
Abstract
This chapter presents the fundamentals, the experimental setups and the applications of temperature-programmed desorption (TPD), method used to investigate the events that take place at the surface of solid material while its temperature is changed in a controlled manner. At the beginning, fundamental principles of adsorption and desorption phenomena, as well as the data concerning first experimental setups are given. Further, important information related to the construction of nowadays used equipment and the organization of common experiments are underlined. The significance of data directly obtained from temperature-programmed experiment—TPD profile, which are the area under it and the position of peak maximum, are highlighted. Particular attention is given to the results that can be derived from these data—characterization of active sites that can be found on the surface of solid material and determination of kinetic and thermodynamic parameters of desorption process. In this regard, the influence of important experimental parameters on derived values is explained. Besides, the distinctions between TPD experiments performed in ultra-high vacuum and in the flow systems (differences in experimental setups and in the derivation of kinetic and thermodynamic parameters) are explained. Also, the modification of temperature-programmed techniques, known as temperature-programmed oxidation and temperature-programmed reduction are shortly explained and compared with temperature-programmed desorption method. In the end, a brief comparison of the TPD and adsorption calorimetry, two most widely used techniques for the study of acid/base properties of catalysts, is given.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Temperature-programmed desorption (TPD) belongs to the group of techniques in which an event that takes place at the surface of solid substance is monitored, while the temperature of investigated sample is changed with a temperature program \(\beta \)(t) \(=\) dT/dt. When the technique is applied to a system in which the adsorption process is (at least in part) irreversible and surface reaction takes place, this technique is often known as temperature-programmed reaction spectroscopy (TPRS). Although most often investigated surface process is desorption, reactions such as reduction, oxidation or sulfidation can be monitored using the techniques named temperature-programmed reduction (TPR), temperature-programmed oxidation (TPO) or temperature-programmed sulfidation (TPS), respectively. Usually, temperature T is a linear function of the time t; in that case the heating rate \(\beta \) is constant value.
In general, temperature-programmed methods are applicable for the investigation of both porous materials (such as real catalysts) and well-defined surfaces of single-crystalline samples. In addition, their application is experimentally simple and inexpensive, what explains their wide application in several scientific domains.
Temperature-programmed desorption originates from so-called flash desorption, which was originally developed in early fifties of twentieth century to quantitatively investigate the kinetics of molecular desorption from well-defined single crystal surfaces in high vacuum [1]. Flash desorption involved the adsorption of a known gas on the sample (in the form of a ribbon or wire, rigorously cleaned previously) while kept in vacuum. Subsequently, desorption was provoked by heating while the pressure in the system was recorded: as the temperature increased, certain previously absorbed species had enough energy to desorb from the surface and would be detected as a rise in pressure. The resulting pressure-time curve was referred to as a “desorption spectrum” [1, 2]. In flash desorption, temperature was raised very quickly (from 1 to \(1000~\mathrm{Ks}^{-1})\). Two heating schedules were applied: a linear variation of sample temperature with time: \(\mathrm{T} = \mathrm{T}_{0}\,+\,\beta \)t and its reciprocal variation \(1/\mathrm{T} = 1/\mathrm{T}_{0}\,-\,\alpha \)t (T\(_{0}\) is starting temperature, \(\alpha \) and \(\beta \) are constants) [1]. From the beginning, the possibility to extract the heat of adsorption from the obtained results was demonstrated.
Flash desorption was mainly applied for the investigations of low-surface area substances such as metals. The method was later adapted for the investigations of high-surface area materials, under carrier gas and ambient pressure. In the domain of catalysis, the pioneering work in this direction was done by Amnenomiya and Cvetanović [3], who studied the catalysts’ active sites, and needed the methodology that enabled conditions similar to those ordinarily used in catalytic reaction. Their apparatus allowed the pre-treatment of solid material by heating and evacuation, its exposure to molecular species of interest (at low temperature) and subsequent programmed desorption performed by heating in a controlled manner with the possibility to detect the desorbed gas in the carrier. This equipment consisted of two important parts: a temperature programming controller and a thermistor type thermal conductivity cell for measuring the rate of desorption of pre-adsorbed molecules from the surface. In this way, it was enabled to increase the temperature in a controlled manner, slowly (about 1 to tenth degrees K per minute), in a manner similar to those applied in real catalytic reactions.Footnote 1 An additional important feature of their modified technique was that it permitted simultaneous study of a chemisorption process and the surface reaction which accompanied it. Applying the “temperature-programmed desorption” (the name they used for this modification of flash desorption), Amnenomiya and Cvetanović discovered the existence of two different active sites for the adsorption of ethylene on alumina [4]. Importantly, they employed the method to calculate the values of energies for ethylene desorption.
Since that time, the method is widely developed; experimental setups are improved and adjusted to many different purposes (e.g. for the investigations of oxidation and reduction reactions). Today, two main types of equipment are available: those operating under ultrahigh vacuum and so-called “flow” systems. Well-defined surfaces of single-crystalline samples are investigated in a continuously pumped ultra-high vacuum (UHV) chamber (this technique is often referred to as thermal desorption spectroscopy—TDS [5]). The equipment that is constructed to allow adsorption–desorption in the gas flow are most often used for the investigation of porous materials (catalysts, for example). Vacuum setups are customarily used for surface science studies, but they can be also useful for the characterization of porous materials.
Generally, TPD can be described as the measurement of the rate of desorption of adsorbed molecules, as a function of temperature. Therefore, this method can be useful in the extraction of very important information. It can be used in the identification and characterization of sites active in adsorption and catalytic reactions, in the study of adsorption states, binding energies, surface concentration and desorption kinetics. To summarize, this method is nowadays very important and often applied for the characterization of materials used as catalysts. In this domain, two main areas of applications are: the characterisation of acid/base properties of solid materials, what is essential for understanding their reactivityFootnote 2; and the determination of kinetic and thermodynamic parameters of desorption processes or decomposition reactions.
In the following sections, the basic concepts of temperature-programmed methods (primarily temperature-programmed desorption, but also temperature-programmed reactions) are outlined. At the beginning, fundamental principles of adsorption and desorption—their thermodynamic and kinetic aspects, are presented. Furthermore, the descriptions of experimental setups, the data that can be obtained from the experiments and their interpretation are given. The possibilities to extract the adsorption energies and kinetic parameters from experimental results are discussed. Finally, the examples of possible applications and the comparison of results obtained by TPD with those obtained from adsorption calorimetry, are presented.
2 Adsorption–Desorption; Fundamental Principles
Adsorption is defined as the enrichment of gas or liquid (adsorbate) at the surface of a solid material (adsorbent); or as the increase in the density of the fluid in the vicinity of an interface. Adsorption takes place at the active sites—specific points on the solids’ surface that possess affinity toward the particles coming from the gas or liquid phase; it happens when an attractive interaction between a particle from adsorbate phase and a surface is strong enough to overcome the disordering effect of thermal motion [6, 7].
In discussing the fundamentals of adsorption it is usual to distinguish between physisorption, which takes place when the attractive interactions are essentially the result of weak intermolecular forces; and chemisorption, which involves the overlapping between the molecular orbitals of the adsorbed particle and the surface atoms (i.e. electron transfer which leads to the formation of chemical—covalent bond between adsorbate molecule and the active site at the surface). Although this distinction is very useful, it has to be pointed out that there are many intermediate cases, and that it is not always possible to categorize a particular event at the surface as physi- or chemisorption. However, there are some general characteristics which distinguish these two possible types of adsorption. Chemisorption is highly specific, while quite contrary, physisorption is non-specific; chemisorption may involve dissociation, it is possible over a wide range of temperature and a monolayer is formed; while physisorption takes place only at relatively low temperatures, multilayer can be formed, dissociation of adsorbed species does not happen. However, the most important distinction is the amount of heat that is associated to either one of these two general types of adsorption: physisorption is characterized by low heat of adsorption (below approximately 50 kJ mol\(^{-1})\); while chemisorption is characterized by high heat of adsorption, typically exceeding 50 kJ mol\(^{-1}\). Hence, as a result of molecular chemisorption, the weakening of intramolecular bonds inside the adsorbed molecule can happen, which may lead to its dissociation [6–8]. The differences in potential energies curves that present processes of physisorption and chemisorption (dissociative and non-dissociative) are presented in Fig. 4.1.Footnote 3

