
Abstract
The clinical diagnosis of cutaneous tumors in a child may be a challenge not only due to the rarity of the diseases but also because benign lesions may have alarmingly malignant tumor-like features as well as malignant neoplasms may present with benign characteristics (i.e., pedunculated or amelanotic melanoma simulating a pyogenic granuloma). The same can be said for the pathological diagnosis. For some histotypes, the histological diagnosis of a tumor of the skin in a child can be very difficult for many reasons. For example, the diagnosis of melanoma in pediatric age has often been a problem for pathologists. Melanoma is a very rare entity in children, so pathologists are always psychologically tempted to find an alternative diagnosis. Actually, pediatric melanomas do exist and deserve a distinct treatment. In pediatric age, the histological characteristics of melanoma are mimicked by other more frequent neoplasms, i.e., Spitz tumors, for which a spectrum of aggressiveness is reported from benign lesions (Spitz nevi) through the so-called atypical lesions (atypical Spitz tumors, with a risk ranging from low to high) up to the Spitzoid melanoma.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Infantile Hemangioma
- Pyogenic Granuloma
- Melanocytic Lesion
- Epithelioid Hemangioendothelioma
- Superficial Spreading Melanoma
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Differential Diagnosis of Cutaneous Tumors
The clinical diagnosis of cutaneous tumors in a child may be a challenge not only due to the rarity of the diseases but also because benign lesions may have alarmingly malignant tumor-like features as well as malignant neoplasms may present with benign characteristics (i.e., pedunculated or amelanotic melanoma simulating a pyogenic granuloma). The same can be said for the pathological diagnosis. For some histotypes, the histological diagnosis of a tumor of the skin in a child can be very difficult for many reasons. For example, the diagnosis of melanoma in pediatric age has often been a problem for pathologists. Melanoma is a very rare entity in children, so pathologists are always psychologically tempted to find an alternative diagnosis. Actually, pediatric melanomas do exist and deserve a distinct treatment. In pediatric age, the histological characteristics of melanoma are mimicked by other more frequent neoplasms, i.e., Spitz tumors, for which a spectrum of aggressiveness is reported from benign lesions (Spitz nevi) through the so-called atypical lesions (atypical Spitz tumors, with a risk ranging from low to high) up to the Spitzoid melanoma. Moreover, the borders between these entities are not so sharply defined. The misdiagnosis of pigmented lesions may be common in pediatric age. As an example, a study conducted by the European Organisation for Research and Treatment of Cancer (EORTC) reported that, among 102 lesions originally diagnosed as melanoma in children, only 60 were confirmed as being malignant at histological review, while 42 were reclassified as benign (Spatz et al. 1996). In this very complex scenario, a further issue is the attitude of dermatopathologists of adults to apply adult-type histological criteria and classify pediatric lesions into adult-type categories. In the end, on one side, there is the risk of underdiagnosing malignant tumors due to the conviction that melanoma and other malignant diseases are virtually nonexistent in the young, and patients may pay the price of this in terms of uncorrected treatment approach, advanced disease at the time of the diagnosis, and – ultimately – in terms of survival. On the other side, there is the risk of overdiagnosis and therefore overtreatment of benign lesions. All these facts underline the importance of referring suspected cases to expert physicians who are professionally dedicated to skin tumors – also in the light that early diagnosis remains the most reliable way to cure melanoma and other cutaneous tumors. For clinicians, it is not so much a matter of pediatric oncologists being capable of diagnosing melanoma but of their knowing that it does occur in children and referring any suspected cases to experts dedicated to melanoma. For pathologists, pediatric melanocytic lesions of the skin should be diagnosed by a pathologist aware of pediatric skin pathology. The creation of panel of experts, the building of network of cooperation, the centralized review, and the option of second opinion are possible suggestions to improve the quality of pathological diagnosis of childhood skin tumors.
2 Cutaneous Melanoma
Melanoma in children and adolescents is rare, accounting for less than 3% of cancers in patients under 20 years of age and for 7% of all cancers in 15–19-year-olds (Fig. 43.1) (Ries et al. 1999). The melanoma age-adjusted specific incidence rate in patients under 20 years is 5.8 per million (http://seer.cancer.gov/csr/1975_2007/results_merged/sect_29_childhood_cancer_iccc.pdf), and current estimates suggest that there are approximately 427 new cases of melanoma diagnosed each year in the United States in patients under the age of 20 (Bleyer et al. 2006). Melanoma is more common in young females, and males predominate after the age of 40 years. In a review by Strouse, the incidence of pediatric melanoma was reported to have increased at a rate of 2.9% per year over a 28-year period after adjusting for age, race, sex, and ambient UV radiation (Strouse et al. 2005). In this study, adolescents and young adults had higher increases in incidence rates when compared to younger patients. Epidemiological studies have demonstrated that the melanoma incidence increases with age; over 90% of cases occur in patients older than 10 years of age, and 74% are seen in those aged 15–19 years (Bleyer et al. 2006). Melanoma most commonly affects fair-skinned individuals; in two epidemiological studies, more than 90% of patients over the age of 10 years were white (Strouse et al. 2005; Lange et al. 2007). Major increases were seen in males between 1975 and 1979 as well as 1975 and 1999 and also in whites, particularly in ages 5–9 and ages 15–19 years.

