Nampt, Alas1) are regulated by the core clock genes.
Some CCGs are transcription factors, such as albumin D-box-binding protein (DBP), RORα, and REV-ERBα, which can then regulate cyclic expression of other genes. DBP binds to D-boxes [TTA(T/C)GTAA], whereas RORα and REV-ERBα bind to the Rev-Erb/ROR-binding element, or RRE [(A/T)A(A/T)NT(A/G)GGTCA]. Approximately 10 % of the transcriptome displays robust circadian rhythmicity (Akhtar et al. 2002; Panda et al. 2002). Interestingly, most transcripts that oscillate in one tissue do not oscillate in another (Akhtar et al. 2002; Miller et al. 2007; Panda et al. 2002).
2 Epigenetics and the Circadian Clock
“Epigenetics” literally means “above genetics.” It is defined as the study of heritable changes in gene expression that does not involve any change to the DNA sequence. Such changes in gene expression can be brought about by a variety of mechanism that involves a combination of posttranslational modifications of histones, remodeling of chromatin, incorporation of histone variants, or methylation of DNA on CpG islands. Histone acetylation is a mark for activation of transcription, which is achieved by remodeling the chromatin to make it more accessible to the transcription machinery (Jenuwein and Allis 2001). Histone methylation, on the other hand, acts as a signal for recruitment of chromatin remodeling factors which can either activate or repress transcription. DNA methylation leads to compaction of the chromatin and causes gene silencing. Many of these epigenetic events are crucial in regulation of cellular metabolism and survival.
Genes encoding circadian clock proteins are regulated by epigenetic mechanisms, such as histone phosphorylation, acetylation, and methylation, which have been shown to follow circadian rhythm (Crosio et al. 2000; Etchegaray et al. 2003; Masri and Sassone-Corsi 2010; Ripperger and Schibler 2006). The first study demonstrating that chromatin remodeling is involved in circadian gene expression reported that exposure to light causes rapid phosphorylation of histone 3 on serine 10 (H3-S10) in the SCN (Crosio et al. 2000). This phosphorylation parallels induction of immediate early genes such as c-fos and Per1, thereby indicating that light-mediated signaling can regulate circadian gene expression by remodeling the chromatin (Crosio et al. 2000).
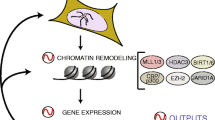
CLOCK-BMAL1-mediated activation of CCGs has been shown to be coupled to circadian changes in histone acetylation at their promoters (Etchegaray et al. 2003). The central element of the core clock machinery, the transcription factor CLOCK, also possesses intrinsic histone acetyltransferase (HAT) activity (Doi et al. 2006). Since CLOCK binds to E-box regions of DNA, the HAT activity of CLOCK can selectively remodel chromatin at the promoters of CCGs and is essential for circadian gene expression (Fig. 1). The enzymatic activity of CLOCK also allows it to acetylate nonhistone substrates such as its own binding partner, BMAL1 (Hirayama et al. 2007). CLOCK specifically acetylates BMAL1 at a conserved residue, an event that facilitates CRY-dependent repression.

Epigenetic regulation of gene expression by circadian clock CLOCK can acetylate histones to induce gene expression. CLOCK interacts with MLL1 (a histone methyltransferase) and SIRT1 (a deacetylase). These epigenetic regulators can modify the chromatin according the environmental stimuli, such as nutrient availability. Furthermore, REV-ERBα, a clock-controlled gene, can cause recruitment of HDAC3 and deacetylate histones. Circadian regulation of either the expression or the activity of these epigenetic regulators determines whether the gene gets turned “ON” (green arrows) or “OFF” (red arrows)
Histone methylation is also important for circadian gene expression. Mixed lineage leukemia 1 (MLL1), a methyltransferase that methylates histone H3 at lysine 4 (H3K4), associates with CLOCK and is recruited to promoters of CCGs in a circadian manner (Fig. 1) (Katada and Sassone-Corsi 2010). H3K4 methylation at these promoters also displays rhythmicity (Katada and Sassone-Corsi 2010). H3K4 methylation has been intimately linked to transcriptional activation. Lysine residues can be mono-, di-, or trimethylated at the ε-amino group, with each state correlating with a distinct functional effect. Dimethylated H3K4 (H3K4me2) occurs at both inactive and active euchromatic genes, whereas H3K4me3 is present prominently at actively transcribed genes and is widely accepted as a unique epigenetic mark that defines an active chromatin state in most eukaryotes. It is thereby noteworthy that MLL1 is specifically involved in trimethylation (Katada and Sassone-Corsi 2010). Notably, H3K4 methylation has often been shown to be associated with specific H3 Lys9 (H3K9) and Lys14 (H3K14) and H4 Lys16 (H4K16) acetylation, and these are all “marks” associated with active gene expression (Ruthenburg et al. 2007).