Potential energy curves for (1) physical and (2) chemical adsorption: (a) non-activated (b) activated. \(\mathrm{E}_{\mathrm{pot}}\) - potential energy, \(\mathrm{Q}_{\mathrm{c}}\) - heats of chemisorption, \(\mathrm{Q}_{\mathrm{p}}\) - heats of physisorption, \(\mathrm{E}_{\mathrm{ad}}\) - energy of activation for desorption, \(\mathrm{E}_{\mathrm{diss}}\) - dissociation energy for the diatomic molecule. The sum: \(\Delta \mathrm{E}_{\mathrm{des}} = \mathrm{E}_{\mathrm{ad}} + \mathrm{Q}_{\mathrm{c}}\) is the the heat of hemisorption, in the activated processes [8]
In addition, chemisorption is often an activated process, which means that the formation of a chemisorptive bond requires overcoming the activation barrier (Fig. 4.1b); it may be slow and irreversible. By contrast, physisorption is rapid, non-activated and reversible process.
2.1 Thermodynamic View
Both physisorption and chemisorption are exothermic processes, what can be concluded from a simple thermodynamic consideration. The adsorbed molecule has at most two degrees of translational freedom on the surface; in addition, its rotational freedom must always be less than that of the gas phase molecule. In total the adsorbed molecule possesses less degree of freedom than the same molecule in the gas phase. Consequently, the entropy change of adsorption \(\Delta \)S\(_{ads}\) \(=\) S\(_{ads}\) \(-\) S\(_{gas}\) is obligatory negative. However, the adsorption is spontaneous process, which means that the free energy change (\(\Delta \)G \(=\) \(\Delta \)H \(-\)T\(\Delta \)S) must be negative. Negative value of entropy change \(\Delta \)S\(_{ads}\) means that the second term in previous relation is positive (\(-\)T\(\Delta \)S\(_{ads})\), what requires the value of \(\Delta \)H necessarily negative. Hence, adsorption is always exothermic process [6].
The heats of adsorption provide a direct measure of the strength of the bonding between adsorbate and the active site at the surface of solid substance. Therefore, it is of importance to estimate these values, particularly in the domain of catalysis where the strength of active sites determines the mechanism and the yield of certain process. One possible way to determine the heat of adsorption is to apply calorimetry, experimental technique which provides the heats of adsorption as a function of the adsorbed amount (\(-\) \(\Delta \)H \(=\) f(n\(_{a})\), where \(n_{a}\) is the adsorbed amount and \(-\) \(\Delta \)H is, in that case, so-called differential heat) [9]. The heats of adsorption can be derived also from the variation of adsorption with temperature. In that case, Clausius-Clapeyron equation and the data from isosteric measurements are used (in that way, so-called isosteric enthalpy of adsorption can be obtained).
The extent of surface coverage (or simply surface coverage), reached as a result of adsorption, is usually denoted as \(\theta \). It is a ratio between adsorbed particles number (N\(_{ads})\) and the number of adsorption sites available at a surface (usually denoted as active sites \(-\)N\(_{surf})\): \(\theta \) \(=\) N\(_{ads}\)/ N\(_{surf}\). The chemical equilibrium between adsorbed species and gas phase particles is reached when chemical potentials of adsorbate particles in both phases are equal (the rates of adsorption and desorption are equal); and it is characterized by constant value of surface coverage \(\theta \). The temperature dependence of the gas pressure p required for equilibrium between the adsorption and desorption can be calculated from the Clausius-Clapeyron equation [6]. Neglecting the volume of the condensed surface phase, this relation becomes:
where \(q_{isost}\) denotes so-called isosteric heats of adsorption. Evidently, these values can be calculated from the temperature dependence of the adsorption isotherms, i.e. from the isosteres, for each average temperature:
where T is the absolute temperature, R the gas constant and \(n_{r}\) the number of reversibly adsorbed molecules.Footnote 4
Third possibility to obtain the adsorption heats is to extract them from the data acquired from temperature-programmed desorption experiments. This possibility will be exposed in detail later, in the Sect. 4.5.2. However, for that purpose, it is obligatory to know some basic postulates about the kinetics of adsorption and desorption; what is given in the following section.
2.2 Kinetics of Adsorption and Desorption
In the case of gas-phase adsorbate, the surface coverage \(\theta \) is dependent on the gas pressure. Adsorption isotherms relay the surface coverage and the gas pressure (at constant temperature); the most known equation of this type is Langmuir adsorption isotherm. It is based on the following assumptions [6]:
-
one adsorbed particle interacts with one active site at the surface (once adsorbed, it is immobile on the surface);
-
the surface of adsorbent is saturated when all adsorption sites are covered with the particles of adsorbate, i.e. the adsorption goes on until monolayer of adsorbate particles is spread over the solid (then, \(\theta \) equals 1);
-
there are no interactions between the adsorbed particles.
The rates of adsorption (\(r_{ads})\) and desorption (\(r_{des})\) are proportional to the numbers of empty or occupied active sites, \(\theta \) or (1 – \(\theta \)), respectively:
where \(r_{ads}\) and \(r_{des}\) are the rates of adsorption and desorption, respectively, \(A_{n}\) and \(B_{n}\) are constants, while x is the kinetic order of surface event (x can be 1 or 2, although adsorption and desorption are usually considered as the first order events). In the equilibrium, the rates of adsorption and desorption are equal: \({\vert }r_{ads}{\vert } = {\vert }r_{des}{\vert }\), therefore, surface coverage \(\theta \) can be expressed as:
(where \(b_{n}(T)=A_{n}/B_{n}\) = const, x \(=\) 1, 2).
If adsorption is a reversible process (i.e. backward process—desorption, passes through exactly the same states), the rates of both processes can be described using the same equation:
However, in contrast to adsorption which may or may not be activated process, desorption is always activated, with a minimum activation energy denoted as activation energy for desorption (\(\Delta E^{a}_{des})\). The rate constant for desorption can be expressed by Arrhenius equation:
where \(\nu \) (\(\theta \)) is pre-exponential factor, which is in general dependent on surface coverage \(\theta \). Now, the rate of desorption becomes:
The relation (4.7) is customarily used to describe the rate of desorption, and is known as Polanyi-Wigner equation.
Desorption is often explained assuming the existence of transition state:
where (A)\(_{ads}\) is molecule of specific adsorbate adsorbed on the active site at the surface ()\(_{ads}\), A\(_{gas}\) is the gas-phase molecule and (A)\(_{ads}^{\# }\) is the transition complex. The equilibrium between the adsorbate and transition state is defined as:
Equilibrium constant for overall desorption process (A)\(_{ads} \quad \rightarrow \) A\(_{gas}\) + ()\(_{ads}\), (K) can be considered as equal to \(K^{\# }\). The reaction rate constant is related with the equilibrium constant K\(^{\# }\) (hence, with equilibrium constant K), as:
From the other side, thermodynamic functions are related with the equilibrium constant through the known relation:
The rate of desorption is customarily extracted from the experiments of temperature-programmed desorption. Since the rate of desorption is related to the equilibrium constant through the relations (4.5) and (4.10); it is evident that the rate constant for desorption can be expressed as:
Obviously, thermodynamic quantities (\(\Delta H_{des}\), \(\Delta S_{des}\) and \(\Delta G_{des})\) for the activated process such as desorption can be extracted from the data obtained from desorption experiments. Importantly, since the adsorption is spontaneous process (it does not need the energy of activation), the heat of adsorption equals, in general, the activation energy for desorption. Hence, desorption experiments provide also thermodynamic parameters of adsorption.
3 Experimental Setups
The experimental setups for temperature-programmed desorption have evolved with time. As it has been already stated, there are numerous experimental designs that allow the application of this method under the conditions that are the same or very similar to those applied in real catalytic reaction (or any other surface event). Nevertheless, the various equipment used for these experiments, although different, is all constructed to allow two main steps that are common for all thermal desorption methods:
-
1.
The admission of desired gas to the sample.
-
2.
The heating of the sample in a programmed way.
In TPD experiment, gaseous molecules (atoms) of interest are adsorbed at the surface, at constant temperature. The adsorption is very often performed at ambient temperature, but can be sometimes done at sub-ambient or at elevated temperature. In the modifications of technique such as TPO or TPR, gaseous species are consumed while temperature is increased in a programmed manner. In the case of TPD procedure, desorption of adsorbate is monitored while increasing the solid sample temperature in a controlled fashion; while in the case of TPR/TPO, the consumption of active gas is monitored during temperature increase, as explained later in more details (see Sect. 4.4).

(a) Experimental setup for temperature-programmed desorption performed in vacuum. The sample (monocrystal or a thin layer of powder) is exposed at the sample holder, which is connected to the system for temperature control. The pumping speed has to be high enough to allow the monitoring of desorbed species by mass spectrometer. Inset presents the output of one TPD experiment—the TPD profile [5]. (b) In the flow systems, sample is placed in the sample holder, inside the furnace [10]
From previously stated, it follows that temperature-programmed experiments can be performed under ultrahigh vacuum or in the flow of gas. Still, whatever is the experimental design, three main parts of equipment are always necessary to perform this kind of investigations:
-
The system for the controlled admission of (different) gases. The adsorption is commonly performed as isothermal process. Nowadays, it is possible to construct the equipment which enables the adsorption of desired and precisely known amount of adsorbate. In the past, small polar molecules (\(\mathrm{{NH}}_{3}\), CO, \(\mathrm{{CO}}_{2}\), \(\mathrm{{SO}}_{2}\), \(\mathrm{{H}}_{2}\)O) have been usual adsorbates in the TPD studies. More recently, larger molecules (such as hydrocarbons) and non-polar molecules (such as Ar\(_{2}\) or N\(_{2})\) have been applied as adsorbates. Usually, adsorbates are denoted as “probes” or “probe species”. Most often used are the probes customarily applied to titrate acidic or basic surface sites (NH\(_{3}\), pyridine, CO, CO\(_{2}\), and SO\(_{2})\).
-
“Reactor” or sample holder, placed in a heated area (or furnace) where temperature can be controlled. In the case of experimental setup constructed to allow TPD in ultra-high vacuum (schematic presentation shown in Fig. 4.2a) sample is deposited on a sample holder as a single crystal or monolayer, and connected with a system for temperature control. In the systems that are designed to work in the flow of gas, sample is placed in a reactor, which is usually a quartz tube placed in furnace (Fig. 4.2b). By far, the most common approach is to increase the sample temperature linearly with time at constant rates (\(\beta =dT/dt= \mathrm{{const}}\)) that have values between 0.5 K s\(^{-1}\) and 25 K s\(^{-1}\).
-
A system for detection of evolved gases. Heating of the sample provokes the evolution of species from the surface back into the gas phase, which has to be monitored. The detectors used for detection and possible quantification of evolved gases are: thermal conductivity detector (catharometer), flame ionization detector, conductometric titration and mass spectrometer.
-
Catharometer serves for measuring the difference in thermal conductivity between reference gas and the gas that flows through the sample (it is used only in flow systems). This kind of detectors is often used in the variations of temperature-programmed (TP) methods, known as TPO or TPR.
-
Flame ionization detector is in specific use for the detection of organic effluents: effluent enters in a flame obtained by combustion of hydrogen and air, then, the ions that are formed are trapped by two electrodes (with a potential difference between them). As a result, electric current appears and can be detected.
-
Conductometric titration is applied if it is possible to entrap the evolved gas in an aqueous solution; then, the change in conductivity can be detected.
-
In modern implementations of temperature-programmed techniques the detector of choice is a small, quadrupole mass spectrometer (QMS). The application of mass spectrometer enables simultaneous acquisition of single or multiple masses desorbed during heating. In fact, the application of this kind of detector enabled the distinction between different species desorbed in the same time from the surface.
Nowadays, two main techniques that are most often used for detection of effluents are mass spectrometry and thermal conductivity; the whole process is most often controlled by computer.
4 The Design of Temperature-Programmed Experiment; Obtained Data
The equipment used for TPD experiments have to be designed in a way which allows the performance of certain steps that may be necessary in particular experiment. Firstly, sample is placed in the sample holder (reactor) and pre-treated in the appropriate way (in vacuum or in the flow of desired gas, at desired temperature); the pre-treatment procedure depends on the characteristics of investigated material and the purpose of TPD experiment. Afterwards, the sample is exposed to the adsorbate. Usually, the adsorption is performed isothermally, at appropriate temperature (frequently at 300 K, but also at temperatures higher then this one, or even at sub-ambient temperatures). Subsequently, physisorbed part of adsorbed gas is removed from the surface, either by the evacuation, or by inert gas flow. The residual chemisorbed adsorbate is desorbed by heating the sample in a controlled manner, preferably in a way to give a linear temperature ramp; the analysis of the evolved gas (gases) is performed to establish its identity and the amount, using the appropriate detection system. The whole procedure is performed in situ.

Experimental setups for temperature-programmed desorption, reduction and oxidation. (a) The reactor is placed inside the furnace which is connected with temperature programmer. Detection of evolved gas(es) is performed by monitoring the variations in thermal conductivity of gas mixture. (b) The TPD apparatus equipped with mass spectrometer as a detector [5]

(a) Time dependence of adsorbate concentration upon exposing the solid sample. (b) An example of TPD profile, drown as a signal of detector versus temperature. Common TPD profile is a complex shaped curve. Figure presents the interaction (adsorption and desorption) of CO with CoY zeolite [11]
Schematic presentation of one experimental setup that enables realisation of all mentioned steps in vacuum is shown in Fig. 4.2, while Fig. 4.3 shows two typical constructions designed for the experiments in the flow of appropriate gases.
The data obtained from one temperature-programmed experiment are presented as the variation of a detector signal intensity (presented at y axis) as a function of time (or temperature, presented at x axis). Consequently, as detector signal is proportional to the concentration of the species desorbed from the surface, y axis values are proportional to the rate of desorption (r \(_{des})\).
It is important to point out that in the case of flow systems, temperature-programmed techniques can be used for quantitative measurements. For that purpose, the gas flow has to be controlled and constant in time. If this condition is achieved, the detector signal can be properly calibrated by using dilute gas streams of known concentrations: the signal of such a known mixture is passed through the empty reactor (or bypass reactor) and the signal intensity is monitored. Furthermore, the intensity of signal obtained as a result of the same gaseous species evolved from the sample can be considered as proportional to the value obtained for the known mixture. Once calibrated in that way, the intensity of detector signal can be given in concentration units (Fig. 4.4), and the measurement of evolved gas concentration becomes possible. The calibration of a detector signal enables the precise determination of both adsorbed and desorbed amounts.