Incidence of pediatric melanoma
A study from the Italian TREP project (Tumori Rari in Età Pediatrica [rare tumors in pediatric age]) analyzed the incidence of rare tumors included in the TREP list on the basis of a definition of annual incidence of less than two cases per million population. Using the data from the population-based cancer registries AIRTUM (that covers 33% of the Italian resident population in the 0–14-year-old age group and 27% of the 15–19-year-old age group), the study reported that annual incidence of melanoma was less than 2 per million population among 0–10-year-olds (0.64 for 1–4- and 0.30 for 5–9-year-olds) but was higher than 2 per million population in the age ranges over 10 years, being, in particular, 2.38 for 10–14- and 8.78 for 15–17-year-olds. In other words, melanoma would be a “rare pediatric tumors” – as defined by the TREP group – in children < 10 years, but not in older ones and, in particular, in adolescents (Pastore et al. 2009).
2.1 Risk Factors
Several risk factors for the development of adult melanoma have been observed in children. These include light pigmentary traits, tendency to freckle, and increased number of melanocytic nevi (Youl et al. 2002). However, a subset of pediatric patients has unique risk factors that predispose them to the development of malignant melanoma, and these include:
-
1.
Xeroderma pigmentosum. This autosomal recessive disorder is characterized by extreme photosensitivity to ultraviolet radiation and mutations of the nucleotide excision repair complementation groups. Most of the skin cancers develop during the first decade of life and preferentially affect the head and neck area (Kraemer et al. 1994). Early recognition of sun sensitivity with molecular testing is essential in establishing the diagnosis. Sun avoidance is crucial, and use of high-dose oral isotretinoin has been shown to be effective in preventing new cancers in patients with multiple skin cancers (Kraemer et al. 1988).
-
2.
Retinoblastoma. Survivors of hereditary retinoblastoma are at increased risk for developing melanoma, and this risk is seen in both irradiated and nonirradiated patients (Kleinerman et al. 2005).
-
3.
Werner syndrome. Patients with Werner syndrome are at increased risk for developing various malignancies, including melanoma (Goto et al. 1996). This autosomal recessive syndrome is characterized by the onset of premature features associated with aging and is due to mutations of the WRN gene, a member of the human RecQ family of DNA helicases (Muftuoglu et al. 2008).
-
4.
Congenital melanoma is the most common transplacentally acquired malignancy. Of six cases reported in the literature, only one is a long-term survivor. It is recommended that the placentas of all women with suspected melanoma be evaluated for the presence of the disease and that the neonate be screened for signs of disease for 24 months postpartum (Alexander et al. 2003).
-
5.
Melanoma can arise from medium-sized and large congenital nevi and can more commonly involve the scalp (Fig. 43.2). Even though the overall risk for developing melanoma in a congenital nevus is only 0.7%, it raised with the size of the nevus. The melanoma risk is highest for those nevi designated as garment nevi.
Fig. 43.2 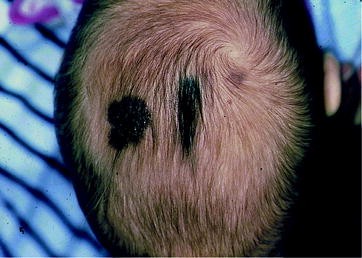
Congenital melanoma arising in a medium-sized nevus
-
6.
Giant congenital melanocytic nevi (Fig. 43.3) affect less than 1 in 20,000 newborns. Patients with these lesions have about a 5% lifetime risk of developing melanoma, and most develop early in life (Hale et al. 2005). Patients with larger-sized nevi and increased numbers of satellite nevi are at increased risk for developing melanoma.
Fig. 43.3 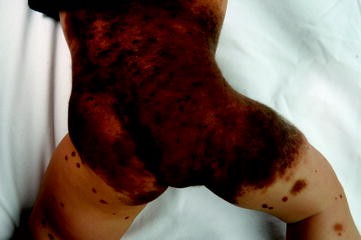
Giant congenital melanocytic nevi
-
7.
Neurocutaneous melanosis is an extremely rare disorder characterized by large or multiple congenital nevi in association with meningeal melanoma or melanosis (Makkar and Frieden 2004). The likelihood of developing neurocutaneous melanosis in giant congenital nevi ranges from 2.5% to 12%, and larger nevus size and multiple satellites appear to increase the risk of this complication (Ceballos et al. 1995). Most symptomatic patients present with neurological manifestations, such as increased intracranial pressure within the first 2 years of life. Central nervous system (CNS) melanoma develops in up to 64% of patients with symptomatic disease, and the prognosis is extremely poor, with a median survival of about 6 months. Approximately 25% of asymptomatic patients with large congenital nevi have radiographic evidence by MRI of CNS melanosis. In one series, only 1 of 20 children studied developed symptomatic disease (Ceballos et al. 1995).
-
8.
Immunosuppression. Patients with immune deficiencies and those that have undergone solid organ and bone marrow transplantation are at increased risk for developing melanoma (Curtis et al. 1997; Euvrard et al. 2003). Survivors of childhood cancer also have an increased risk of developing melanoma (Friedman et al. 2010).
-
9.
Genetic factors. It is estimated that about 8%–10% of melanoma cases have a family history of the disease. Germline mutations in CDKN2A and CDK4 susceptibility genes have been identified in only 10–25% of familial melanoma cases, and therefore, additional undefined high-penetrance predisposition genes probably exist (e.g., the locus on chromosome band 1p22) (Hayward 2003; Whiteman et al. 1997). A recent Italian study on 21 pediatric melanoma samples was able to identify not only some genetic traits in common with melanoma of adulthood (i.e., BRAF oncogene activation and a frequent loss of the CDKN2A gene, suggesting that the tumor’s pathogenesis partially coincides with that of adult melanoma) but also other traits peculiar to the younger age group (frequent c-Kit gene alterations), hinting at the involvement of novel genetic networks (Danietti et al. 2009). When known melanoma susceptibility genes were analyzed, a particular pattern emerged from the comparison with familial melanoma cases, featuring the lack of any CDKN2A germline mutations in the absence of a family history of melanoma and a marginal role for MC1R variants (suggesting a prevalent role for other genes affecting pigmentation) (Uribe et al. 2005). So, other genetic factors may play a part in sporadic childhood melanoma, probably including high-penetrance predisposition genes as well as low-penetrance polymorphic variants. A particular pattern involving the loss of heterozygosity and microsatellite instability has also been described in childhood melanoma (Uribe et al. 2005). Finally, it is worth of being quoted the genome-wide association studies (GWAS) conducted in melanoma and in other common cancer types; these studies were able to identify novel disease loci not previously suspected to be related to carcinogenesis, confirming that tumor susceptibility is polygenic and pointing to new disease mechanisms (Easton and Eeles 2008).
-
10.
Environmental factors. A history of sunburns in early life and tanning bed exposure also confers and increases risk for developing melanoma (Lazovich et al. 2010). An increase in the incidence of melanoma in pediatric ages has been recently observed also in Northern Europe (Karlsson et al. 1998; Pearce et al. 2003) and, particularly, in Australia (Whiteman et al. 1995; Milton et al. 1997) where the overall incidence of melanoma is 5 times more than that in Europe. In particular, melanoma in Queensland accounts for 6% of all pediatric tumors and is the seventh most frequent malignancy in children: incidence rates rise from 1 per million in the 0–4 age group to 30 per million in the 10–14 age group (Whiteman et al. 1995). This data are undoubtedly related to UV exposure, latitude, and skin type and pigmentation (fair-skinned individuals, with red hair, are more likely to develop melanoma than darkly pigmented individuals). The great awareness of physicians and parents, and the improved accuracy in histological diagnosis (e.g., diagnosis of melanoma versus atypical Spitz nevus), may play a role in the increased incidence, but the main role is played by the increased cumulative UV exposure during childhood and adolescence (e.g., increased risk of melanoma in lower extremities of girls). This data have been measured in various studies by the number of blistering sunburns and reported time spent outdoors and would confer a two to fivefold increased risk of melanoma in case-control studies of adults (Fears et al. 2002; Loria and Matos 2001; Gilchrest et al. 1999).


2.2 Clinical Manifestations
Melanoma in prepubertal children is a challenge even for clinicians who see pigmented skin lesions in children on a daily basis because of the alarming melanoma-like features of some benign nevi (Spitz nevi, Reed nevi, and junctional nevi) and because prepubertal melanoma often does not appear with the typical appearance of the adult forms. The disease often presents as a nodular, or a pedunculated, and/or an amelanotic lesion, sometimes simulating pyogenic granulomas (Ferrari et al. 2005; Ceballos et al. 1995; Handfield-Jones and Smith 1996; Mones and Ackerman 2003; Sybert 1991; Zuckerman et al. 2001; Sander et al. 1999; Tate et al. 1993). The commonly used ABCD clinical rule (Asymmetry – Border irregularity – Color variability – Dimension > 6 mm) may be useless and even misleading in childhood melanoma (Bono and Ferrari 2005). Regarding tumor size, for instance, is noteworthy since benign nevi normally grow up to relatively large size as the child grows, whereas, at the same time, melanomas may be frequently detected at a very small size (Bono 2001). When melanoma arises in a congenital nevus (particularly, in the case of “bathing trunk nevus,” congenital darkly pigmented and often disfiguring melanocytic lesion, sometimes associated with neurofibromatosis, lipomas, and spina bifida), diagnosis is even more very difficult because malignant transformation often evolves in its deeper components and surface alterations may be a late manifestation.
Presenting features such as bleeding, ulceration, increasing mole size, itching, and a palpable mass, may be present in pediatric cases as (Kaste et al. 1996). The diagnosis of melanoma in children is often unsuspected, and in various series, misdiagnosis and diagnostic delays were reported (in up to 60% of patients) (Saenz et al. 1999; Melnik et al. 1986). In two large series of pediatric melanoma from the SEER and National Cancer databases, female sex predominated, and two thirds of patients presented with localized disease; less than 5% of patients presented with distant metastatic disease (Strouse et al. 2005; Lange et al. 2007). In these series, over 90% of children were white, and the most common histologic subtype was superficial spreading melanoma. Thin and intermediate-thickness lesions were more commonly observed, and thick lesions, defined as those >4 mm in thickness, were seen in only 2% of cases. In both series, younger patients were more likely to be nonwhite, present with disseminated disease, and have nodular histology, head and neck primaries, history of cancer in the family, thicker lesions, and an inferior clinical outcome (Strouse et al. 2005; Lange et al. 2007). Others reported better outcome for younger patients compared to adolescents and adults (Ferrari et al. 2005).
The clinical characteristics of melanoma in children were studied in an evaluation of the database of the Central Malignant Registry in Germany. During the time from 1983 to 2004, 60 dermatologic departments contributed to this database, and 54,033 patients with cutaneous melanomas were documented. Only 316 patients (0.6%) were found with an age up to 18 years. The age distribution of these patients is given in Fig. 43.4. There were only very few cases up to the age of 12 years. In the age of 13–18 years, the prevalence of melanoma patients increased rapidly. As this kind of data collection is based on the histopathologic reports of the documenting centers, there are no clear data on how many misdiagnoses are among these melanoma reports for children.