2.1 Role of SIRT1 in Regulation of Circadian Rhythms
The finding of a circadian HAT opened the search for a counterbalancing histone deacetylase (HDAC). Recently, SIRT1 was identified to be a modulator of the circadian clock machinery (Asher et al. 2008; Nakahata et al. 2008). SIRT1 belongs to the family of sirtuins, which constitutes the so-called class III of HDACs. These are HDACs whose enzymatic activity is NAD+ dependent and that has been directly linked to the control of metabolism and aging (Bishop and Guarente 2007). SIRT1 plays crucial roles in metabolism by (a) deacetylating several proteins that participate in metabolic pathways and (b) regulating gene expression by histone deacetylation. Since the NAD+/NADH ratio is a direct measure of the energy status of a cell, the NAD+ dependence of SIRT1 directly links cellular energy metabolism and deacetylation of target proteins (Imai et al. 2000). Recently, two independent studies identified SIRT1 to be a critical modulator of the circadian clock machinery (Asher et al. 2008; Nakahata et al. 2008). While Asher et al. observed oscillations in SIRT1 protein levels (Asher et al. 2008), Nakahata et al. demonstrated that SIRT1 activity, and not its protein levels, oscillates in a circadian manner (Nakahata et al. 2008). Circadian oscillations in NAD+ levels were later shown to drive SIRT1 rhythmic activity (Nakahata et al. 2009). SIRT1 modulates circadian rhythms by deacetylating histones (histone H3 Lys9 and Lys14 at promoters of rhythmic genes) and nonhistone proteins (BMAL1 and PER2). The CLOCK-BMAL1 complex interacts with SIRT1 and recruits it to the promoters of rhythmic genes (Fig. 1). Importantly, circadian gene expression and BMAL1 acetylation are compromised in liver-specific SIRT1 mutant mice (Nakahata et al. 2008). While BMAL1 acetylation acts as a signal for CRY recruitment (Hirayama et al. 2007), PER2 acetylation enhances its stability (Asher et al. 2008). These findings led to the concept that SIRT1 operates as a rheostat of the circadian machinery, modulating the amplitude and “tightness” of CLOCK-mediated acetylation and consequent transcription cycles in metabolic tissues (Nakahata et al. 2008).
Circadian oscillation of SIRT1 activity suggested that cellular NAD+ levels may also oscillate. Circadian clock controls the expression of nicotinamide phosphoribosyltransferase (NAMPT), a key rate-limiting enzyme in the salvage pathway of NAD+ biosynthesis (Nakahata et al. 2009; Ramsey et al. 2009). The rhythmicity in the expression of this enzyme drives the oscillation in NAD+ levels (Nakahata et al. 2009; Ramsey et al. 2009). CLOCK, BMAL1, and SIRT1 are recruited to the Nampt promoter in a circadian time-dependent manner (Fig. 2). The oscillatory expression of Nampt is abolished in Clock/Clock mice, which results in drastically reduced levels of NAD+ in MEFs derived from these mice (Nakahata et al. 2009). These results make a compelling case for the existence of an enzymatic/transcriptional feedback loop, wherein SIRT1 regulates the levels of its own cofactor. Interestingly, mice deficient of NAD+ hydrolase CD38 displayed altered rhythmicity of NAD+. Very high levels of NAD+ in tissues such as the brain and liver have been reported in the CD38-null mice (Aksoy et al. 2006). The high, chronic levels of NAD+ result in several anomalies in circadian behavior and metabolism (Sahar et al. 2011). CD38-null mice display a shortened period length of locomotor activity and alteration in the rest–activity rhythm (Sahar et al. 2011).