Top TPD profiles of ammonia (m/z \(=\) 17) and hydrocarbons (\(\mathrm{C}_{x}\mathrm{H}_{y}\), m/z \(=\) 26) obtained as a result of thermal degradation of 1,2-diaminopropane entrapped inside a zincophosphate structure-ZnPO-HDAP. Bottom Thermogravimetric and differential thermogravimetric signal of this decomposition [12]
The precise amount of adsorbed gas can be obtained by passing it through “bypass” reactor, and subsequently, through the sample (Fig. 4.4a). If the detector signal has been previously properly calibrated, the monitoring of two signals (bypass and through the sample) provides the true adsorption amount of respective gas at a given temperature, which can be derived from the surface in between these two signals. Precise amount of desorbed gas can be obtained from the surface under the TPD profile (Fig. 4.4b).
It is very important to keep in mind that many different chemical species can evaporate from the sample in the same temperature region (particularly in the case of real catalytic systems). In the old versions of TPD setups, the overall desorbed amount would be recorded as a rise in the pressure. It is especially important to point out that the incorporation of mass spectrometer as a detector enabled the discrimination of different products, desorbed in the same time (in the same temperature region).
Figure 4.5 shows one example: evidently, the evolution of ammonia and hydrocarbons happens in the same temperature region (from \(\sim \)370\(\,^{\circ }\)C up to \(\sim \)440\(\,^{\circ }\)C), during the thermal decomposition of 1,2-diaminopropane entrapped inside a zincophosphate structure.
The insight in the typical TPD profiles presented in Figs. 4.4 and 4.5 reveals what information can be obtained from one TPD experiment. There are two main classes of data that we can “read” from the desorption profile:
-
1.
The area under the TPD profile, which is proportional to the amount of adsorbate originally adsorbed, in other words, to the surface coverage \(\theta \). Under particular circumstances, id est, if the limitations such are diffusion and/or readsorption can be neglected (see later, in the Sect. 4.5.2.2), temperature-programmed technique can be employed as an excellent tool for determination of surface coverage.
-
2.
The position of the peak maximum (along the temperature scale, \(T_{max}\)), which is related to the activation energy for desorption. Generally, the higher the value of temperature of peak maximum (\(T_{max})\) is, desorption is more difficult, which is an indication of stronger interaction between the adsorbate species and the active site on the surface. As it has been explained previously, the adsorption is spontaneous process, therefore, the heat of adsorption equals, in general, the activation energy for desorption. Hence, the value of \(T_{max}\) is related to the heat (enthalpy) of adsorption; in other words, to the strength of adsorbate binding to the surface. The methods for deriving the activation energy for desorption from the position of \(T_{max}\) will be discussed latter. However, it is very important to note here that a simple TPD profile that possesses only one peak (and one \(T_{max}\), like a desorption profile of mass 17, presented in Fig. 4.5) is not a common case that can be found for the majority of investigated systems. In fact, very often there is more than one binding state for the adsorbate molecules on a surface, which express significantly different adsorption enthalpies (consequently, significantly different activation energies for desorption); therefore, this will give rise to multiple peaks in the TPD spectrum (one example is desorption profile presented in Fig. 4.4b). In that case, the determination of \(T_{max }\) positions demands particular attention.
4.1 The Design of TPR/TPO Experiments; Obtained Data
In the modification of temperature-programmed methods known as temperature-programmed reduction (TPR), a reductive gas (usually H\(_{2}\) mixed with an inert gas) is consumed by the sample, while the temperature is increased with a constant heating rate \(\beta \). In the case of temperature-programmed oxidation, the gas or gas mixture which can perform oxidation (O\(_{2}\) mixed with an inert gas, air, N\(_{2}\)O, etc.) is passed through the sample. The reductive or oxidant gas consumption is monitored either by mass spectrometer or by catharometer. If reduction is done by hydrogen, the analysis is usually performed using a catharometer as a detector; the same stands for TPO performed with oxygen as oxidative gas.Footnote 5 The experiments of temperature-programmed reactions (reduction, oxidation, or any other reaction, such is sulfidation, for example) have to be performed in the systems that allow the calibrations of detector signals [14]. Therefore, these experiments can be performed only using the equipment that allows flow of different gases (as those setups presented in Fig. 4.3).
TPR and TPO profiles give information concerning the reduction or oxidation (red-ox) state of the solid which is analyzed. These features are very important data for commercial catalysts; what explains the vast application of TPR and/or TPO in the characterization of solid materials for industrial applications. For example, reduction is an inevitable step in the preparation of metallic catalysts [5, 15]. In addition, it is often a critical step—if it is not performed correctly the catalyst may sinter or may not reach its optimum state of reduction.
Similarly to the case of TPD, the data obtained from TPR or TPO experiments are presented as the variation of detector signal intensity as a function of time (or temperature). Generally, the data that can be obtained from temperature-programmed reduction or oxidation are:
-
1.
The difference in reduction (oxidation) temperature between different materials; these differences are recognized from different positions of \(T_{max}\);
-
2.
The profile of TPR (TPO) pattern, which indicates the presence of one (or more) species that can be reduced (oxidized);
-
3.
The area under a TPR or TPO curve represents the total hydrogen (oxygen) consumption, and is commonly expressed in moles of reductive (oxidative) gas consumed per mole of metal atoms. Hence, the calculation of exact amounts of those species which were reduced (oxidized) at the surface is possible.
-
4.
The activation energy of the reduction can be estimated from the temperature \(T_{max}\) at which the reduction rate is maximal by using appropriate equations.
The organization of one TPR or TPO experiment is somehow different in comparison with that one applied for temperature-programmed desorption. For example, the sample has to be purged with inactive gas, before exposure to active (reductive or oxidative) gas.Footnote 6 These differences can be seen in Table 4.1, which presents the organisation of both TPD and TPR/TPO experiments.
5 The Interpretation of Results Obtained from Temperature-Programmed Desorption Experiments
Temperature-programmed desorption technique offers very useful and important methodology which can be applied for the characterization of materials used as catalysts. There are two main fields of applications:
-
1.
The characterisation of active sites of solid materials. It is of outmost importance to determine and understand the acid/base character of solid catalysts, because these features are essential for their reactivity [16–20]. There are several groups of techniques developed and particularly adapted for the investigation of acidity/basicity of solid catalysts; most of these methods are based on the adsorption of gas-phase probe molecules, which are chosen on the basis of their reactivity, molecular shape and size [13, 21–26]. Among the other techniques, TPD is of particular importance because its experimental conditions can be organized in the same (or very similar) way as the conditions of real catalytic reaction. The investigation of acid/base character of solids is perhaps the most common application of TPD. For that purpose, many different chemical species can be used as adsorbates (probes). In addition, the strength and the population of specific active sites can be estimated, using the appropriate probes and applying appropriate experimental conditions.
-
2.
The determination of kinetic and thermodynamic parameters of desorption processes, decomposition or other reactions. The interpretation of experimentally obtained data and derivation of kinetic and thermodynamic parameters from TPD results depends on the type of TP experiment: specific experimental conditions have influence on the overall TP profile and on the position of \(T_{max}\) obtained either in ultra-high vacuum or in the flow system.
The details that explain more closely how the data obtained from TPD experiment can be used to get the information concerning the characterization of active sites, kinetic and thermodynamic parameters, are given in the following sections.
5.1 The Application of Temperature-Programmed Desorption in Active Sites Characterisation
The characterization of active sites of solid catalysts includes the determination of active sites nature, the estimation of their density (or population, i.e. the number of active sites per unit of mass or per unit of surface area), their strength and strength distribution. Active sites can be acidic, basic and, in certain cases, amphoteric. All mentioned characteristics are very important for catalysts functionality; therefore, many experimental techniques are invented and adapted for their investigation. Among others, mainly spectroscopic methods (like NMR, IR, XPS, XRF...), temperature-programmed desorption is particularly important because it can be useful in the characterization of all mentioned features.
The strategy which is employed in order to get the above mentioned features of catalysts’ active sites is the adsorption of appropriate gas phase probe, under the specific experimental conditions (that are chosen in a way to be similar to those applied in the particular catalytic reaction), followed by subsequent desorption, monitored with appropriate detector. One experiment, in which the characterisation of acid/base properties of solid material is performed, is designed as follows:
-
The sample is pre-treated in situ, in desired atmosphere (or in vacuum), at appropriate temperature, and during the appropriate time. Usually, the purpose of pre-treatment is to remove water (eventually, some impurities) and/or to perform degasification;
-
The probe gas is admitted and adsorbed on the solid surface up to some specific surface coverage or up to the saturation;
-
Desorption process is performed.
According to the Lowry-Brönsted theory, a Brönsted acid is a proton donor, while a Brönsted base is a proton acceptor. In Lewis’ concept, acid acts as electron-pair acceptor, while base is electron donor (such as molecules possessing electron lone pairs). Hence, a Lewis base is in practice equivalent to a Brønsted base. However, the concepts of acidity are markedly different [27].
In the case of solid catalysts, any atomic (ionic) group at the surface that can donate a proton is a Brönsted acid; while any place where one empty electron orbital exists is Lewis acid. For example, in the case of zeolites, Brönsted acid site is a part of microporous aluminosilicate framework—a bridging [\(\equiv \) Si \(\cdot \cdot \cdot (\mathrm{OH}) \cdot \cdot \cdot \) Al \(\equiv \)] configuration which is able to donate a proton to an acceptor; while Lewis acid site is either tri-coordinated Al atom or charge-balancing cation Me\(^{z+}\) which are able to accept the electron pair. Accordingly to the same theories, any place at the solid surface which can accept proton is a Brönsted base; while any place which can donate electron(s) is a Lewis basic site. For example, in the case of MeO\(_{x}\) (metallic oxides), the oxygen ions (O\(^{z-})\) behave as Brønsted bases (because they are proton acceptors); while cations at the surface possess Lewis acidity (they are electron acceptors) [27, 28].
The probe molecules that are used to investigate surface acidity should be chosen accordingly to their ability to accept proton from the surface active site, or to donate electron pair to the solid surface. The molecules that fulfil these demands are, for example, ammonia, pyridine, or hydrocarbons. Similarly, the probe molecules that can be used to “trace” the basic site of solid catalysts must be able either to donate a proton or to accept electron(s). Importantly, many species (that even do not contain hydrogen in their formula, which is a demand according to Lowry-Brönsted theory) can function as Lewis acid, accepting electron pair. Hence, the molecules that could be chosen to investigate surface basicity are, for example, dioxides of carbon or sulphur.
However, acidity or basicity of a gas-phase adsorbate is not a sole criterion for its choice as a probe molecule. Firstly, the strength of an acidic or basic probe should be distinguished accordingly to its acid- or base-dissociation constant (\(K_{a}\) or \(K_{b})\). In addition, very important feature of probe molecule is its radius. If there is a need to locate all active sites in the structure of microporous solid material, the radius of probe molecule has to be smaller then the diameter of pore(s) opening(s). In other words, probe molecules have to be of appropriate size, so the entrance in the micropores of the solid and the access of adsorbate to each active site become possible. For example, ammonia, which is frequently used to reveal the acidic property of solids, is selected as a probe due to its basicity and due to the size of the molecule. Its molecule is smaller than the diameter of the pores in the zeolites’ structures, and also in many other solids. The other probe often used for investigation of solids’ acidity is pyridine; however, the application of other chemical species is also possible.
As it has been already stated, the value of \(T_{max}\) is the indication of the strength of the interaction adsorbate—active site. The stronger the active site is, the stronger the interaction with probe molecule is, which causes more difficult desorption: higher \(T_{max}\) indicates that desorption is more difficult. The energy that have to be consumed for desorption is related with the bond energy between surface active site and adsorbate; hence, the position of peak maximum provides information on the strength of this bond. Solid materials possess active sites of different strength, i.e. they express energetic heterogeneity. The origin of active sites strengths heterogeneity is usually the consequence of the solids’ structure, or it can be result of different topologies and chemical environments of active sites.
In the case of energetically heterogeneous surface, TPD curves are generally complex-shaped profiles. Figure 4.6 presents two typical cases. Sometimes, desorption profile is composed of well resolved peaks, like upper TPD curve in the Fig. 4.6, where two desorption peaks are denoted as low (l) and high (h), accordingly to the temperature region of appearance. More often, desorption of probe molecules takes place simultaneously from different sites, what gives more or less pronounced overlapping of the peaks (bottom TPD profile in Fig. 4.6, TPD profiles already presented in the Figs. 4.4 and 4.5).