Age distribution of patients with cutaneous melanoma in childhood and adolescence (German Central Malignant Registry, 1983–2004; 54,033 patients, 316 patients <18 years old)
Regarding the melanoma subtypes, mainly superficial spreading melanomas and nodular melanomas were reported. As expected, there are a very low number of cases with lentigo maligna melanoma or acral lentiginous melanoma. Likewise, there are very few reports on melanomas on large or small congenital nevi (Fig. 43.5). The body sites where melanoma developed differed between male and female gender, but they were equally distributed in children as in adults. While males developed melanomas mainly on the trunk, a predilected site in females was the lower extremity. In children, however, there were, in females, more melanomas on the trunk and, in males, more melanomas on the lower extremity (Fig. 43.6).

Histopathological subtypes of cutaneous melanomas in children up to the age of 18 (German Central Malignant Registry, 1983–2004)

Site distribution of melanomas in males (a) and females (b) (German Central Malignant Registry, 1983–2004)
The tumor thickness of melanomas in children was similarly distributed as in adults. Most melanomas in children were diagnosed with a tumor thickness of less than 1.0 mm. In children, even a higher percentage of patients were diagnosed with thin melanomas (Fig. 43.7). In children, tumor thickness was found to be the most important prognostic factor, and 10-year survival was best in thin melanomas, and the 10-year survival probability in children with thick melanoma with more than 4 mm in tumor thickness was around 70%. These data show that melanomas in children and adults behave very similarly and show rather equal clinical characteristics.

Distribution of tumor thickness in children up to the age of 18 and in adults over 18 years of age (German Central Malignant Registry, 1983–2004)
2.3 Establishing the Diagnosis of Pediatric Melanoma
Back in 1948, Sophie Spitz published a landmark paper defining the term “juvenile melanoma,” which described a benign nevus in childhood histologically mimicking malignant melanoma (Spitz 1948). Later on, the term “juvenile melanoma” was replaced by “Spitz nevus” to emphasize the clinical benign behavior of these lesions. Establishing the diagnosis of pediatric melanoma can be challenging, and other lesions can be confused with this diagnosis in the pediatric population. In a study of histopathologic diagnosis of malignant melanoma in childhood, a poor reliability even among experts was detected (Wechsler et al. 2002). Additionally, it seems that in retrospective reevaluations of histological specimens, knowing the clinical course of the children, the initial diagnosis of prepuberal melanoma had to be revised to benign melanocytic lesions (Leman et al. 2005). Therefore, it is likely that several documented cases of malignant melanoma in childhood in cancer registries are benign Spitz nevus, leading to an overestimation of incidence rates in such registries.
Furthermore, the emergence of purely descriptive entities, such as Spitzoid melanoma, atypical Spitzoid lesions, melanocytic lesions or tumors of unknown metastatic potential (MELTUMP), and atypical Spitz nevus, is confusing and ill defined (Mones and Ackerman 2004), and their diagnosis places a significant responsibility on the treating clinician who is ultimately responsible for making therapeutic decisions, such as the appropriateness of complete lymph node dissections or administration of adjuvant therapies with interferon. It is important to remember that although these interventions have been well studied in adults, their long-term impact in children is unknown (Su et al. 2003; Lohmann et al. 2002; Busam et al. 2009). In one publication of 57 MELTUMP, an expert panel of dermatopathologists reviewed these lesions and matched their interpretation with the actual clinical behavior of the lesion. Only half of the participants were able to diagnose clinically favorable lesions as benign, and 73%, with unfavorable behavior as malignant. These lesions are being increasingly recognized and are biologically different from conventional melanomas or benign melanocytic nevi (Cerroni et al. 2010).
Several investigators are now relying on molecular tests to better classify the diagnosis of melanocytic tumors in children. The use of comparative genomic hybridization (Fig. 43.8a, b) and FISH has identified multiple chromosomal aberrations in melanoma that most commonly involve regions at 6p25, 6q23, 9p, 10q, 8p 1q, and 11q13 (Gerami et al. 2009; Bastian et al. 1999, 2000). These findings are not seen in patients with Spitzoid lesions, which usually have a normal chromosomal complement and occasional gains of chromosome 11p. Additionally, the detection of BRAF mutations can be helpful in differentiating between melanoma and Spitz nevi. Whereas BRAF mutations are rare in patients with Spitzoid lesions, a significant number of melanomas have a mutation in this gene (Gill et al. 2004; Brose et al. 2002). In a recent Italian paper, analysis of 21 pediatric patients with melanoma revealed CDKN2A and MC1R gene variants in 2/21 and 12/21 patients, respectively. At the somatic level, 9/14 lesions had CDKN2A locus homozygous deletions and a null p16 immunophenotype. Loss of KIT protein expression was seen in 7/14 cases, and BRAF (V600E) mutations were seen in 5/10 cases (Danietti et al. 2009).