SIRT1: a circadian regulator The circadian clock controls the expression of nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in mammalian NAD+ biosynthesis from nicotinamide. NAMPT catalyzes the transfer of a phosphoribosyl residue from 5-phosphoribosyl-1-pyrophosphate (PRPP) to nicotinamide to produce nicotinamide mononucleotide (NMN), which is then converted to NAD+ by nicotinamide mononucleotide adenylyltransferases (there are three Nmnat genes). Oscillation in NAMPT results in circadian variations in NAD+ levels, which determines the activity of SIRT1. Thus, SIRT1 determines the oscillatory levels of its own coenzyme, NAD+. SIRT1 can also deacetylate and regulate proteins involved in metabolism and cell proliferation
SIRT1 also deacetylates and thereby regulates several proteins involved in the regulation of metabolism and cell proliferation (Fig. 2). For example, SIRT1 regulates gluconeogenesis by deacetylating and activating PPARγ-coactivator α (PGC1α) and Forkhead box O1 (FOXO1) (Schwer and Verdin 2008). FOXO1 directly regulates expression of several gluconeogenic genes (Frescas et al. 2005), whereas PGC1α coactivates glucocorticoid receptors and hepatic nuclear factor 4-alpha (HNF-4α) to induce the expression of gluconeogenic genes (Yoon et al. 2001). SIRT1 also regulates cholesterol metabolism by deacetylating, and thus activating, Liver X receptor (LXR) (Li et al. 2007) (Fig. 2). LXR regulates cholesterol metabolism by inducing the expression of the ATP-binding cassette transporter A1 (Abca1), which mediates cholesterol efflux from peripheral tissues to the blood. Furthermore, it seems evident that SIRT1 may promote or prevent cancer depending on the specific function of its substrate. By deacetylating and thereby inactivating β-catenin, SIRT1 may lead to reduced cell proliferation (Firestein et al. 2008). SIRT1 deacetylates p53 and thus inhibits its activity (Vaziri et al. 2001), resulting in reduced apoptosis after genotoxic stress (Fig. 2). Since SIRT1 activity is regulated in a circadian manner, it would be interesting to determine if the acetylation of other SIRT1 targets oscillates in a circadian manner.
2.2 The Complexity of the Circadian Epigenome
Accumulating evidence shows that a variety of chromatin remodelers contribute to various aspects of the circadian epigenome (Masri and Sassone-Corsi 2010). In addition, the circadian machinery appears to occupy a pivotal position in linking metabolism to epigenetics (Katada et al. 2012). Histone deacetylase 3 (HDAC3) is a deacetylase that has recently been shown to modulate histone acetylation of circadian genes, particularly those that are responsible for lipid metabolism. The regulatory function of REV-ERBα is controlled by the nuclear receptor corepressor 1 (NCoR1), a corepressor that recruits HDAC3 to mediate transcriptional repression of target genes, such as Bmal1. When the NCoR1-HDAC3 association is genetically disrupted in mice, circadian and metabolic defects develop (Alenghat et al. 2008). These mice demonstrate a shorter period, increased energy expenditure and are resistant to diet-induced obesity (Alenghat et al. 2008). HDAC3 recruitment to the genome was recently shown to be rhythmic in liver (high during the day and low at night) (Feng et al. 2011). At these HDAC3 binding sites, REV-ERBα and NCoR1 recruitment were in phase with HDAC3 recruitment, whereas histone acetylation and RNA polymerase II recruitment were anti-phasic. Depletion of either HDAC3 or REV-ERBα was shown to cause fatty liver phenotype, such as increased hepatic lipid and triglyceride content (Feng et al. 2011).
HDAC1 was also shown to form a complex with SIN3A (a protein that modulates transcription by interacting with transcription repressors) and PER2, and it is known to be recruited to Per1 promoter. HDAC1 can then deacetylate the histones and repress transcription of Per1. Depletion of SIN3A from synchronized fibroblasts caused a shortening of circadian period length (Duong et al. 2011).
Although the identity of a circadian histone demethylase is currently unknown, JARID1a, a histone demethylase, has been recently shown to regulate circadian gene expression (DiTacchio et al. 2011). Surprisingly, the histone demethylase activity of this enzyme is not required for its regulation of circadian rhythms.
Altogether, these findings underscore the importance of epigenetic mechanisms in circadian regulation and reveal the molecular pathways by which such essential control is achieved.
3 Circadian Disruption and Disease: Cancer and Metabolic Disorders
Circadian control of physiology and behavior is required for a healthy life. Disruption of circadian rhythms has been considered as a causative factor for development of several diseases. As discussed below, mutation in circadian clock proteins that either have histone-modifying ability (such as CLOCK) or associate with histone modifiers (such as BMAL1, PER2, and REV-ERBα) has been linked to cancer and metabolic syndrome (Sahar and Sassone-Corsi 2012).
3.1 Mutations in the Clock Machinery and Cancer Association
A number of epidemiological studies have linked defects in circadian rhythms to increased susceptibility to develop cancer and poor prognosis. This evidence is supported by gene expression studies. For example, the expression of all three Per genes is deregulated in breast cancer cells (Chen et al. 2005). PER1 expression is downregulated in most patients, possibly due to methylation of its promoter. Mutations in NPAS2 have been associated with increased risk for breast cancer and non-Hodgkin’s lymphoma (Hoffman et al. 2008). More importantly, a number of studies using mouse models have established convincing links between some clock genes and tumorigenesis. Specifically, Per1 and Per2 appear to act as tumor suppressors in mice (Fu et al. 2002; Gery et al. 2006). Targeted ablation of Per2 leads to the development of malignant lymphomas (Fu et al. 2002), whereas its ectopic expression in cancer cell lines results in growth inhibition, cell cycle arrest, apoptosis, and loss of clonogenic ability (Gery et al. 2005). Interestingly, Per2 mRNA levels are downregulated in several human lymphoma cell lines and acute myeloid leukemia patients (Gery et al. 2005). Overexpression of Per1 can also suppress growth of human cancer cell lines (Gery et al. 2006). Furthermore, PER1 mRNA levels are also downregulated in non-small cell lung cancer tissues compared to matched normal tissues (Gery et al. 2006). In addition, knockdown of CK1ε induces growth inhibition of cancer cells, and CK1ε expression is increased in various human cancers, such as leukemia and prostate cancer (Yang and Stockwell 2008). These results consistently point toward a direct link between the dysfunction of key circadian regulators and cancer (Sahar and Sassone-Corsi 2007).