TPD profiles of ammonia obtained from H-mordenite \((\mathrm{SiO}_{2}/\mathrm{Al}_{2}\mathrm{O}_{3} = 15.0)\) and H-ZSM-5 \((\mathrm{SiO}_{2}/\mathrm{Al}_{2}\mathrm{O}_{3} = 23.8)\) [54]

TPD signals obtained after pyridine was adsorbed at Y-type zeolite. The applied \(T_{ads}\): (a) \(150\,^{\circ }\mathrm{C}\) (b) \(200\,^{\circ }\mathrm{C}\) (c) \(250^{\circ }\mathrm{C}\) (d) \(300\,^{\circ }\mathrm{C}\) [29]
It is important to notice the influence of adsorption temperature (T\(_{ads})\) on the shape of TPD profile. Desorption takes part consequently to adsorption as a result of thermal motion which kinetic energy is high enough to break the bond between the adsorbate and weak active sites. If adsorption is performed at high temperature, TPD profiles are either single-peak shaped, or overlapping of peaks is less pronounced. By contrast, low temperature of adsorption allows the bonding of adsorbate with all active sites present in the investigated structure. In that case, complex-shaped TPD profile is obviously obtained, in the case of heterogeneous solid surface. The adsorption temperature is apparently very important experimental condition: its influence on the shape of TPD profile (hence, on the conclusions that can be derived from TPD experiment) is illustrated by the example shown in Fig. 4.7. Evidently, the lower the \(T_{ads}\) value is, the more complex TPD profile is obtained, and vice versa. Of course, it has to be kept in mind that the terms “low” or “high temperature” are relative—the choice of adsorption temperature depends on the particular investigated system (on the pair adsorbate—solid surface). In addition, it is important to notice that, the higher the \(T_{ads}\) value is, the overlapping of peaks is less visible, while desorption profile is shifted more to high-temperature region.
Usually, sites denoted as “weak”, “medium”, “strong” or “very strong” are recognized from temperature-programmed desorption experiments, accordingly to the temperature region of appearance.
Apparently, the overlapping of desorption peaks that has its origin in the surface heterogeneity imposes the necessity to resolve the complex TPD curve and to assign particular desorption profiles to the sites of definite strength. Generally, there are two possible methods that can be applied in order to get information concerning the presence and population of particular active sites on the surface of solid material.
Firstly, if a complex desorption profile has been obtained as result of probe adsorption, the mathematical procedure of deconvolution can be applied. Usually, deconvolution is based on the assumption that the desorptions from different sites are parallel and independent events of first order (in surface coverage) [31, 33]. Then, desorption from the sites of same type and same strength would give symmetric desorption profile, with well-defined single temperature of maximum. In that way, certain number of symmetric desorption traces can be obtained, their sum should give the overall TPD profile obtained from experiment. This procedure enables to get information about the presence of different active sites in the investigated system (from the number of single-peak symmetrical curves), their population (from the percent with which each of these curves contribute in the area of overall desorption profile) and their strength (from the positions of \(T_{max}\)). The procedure of deconvolution can be performed relatively simply: the \(T_{max}\) positions could be recognized from the experimental TPD profile, the whole numerical procedure should be performed as to choose the set of parameters in such a way to enable minimization of standard deviations (in comparison between the linear sum of single desorption profiles and the overall complex experimental curve). However, even though this numerical procedure can be performed to give a unique deconvolution of the experimental curves, it is recommended to compare the results obtained by deconvolution with the information provided by other experimental techniques (for example, the adsorption-desorption studied by FTIR spectroscopy).
Another possibility to investigate desorption from a heterogeneous surface and to recognize the presence of some particular active sites is to perform step-wise filling of the surface with the probe. When the active (probe) gas is admitted to the solids’ surface, the first interactions would be those between the strongest active sites and the probe molecules. Therefore, in this approach, the main idea is to admit small quantities of probe gas to enable the adsorption on the most active (the strongest) sites separately, and to continue with the filling of surface, step by step. This task can be fulfilled in two ways:
-
The usage of experimental setup which enables the admission of controlled amounts of a gas-phase probe;
-
The variation of adsorption temperature in order to start the adsorption at the highest possible temperature.
It is evident from the example presented in Fig. 4.7 that when the lower the temperature of adsorption is applied more complex TPD profiles are obtained. By contrast, the higher the adsorption temperature is, less complicated TPD profile is obtained. Hence, if high enough temperature is applied, the interaction with one single type of energetically homogeneous centres should be expected; what should give a symmetric, single-peak desorption curve. Figure 4.8 presents more obvious example.

TPD signals, obtained after pyridine was adsorbed on \(\mathrm{AlPO}_{4}-\mathrm{Al}_{2}\mathrm{O}_{3}\). The step-wise filling of the surface is achieved by applying different \(T_{ads}\): (a) \(50\,^{\circ }\mathrm{C}\) (b) \(100\,^{\circ }\mathrm{C}\) (c) \(150\,^{\circ }\mathrm{C}\) (d) \(200\,^{\circ }C\) (e) \(300\,^{\circ }\mathrm{C}\) [30]

(a) TPD profiles of ammonia obtained from HY and dealuminated HY zeolites. (b) TPD profiles of pyridine obtained from HY and dealuminated HY zeolites. Overall TPD profile is presented by spotted line; dashed lines, obtained by deconvolution, present desorption from the acid sites of different strength [31]

TPD profiles of argon, obtained from different solid acids. \(T_{ ads}\) \(=\) 113 K [32]
Evidently, step-wise filling of AlPO\(_{4}-\mathrm{Al}_{2}\mathrm{O}_{3}\) surface with pyridine has enabled more information about the population and strength distribution of active sites of this solid catalyst. For example, the adsorption of pyridine performed at high temperature (\(300\,^{\circ }\mathrm{C}\)) enabled to reveal the existence of some very strong active sites, which population is low.
This presentation of active sites characterisation by TPD has been started with the statement that the applied strategy in investigation of active sites’ characteristics is the adsorption of appropriate gas phase probe, under the specific experimental conditions. Evidently, the first criterion that has to be applied in order to choose the gas-phase probe is its acidity/basicity. Through this text, the importance of gas-phase nature, the size of its molecules and the temperature of adsorption, has been considered. At this place, the importance of the nature of gas-phase probe will be underlined. Figure 4.9 presents the example in which the investigation of same solids has been performed using two different probes: HY and dealuminated HY zeolites have been investigated using ammonia and pyridine, respectively. It is possible to see from Fig. 4.9a that the adsorption of ammonia revealed the existence of three or four different types of acid centres (in the case of HY and dealuminated HY zeolite, respectively). However, the same solids possess only two different types of sites for pyridine adsorption, Fig. 4.9b. Apparently, pyridine molecule could not reach all active sites in microporous zeolitic structure. Evidently, not only the basicity of gas-phase molecule is important for its application as a probe, the diameter of its molecule seems to be decisive in the example presented in Fig. 4.9.
It should be emphasized that the choice of a probe molecule should be done by taking into account all relevant parameters, and having in mind the features of solid material at which surface this probe should be adsorbed. In fact, the solid surface and the gas which is chosen as a probe for the characterization of its active sites should be considered as a pair. Very often, the separate adsorption-desorption experiments of more then one gas-phase probe is necessary in order to obtain reliable information concerning all active sites for particular solid material. The adsorption-desorption of more than one probe molecule should complete the picture about the catalysts’ active sites, particularly in the case of complex systems, where different types of active sites and energetic heterogeneity could be expected.
It is worth noting that the improvement of equipment available for temperature-programmed desorption, made from the first experiments in the field until nowadays, has enabled the application of many gases as probes. In fact, the possibility to perform adsorption at low temperatures (even sub-ambient) and the improvements in the detection systems allowed to introduce the gases which molecules are poorly polar (such as CO), or even non-polar (such are inert gases and saturated hydrocarbons). The interactions of these probes with the active sites are often realised through dispersion forces. Applied as probes, these kinds of molecules enable the recognition and “titration” of very weak active sites. Being on the level of dispersion forces, the interactions of inert gases with the active sites can be considered as specific, in some cases. In Fig. 4.10, the application of argon as probe gas is shown.
The application of same probe for the characterization of different solids revealed important differences in the strength and strength distributions of acid sites. It can be seen from the results presented in Fig. 4.10 that \(T_{max}\) positions and the shapes of desorption profiles differ for all investigated systems; while the strongest sites and the most pronounced heterogeneity are found for two specific solids (mordenite-type zeolite and Cs-salt of heteropolyacid). It is important to note that if some less specific probe would be used (such are ammonia or pyridine) all investigated solids should express significant acidity and heterogeneity. In that case, the differences among these catalysts would not be noticed.