Comparative genomic hybridization: (a) finding of an 11p gain typical of Spitz nevi in a 14-year-old girl; (b) histopathologically diagnosed as MELTUMP in a 10-year-old boy. Complex copy number aberrations on chromosomes 9 and 11. The narrow gains on chromosome 11 include cyclin D1. The genetic instability is in favor of a melanoma
2.4 Histological Diagnosis: MELTUMP
MELTUMP is a term proposed in 2004, encompassing a group of lesions that pose problems in the definition of their malignant potential as defined by the classical histological parameters. In these lesions, the application of the usual histological criteria of malignancy of conventional melanocytic lesions does not carry the same risk of malignancy. MELTUMP should not be a wastebasket for any difficult lesion. MELTUMP are tumorigenic atypical deep compound and dermal melanocytic tumors. They exhibit one or several features indicative of possible malignancy, such as nuclear atypia, macronucleoli, mitotic activity, necrosis, or ulceration, but they exhibit these features in number or degree insufficient to justify a diagnosis of malignancy. The term “MELTUMP” should be used for “lesions that do not display all of the characteristics that permit a diagnosis of vertical growth phase melanoma and whose capacity to metastasize is indeterminate or uncertain.” This term implies a risk to progress upon incomplete excision and potential for metastasis after complete excision. MELTUMP are a heterogeneous group of lesions encompassing atypical Spitz tumors (AST), cellular and atypical epithelioid/spindled blue nevi (ABN), and minimal deviation melanomas. Also, pigmented epithelioid melanocytoma is included. In particular, presence of deep dermal mitoses in AST and of mitoses irrespective of location in ABN is considered as a marker of risk, enough to include these two entities among MELTUMP (Mones and Ackerman 2004; Busam et al. 2009; Cerroni et al. 2010).
2.5 Histological Diagnosis: Melanoma
To summarize, the histological diagnosis of melanoma in childhood is difficult due to many factors:
-
It is a rare tumor in pediatric age, but it still exists.
-
Childhood melanoma can show histological features not present in adulthood melanoma.
-
Proliferative nodules in congenital nevi should be carefully distinguished from melanoma.
-
Spitzoid lesions are particularly problematic, and many of them fall within MELTUMP.
The five major subtypes of melanoma in adulthood are superficial spreading melanoma, nodular melanoma, lentigo maligna melanoma, acral lentiginous melanoma, and mucosal lentiginous melanoma. These histotypes can be found in children. Though, in childhood, many lesions are composed by epithelioid and spindle cells with Spitzoid characteristics, and a careful differential diagnosis in the range of atypical Spitzoid tumors is required. Also, there are melanomas arising in congenital nevi that are to be carefully differentiated from proliferative nodules.
A series of features should alert the pathologist to take a diagnosis of melanoma into consideration: size larger than 7 mm, asymmetry, poorly defined borders, ulceration, marked pagetoid spread, pleomorphism, expansile dermal growth, high mitotic count (>4/mm2), deep mitoses, atypical mitoses, absence of maturation, lymphatic/vascular invasion, and perineural diffusion. The pathologist should take into account all these features of the lesion. The presence of a single factor is not significant per se.
When a melanoma is diagnosed, the pathologist should apply the same histological parameters as in the adult case, even if their true prognostic significance could not always be superimposable in childhood. In particular, a diagnosis of primary cutaneous melanoma should include histological type, Clark level, thickness, mitoses/mm2, type of growth phase, presence of microscopic satellites, presence of regression, presence of ulceration, tumor-infiltrating lymphocytes (TIL), neuro and vascular diffusion, associated nevus, and state of resection margins (Cerroni et al. 2010; Brenn and McKee 2008).
2.6 Staging
Staging guidelines have not been developed for patients with pediatric and adolescent melanoma, and therefore, most of the reported series have incorporated the adult American Joint Committee on Cancer (AJCC) classification of melanoma. In the new AJCC classification, which was modified in 2009, patients are stratified into four different groups (Table 43.1). Patients with localized disease can be classified as stage I and II based on the thickness of the primary tumor, presence or absence of ulceration, and mitotic rate. Patient with stage III disease have nodal involvement, and those with stage IV disease have distant metastatic disease. As in adults, the stage of the disease is predictive of outcome in pediatric melanoma, and thicker lesions have a higher incidence of nodal involvement (Strouse et al. 2005; Rao et al. 1990; Paradela et al. 2010) (Fig. 43.9). However, although the incidence of nodal involvement appears to be higher, particularly amongst patients with MELTUMP (Busam et al. 2009), the outcome of these patients does not appear to be significantly affected. The use of routine imaging to detect metastatic disease in children with melanoma has not been studied extensively, but one publication suggests that patients who present with thick primary lesions or lesions with an unknown primary have an increased incidence of clinically unsuspected metastases and should therefore be imaged routinely (Kaste et al. 1996).

Survival for patients with melanoma stages I–IV (reproduced with permission. JCO 27:6199, 2009)
2.7 Treatment
There is a general consensus that the therapeutic recommendations for childhood melanoma should remain the same as for adults. Surgical resection remains the mainstay of therapy for pediatric melanoma. When possible, we advocate following the same surgical guidelines that have been developed for adults: lesions ≤1 mm should be resected with a 1-cm margin; 1–4 mm lesions, with a 2-cm margin; and those more than 4 cm, with at least a 2-cm margin. Patients with lesions ≤1 mm and no ulceration or mitotic rate <1 mm2 can be treated with surgical resection alone and observation. Lesions with more adverse features, such as thickness ≥0.75–1 mm, increased mitotic rate, and evidence of ulceration, including lymphovascular invasion, should undergo sentinel node sampling (Shah et al. 2006; Butter et al. 2005; Roaten et al. 2005). If the sentinel node is positive, patients should be offered a complete lymph node dissection with the caveat that only 15–20% of patients will have subsequent positive nodes and that this procedure may not impact survival (Morton et al. 2006) (Table 43.2).
The issue of medical treatment in children with melanoma is problematic. Because of the rarity of the disease and the lack of experience of pediatric oncologists in treating these tumors, pediatric patients should be included – when possible – in adult therapeutic trials (i.e., immunotherapy, immunochemotherapy, vaccinotherapy). Unfortunately, most of clinical trials are not open to children. Alpha interferon (Kirkwood et al. 2004) has shown to improve relapse-free survival in patients with high-risk resected melanoma. It has been approved in North America by the Food and Drug Administration for high-risk melanoma, and it is virtually the only adjuvant treatment currently available for children. The use of this therapy is well tolerated in children, but its efficacy has not been tested, given the small numbers of patients that have been treated in limited institutions over the years (Shah et al. 2006; Navid et al. 2005; Chao et al. 2005). For patients with metastatic disease, limited reports suggest that dacarbazine-based chemotherapy has similar limited efficacy. The use of interleukin-2 has been studied in limited numbers, and although tolerable, its efficacy has not been assessed (Ribeiro et al. 1993; Bauer et al. 1995). Newer therapies, such as ipilimumab and the BRAF inhibitor PLX4032, have produced promising responses in adults with metastatic melanoma, but its use in pediatrics has not yet been studied (Flaherty et al. 2010; Hodi et al. 2010).
2.8 Survival Prognosis
The population-based analysis from the SEER reported a 5-year melanoma-specific survival of 93.6% (Strouse et al. 2005), which is consistent with European population-based data (80%) (Conti et al. 2001). This result is better than those reported in published hospital-based series (that can be biased by the selection of patients with more advanced disease) and however is better than that usually achieved in adults. Data from the SEER registry showed a 4% survival improvement per year during the last three decades. Other series have described slightly better outcomes in children than in adults (Ferrari et al. 2005; Saenz et al. 1999; Gibbs et al. 2000; Barnhill et al. 1995; Hamre et al. 2002; Schmid-Wendtner et al. 2002).
Overall survival rates for childhood melanoma are in excess of 95% for localized disease, around 70% for regional disease, while for metastatic cases, survival rates range from 10% to 50% (Lange et al. 2007) (Figs. 43.10–43.14).