An interesting link between circadian clock and breast cancer was established in a study demonstrating that PER2 can bind to and destabilize estrogen receptor α (ERα) (Gery et al. 2007), a key transcription factor that promotes growth of mammary epithelial cells and whose dysregulated activity is known to cause breast cancer (Green and Carroll 2007). Consequently, Per2 overexpression leads to reduced ERα protein levels and transcriptional activity.
It is important to note that mutation of one or more core clock genes is itself not necessarily sufficient to elicit enhanced tumor incidence. In addition, there is no apparent correlation between the disruption of circadian behavior and increased tumorigenesis in mouse models of circadian rhythms. Indeed, Cry1 −/− Cry2 −/− mice (Gauger and Sancar 2005) or Clock/Clock mutant mice (Antoch et al. 2008), whose circadian rhythms are highly compromised, do not show a predisposition to cancer upon irradiation. Moreover, MEFs derived from Clock/Clock mutant mice display reduced DNA synthesis and cell proliferation compared to wild-type MEFs (Miller et al. 2007). Somewhat unexpectedly, ablation in the mouse of both Cry genes in a p53 −/− background delays the onset of cancer (Ozturk et al. 2009). These notions may suggest that other regulatory features intrinsic to clock regulators, independent of their circadian function, could participate in carcinogenesis. It seems that individual core circadian clock proteins (such as PER1, PER2) might have acquired multiple roles and hence can control both rhythms and cell cycle. Also, the consequence of circadian disruption on cancer predisposition might be dependent on how the rhythm is disrupted.
The molecular mechanism of how circadian clock influences cancer development and progression could be explained by its regulation of cell cycle, DNA damage response, and cellular metabolism (Hunt and Sassone-Corsi 2007; Antoch and Kondratov 2013). Circadian regulation of genes encoding key cell cycle regulators, such as Wee1 (G2/M transition) (Matsuo et al. 2003), c-myc (G0/G1 transition) (Fu et al. 2002), and Cyclin D1 (G1/S transition) (Fu et al. 2002), has been demonstrated in mammals, and light induces the expression of Wee1 in zebra fish (Hirayama et al. 2005). WEE1 is a kinase that phosphorylates and inactivates the CDC2/cyclin B1 complex to control G2/M transition during mitosis. Wee1 displays robust CLOCK-BMAL1 dependent circadian oscillations in the mouse liver (Matsuo et al. 2003). Furthermore, partial hepatectomy-induced liver regeneration is impaired in Cry-deficient arrhythmic mice, which also show deregulated expression of Wee1 (Matsuo et al. 2003). These studies indicate that WEE1 may function as a key molecular link between circadian and cell cycles.
Damage to cellular DNA, either by intracellular agents (such as metabolic by-products) or external agents (such as ionizing radiations), can cause cancer. However, cells have evolved several mechanisms to repair the damaged DNA. Recent results suggest that one such repair mechanism, the nucleotide excision repair pathway, displays circadian oscillation in mouse brain, possibly through oscillation in the expression of the DNA damage-recognition protein xeroderma pigmentosum A (XPA) (Kang et al. 2009). XPA levels also oscillate in mouse liver (Kang et al. 2009), suggesting that the circadian nucleotide excision repair might also be operating in peripheral tissues. Confirming this notion, a recent study found that XPA protein levels and the rate of excision repair oscillate in a circadian manner in mouse skin (Gaddameedhi et al. 2011). Consequently, mice are more susceptible to skin cancer when exposed to ultraviolet radiation in the morning when the rate of DNA repair is lower (Gaddameedhi et al. 2011).
Finally, circadian clock proteins, such as PER1 and Timeless (TIM), interact with key checkpoint proteins (Gery et al. 2006; Unsal-Kacmaz et al. 2005). It is conceivable that uncoupling of this delicate balance could induce DNA damage, predisposing cells to tumorigenesis.