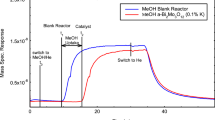
TPD profiles of different saturated hydrocarbons obtained from single-crystal Rh (111) surface [34]
The adsorption of non-polar gases offers the possibility to find an appropriate probe which would allow desorption in the desired range of temperature. Figure 4.11 presents one example of hydrocarbons’ application as probe gases. Single crystals possess energetically homogeneous surface for adsorption-desorption of these molecules. Figure 4.11 shows that, in the case of adsorption on Rh(111), progressively higher peak desorption temperature is noticed with increasing molecular weight of adsorbate (saturated hydrocarbon).
At the end of this section, it can be concluded that in the domain of active sites characterisation, there is a large body of methods and techniques that are developed for the determination of nature, population, strength and strength distribution of active sites. Among them, temperature-programmed desorption, a well-established and simple technique, continues to be a very useful and often applied in active sites characterisation. It is important to keep in mind that, although TPD of appropriate probe can be successfully used for distinction between the sites of different strength, it does not enable to distinguish their nature. From the interaction with the probe gas it can be concluded whether some active sites are acidic or basic; but, it can not be concluded if they belong to Brönsted or Lewis type. In order to get precise data on the active sites’ nature, the application of other techniques is needed.
Despite the evident richness of data derived from TPD experiment, there are several significant limitations of this technique. A short summary of data that can be obtained from TPD experiments and the limitations of technique are given in Table 4.2.
5.2 The Application of TPD in the Determination of Kinetic and Thermodynamic Parameters of Desorption Processes
Temperature-programmed desorption is by far the most often used technique for determination of kinetic parameters on both model and real systems. From these experiments, kinetic and thermodynamic information can be extracted under the conditions of variable temperature. In the following section, the procedures of evaluation of these important parameters will be presented.
Most often, the estimation of kinetic parameters is based on the assumption of independent desorption that takes part from different active sites as the first order event. In the interpretation of data obtained from TPD experiments, it is also assumed that desorption is the sole surface event. However, in reality the readsorption and diffusion of probe molecules take part, as events consecutive to desorption. These effects are particularly prominent in the case of microporous solids, and in the experiments performed in the flow systems. In those cases, the results obtained in TPD experiment can be often misinterpreted. The readsorption and diffusion can be avoided by adjusting some important experimental parameters. First of all, high heating rate would not favour these processes. However, the choice of very high heating rate is not recommended, particularly in modern implementations of TPD; where the investigation of solid materials used in real catalytic systems is the most common application.
If readsorption is important part of surface event, its influence on temperature-programmed desorption is that TPD profile broadens towards higher desorption temperatures; the same stands for diffusion limitations. Therefore, the task to neglect or minimize the effects of these processes is imposed; and it is relatively easy if TPD experiments are performed in UHV setups. The pumping speed should be sufficiently high to prevent readsorption of the desorbed species back onto the surface. However, in the case of flow systems there are many experimental conditions that have to be adjusted; even though, the interpretation of data obtained from TPD experiment is not simple—in the estimation of kinetic parameters, those experimental conditions have to be taken into consideration. Therefore, the derivation of kinetic and thermodynamic parameters from the results obtained in UHV and in the flow system will be discussed separately.
5.2.1 The Interpretation of Data Obtained in Ultra-High Vacuum Systems
If the pumping speed is high enough, readsorption may be ignored and the rate of desorption, defined as the change in adsorbate coverage per unit of time, is given by Eq. (4.7). In a TPD experiment temperature (T) is usually increased linearly with time from some initial temperature \(T_{0}\), with the heating rate \(\beta \):
where all symbols have the same meaning as previously stated (see Sect. 4.2.2).
The intensity of the desorption signal, I(T), which is proportional to the rate at which the surface concentration of adsorbed species is decreasing, i.e. to the rate of desorption, can be expressed by combining Eqs. (4.7) and (4.13):
Molecular adsorption and desorption are often the first order events (x \(=\) 1). The maximum desorption signal will occur when the first derivative of signal intensity with temperature equals 0 (dI/dT \(=\) 0):
Since surface coverage changes with temperature, i.e. \(\theta \) \(=\) f(T), this derivative is:
The derivative of surface coverage with temperature (\(d\theta /dT\)) can be substituted from Eq. (4.14). In that way, Eq. (4.16) is transformed to:
The solution of Eq. (4.17) can be obtained by setting the expression in square brackets to be equal to zero; from where the relation between the temperature at which the desorption maximum (\(T_{max})\) appears and \(E^{a}_{des}\) is obtained as:
These equations give the relations between the temperature \(T_{max }\) at which the desorption maximum appears and the activation energy for desorption. Hence, a simple approach to obtain the value \(E_{des}^a \) should be to analyze the TPD curve in order to get easily accessible parameter such as the temperature \(T_{max}\). Unfortunately, the differential equation in (4.7) and (4.14) can not be solved analytically, so the value \(E_{des}^a \) can not be obtained simply by substituting \(T_{max}\) value in Eq. (4.18). Therefore, the derivation of kinetic parameters can be rather complicated task, in particular because the kinetic parameters usually depend on surface coverage. However, we can note that several facts can be stated from each temperature-programmed experiment:
-
as the activation energy for desorption increases peak temperature \(T_{max}\) increases;
-
the peak temperature is not dependent upon, and consequently, does not change with the initial coverage, \(\theta _{t=0}\);
-
after the desorption maximum, the shape of the desorption peak tend to be asymmetric, with the signal which decreases rapidly.
Consequently, the values of \(T_{max}\) are evident from the experimental result. The procedure that can be applied to derive kinetic parameters is to solve Eq. (4.18) iteratively, applying a suitable choice for pre-exponential factor \(\nu (\theta )\) (for chemisorption this value is typically 10\(^{13 }\) s\(^{-1})\). The procedure is to read \({ T}_{max}\) from the measurement, to insert an estimated value for \(E_{des}^a \) in the right-hand side of Eq. (4.18) and to calculate the resulting \(E_{des}^a \) value. The obtained value has to be fed back into Eq. (4.18) to yield an improved value. The iterations should be done until the difference between two subsequent iterations becomes negligible [5].
In the case of second-order desorption, a similar, although more complicated expression exists for second-order desorption kinetics. In this case, the maximum desorption signal will occur when the second derivative of surface coverage is equal to zero:
From Eq. (4.19) the relation analogue to (4.18) can be derived:
Again, iterations are necessary in order to estimate the desorption energy. The insight in the Eq. (4.20) reveals evidence that \(\theta \) has to be known (or estimated) at the point where \(T_{max}\) is reached.
Apart from this approach which implies the evidence of \(T_{max}\), there is another which includes the value of peak width in the analysis. Also, many authors rely on the application of other, even more simplified methods that enable the calculation of kinetic parameters. Particularly popular among surface scientists are the Redhead’s and Kissinger’s methods.
From all previously stated, it can be inferred that the starting point for extraction of kinetic parameters from thermodesorpion profiles is desorption rate equation proposed by Polanyi and Wigner (Eqs. (4.7) and (4.14)) [6]. However, it has to be kept in mind that the term \(\theta ^{n}\) is just one particular case of one general function f(\(\alpha )\), where \(\alpha \) denotes the reacted (desorbed) fraction (the degree of surface event) and f(\(\alpha )\) is the reaction kinetic model. Therefore, generalized form of Eq. (4.14) can be written as:
The methods that are derived for the calculation of kinetic parameters from TPD profiles can bee divided in two big groups, shortly presented by following text.
-
(i)
Integral methods are based on the temperature of desorption rate maximum (\(T_{max})\) and/or peak half-widths. These methods assume that pre-exponential factor, reaction order and activation energy are coverage independent values. The most known is Redhead method [1], where Eq. (4.7) is solved in order to find the temperature at which desorption rate expresses its maximum. For the first-order desorption (x \(=\) 1), the relation between the temperature of peak maximum (\(T_{max})\), activation energy, heating rate and pre-exponential factor is:
$$\begin{aligned} \frac{E}{RT_{\max }^2 }=\frac{\nu _1 }{\beta }\exp \left( {-\frac{E_{des}^a }{RT_{\max } }} \right) \end{aligned}$$(4.22)where \(\nu _{1}\) is pre-exponential factor for the first-order desorption. The relation between activation energy and \(T_{max}\) is almost linear; therefore for \(\nu /\beta \) values which are between 10\(^{8}\) and 10\(^{13}\ {^{\circ }}\), Eq. (4.22) can be written as:
$$\begin{aligned} \frac{E}{RT_{\max }^2 }=\ln \frac{\nu _1 T_{\max } }{\beta }-3.46 \end{aligned}$$(4.23)The activation energy can be determined by varying heating rate \(\beta \) and plotting ln(\(T_{max})\) values against \(ln\beta \), without assuming the value of rate constant. For the second-order desorption (x \(=\) 2), the relation analogue to (4.23) is:
$$\begin{aligned} \frac{E_{des}^a }{RT_{\max }^2 }=\frac{2\theta _{\max } \nu _2 }{\beta }\exp \left( {-\frac{E_{des}^a }{RT_{\max } }} \right) \end{aligned}$$(4.24)where \(\nu _{2}\) is pre-exponential factor for second-order desorption, \(\theta _{max}\) is the adsorbate coverage at T \(=\) \(T_{max}\), and it is assumed that \(\theta _{max}=\theta _{0}\)/2, with \(\theta _{0}\) being the initial surface coverage. Equation is approximately correct for the first-order desorption and for values of \(\nu /\beta \) between 10\(^{8}\) and 10\(^{13}~{^{\circ }}\). It is very often applied to determine \(E_{des}\) from a single TPD spectrum.
-
(ii)
Differential methods are based on the assumption that at the temperature of maximum, the second derivative of desorption rate (Eqs. (4.7), (4.14) or (4.21)) is equal to zero. The most known is Kissinger method [40]. If reaction rate is expressed by (4.21), the second derivative is:
$$\begin{aligned} \frac{d^{2}\alpha }{dt^{2}}=\left( {\frac{E_{des}^a \beta }{RT_{\max }^2 }+\nu f^{{\prime }}\left( {\alpha _{\max } } \right) \exp \left( {-\frac{E_{des}^a }{RT_{\max }}} \right) } \right) \left( {\frac{d\alpha }{dt}} \right) _{\max } =0 \end{aligned}$$(4.25)where \(\alpha _{max}\) and (d\(\alpha /dt)_{max}\) are reacted fraction and reaction rate at the maximum; while heating rate \(\beta \) should be constant. From (4.25) it follows:
$$\begin{aligned} \frac{E\beta }{RT_{\max }^2 }=-\nu f^{{\prime }}\left( {\alpha _{\max } } \right) \exp \left( {-\frac{E_{des}^a }{RT_{\max } }} \right) \end{aligned}$$(4.26)Equation (4.26) can be rearranged after taking logarithms:
$$\begin{aligned} \ln \left( {\frac{\beta }{T_{\max }^2 }} \right) =\left( {-\frac{\nu R}{E_{des}^a }f^{{\prime }}\left( {\alpha _{\max } } \right) } \right) -\frac{E_{des}^a }{RT_{\max } } \end{aligned}$$(4.27)Evidently, for the first order reaction, f’(\(\alpha ) = -1,\) and (4.27) becomes:
$$\begin{aligned} \ln \left( {\frac{\beta }{T_{\max }^2 }} \right) =\ln \frac{\nu R}{E_{des}^a }-\frac{E_{des}^a }{RT_{\max }} \end{aligned}$$(4.28)
The procedure of extracting the activation energy for desorption is to analyse a set of TPD profiles measured with different constant heating rates \(\beta \), and to plot graphs of left hand side of (4.28) versus \(1/T_{max}\), what should lead to a straight line whose slope gives the activation energy, independently of the value of reacted fraction, \(\alpha _{max}\), at this point.
Evidently, for application of either integral or differential methods, the values of \(T_{max}\) have to be detectable. In case of poorly resolved TP profiles, their application would not provide reliable kinetic parameters. In those cases, either deconvolution of complex desorption profiles should be performed or desorption would be done under different experimental conditions, so the resolving of simple desorption profiles becomes possible.
5.2.2 The Interpretation of Data Obtained in the Flow Systems
Previously presented procedures for evaluation of kinetic parameters would give reliable values only if desorption is a lone surface process that takes part as a result of temperature increase. In the case of UHV systems, where samples are usually spread in a thin layer, diffusion takes part in a very limited extent; while readsorption can be avoided using sufficiently high pumping speed. However, if experiment is organized in a flow of a gas, in one usual physical situation, desorption and readsorption are occurring simultaneously with diffusion. In these systems, the construction of sample holders (Fig. 4.2b) does not allow neglecting mass transfer and readsorption limitations. These effects are particularly significant in the case of porous samples.
Therefore, the interpretation of results obtained in the systems designed to allow temperature-programmed experiments in the flow of gas requires consideration of all parameters that may induce mass transfer and readsorption limitations. The parameters that have to be considered are related to the gas (carrier or adsorbate), the geometry of furnace and sample holder, and the features of the sample.
The nature of both carrier and probe gas should be important, as well as their purities and flow rate. High gas flow can provoke desorption of weakly bound species. High amount of desorbed species that arrive in carrier gas can change its purity, so the sensitivity of experiment can be reduced. The consequences of low flow are: diffusion and readsorption effects become more probable, the time between desorption and detection is longer. In addition, appropriate gas flow has to be chosen to avoid concentration gradients within the catalyst particles and along the length of the bed. Hence, a compromise between low and high flow must be found.
The characteristics of furnace and sample holder that may influence the desorption profile are bed length, diameter and porosity, while the characteristics of the sample that could be important are its weight, particle size radius, sample density, particle porosity and number of active sites. In order to avoid temperature gradients, the reactor can not be of big size; hence, mass of the sample is limited by its size and geometry. Diameters of sample particles are another important factor - small particles decrease the possibility of intra-granular diffusion.
Some additional parameters such are temperature range, heating rate, the temperature detection and monitoring, and distance between sample holder and detector may influence the shape of temperature-programmed profile. Distance between reactor and detector has to be the smallest possible, so the answer of detector is instantaneous. Thermocouple has to be precise enough to enable the time of furnace response appropriate, so the temperature rise is absolutely linear. In addition, it has to be kept in mind that temperature of desorbed gas (which has to be analysed) can change during the experiment, what can cause a so-called “apparent” concentration. This effect can be minimized if high flow of gas, low mass of the sample and the equipment situated in the constant temperature area, are employed.
Evidently, in order to calculate reliable kinetic parameters, the temperature of peak maximum \(T_{max}\) has to be exactly the temperature at which the rate of desorption is maximal. However, additional surface events (diffusion and readsorption) can influence the TPD profile. The coupling of readsorption and mass transfer effects with desorption can shift the peak of TPD curve significantly.
Therefore, it is necessary to select suitable operating conditions that enable to avoid effects that could have influence on temperature-programmed profile. The ways how to find experimental conditions required to obtain reliable activation energies have been discussed in the literature. The recommendations that help to find appropriate sets of experimental parameters for experiments of temperature-programmed desorption [41–43] or other tempetrature-programmed techniques can be found [14, 44]. Once limitations that arrive from diffusion and readsorption are minimized, simplified procedures can be applied to evaluate kinetic parameters.
In the case of desorption which takes part in the flow of gas, material balance can be obtained from the assumption of equilibrium which is reached in a time t, between the adsorbed and the gas-phase molecules. In the absence of diffusion, this equilibrium can be expressed by following relations:
where \(C_{sm}\) is concentration of adsorbed molecules (in mol kg\(^{-1}\), for \(\theta \) \(=\) 1) and \(C_{g}\) is concentration of adsorbate molecules in the gas phase (in mol dm\(^{-3})\); while \(k_{des}\) and \(k_{ads}\) are rate constants of desorption and adsorption. If temperature increase is linear, with constant heating rate (\(\beta \)), previous relation is transformed:
If the gas flow (F, dm\(^{3}\mathrm{s}^{-1})\) is constant in time and the mass of adsorbent is known (W, kg), the same equilibrium can be expressed as:
From Eq. (4.31), the concentration of adsorbate molecules in the gas phase can be obtained from:
The same value can be related with the rate of desorption (\(d\theta /dT\)) through the equation obtained from (4.30) and (4.31):
Readsorption is negligible if the gas flow is high enough (\(F>> { Wk}_{\mathrm{ads}} (1-\theta )\)). In that case the concentration of adsorbate molecules in the gas phase can be obtained from:
At the temperature of peak maximum (\(T_{max})\), the concentration of adsorbate molecules in gas phase reaches its maximum, so \(dC_{g}/dT=0\). Since, the rate of desorption is expressed by Eq. (4.7), for the first order desorption which takes part without readsorption and diffusion limitations, the value of rate constant reached at \(T_{max},(k_{des})_{m}\), is related with \(T_{max}\) value as:
The relation between \(T_{max }\) and activation energy for desorption is given by equation identical to (4.27):
It can be seen that for constant heating rate \(\beta \), the value \(T_{max}\) is independent on surface coverage \(\theta \). The activation energy for desorption can be obtained from the slope of a plot: \(\ln \left( {\frac{T_m^2 }{\beta }} \right) =f\left( {1/T_{\max } } \right) \).
If a flow of carrier gas is not high enough (\(F << {Wk}_{ads}(1-\theta )\)) Eq. (4.32) is transformed to:
where \(K=\exp \left( {\frac{\Delta S_{des} }{R}} \right) \cdot \left( {-\frac{\Delta H_{des}}{RT}} \right) \). Again, having in mind that at the temperature of peak maximum \(dC_{g}/dT\) is equal to zero, the relation between \(T_{max}\) and the heat of adsorption can be obtained:
Evidently, the heat of adsorption can be obtained from the slope of the same plot as previously. Similarly, it can be shown that for desorption which takes part as a second-order surface event, the activation energy for desorption (in the case of negligible readsorption) or the heat of adsorption (in the case of significant readsorption) could be obtained from the slope of the same plot. Hopefully, even in the cases when desorption is significantly affected by side effects such are readsorption or diffusion, kinetic parameters can be obtained using relatively simple procedures.
After everything that has been said about temperature-programmed methods, few examples will be considered further in the text. The two experimental techniques most commonly used for the study of acid/base properties of porous solid materials are temperature-programmed desorption (TPD) and adsorption calorimetry. Application of these techniques for characterisation of several different classes of materials will be presented as well as comparison of data obtained by both techniques.
6 The Examples of TPD Application; the Comparison with Data Obtained by Adsorption Calorimetry
As discussed in details previously, in TPD experiments, temperature increases linearly and the concentration of desorbed gas is recorded as a function of temperature, whereas calorimetry involves the adsorption of gases onto the sample’s surface while it is kept at a constant temperature and a heat-flow detector emits a signal proportional to the amount of heat transferred per unit time. The peak maxima temperatures in the TPD spectra are influenced by the active site strength, the number of active sites, the structure of investigated material and the heating rate [45, 46]. In particular, adsorption microcalorimetry gives access to the number, strength, and strength distribution of the acid sites in a single experiment [21]. This information is of outmost importance for design of catalysts with high activity and selectivity.
Every micro or mesoporous material can be investigated by these two techniques. Among many, few examples will be presented for the most often investigated catalysts; such are zeolite, oxides and metals.
6.1 Zeolites
Zeolites are known to be important catalysts for a number of industrially important reactions. A question of basic interest, which provides opportunity for development of catalyst with suitable and tailored characteristics, is to determine the correlation between number, strength and strength distribution of active sites and the promotion of catalytic activity. Therefore, the investigation of acid sites, both Lewis and Brönsted type, is very important subject. Properties of zeolites as catalysts will depend on many factors: the adsorption or desorption temperature of the probe, pretreatment of the sample, proton exchange level, influence of coking as well as Si/Al ratio and dealumination and influence of exchanged cations [47].
6.1.1 Influence of the Si/Al Ratio and Dealumination
The Si/Al ratio plays a significant role, since the aluminum atom is directly related to the acidic site. Dealumination processes can promote porous structure modifications, which may improve some interesting properties of zeolites, like thermal and hydrothermal stability, acidity, catalytic activity, resistance to aging and low coking rate, and matter transfer. However, a severe dealumination may also cause a loss of crystallinity [47].
Different dealumination processes have been proposed, namely steaming and acid treatments, as well as reactions with SiCl\(_{4}\) or SiF\(_{6}^{2- }\) [47]. The removal of aluminum from zeolite crystals leads to products with high framework Si/Al ratios. Some of the aluminum atoms are released from the framework and form non-framework aluminum-containing species. The non-framework aluminum species can be eliminated by treatment with diluted hydrochloric acid. Dealumination generally brings a decrease in the acid site concentration. However, the extent of the indicated decrease varies with the kind of base probe, and a significant change was observed by Mitani et al. [48] in the ratio of acid site concentrations when titrated with pyridine instead of ammonia. An important increase of the initial heat values and of the site strength heterogeneity was observed for samples presenting many extra-framework aluminum species. Samples subjected to a moderate dealumination and nearly total removal of the extra-framework aluminum displayed a homogeneous acid strength [49].