Superficial spreading melanoma in a 17-year-old boy

Superficial spreading melanoma in a 18-year-old boy

Nodular melanoma in a 17-year-old girl

Spitz nevus initially classified as malignant melanoma

Melanoma in a 15-year-old girl, distant metastases 14 months after diagnosis
3 Skin Carcinoma
Squamous cell carcinoma of the skin (SCC) is rarely diagnosed in childhood and adolescence. It is a malignant locally destructive epithelial tumor developing from the keratinocytes of the skin, which uncommonly metastasizes in nonimmunosuppressed patients.
Most cases are described in children suffering from xeroderma pigmentosum, an autosomal recessive disorder with mutations in the nucleotide excision repair complementation groups, resulting in extreme photosensitivity to ultraviolet radiation. It affects boys and girls in equal frequencies. In a series of 830 published cases, 45% of the patients had basal cell carcinoma or SCC. The median age of first nonmelanoma skin cancer among these patients was 8 years (Kraemer et al. 1987).
Another risk factor for developing SCC is iatrogenic chronic immunosuppression, e.g., after solid organ transplantation, whereas the risk of posttransplant cutaneous SCC is related to the kind of immunosuppression (especially cyclosporine), the duration of the immunosuppressive therapy, and the amount of cumulative UV exposure (Jensen et al. 1999). Also, sporadic chronic immunosuppression, e.g., in case of AIDS, is a risk factor for the development of SCC (Godfrey et al. 2003).
SCC is also reported to develop in children with interferon-gamma receptor two deficiency (Toyoda et al. 2010), in patients suffering from dystrophic epidermolysis bullosa (DEB), a genodermatosis resulting from mutations in COL7A1 and encoding type VII collagen (Horn and Tidman 2002), and in children with systemic sclerosis or pansclerotic morphea (Wollina et al. 2002) or chronic infections and wounds (Kassi et al. 2010). Single cases of a SCC arising in a nevus sebaceous of Jadassohn were also reported in the literature (Ball et al. 2005).
Treatment of choice is the complete excision of the tumor with three-dimensional histological visualization and evaluation of excision margins (Moehrle et al. 2007). In selected cases (deep infiltrating tumors), a sentinel node biopsy can be applied.
4 Other Rare Tumors of the Skin and Subcutaneous Tissue
4.1 Dermatofibrosarcoma Protuberans
Dermatofibrosarcoma protuberans (DFSP) is a fibrohistiocytic tumor of intermediate malignancy that accounts for about 1–2% of all soft tissue sarcomas and most commonly affects young adults (McArthur 2007; Terrier-Lacombe et al. 2003). This malignancy has been described in children and can be congenital (Checketts et al. 2000; Chien et al. 2007; Jafarian et al. 2008; Pappo et al. 1997). In one series, pediatric DFSP accounted for 6% of all of this diagnosis seen at a single institution (McKee and Fletcher 1991). This tumor most commonly affects males and typically arises in the trunk and extremities. DFSP usually presents as a slow growing vascular-appearing macule or plaque that later develops into a nodular cutaneous mass. In a review of over 25 pediatric DFSP, the median age at presentation was 12 years. In another review of over 150 cases of pediatric DFSP, the most common sites of involvement were the trunk and extremities; the former location is preferentially seen in patients with congenital DFSP (Checketts et al. 2000; Chien et al. 2007; Jafarian et al. 2008; Pappo et al. 1997). Histologically, this tumor is characterized by a monotonous proliferation of fibroblasts arranged in a storiform pattern. A small number of DFSP have sarcomatous changes, a feature that has rarely been reported in the pediatric literature but that has been reported to carry an increased risk for the development of local recurrence and distant metastases (Abbott et al. 2006; Mentzel et al. 1998). DFSP is characterized by a common translocation t (17;22)(q22;q13) which fuses the COL1A1 and PDGFB genes (McArthur 2007). This fusion protein is processed to a mature PDGFB and interacts with the PDGFB receptor present on the cell surface of DFSP in an autocrine or paracrine fashion (Rubin et al. 2002). The primary treatment of DFSP is wide surgical resection with negative margins (Fiore et al. 2005); Mohs micrographic surgery has also been used in selected cases (Sondak et al. 1999). Using this primary surgical approach, cause-specific mortality in adults has been reported to be 3% in 10 years, and local recurrence rates have been documented in less than 5% of cases (Fiore et al. 2005). For adults and children with unresectable or metastatic DFSP, the administration of imatinib mesylate has produced promising clinical responses (Rutkowski et al. 2010; Price et al. 2005; Gooskens et al. 2010).
4.2 Giant Cell Fibroblastoma
Giant cell fibroblastoma (GCF) is a rare nonmetastasizing subcutaneous fibrohistiocytic neoplasm that occurs predominately in younger patients. In one series of 86 GCF, the median age was 6 years, and nearly two thirds of the patients were younger than 10 years of age. This tumor most commonly presents as painless mass located in the dermis or subcutis and most commonly arises in the trunk. GCF has been referred as a juvenile form of DFSP, and both entities share the t (17;22)(q22;q13) (Jha et al. 2007). Histologically, GCF is characterized by the presence of spindle cells that infiltrate adnexal structures and form pseudovascular spaces that are lined with discontinuous multinucleated giant tumor cells (Billings and Folpe 2004) Although both GCF and DFSP have similar cytogenetic findings, adult DFSP is often characterized by the presence of ring chromosomes, a feature that is notably absent in pediatric DFSP and GCF. The clinical behavior of GCF is similar to DFSP, and surgical resection with negative margins is the treatment of choice.
4.3 Angiomatoid Fibrous Histiocytoma
Angiomatoid fibrous histiocytoma (AFH) is a rare subcutaneous fibrohistiocytic tumor of intermediate malignancy that accounts for about 0.3% of all soft tissue sarcomas (Thway 2008). AFH was initially described by Enzinger in 1979 as a soft tissue tumor related to malignant fibrous histiocytoma that affected the superficial tissues of younger patients (Enzinger 1979). More recent studies have demonstrated that this entity has an excellent prognosis following surgical resection alone, and thus the original nomenclature of “malignant” has been removed from its name (Thway 2008). In one series of 108 patients, the median age at presentation was 14 years, females were slightly more commonly affected, and the tumor most commonly arose in the extremities (65%) and trunk (28%) (Costa and Weiss 1990). These tumors have an indolent clinical presentation, and most are located superficially; in one series, 60% of tumors arose within areas of normal lymphoid tissue (Fanburg-Smith and Miettinen 1999; Antonescu et al. 2007). In one series, systemic symptoms, such as anemia, weight loss, and fever, were seen in 14%, 4%, and 2% of patients (Fanburg-Smith and Miettinen 1999). Histologically, AFH is characterized by the presence of a fibrous pseudocapsule, a round or spindled histiocyte-like proliferation of cells, a plasma lymphocytic response, and pseudovascular spaces (Antonescu et al. 2007). Cytogenetic studies have documented translocations involving the FUS gene on chromosome 12q13 with either the EWSR1 gene on chromosome 22q12 or the FUS on 16p11. More recently, a translocation between EWSR1 and CREB1 on chromosome 2q34 has been identified as the predominant fusion product in AFH. The treatment of choice for this tumor is complete surgical excision. Local recurrences have been reported to occur in up to 12% of patients after incomplete excision, but reexcision often renders patients disease-free. In one series, 85 of 86 patients were alive and disease-free, and only one patient developed nodal metastases. In another series, only 1 of 94 patients developed distant metastases (Fanburg-Smith and Miettinen 1999; Antonescu et al. 2007).
4.4 Plexiform Fibrohistiocytic Tumor
Plexiform fibrohistiocytic tumor is a rare neoplasm with features that resemble fibrous histiocytoma and fibromatosis and mainly affects children and young adults. The median age at diagnosis in two large series was 14.5 and 20 years, respectively (Moosavi et al. 2007; Enzinger and Zhang 1988). This tumor occurs slightly more often in females and presents as a painless slow growing nodule located in the dermis or subcutis (Luzar and Calonje 2010). The tumor most often involves the upper extremity, with the fingers, hands, and the wrist being more commonly affected (Taher and Pushpanathan 2007). The lesions are usually small and contain a mixture of histiocyte-like and spindle fibroblast-like cells. On low-power microscopy, the dermis and subcutaneous tissue are infiltrated with multiple nodules that contain multinucleated osteoclast-like giant cells (Luzar and Calonje 2010). Immunocytochemistry reveals CD68 positivity on the histiocyte and osteoclast-like cells, whereas the fibroblast-like cells show focal positivity for smooth muscle actin. Cytogenetic analysis in two cases has revealed numerous deletions and a 46,XY,t(4;15)(q21;q15) translocation (Luzar and Calonje 2010). Surgical excision is the treatment of choice, but local recurrences have been documented in up 40% of cases within the first 2 years from diagnosis. Recurrences are usually successfully treated with surgical reexcision. Metastases to lymph nodes and lung have been rarely described (Salomao and Nascimento 1997).
4.5 Dermoid Cysts
Dermoid cysts are a subset of benign heterotopic neoplasms termed choristomas, probably deriving from dermal and epidermal tissues trapped in the cranial fusion lines as the neural tube closes in embryogenesis. Histologically, they may have a lining of squamous epithelium with dermal elements, such as hair follicles, sebaceous, and sweat glands: within the cyst, mature skin complete with hair follicles and sweat glands, sometimes clumps of long hair, and often pockets of sebum, blood, fat, bone, nails, teeth, eyes, cartilage, and thyroid tissue can be found.
Dermoid cysts can be deep and superficial (the former being more frequent in teenagers; the latter, in early childhood) and may occur as soft tissue swelling in three primary locations in the head and neck: the frontotemporal region, the periorbital region, and the nasoglabellar region.
Complete surgical excision – preferably in one piece and without any spillage of cyst contents – is curative, but dermoid cysts can recur if not completely excised. Lesions invading deeply within the orbit may require a more aggressive approach. Craniotomy and neurosurgical involvement may be required for intracranial extension (Ahuja and Azar 2006; Bartlett et al. 1993; Pryor et al. 2005).
5 Vascular Tumors
Vascular tumors are the most common subcutaneous neoplasms in children. The tumor cells in vascular tumor are the endothelial cells that show dedifferentiation, transformation, and uncontrolled growth, leading to a benign or malignant neoplastic phenotype. Genetic alterations as well as molecular biological characteristics are responsible here for, but in most vascular tumor types this is yet not clear. The pathogenesis of vascular tumor and the possible role of genetic alterations are still unclear.
The most frequent vascular tumor in childhood is infantile hemangioma (IH), which shows a special biology with continuous growth during the first year of life but spontaneous regression thereafter. Cutaneous IH are very frequent. Variants of IH have been described, from capillary IH and cavernous to segmental and congenital IH. IH may arise anywhere, but the head and neck region is the most frequent location. IH are connected to the circulatory system and filled with blood. Clinically, superficial IH appears as a cutaneous lesion or a bluish-red, rubbery or firm, well-circumscribed mass (Coffin and Dehner 1993). Clinical complications are related to ulceration and hemorrhages, or in case of impingement upon vital structures, as larynx or mediastinum. In most cases, IH hemangiomas will disappear over time; however, treatment is required when the tumor interferes with vision and breathing or threatens significant cosmetic injury (Haggstrom et al. 2006). Different treatment approaches have been proposed over the years, i.e., surgery, oral corticosteroids, injection of corticosteroid directly into the lesion, pulsed dye laser (Rizzo et al. 2009), and systemic therapy with interferon and vincristine (Wilson et al. 2007). A recent and extremely promising treatment option is represented by beta-blocker propranolol (Léauté-Labrèze et al. 2008).
IH can easily be confounded with other benign vascular tumors, such as tufted angioma or kaposiform hemangioendothelioma. However, in contrast to IH, they do not show spontaneous regression. In most cases, only clinical evolution or histology can discriminate these benign vascular tumors from IH.
On the other side of the spectrum, malignant vascular tumors are extremely rare and show aggressive behavior.
Latest basic research achievements have opened the possibility to discriminate between blood and lymph vessels: blood and lymph endothelial cells show special biological characteristics. Most of the vascular tumors are formed by blood endothelial cells; only rarely, lymph vessels can be found. In fact, most lymphangiomas are really malformations that arise from lymphatic tissue sequestered during embryologic development. However, some are probably true neoplasms with invasive growth pattern. In the future, these two vessel systems will be further characterized in more detail in vascular tumors, as it has already been the case in vascular malformations. The classification of the International Society on the Studies of Vascular Anomalies (ISSVA) for vascular anomalies clearly separates vascular tumors from vascular malformations that are inborn defects of vasculogenesis (Table 43.3).
5.1 Infantile Hepatic Hemangiomas
Infantile hepatic hemangiomas (IHH) can be found in the context of disseminated or diffuse neonatal hemangiomatosis that is characterized by multiple cutaneous IH, usually of small size, associated with visceral involvement. IHH are classified into three categories: focal, multifocal, or diffuse, according to the number of lesions. The clinical presentation can be asymptomatic or associated with hepatomegaly, jaundice, or liver dysfunction. Ultrasound or magnetic resonance imaging (MRI) can identify the vascular tumors with increased perfusion in the duplex ultrasound and hyperintensive signals in T2 (Fig. 43.15). Diffuse IHH can be life-threatening since they may induce congestive heart failure associated with high-volume vascular shunting. Hypothyroidism secondary to overproduction of type III iodothyronine deiodinase may be present.