3.2 Cancer Chronotherapy
Chronotherapy refers to the administration of drugs at a certain time of the day when its efficacy is the highest and the side effects are the lowest (see also Ortiz-Tudela et al. 2013). An example of successful chronotherapy is the use of the cholesterol-lowering drugs statins. Statins inhibit HMG-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis. The expression of HMG-CoA reductase displays circadian rhythmicity, being highest at night. Hence, statins are most effective when administered before bedtime. Chronotherapy has also shown promise in treating cancer. It is widely accepted that cells enter various phases of cell cycle in a circadian manner. Fast-growing or advanced tumors become asynchronous with the host cells and display ultradian (less than 24 h) rhythms (Lis et al. 2003). In an elegant experiment, Klevecz et al. demonstrated that the proliferation of tumor and non-tumor cells from ovarian cancer patients significantly differed in their peak S phase (Klevecz et al. 1987). Similar observations in other types of cancer [such as non-Hodgkin’s lymphoma (Smaaland et al. 1993)] suggest that there is a possible window of time when a cytotoxic drug would kill the tumor cells more effectively than the noncancerous host cells. More than 30 anticancer drugs have been found to vary in toxicity and efficacy by more than 50 % as a function of time of administration in various experimental models (Levi et al. 2007). In clinical studies several anticancer drugs, such as 5-fluorouracil (5-FU) and platinum complex analogs that are specifically toxic to replicating cells, have been shown to be more efficacious and less toxic when administered at a specific circadian time (Levi et al. 2007). For example, chronotherapy using doxorubicin and cisplatin showed significant improvement in survival rate of patients with ovarian cancer when doxorubicin was administered in the morning followed by cisplatin 12 h later (Kobayashi et al. 2002). Further studies are needed to identify the molecular mechanisms responsible for the beneficial effects of circadian administration of anticancer drugs. Another report demonstrated that sensitivity to cyclophosphamide, an anticancer drug, varies greatly in wild-type mice depending upon the time of administration (Gorbacheva et al. 2005). However, Clock/Clock mutant mice and Bmal1 −/− mice are more sensitive and did not display variation in sensitivity at different times, indicating dependency on clock components, whereas Cry1 −/− Cry2 −/− mice are more resistant to cyclophosphamide. These results suggest that activities of the core clock components have direct manifestation in response to genotoxic stress induced by anticancer drugs.
3.3 Circadian Disruption and Metabolic Disorders
Shift work, and accompanying light exposure at night, has been implicated in the development of metabolic syndrome and cardiovascular diseases (De Bacquer et al. 2009; Karlsson et al. 2001; Marcheva et al. 2013). A recent study showed that mice exposed to light at night gained more weight, had reduced glucose tolerance, and ate more during the light phase. Interestingly, when food was restricted to the dark phase, weight gain was prevented (Fonken et al. 2010). In another study, mice fed a high-fat diet only during the light phase gained more weight when compared to mice that ate the same high-fat diet but only during the dark phase, an observation that highlights the importance in the timing of food intake (Arble et al. 2009). These results raise an interesting question: Could adjusting our food intake exclusively during the active phase (daytime for humans) be an effective way of weight control? Since humans have evolved for thousands of years without artificial light, our internal clock still functions the best when natural light is the only source of light, so it is conceivable that restricting food intake to daytime may help control weight gain.
Not just the timing but the quality of the diet might also affect the clock. Mice fed a high-fat diet had altered circadian rhythms and displayed a lengthening of the period of locomotor activity (Kohsaka et al. 2007). Interestingly, these mice also consumed a higher-than-normal percentage of food during the light phase. Moreover, the expression of core clock genes and the clock-controlled genes (CCGs) was altered in the mice that were fed a high-fat diet (Kohsaka et al. 2007). These studies have clearly established that metabolism can also control peripheral clocks.
If the circadian machinery is critical for metabolic homeostasis, deletion or mutation of individual core clock components or of CCGs should lead to metabolic disorders. This is indeed the case as illustrated by examples discussed below.
3.3.1 CLOCK and BMAL1
Loss of function of CLOCK and BMAL1, the central transcription factors that regulate circadian rhythms, leads to several metabolic anomalies. Clock/Clock mutant mice, which are arrhythmic when placed in constant darkness, become hyperphagic and obese and develop classical signs of “metabolic syndrome” such as hyperglycemia, dyslipidemia, and hepatic steatosis (fatty liver) (Turek et al. 2005). In addition, the mRNA levels of the neuropeptides orexin and ghrelin—both involved in the neuroendocrine regulation of food intake (Adamantidis and de Lecea 2009; Saper et al. 2002)—are also reduced in these mice. Furthermore, renal sodium reabsorption is compromised and arterial blood pressure is reduced in the Clock −/− mice (Zuber et al. 2009). Loss of BMAL1, which renders mice completely arrhythmic (Bunger et al. 2000), also leads to disruption of oscillations in glucose and triglyceride levels (Rudic et al. 2004). To address the question of whether the metabolic defects are due to a loss of rhythmicity in the SCN or in the peripheral clocks, mice with tissue-specific deletion of Bmal1 in the liver or pancreas have been generated. Even though these mice show normal locomotor activity, they display disturbances in the maintenance of blood glucose levels. In liver-specific Bmal1 KO mice, the circadian expression of key metabolic genes, such as glucose transporter 2 (Glut2), is abolished. This results in mice being hypoglycemic during the fasting phase of the feeding cycle (Lamia et al. 2008). Further illustrating the importance of peripheral circadian clocks, deletion of BMAL1 in the pancreas leads to diabetes (Marcheva et al. 2010; Sadacca et al. 2011). These mice display elevated blood glucose levels, impaired glucose tolerance, and decreased insulin secretion (for a review see Marcheva et al. 2013).