Influence of dealumination on \(\mathrm{NH}_{3}\)-\(\mathrm{TPD}\) profiles of HY zeolites [14]
The effect of dealumination on NH\(_{3}\)-TPD profiles of HY zeolites is shown in Fig. 4.12 [14]. Evidently l-peak became smaller upon dealumination, while the h-peak increased up to a maximum for Si/Al \(=\) 15 and then decreased upon further dealumination.

NH3-TPD profiles observed on H-MFI with Si/Al ratios: (a) 11.9 (b) 12.5 (c) 19 (d) 38 (e) 50 [35]

Differential heats of ammonia adsorption at 393 K on H-ZSM-5 zeolites with Si/Al ratios: (\(\bullet \)) 14 \((\circ )\) 25 (\(\Delta \)) 37.5 (\(\square \)) 50 (\(\blacksquare \)) 75 [36]

Differential heat of adsorption versus \(\mathrm{{NH}}_{3}\) uptake on HZSM-5 and ion exchanged ZSM-5 zeolites [37]

\(\mathrm{NH}_{3}-\mathrm{TPD}\) profiles for HZSM-5 and ion exchanged ZSM-5 zeolites [37]
Figure 4.13 shows the effect of varying the Si/Al ratio of a MFI sample on its TPD profile. As shown at the figure, both the l- and h-peaks became smaller upon dealumination. A curve-fitting analysis led to the determination of average adsorption heats that were almost constant for all investigated MFI samples, ca. 130 kJ mol\(^{-1}\) [35].
The acidity of H-ZSM-5 zeolites synthesized with different Al contents has been characterized by microcalorimetric measurements of the differential heats of adsorption of ammonia [50]. The strength of the strongest acid sites increased with the Al content to a maximum for Si/Al \(=\) 17.5 and then decreased notably. The total acidity increased regularly with Al content. The importance of selecting appropriate Si/Al ratios for specific catalytic applications is therefore obvious.
Figure 4.14 represents the differential heats of adsorption on dealuminated H-ZSM-5 zeolite samples at 393 K versus the adsorbed amount of ammonia [36]. It appears that the adsorption heat decreases gradually with the amount of adsorbed ammonia and exhibits various steps, attributed to populations of sites of different strengths. A heterogeneous distribution of sites is clearly evidenced by the shape of the curves and can be attributed to the presence of extra-framework aluminium spices. The samples adsorb ammonia with initial differential heats that vary between 160 and 177 kJ mol\(^{-1}\). Karge et al. [51] have reported that, in first approximation, the sites evolving Q\(_{diff} > 150\,\mathrm{kJ}\,\mathrm{mol}^{-1}\) can be assigned to strong Lewis sites, while the sites with Q\(_{diff.} = 150-100\,\mathrm{kJ}\,\mathrm{mol}^{-1}\) typically correspond to strong Brönsted acid sites. This assumption is supported by a comparison of the numbers of acidic sites obtained from infrared and microcalorimetric measurements. The initial heat of adsorption for the zeolite with Si/ Al \(=\) 75 is lower than that of the other zeolites except for Si/Al \(=\) 14, and the decrease in heat with coverage is steeper. The heat evolved falls abruptly from an initial value of 150 kJ mol\(^{-1}\) to 70 kJ mol\(^{-1}\) at around 50 % coverage.
6.1.2 Influence of Substitution by Other Cations
The nature of the exchanged cation is one of the key points that determine acidity in zeolites. It is very important to use an acidic probe able to distinguish the alkali cations from the basic sites.
Differential heats of ammonia adsorption and NH\(_{3}\)-TPD profiles of HZSM-5 zeolite as well as FeZSM-5, Cu-ZSM-5 and MnZSM-5 zeolites are presented in Figs. 4.15 and 4.16. As can be seen in Fig. 4.15, the overall acidity of investigated samples was not significantly modified by ion-exchange procedure. However, changes in the Q\(_{diff}\) versus NH\(_{3}\) uptake profiles, particularly in their middle parts (140 – 65 kJ mol\(^{-1})\), indicate that the distribution of strength of acid sites was affected by ion exchange. Ion exchange with Cu and Mn resulted in enhanced heterogeneity of the acid site strength, as confirmed by the NH\(_{3}\)-TPD profiles, which present poorly separated desorption peaks and even a single very broad peak for sample MnZSM-5 (Fig. 4.16) [37].
This example clearly shows that TPD and adsorption microcalorimetry are complementary techniques, and combination of these two techniques provides very good characterisation of active sites number, strength and distribution on particular solid material.
6.2 Metal Oxides
Metal oxides, either bulk, doped, supported or mixed, are widely used as catalysts in chemical industry. Catalytic behavior of these materials, in terms of activity and selectivity, is related to their acid/base properties.
Metal oxide surfaces react with gases or solutions; they can be used as active phases or as supports for catalysts. The behavior of metal oxide surfaces is controlled by: (i) coordination—sites of low coordination are in general more reactive than sites of high coordination; (ii) acid/base properties—clean and anhydrous metal oxide surfaces present two different types of active sites, cations and anions (acid/base pairs) which determine reactivity towards gas-phase adsorbates; (iii) the redox mechanism—when the oxide deviates from the stoichiometry due to the presence of defects such as vacancies or adatoms, the oxidation state of surface atoms varies [52].
Molecular and dissociative adsorption can be understood as acid/base processes. Molecules adsorbing without dissociation always bind to one or several metal cations. Ammonia and pyridine are the most commonly used probes for determining the acid site strength of oxides.
In the case of supported metal oxide catalysts, the role of the support is to disperse the active phase and to create new active surface species by host (active phase)– guest (support) interaction. The dispersion of the active phase plays a fundamental role, and very often a maximum of strength of the active sites is observed when the monolayer coverage is reached. The pure oxides, such as Al\(_{2}\)O\(_{3}\), ZrO\(_{2}\), TiO\(_{2}\) (the most frequently used catalyst supports) carry both basic and acidic Lewis sites on their surface; depending on the probe molecules used (CO\(_{2}\) or NH\(_{3})\), they can exhibit either acidic or basic character. Excess negative or positive charges can be induced, and therefore acidity (Brönsted or Lewis) or basicity can be generated by mixing oxides. Modifying the surface with a minor anionic, cationic or metallic component enhances or decreases the acidic or basic strength of the sites. For example, the incorporation of chloride, fluoride or sulfate ions increases the acidity of carrier oxides (Al\(_{2}\)O\(_{3}\), ZrO\(_{2}\), TiO\(_{2})\), while alkali cations enhance the basic strength of alumina or silica [53].
Silica-aluminas (amorphous aluminosilicates) are widely used as catalyst supports due to their high acidity and surface area. The behavior of silica-alumina surfaces is similar to that of zeolites concerning the initial differential heats of ammonia and pyridine, but the total number of acidic sites varies with the preparation method and the Si/Al ratio.
Auroux et al. [38] have studied acid properties of silica, alumina and multilayers of silica on alumina (SA) and alumina on silica (AS), obtained by grafting. The surface acidity of the pure oxides and samples obtained by grafting, SA and AS, of both Lewis and Brönsted type, has been investigated by TPD and microcalorimetry, using pyridine as probe molecule.