Infantile hepatic hemangiomas in a newborn child with numerous skin hemangiomas. (a) Duplex ultrasound detects multiple hepatic lesions with perfusion signal. (b) In T2-weighted MRI, the lesions are hyperintense. (c) Shows an immunohistological staining of a biopsy with alpha-actin antibodies. Multiple blood vessels are seen, confirming the diagnosis of infantile hepatic hemangiomas
Severe and complicated IHH require therapy. First-line therapy of IHH with hemodynamically significant shunting used to be administration of systemic corticosteroids. However, recently, dramatically clinical improvement and decrease in size of IHH on propranolol were reported. This new therapeutic option may replace embolization; other pharmacological agents, such as vincristine and alfa-2a interferon; hepatic artery ligation; resection; or liver transplantation that have been reported in literature before (Léauté-Labrèze et al. 2008; Mazereeuw-Hautier et al. 2010; Marsciani et al. 2010).
5.2 Pyogenic Granuloma
Pyogenic granuloma (PG) is a benign nonneoplastic mucocutaneous lesion. It is usually a small red, oozing, and bleeding bump (Fig. 43.16). PG can result to constant minor trauma and might be related to hormonal changes. PG preferentially affects the gingiva but may also occur on the lips, tongue, cheek or oral mucosa, and palate. In the mouth, PG is manifested as a sessile or pedunculated, resilient, erythematous, exophytic, and painful papule or nodule with a smooth or lobulated surface that bleeds easily. The most common treatment is surgical excision or combinations of curettage, shave, and cautery (Tay et al. 1997; Matsumoto et al. 2001; Giblin et al. 2007).

Pyogenic granuloma at the cheek of a 4-year-old girl. The reddish nodule developed in the last 4 weeks and shows intermitted bleeding
5.3 Tufted Angioma
Tufted angiomas (TA) are rare locally aggressive vascular tumors of unknown pathogenesis. Most appear during childhood: approximately 25% of cases are congenital, and 50% appear in the first year of life. TA that are present at birth (congenital TA) or in the first year of life (acquired TA) have a greater tendency to spontaneously regress than those that appear later in life (Alberola et al. 2010). The clinical presentation of TA is nonspecific and characterized by bluish-erythematous plaques (Fig. 43.17a) or nodules. There is a well-recognized association with the Kasabach–Merritt phenomenon. The differential diagnosis includes IH, KHE, and vascular malformations.

(a) Tufted angioma in a 1-year-old girl. The red lesion showed continuous growth during the first year of life. A biopsy confirmed the diagnosis of tufted angioma. After birth, an accompanying Kasabach–Merritt phenomenon was treated by corticosteroids. (b) Laser therapy led to brightening of the vascular tumor
TA have a characteristic histology consisting of a proliferation of endothelial cells forming lobules with the typical “shotgun” distribution. TA are considered neoplasms of intermediate malignancy because of infiltrative growth, local aggressiveness, and variable prognosis. To date, definitive treatment for TA has had limited success. Local treatment with laser (Fig. 43.17b) and steroids, vincristine, or experimental antiangiogenic drugs has been reported (Fahrtash et al. 2010).
5.4 Kaposiform Hemangioendothelioma
Kaposiform hemangioendothelioma (KHE) is a locally aggressive, immature vascular neoplasm typically presenting in infancy and the first decade. KHE may involve various organs: generally, it originates on the skin, affecting deeper tissue by infiltrative growth (Fig. 43.18), and the retroperitoneum. KHE tend to be locally invasive but are not known to produce distant metastases. Visceral involvement is uncommon, but several cases with bone, retroperitoneal, mediastinal, or choledochus involvement have been described (Terui et al. 2010). KHE appears as one or multiple masses and may involve the underlying bone and rarely regional lymph nodes. There is no known association with HIV or HHV 8 infection. In most cases, KHE can be associated to the Kasabach–Merritt phenomena. The development of KHE in adolescents or in adults is very rare, but cases have also been described.