3.3.2 REV-ERBα
REV-ERBα was originally identified as a nuclear receptor that regulates lipid metabolism and adipogenesis (Fontaine et al. 2003). Thus, the role of Rev-Erbα in controlling Bmal1 expression—a function that provides robustness to circadian oscillations (Preitner et al. 2002)—established a critical link between the molecular machinery that regulates circadian oscillations and metabolism. Although Rev-Erbα −/− mice are not arrhythmic, the rhythmicity in their locomotor activity is altered (a shorter period length under constant light or constant dark conditions) (Preitner et al. 2002).
REV-ERBα appears to act downstream of PPARγ, a key regulator of fat metabolism and adipocyte differentiation (Fontaine et al. 2003). Genes involved in lipid metabolism in the liver also appear to be major targets of REV-ERBα. Depletion of REV-ERBα was shown to cause fatty liver phenotype, such as increased hepatic lipid and triglyceride content (Feng et al. 2011).
4 Conclusion
The importance of epigenetic control is becoming clear in the regulation of circadian rhythms. Current data suggests that many epigenetic regulators themselves are regulated in a circadian manner, at least in some tissues. The challenge ahead is to understand whether these epigenetic events follow a rhythmic pattern in tissues that are involved in diverse physiologies, such as process of learning and memory (e.g., hippocampus, cortex, and amygdala) and metabolism (e.g., liver, adipose tissue, kidney). As more data accumulates describing specific mechanistic roles of clock genes in regulating cellular proliferation and metabolic pathways, new therapeutic targets are emerging. As the pharma industry is converging on epigenetic regulators as promising targets for therapy, it is conceivable that drugs that modulate the clock function may result effective in specific strategies against certain types of cancer and metabolic disorders.
References
Adamantidis A, de Lecea L (2009) The hypocretins as sensors for metabolism and arousal. J Physiol 587:33–40
Akhtar RA, Reddy AB, Maywood ES, Clayton JD, King VM, Smith AG, Gant TW, Hastings MH, Kyriacou CP (2002) Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol 12:540–550
Aksoy P, White TA, Thompson M, Chini EN (2006) Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun 345:1386–1392
Alenghat T, Meyers K, Mullican SE, Leitner K, Adeniji-Adele A, Avila J, Bucan M, Ahima RS, Kaestner KH, Lazar MA (2008) Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature 456:997–1000
Antoch MP, Kondratov RV (2013) Pharmacological modulators of the circadian clock as potential therapeutic drugs: focus on genotoxic/anticancer therapy. In: Kramer A, Merrow M (eds) Circadian clocks, vol 217, Handbook of experimental pharmacology. Springer, Heidelberg
Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, Lee C, Nikitin AY (2008) Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle 7:1197–1204
Arble DM, Bass J, Laposky AD, Vitaterna MH, Turek FW (2009) Circadian timing of food intake contributes to weight gain. Obesity 17:2100–2102
Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U (2008) SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134:317–328
Bishop NA, Guarente L (2007) Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat Rev Genet 8:835–844
Brown SA, Azzi A (2013) Peripheral circadian oscillators in mammals. In: Kramer A, Merrow M (eds) Circadian clocks, vol 217, Handbook of experimental pharmacology. Springer, Heidelberg
Buhr ED, Takahashi JS (2013) Molecular components of the mammalian circadian clock. In: Kramer A, Merrow M (eds) Circadian clocks, vol 217, Handbook of experimental pharmacology. Springer, Heidelberg
Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA (2000) Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103:1009–1017
Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ, Chang JG (2005) Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis 26:1241–1246
Crosio C, Cermakian N, Allis CD, Sassone-Corsi P (2000) Light induces chromatin modification in cells of the mammalian circadian clock. Nat Neurosci 3:1241–1247
De Bacquer D, Van Risseghem M, Clays E, Kittel F, De Backer G, Braeckman L (2009) Rotating shift work and the metabolic syndrome: a prospective study. Int J Epidemiol 38:848–854
DiTacchio L, Le HD, Vollmers C, Hatori M, Witcher M, Secombe J, Panda S (2011) Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science 333:1881–1885
Doi M, Hirayama J, Sassone-Corsi P (2006) Circadian regulator CLOCK is a histone acetyltransferase. Cell 125:497–508
Duong HA, Robles MS, Knutti D, Weitz CJ (2011) A molecular mechanism for circadian clock negative feedback. Science 332:1436–1439
Etchegaray JP, Lee C, Wade PA, Reppert SM (2003) Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 421:177–182
Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, Liu XS, Lazar MA (2011) A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 331: 1315–1319
Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, Hahn WC, Guarente LP, Sinclair DA (2008) The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 3:e2020
Fonken LK, Workman JL, Walton JC, Weil ZM, Morris JS, Haim A, Nelson RJ (2010) Light at night increases body mass by shifting the time of food intake. Proc Natl Acad Sci USA 107: 18664–18669
Fontaine C, Dubois G, Duguay Y, Helledie T, Vu-Dac N, Gervois P, Soncin F, Mandrup S, Fruchart JC, Fruchart-Najib J, Staels B (2003) The orphan nuclear receptor Rev-Erbalpha is a peroxisome proliferator-activated receptor (PPAR) gamma target gene and promotes PPARgamma-induced adipocyte differentiation. J Biol Chem 278:37672–37680
Frescas D, Valenti L, Accili D (2005) Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem 280: 20589–20595
Fu L, Pelicano H, Liu J, Huang P, Lee C (2002) The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111:41–50
Gaddameedhi S, Selby CP, Kaufmann WK, Smart RC, Sancar A (2011) Control of skin cancer by the circadian rhythm. Proc Natl Acad Sci USA 108(46):18790–18795
Gauger MA, Sancar A (2005) Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res 65:6828–6834
Gery S, Gombart AF, Yi WS, Koeffler C, Hofmann WK, Koeffler HP (2005) Transcription profiling of C/EBP targets identifies Per2 as a gene implicated in myeloid leukemia. Blood 106:2827–2836
Gery S, Komatsu N, Baldjyan L, Yu A, Koo D, Koeffler HP (2006) The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol Cell 22: 375–382
Gery S, Virk RK, Chumakov K, Yu A, Koeffler HP (2007) The clock gene Per2 links the circadian system to the estrogen receptor. Oncogene 26:7916–7920
Gorbacheva VY, Kondratov RV, Zhang R, Cherukuri S, Gudkov AV, Takahashi JS, Antoch MP (2005) Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK-BMAL1 transactivation complex. Proc Natl Acad Sci USA 102:3407–3412
Green KA, Carroll JS (2007) Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat Rev Cancer 7:713–722
Hirayama J, Cardone L, Doi M, Sassone-Corsi P (2005) Common pathways in circadian and cell cycle clocks: light-dependent activation of Fos/AP-1 in zebrafish controls CRY-1a and WEE-1. Proc Natl Acad Sci USA 102:10194–10199
Hirayama J, Sahar S, Grimaldi B, Tamaru T, Takamatsu K, Nakahata Y, Sassone-Corsi P (2007) CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 450:1086–1090
Hoffman AE, Zheng T, Ba Y, Zhu Y (2008) The circadian gene NPAS2, a putative tumor suppressor, is involved in DNA damage response. Mol Cancer Res 6:1461–1468
Hunt T, Sassone-Corsi P (2007) Riding tandem: circadian clocks and the cell cycle. Cell 129: 461–464
Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795–800
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293:1074–1080
Kang TH, Reardon JT, Kemp M, Sancar A (2009) Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci USA 106(8):2864–2867
Karlsson B, Knutsson A, Lindahl B (2001) Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup Environ Med 58:747–752
Katada S, Sassone-Corsi P (2010) The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat Struct Mol Biol 17:1414–1421
Katada S, Imhof A, Sassone-Corsi P (2012) Common threads: metabolism and epigenetics. Cell 148:24–28
Klevecz RR, Shymko RM, Blumenfeld D, Braly PS (1987) Circadian gating of S phase in human ovarian cancer. Cancer Res 47:6267–6271
Kobayashi M, Wood PA, Hrushesky WJ (2002) Circadian chemotherapy for gynecological and genitourinary cancers. Chronobiol Int 19:237–251
Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, Turek FW, Bass J (2007) High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab 6:414–421
Lamia KA, Storch KF, Weitz CJ (2008) Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci USA 105:15172–15177
Levi F, Focan C, Karaboue A, de la Valette V, Focan-Henrard D, Baron B, Kreutz F, Giacchetti S (2007) Implications of circadian clocks for the rhythmic delivery of cancer therapeutics. Adv Drug Deliv Rev 59:1015–1035
Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L (2007) SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28:91–106
Lis CG, Grutsch JF, Wood P, You M, Rich I, Hrushesky WJ (2003) Circadian timing in cancer treatment: the biological foundation for an integrative approach. Integr Cancer Ther 2:105–111
Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, Lopez JP, Philipson LH, Bradfield CA, Crosby SD, JeBailey L, Wang X, Takahashi JS, Bass J (2010) Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 466:627–631