TPD curve for \(\gamma \)-alumina, using pyridine as probe molecule [38]
Figure 4.17 shows the TPD curve of pure alumina; a semi-quantitative estimation of the number of acid site can be derived from the TPD plots such as this one. Three classes of acid sites have been considered, depending on the desorption temperature: weak, (T\(_{D} <\) 523 K), medium (523 \(<\) T\(_{D} <\) 673 K) and strong, (T\(_{D}>\) 673 K) acid sites.
Calorimetric results are presented in Fig. 4.18 where the differential heat of adsorption (\(Q_{\mathrm{diff}}\)) of pyridine is plotted versus coverage. From the calorimetric data, the number of sites and their distribution, according to the adsorption energies, can be determined. Analyzing calorimetric profile of differential heats vs. surface coverage, following sites active for adsorption can be distinguished: weak acid sites, (\(90\le Q_{\mathrm{diff}} {<}120 kJ mol^{-1})\), medium acid sites, m (\(120\le Q_{diff} {<} 150 kJ mol^{-1})\) and strong acid sites, s (\(Q_{\mathrm{diff}} \ge 150 kJ mol^{-1})\).

Differential heats of pyridine adsorption as function of the coverage degree [38]
From Fig. 4.18 it can be seen that calorimetric curve for pure alumina shows three type of sites with different strength, in accordance with TPD result. Also, it can be seen that the grafted mixed oxides SA and AS have acidic properties different from those of the pure alumina and silica supports used as starting materials, i.e. properties of catalysts depend on preparation procedure.
It can be noticed that some differences exist between results obtained from calorimetric measurements and those revealed from TPD results, with regard to the sites strength distribution: the percent of medium acid sites, derived from TPD results is always remarkably higher than one found using microcalomitery. This is due to the fact that strong acid sites having Q\(_{diff} > 180\) kJ mol\(^{-1}\) might not completely release the organic base at 693 K and therefore are not disclosed by the TPD technique. This implies importance of knowing limitations of techniques used for characterizations as well as operative conditions adopted during experiments [38].
6.3 Metals
TPD and microcalorimetric methods provide an effective means for measuring the strengths of adsorbate-surface interactions, not only on clean metal surfaces, but also on metal surfaces that have been exposed to reaction conditions. Many recent studies use H\(_{2}\), CO, O\(_{2}\) and hydrocarbons as probe molecules since they are involved in numerous commercial catalytic processes.
Bimetallic catalysts have been the subject of great interest for a long time because of their exceptional properties compared to the monometallic catalysts, yet the reason behind their improved activity is still a question of debate and they are subject of many recent studies. Tanskale et al. [39] have studied the promoting effect of Pt and Pd in bimetallic Ni–Pt and Ni–Pd catalysts supported on alumina nanofibre (Alnf) for the liquid phase reforming of sorbitol to produce hydrogen. Fig. 4.19 shows TPD profiles for CO desorption for several monometallic and bimetallic catalysts dispersed on alumina nanofibre.

Results from CO-TPD of monometallic and bimetallic catalysts over alumina nano-fibre support; rate of CO desorption. Pt/Alnf \((\blacktriangledown )\), Ni/Alnf (\(\bullet \)), Pd/Alnf (\(\blacktriangle \)), Ni/Pd/Alnf (\(\square \)), Ni-Pd/Alnf(\(\lozenge \)) [39]