(a) Kaposiform hemangioendothelioma at the lumbosacral region. (b) Infiltrative growth in the subcutaneous region has been confirmed by MRI
Microscopically, KHE is characterized by nodules containing fascicles of spindle cells interspersed among small vessels blending with slit-like vascular spaces. Glomeruloid nests of endothelial cells are found at the periphery. Fibrous bands surround nodules and contain dilated lymphatic vessels. Small biopsies, including capillary blood vessel biopsy, may mimic a capillary hemangioma. The spindle cells are usually nonreactive for factor VIII–related antigen and stain for smooth muscle actin. Endothelial markers CD31 and CD34 are expressed in the capillary vessels. GLUT-1, expressed in infantile hemangioma, is typically negative.
Gradual regression of KHE has been observed. However, the tumor can cause potentially life-threatening complications. Several factors are associated with the outcome of patients with KHE: accessibility to surgical excision, location (cutaneous versus visceral), size of tumoral mass, and the Kasabach–Merritt phenomena. Death related to the Kasabach–Merritt syndrome or to the progressive failure of the infiltrated organ(s) has been described. Huge visceral tumor may lead to a 40–50% mortality rate. Various therapeutic options have been recommended according to the site, extent, and behavior of the disease: steroid, interferon-alpha, and vincristine – also in combination – are often used (Fahrtash et al. 2010).
5.5 Epithelioid Hemangioendothelioma
Epithelioid hemangioendothelioma (EH) was first described by Weiss and Enzinger in 1982 (Weiss and Enzinger 1982). It is a vascular tumor occurring at any age and may be either superficially or deeply located, arising at a variety of sites, including the skin and subcutis, the skeleton (Fig. 43.19), the lung, liver, and central nervous system. Microscopically, it arises from a vessel and extends into the surrounding soft tissue. The tumor is composed of epithelioid endothelial cells arranged in short cords or solid nests, with a prominent myxoid–jaline matrix. The diagnostic key feature is represented by epithelioid cells with a cytoplasmic vacuole representing a miniature lumen. It shows positive staining for endothelial markers (CD31, CD34) and cytokeratin.