Marcheva B, Ramsey KM, Peek CB, Affinati A, Maury E, Bass J (2013) Circadian clocks and metabolism. In: Kramer A, Merrow M (eds) Circadian clocks, vol 217, Handbook of experimental pharmacology. Springer, Heidelberg
Masri S, Sassone-Corsi P (2010) Plasticity and specificity of the circadian epigenome. Nat Neurosci 13:1324–1329
Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H (2003) Control mechanism of the circadian clock for timing of cell division in vivo. Science 302:255–259
Merrow M, Spoelstra K, Roenneberg T (2005) The circadian cycle: daily rhythms from behaviour to genes. EMBO Rep 6:930–935
Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, Antoch MP, Walker JR, Esser KA, Hogenesch JB, Takahashi JS (2007) Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci USA 104:3342–3347
Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P (2008) The NAD+−dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134:329–340
Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P (2009) Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324:654–657
Ortiz-Tudela E, Mteyrek A, Ballesta A, Innominato PF, Lévi F (2013) Cancer chronotherapeutics: experimental, theoretical and clinical aspects. In: Kramer A, Merrow M (eds) Circadian clocks, vol 217, Handbook of experimental pharmacology. Springer, Heidelberg
Ozturk N, Lee JH, Gaddameedhi S, Sancar A (2009) Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci USA 106(8):2841–2846
Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB (2002) Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109:307–320
Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U (2002) The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110:251–260
Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, Takahashi JS, Imai S, Bass J (2009) Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324:651–654
Ripperger JA, Schibler U (2006) Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat Genet 38:369–374
Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, Fitzgerald GA (2004) BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol 2:e377
Ruthenburg AJ, Li H, Patel DJ, Allis CD (2007) Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 8:983–994
Sadacca LA, Lamia KA, deLemos AS, Blum B, Weitz CJ (2011) An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia 54:120–124
Sahar S, Sassone-Corsi P (2007) Circadian clock and breast cancer: a molecular link. Cell Cycle 6: 1329–1331
Sahar S, Sassone-Corsi P (2009) Metabolism and cancer: the circadian clock connection. Nat Rev Cancer 9:886–896
Sahar S, Sassone-Corsi P (2012) Regulation of metabolism: the circadian clock dictates the time. Trends Endocrinol Metab 23:1–8
Sahar S, Nin V, Barbosa MT, Chini EN, Sassone-Corsi P (2011) Altered behavioral and metabolic circadian rhythms in mice with disrupted NAD+ oscillation. Aging 3:794–802. doi:100368 [pii]
Saper CB, Chou TC, Elmquist JK (2002) The need to feed: homeostatic and hedonic control of eating. Neuron 36:199–211
Schibler U, Sassone-Corsi P (2002) A web of circadian pacemakers. Cell 111:919–922
Schwer B, Verdin E (2008) Conserved metabolic regulatory functions of sirtuins. Cell Metab 7: 104–112
Slat E, Freeman GM, Herzog ED (2013) The clock in the brain: neurons, glia and networks in daily rhythms. In: Kramer A, Merrow M (eds) Circadian clocks, vol 217, Handbook of experimental pharmacology. Springer, Heidelberg
Smaaland R, Lote K, Sothern RB, Laerum OD (1993) DNA synthesis and ploidy in non-Hodgkin’s lymphomas demonstrate intrapatient variation depending on circadian stage of cell sampling. Cancer Res 53:3129–3138
Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J (2005) Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308:1043–1045
Unsal-Kacmaz K, Mullen TE, Kaufmann WK, Sancar A (2005) Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol 25:3109–3116
Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107:149–159
Yang WS, Stockwell BR (2008) Inhibition of casein kinase 1-epsilon induces cancer-cell-selective, PERIOD2-dependent growth arrest. Genome Biol 9:R92
Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413:131–138
Zuber AM, Centeno G, Pradervand S, Nikolaeva S, Maquelin L, Cardinaux L, Bonny O, Firsov D (2009) Molecular clock is involved in predictive circadian adjustment of renal function. Proc Natl Acad Sci USA 106:16523–16528
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Sahar, S., Sassone-Corsi, P. (2013). The Epigenetic Language of Circadian Clocks. In: Kramer, A., Merrow, M. (eds) Circadian Clocks. Handbook of Experimental Pharmacology, vol 217. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-25950-0_2
Download citation
DOI: https://doi.org/10.1007/978-3-642-25950-0_2
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-25949-4
Online ISBN: 978-3-642-25950-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)