Differential heats of CO adsorption as a function of coverage, adsorption temperature \(3\,^{\circ }\mathrm{C}\). Pt/Alnf (\(\blacktriangledown \)), Ni/Alnf (\(\bullet \)), Pd/Alnf (\(\blacktriangle \)), Ni/Pd/Alnf (\(\square \)), Ni-Pd/Alnf (\(\lozenge \)) [39]
Results obtained by temperature-programmed desorption suggested that in the case of bimetallic catalysts there was a reduction in the number of strong CO-adsorption sites. This finding allows conclusion that the alloying effect of these systems leads to the lowering of the CO heat of adsorption. This finding was confirmed by direct measurement of differential heat of CO chemisorption in the microcalorimetry experiment (see Fig. 4.20).
The differential heat of adsorption for Ni–Pt/Alnf was reduced to 111.28 kJ mol\(^{-1}\), which was 11.45 and 5.51 kJ mol\(^{-1}\) lower than Pt/Alnf and Ni/Alnf, respectively. A similar result was obtained for the Ni–Pd bimetallic catalyst. This was interesting because the atomic ratio of Pt:Ni and Pd:Ni was only 1:33 and 1:18, hence even small amount of surface concentration of solute in the alloy catalysts is sufficient to significantly alter the properties of constituent monometallic catalysts.
The differential heat profiles are characterised by a plateau of nearly constant heat of adsorption at low coverage (15–20 \(\mu \)mol g \(\mathrm{m}^{-1})\) followed by an abrupt decrease as the surface saturation limit is reached. In the low coverage plateau region the adsorption of CO can be considered to be strongly bonded and at high coverage the differential heat of CO adsorption represents an average heat from the various adsorption sites on the surface of a given catalyst particle. Therefore, one can compare only the initial heat of adsorption when the CO chemisorption may be considered to be equilibrated. The initial heat of CO adsorption on Pd/Alnf was found to be the highest (130.09 kJ mol\(^{-1})\) followed by Pt/Alnf (122.73 kJ mol\(^{-1})\) and Ni/ALnf (116.79 kJ mol\(^{-1})\). The initial heat of CO adsorption on Ni–Pt/Alnf was measured as 111.28 kJ mol\(^{-1}\), which is lower than both Pt/Alnf and Ni/Alnf. Similarly, the initial differential heat of Ni–Pd/Alnf (109.60 kJ mol\(^{-1})\) was lower than both the corresponding monometallic catalysts. This indicates that the addition of noble metals, even in small fractions, to the Ni catalyst has a significant but weaker promoting effect on the adsorption of CO. Finding that the CO differential heat of adsorption is lowered in the bimetallic catalysts is substantial because with reduction of the CO binding strength poisoning of the active metal sites can be avoided which will increase its catalytic activity [39].
7 Conclusion
This chapter has presented the fundamentals, the experimental setups and the applications of temperature-programmed desorption (TPD). TPD is widely utilized for characterization of active sites present on the surface of solid materials and determination of kinetic and thermodynamic parameters of desorption processes. It is a powerful technique, even if it does not provide direct information about molecular nature of adsorbed species. Given its relative simplicity and low cost, this technique will continue to find more applications in the future.
Thermal method which is complementary to TPD, adsorption calorimetry, provides tools necessary for measuring the energy of an adsorption system as a function of coverage, allowing precise determination of number of surface active sites as well as their relative populations and strengths. In general, adsorption calorimetry provides a much better description of the surface active site strength distribution than TPD. Combination of these two methods allows detailed characterization of various materials, in particular catalysts, and will surely find more application in different research areas such as hydrogen production and storage or environmental chemistry.
Notes
- 1.
Significant difference in heating rates makes main distinction between “flash desorption”, where the heating rate is very high (the desired temperature is reached in seconds) and temperature-programmed desorption (where the sample is heated in minutes or even hours).
- 2.
TPD is perhaps the most often used for estimation of acid/base properties of solid catalysts.
- 3.
Dissociative chemisorption of a diatomic molecule can also happen through the dissociation in a gas phase and a creation of two gas phase atoms; these two atomic species can be then adsorbed on the surface (this way is almost always non-activated). If the curves describing molecular and atomic adsorption intersect at or below the zero potential energy line, then the precursor physisorbed molecule can experience non-activated dissociation, followed by chemisorption (Fig. 4.1a). In contrast, if the energetic for these two pathways are such that the intersection occurs above the zero energy plane, then chemisorption will be activated with activation energy, E\(_{ad}\), as indicated in Fig. 4.1b.
- 4.
Instead to define equilibrium by constant surface coverage, it is possible to keep constant pressure at the surface; in that case the equilibrium heat of adsorption \(q_{eq}\) is incorporated in Clausius-Clapeyron equation.
- 5.
In those cases, the consumption of either reductive or oxidative gas by the catalyst is derived from the change in thermal conductivity of the gas mixture.
- 6.
If catharometer is used as detector, it is very important to remove traces of water or any other impurities from the gas flows, because they would affect the thermal conductivity measurements.
References
P.A. Redhead, Thermal desorption of gases. Vacuum 12, 203–211 (1962). doi:10.1016/0042-207X(62)90978-8
G. Ehrlich, Modern Methods in surface kinetics: flash desorption, field emission microscopy, and ultrahigh vacuum techniques. Adv. Catal. 14, 255–427 (1963). doi:10.1016/S0360-0564(08)60341-7
R.J. Cvetanović, Y. Amenomiya, Application of temperature-programmed dessorption technique to catalyst studies. Adv. Catal. 17, 103–149 (1967). doi:10.1016/S0360-0564(08)60686-0
R.J. Cvetanović, Y. Amenomiya, A temperature programmed desorption technique for investigation of practical catalysts. Catal. Rev. 6, 21–48 (1972). doi:10.1080/01614947208078690
I. Chorkendorff, J.W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics (Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003)
F. Rouquerol, J. Rouquerol, K. Sing, Adsorption by Powders and Porous Solids, Principles Methodology and Applications (Academic Press, San Diego, 1999)
D.M. Ruthven, Principles of Adsorption and Adsorption Processes (Wiley, New York, 1984)
M.A. Vannice, Kinetics of Catalytic Reactions (Springer Science Business Media, Inc., New York, 2005)
Lj. Damjanović, A. Auroux, Determination of acid/base properties by temperature-programmed desorption (TPD) and adsorption calorimetry, in Zeolite Chemistry and Catalysis: An Integrated Approach and Tutorial, ed. by A.W. Chester, E.G. Derouane (Springer, Heidelberg, 2009)
J.M. Kanervo, T.J. Keskitalo, R.I. Slioor, A.O.I. Krause, Temperature-programmed desorption as a tool to extract quantitative kinetic or energetic information for porous catalysts. J. Catal. 238, 382–393 (2006). doi:10.1016/j.jcat.2005.12.026
V. Rakić, V. Dondur, U. Mioč, D. Jovanović, Microcalorimetry in the identification and characterization of the most reactive active sites of heterogeneous catalysts. Top Catal. 19, 241–247 (2002). doi:10.1023/A:1015328526702
Dj. Stojaković, N. Rajić, V. Rakić, N. Zabukovec Logar, V. Kaučić, Structure and thermal behavior of the layered zincophosphate [\({\rm NH}_{3}{-}{\rm CH}_{2}{-}{\rm CH}({\rm NH}_{3}){-}{\rm CH}_{3}\)](\({\rm ZnPO}_{4})_{2}\). Inorg. Chim. Acta. 362, 1991–1995 (2009). doi:10.1016/j.ica.2008.09.020
B. Brunner, Solid state NMR–a powerful tool for the investigation of surface hydroxyl groups in zeolites and their interactions with adsorbed probe molecules. J. Mol. Struct. 355, 61–85 (1995). doi:10.1016/0022-2860(95)08867-U
D. Delahay, Méthodes en température programmée, in Les matériaux micro et mésoporeux, Charactérisation. ed. by F. Thibault-Starzyk, Groupe français des zéolithes, (EDP sciences, Les Ulis, France, 2004)
S. Bennici, A. Auroux, Thermal analysis and calorimetric methods, in Metal Oxide Catalysis, eds. by S.D. Jackson, J.S.J. Hargreaves (Wiley, New York, 2009)
G.A. Somorjai, Modern surface science and surface technologies: an introduction. Chem. Rev. 96, 1223–1236 (1996). doi:10.1021/cr950234e
W.E. Farneth, R.J. Gorte, Methods for analyzing zeolite acidity. Chem. Rev. 95, 615–635 (1995). doi:10.1021/cr00035a007
A. Corma, Inorganic solid acids and their use in acid-catalyzed hydrocarbon reactions. Chem. Rev. 95, 559–614 (1995). doi:10.1021/cr00035a006
J. Weitkamp, U. Weis, E. Ernst, New aspects and trends in zeolite catalysis, in Catalysis by Microporous Materials, Studies in Surface Science and Catalysis, vol. 94, eds. by H.K. Beyer, H.G. Karge, I. Kiricsi, J.B. Nagy (Elsevier, Amsterdam), p. 363
V. Solinas, I. Ferino, Microcalorimetric characterisation of acid-base catalysts. Catal. Today. 41, 179–189 (1998). doi:10.1016/S0920-5861(98)00048-0
A. Auroux, Microcalorimetry methods to study the acidity and reactivity of zeolites, pillared clays and mesoporous materials. Top Catal. 19, 205–213 (2002). doi:10.1023/A:1015367708955
A. Auroux, Innovation in zeolite materials science, in Proceedings of the International Sumposium on Studies in Surface Science and Catalysis, Nieuwpoort, vol. 37 (Elsevier, Amsterdam, 1988), 13–17 September 1987, eds. by P.J. Grobet, W.J. Mortier, E.F. Vansant, G.G Schulz-Eklo, p. 385
A. Zecchina, S. Bordiga, G. Spoto, L. Marchese, G. Pterini, G. Leofanti, M. Padovan, Silicalite characterization. 2. IR spectroscopy of the interaction of carbon monoxide with internal and external hydroxyl groups. J. Phys. Chem. 96, 4991–4997 (1992). doi:10.1021/j100191a048
M.A. Makarova, K.M. Al-Gefaili, J. Dwyer, Brönsted acid strength in US-Y: FTIR study of CO adsorption. J. Chem. Soc. Faraday Trans. 90, 383–386 (1994). doi:10.1039/FT9949000383
Y. Kuroda, T. Mori, Y. Yoshikawa, S. Kittaba, R. Kumashiro, M. Nagao, What are the important factors determining the state of copper ion on various supports? Analysis using spectroscopic methods and adsorption calorimetry. Phys. Chem. Chem. Phys. 1, 3807–3816 (1999). doi:10.1039/A904754I
E. Garrone, B. Fubini, B. Bonelli, B. Onida, C.O. Arean, Thermodynamics of CO adsorption on the zeolite Na-ZSM-5 A combined microcalorimetric and FTIR spectroscopic study. Phys. Chem. Chem. Phys. 1, 513–518 (1999). doi:10.1039/A806973E
J. Sauer, Acidic sites in heterogeneous catalysis: structure, properties and activity. J. Mol. Catal. 54, 312–323 (1989). doi:10.1016/0304-5102(89)80149-X
P.A. Jacobs, Carboniogenic Activity of Zeolites (Elsevier Scientific Publishing Company, Amsterdam, 1977)
E. Selli, L. Forni, Comparison between the surface acidity of solid catalysts determined by TPD and FTIR analysis of pre-adsorbed pyridine. Microporous Mesoporous Mater. 31, 129–140 (1999). doi:10.1016/S1387-1811(99)00063-3
J.M. Campelo, A. Garcia, D. Luna, J.M. Marinas, A.A. Romero, Characterization of acidity in AlPO\(_{4}\)-Al\(_{2}\)O\(_{3}\) (5–15 wt% Al\(_{2}\)O\(_{3})\) catalysts using pyridine temperature-programmed desorption. Thermochim. Acta. 265, 103–110 (1995). doi: 10.1016/0040-6031(95)02379-G
H.G. Karge, V. Dondur, J. Weitkamp, Investigation of the distribution of acidity strength in zeolites by temperature-programmed desorption of probe molecules. 2. Dealuminated Y-type zeolites. J. Phys. Chem. 95, 283–288 (1991). doi:10.1021/j100154a053
H. Matsuhashi, K. Arata, Temperature-programmed desorption of argon for evaluation of surface acidity of solid superacids. Chem. Commun. 387–388, (2000). doi:10.1039/A909844E
H.G. Karge, V. Dondur, Investigation of the distribution of acidity in zeolites by temperature-programmed desorption of probe molecules. I. Dealuminate mordenites. J. Phys. Chem. 94, 765–772 (1990). doi:10.1021/j100365a047
J. Wilson, H. Guo, R. Morales, E. Podgornov, I. Lee, F. Zaera, Kinetic measurements of hydrocarbon conversion reactions on model metal surfaces. Phys. Chem. Chem. Phys. 9, 3830–3852 (2007). doi:10.1039/B702652H
N. Katada, H. Igi, J.-H. Kim, M. Niwa, Determination of the acidic properties of zeolite by theoretical analysis of temperature-programmed desorption of ammonia based on adsorption equilibrium. J. Phys. Chem. B. 101, 5969–5977 (1997). doi:10.1021/jp9639152
S. Narayanan, A. Sultana, Q.T. Le, A. Auroux, A comparative and multitechnical approach to the acid character of templated and non-templated ZSM-5 zeolites. Appl. Catal A-Gen. 168, 373–384 (1998). doi:10.1016/S0926-860X(97)00368-2
V. Rac, V. Rakić, S. Gajinov, V. Dondur, A. Auroux, Room-temperature interaction of n-hexane with ZSM-5 zeolites. Microcalorimetric and temperature-programmed desorption studies. J. Therm. Anal. Cal. 84, 239–245 (2006). doi:10.1007/s10973-005-7164-z
A. Auroux, R. Monaci, E. Rombi, V. Solinas, A. Sorrentino, E. Santacesaria, Acid sites investigation of simple and mixed oxides by TPD and microcalorimetric techniques. Thermochim. Acta. 379, 227–231 (2001). doi:10.1016/S0040-6031(01)00620-7
A. Tanksale, J.N. Beltramini, J.A. Dumesic, G.Q. Lu, Effect of Pt and Pd promoter on Ni supported catalysts–A TPR/TPO/TPD and microcalorimetry study. J. Catal. 258, 366–377 (2008). doi:10.1016/j.jcat.2008.06.024
K. Kissinger, Reaction kinetics in differential thermal analysis. Anal. Chem. 29, 1702–1706 (1957). doi:10.1021/ac60131a045
R.J. Gorte, Design parameters for temperature programmed desorption from porous catalysts. J. Catal. 75, 164–174 (1982). doi:10.1016/0021-9517(82)90131-2
R.A. Demmin, R.J. Gorte, Design parameters for temperature-programmed desorption from a packed bed. J. Catal. 90, 32–39 (1984). doi:10.1016/0021-9517(84)90081-2
R.J. Gorte, Temperature-programmed desorption for the characterization of oxide catalysts. Catal. Today 28, 405–414 (1996). doi:10.1016/S0920-5861(96)00249-0
D.A.M. Monti, A. Baiker, Temperature-programmed reduction. Parametric sensitivity and estimation of kinetic parameters. J. Catal. 83, 323–335 (1983). doi:10.1016/0021-9517(83)90058-1
J. Shen, A. Auroux, The determination of acidity in fluid cracking catalysts (FCCs) from adsorption microcalorimetry of probe molecules, in Proceedings of International Symposium on Fluid Catalytic Cracking VI, Preparation and Characterization of Catalysts, New York, 7–11 September 2003, eds. by M. Occelli, p. 35. Studies in Surface Science and Catalysis, vol. 149, pp. 35–70 (2004). doi:10.1016/S0167-2991(04)80756-0
A. Corma, From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem. Rev. 97, 2373–2420 (1997). doi: 10.1021/cr960406n
A. Auroux, Acidity and basicity, in Molecular Sieves- Science and Technology, vol. 6. ( Springer, Berlin, 2008), p. 45
Y. Mitani, K. Tsutsumi, H. Takahashi, Direct measurement of the interaction energy between solids and gases. XI. Calorimetric measurements of acidities of aluminium deficient H-Y zeolites. Bull. Chem. Soc. Japan. 56, 1921–1923 (1983). doi:10.1246/bcsj.56.1921
A. Auroux, Y. Ben Taarit, Calorimetric investigation of the effect of dealumination on the acidity of zeolites. Thermochim. Acta. 122, 63–70 (1987). doi:10.1016/0040-6031(87)80105-3
A. Auroux, P.C. Gravelle, J.C. Védrine, M. Rekas, in Proceedings of the 5th International Conference on Zeolite, Naples, 2–6 June 1980, ed. by L.V.C. Rees, L.V. Heyden, p. 433
H.G. Karge, L.C. Jozefowicz, A comparative study of the acidity of various zeolites using the differential heats of ammonia adsorption as measured by high-vacuum microcalorimetry, in Proceedings of the 10th International Conference on Zeolites and Related Microporous Materials: State of the Art 1994, Garmisch-Partenkirchen, Elsevier, Amsterdam, 17–22 July 1994, eds. by J. Weitkamp, H.G. Karge, H. Pfeifer, Hö.W. lderich, p. 685 . Studies in Surface Science and Catalysis, vol. 84, pp. 685–692 (1994). doi:10.1016/S0167-2991(08)64174-9
M. Calatayud, A. Markovits, C. Minot, Electron-count control on adsorption upon reducible and irreducible clean metal-oxide surfaces. Catal. Today 89, 269–278 (2004). doi:10.1016/j.cattod.2003.12.015
Lj. Damjanović, A. Auroux, Heterogeneous catalysis on solids, in The Handbook of Thermal Analysis and Calorimetry. Further advances, techniques and applications, vol. 5, ed. by M. Brown, P. Gallagher (Elsevier, Amsterdam, 2008)
M. Niwa, N. Katada, Measurements of acidic property of zeolites by temperature programmed desorption of ammonia. Catal. Surv. Japan. 1, 215–226 (1997). doi:10.1023/A:1019033115091
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Rakić, V., Damjanović, L. (2013). Temperature-Programmed Desorption (TPD) Methods. In: Auroux, A. (eds) Calorimetry and Thermal Methods in Catalysis. Springer Series in Materials Science, vol 154. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-11954-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-642-11954-5_4
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-11953-8
Online ISBN: 978-3-642-11954-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)