(a) Epithelioid hemangioendothelioma at the right petrosal bone in a 12-year-old boy who presented with facial nerve paresis on the right side. A biopsy confirmed the diagnosis. (b) Signs of skeletal destruction at the CT scan. (c) Early phase of technetium-99 m scintigraphy showing a unifocal lesion at the right petrosal bone
EH of the liver may have peculiar features at enhanced computed tomography (CT) and/or magnetic resonance imaging (MRI) scans, i.e., nodular lesions with a tendency to coalesce, with a peculiar target appearance due to the presence of a central sclerotic zone and a peripheral region of cellular proliferation with high signal intensity; a lollipop sign given by a hepatic or portal vein terminating at or just within the periphery of some of the liver lesions has been described (Scoazec et al. 1988; Lyburn et al. 2003; Kehagias et al. 2000; Alomari 2006).
According to WHO, EH is a malignant vascular tumor because of its high metastatic rate. Metastases to regional lymph nodes, lung, liver, and bone occur in 30% of cases but cause death in half of the patients affected. Cellular atypia, mitotic activity (>1/10 HPF), necrosis, or diffuse spindle cell morphology appear to be associated with metastases (Mentzel et al. 1997). Complete local excision – if possible – is the preferred therapeutic measure. If impossible, sarcoma-based chemotherapy or alpha-2a-interferon is administered (Ferrari et al. 2001). In particular, alpha-2a-interferon is potent inhibitor of angiogenesis and acts by blocking the action of fibroblast growth factor. Many tumors of vascular origin, including EH, are characterized by endothelial cell migration and proliferation in the proliferative phase, and the major stimulus for this proliferation is fibroblast growth factor. The role of alpha-2a-interferon is of particular interest because its mechanism of action is the inhibition of this stimulus (Isowa et al. 2002; Kumar et al. 2003; Roudier-Pujol et al. 1994; Calabrò et al. 2007). Few data are available on the role of radiotherapy for inoperable EH. EH of the liver is characterized by an unpredictable prognosis: in younger patients, a wait-and-see strategy is usually adopted. Orthotopic liver transplantation may be considered, even in the presence of limited extrahepatic disease (Kelleher et al. 1989; Madariaga et al. 1995).
5.6 Kaposi Sarcoma
Kaposi sarcoma is rarely diagnosed in childhood and adolescence in general. It is a locally aggressive vascular neoplasm caused by the human herpesvirus 8 (HHV-8), which can be classified into four different types: classic, iatrogenic (immunosuppression-associated), AIDS-associated, and African/endemic. The classic type predominantly affects older adults of Mediterranean or Eastern European ancestry. Only single cases have been reported in children (Byun et al. 2010; Sahin et al. 2010). The iatrogenic type is linked to immunosuppression (e.g., after solid organ transplantation) and normally resolves with immune restoration. In a series of 326 organ-transplanted pediatric patients, 6 children developed a Kaposi sarcoma (Penn 1994). In contrast to the classic form of Kaposi sarcoma which is mainly located at the extremities, AIDS-associated Kaposi sarcoma usually develops on the head, back, neck, muscular palate, and the area of the gingiva and is seen in patients with low CD4-T-lymphocytes (<400/μL). Typically, Kaposi sarcoma presents with cutaneous lesions in the form of plaques, patches, or nodules. In the classic type, purplish, reddish-blue, or dark-brown macules that can ulcerate are found. In rare cases, internal organs can be affected as well as lymph nodes. In histopathology, the patch stage and the plaque, as well as the nodular stage, can be described. A vascular proliferation is seen with endothelial cells lining perivascular spaces, and new blood vessels can be produced in the lumen of preexisting blood vessels.
In a US series of 4,954 children with AIDS, 8 developed an AIDS-associated Kaposi sarcoma (Biggar et al. 2000). In patients with AIDS-associated Kaposi sarcoma, a highly active antiretroviral therapy (HAART) has been shown to prevent or induce regression of Kaposi sarcoma. Finally, the African/endemic type can be differentiated into the cutaneous and lymphadenopathic type and is seen in pediatric populations in Africa, most of whom develop the clinical disease as a consequence of HIV infection (Ziegler and Katongole-Mbidde 1996).
As most cases of Kaposi sarcoma in children appear in relation to immunosuppression, the major goal in the treatment is to restore immune competence either by changing the immunosuppressant scheme in organ-transplanted children (e.g., to mTOR inhibitors) (Yuksekkaya et al. 2009) or by applying an antiviral therapy in case of HIV infection. Children suffering from the classical type of Kaposi sarcoma can be treated locally by surgery (in case of small single lesions), radiation, or systemic interferon-alpha. Chemotherapy using liposomal anthracyclines, bleomycin, and vincristine is used (Tulpule et al. 1998).
5.7 Angiosarcoma
Angiosarcomas (AS) typically affect adult and elderly patients (it accounts for 1% of all adult soft tissue sarcomas) (Penel et al. 2010) and are extremely rare in children, representing less than 0.3% of pediatric sarcomas overall. Only two series have been reported in pediatric age, respectively, including 15 (Deyrup et al. 2006) and 12 patients (Ferrari et al. 2002).
Histology shows vascular structures with an infiltrative growth pattern and more solid areas with sheets and nests of malignant endothelial cells frequently displaying an epithelioid morphology. Spindled areas can be present as well. Necrosis is found in more than 80% of cases. Tumor cells express CD31 and CD34.
In adults, the majority of AS develop as cutaneous tumors. In children, the majority of AS arise in mediastinum and heart but may involve also visceral organs, breast, and deep soft tissue of abdomen or pelvis. Two risk factors are well established: chronic lymphoedema and previous radiotherapy. Clinical presentations of AS are heterogeneous. Possible output cardiac failure can result in very young patients due to arteriovenous shunting. The lesions can appear as hemorrhagic masses. Hepatic AS are extremely rare and usually present with a rapidly enlarging liver. By the time they are diagnosed, the lesion is often unresectable. AS are highly aggressive tumors; metastases are common, frequently to the lung. The mortality rate is generally high, particularly in hepatic AS in which death usually occurs within the first 6 months. In childhood, published series reported a mortality rate ranging between 62% and 65%.
Large resection followed, if possible, by adjuvant radiotherapy is the cornerstone of curative-intent treatment of localized AS. There are no convincing data supporting the administration of adjuvant chemotherapy, but most protocols currently require adjuvant chemotherapy in high-grade (as AS is) and large tumors, even when resected (see Chap. 44). For metastatic or locally advanced AS, doxorubicin-based chemotherapy or chemotherapy based on weekly paclitaxel (Penel et al. 2008) seems to provide the longer progression-free survival. Three phase II or parts of phase II trials have been published in the last 2 years in adult setting, investigating weekly paclitaxel, sorafenib, and imatinib, demonstrating that clinical trials are feasible for such rare diseases. Biological evidences for the key role of angiogenetic factors have been accumulated during the last years and support the further investigation of antiangiogenetic agents alone and almost combination with chemotherapy in such disease.
5.8 Kasabach–Merritt Phenomenon
Kasabach–Merritt phenomenon (KMP) is defined as thrombocytopenia and hypofibrinogenemia with elevated fibrin split products (D-dimers), suggestive of an active consumptive coagulopathy. Thrombocytes are low, ranging from 6,000 to 98,000, with fibrinogen levels less than 100 mg/dL; whereas, D-dimers are elevated. Prothrombin times (PT) and activated partial thromboplastin time (PTT) can range from normal to significantly prolonged. Additionally, anemia can be present at diagnosis as a consequence of intravascular hemolysis, including red blood cell fragmentation, elevated LDH, and hyperbilirubinemia.
Successful treatment of the underlying vascular tumor is critical to the correction of KMP and to the overall survival of patients. Children with KMP can die of hemorrhage or invasion/compression of vital structures by the vascular tumor. Mortality has ranged from 10% to 30% in most series.
A curative therapy of KMP can only be achieved by treatment of the underlying vascular tumor. However, supportive care to maintain hemostasis is necessary. Platelet transfusions should be reserved for active bleeding or in preparation for surgery or procedures. Aminocaproic acid and local measures may be helpful to reduce the need for platelet transfusions. Antiplatelet agents, such as acetylsalicylic acid and dipyridamole, have been used to reduce platelet aggregation within the vascular tumor. Treatment of hypofibrinogenemia with cryoprecipitate and prolonged PT or PTT with fresh frozen plasma should be a clinical decision rather than correction of a laboratory result. Symptomatic anemia should be treated with red blood cell transfusions.
References
Abbott JJ, Oliveira AM, Nascimento AG (2006) The prognostic significance of fibrosarcomatous transformation in dermatofibrosarcoma protuberans. Am J Surg Pathol 30:436–443
Ahuja R, Azar NF (2006) Orbital dermoids in children. Semin Ophthalmol 21:207–211
Alberola FT, Betlloch I, Montero LC, Nortes IB, Martinez NL, Paz AM (2010) Congenital tufted angioma: case report and review of the literature. Dermatol Online J 16:2
Alexander A, Samlowski WE, Grossman D et al (2003) Metastatic melanoma in pregnancy: risk of transplacental metastases in the infant. J Clin Oncol 21:2179–2186
Alomari AI (2006) The lollipop sign: a new cross-sectional sign of hepatic epithelioid hemangioendothelioma. Eur J Radiol 59:460–464
Antonescu CR, Dal Cin P, Nafa K et al (2007) EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 46:1051–1060
Balch CM, Gershenwald JE, Soong SJ et al (2009) J Clin Oncol 27(36):6199–6206. Epub 2009 Nov 16
Ball EA, Hussain M, Moss AL (2005) Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol 30:259–260
Barnhill RL, Flotte TJ, Fleischli M, Perez-Atayede A (1995) Cutaneous melanoma and atypical Spitz tumors in childhood. Cancer 76:1833–1845
Bartlett SP, Lin KY, Grossman R, Katowitz J (1993) The surgical management of orbitofacial dermoids in the pediatric patient. Plast Reconstr Surg 91:1208–1215
Bastian BC, Wesselmann U, Pinkel D, Leboit PE (1999) Molecular cytogenetic analysis of Spitz nevi shows clear differences to melanoma. J Invest Dermatol 113:1065–1069
Bastian BC, Kashani-Sabet M, Hamm H et al (2000) Gene amplifications characterize acral melanoma and permit the detection of occult tumor cells in the surrounding skin. Cancer Res 60:1968–1973
Bauer M, Reaman GH, Hank JA et al (1995) A phase II trial of human recombinant interleukin-2 administered as a 4-day continuous infusion for children with refractory neuroblastoma, non-Hodgkin’s lymphoma, sarcoma, renal cell carcinoma, and malignant melanoma. A Childrens Cancer Group study. Cancer 75:2959–2965
Biggar RJ, Frisch M, Goedert JJ (2000) Risk of cancer in children with AIDS. AIDS-Cancer Match Registry Study Group. JAMA 284:205–209
Billings SD, Folpe AL (2004) Cutaneous and subcutaneous fibrohistiocytic tumors of intermediate malignancy: an update. Am J Dermatopathol 26:141–155
Bleyer AOLM, Barr R, Ries LA (2006) Cancer epidemiology in older adolescents and young adults 15 to 29 years of age including SEER incidence and survival: 1975–2000. National Cancer Institute, Bethesda
Bono A, Ferrari A (2005) Early diagnosis remains the most reliable way to cure children with melanoma. Pediatr Blood Cancer 45(3):355
Bono A, Maurichi A, Moglia D et al (2001) Clinical and dermatoscopic diagnosis of early amelanotic melanoma. Melanoma Res 11:491–494
Brenn T, McKee PHM (2008) Melanoma in children and adolescents. Diagn Histopathol 14:18–27
Brose MS, Volpe P, Feldman M et al (2002) BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 62:6997–7000
Busam KJ, Murali R, Pulitzer M et al (2009) Atypical spitzoid melanocytic tumors with positive sentinel lymph nodes in children and teenagers, and comparison with histologically unambiguous and lethal melanomas. Am J Surg Pathol 33:1386–1395
Butter A, Hui T, Chapdelaine J et al (2005) Melanoma in children and the use of sentinel lymph node biopsy. J Pediatr Surg 40(5):797–800
Byun M, Abhyankar A, Lelarge V et al (2010) Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med 207:2307–2312
Calabrò L, Di Giacomo AM, Altomonte M et al (2007) Primary hepatic epithelioid hemangioendothelioma progressively responsive to interferon-alpha: is there room for novel anti-angiogenetic treatments? J Exp Clin Cancer Res 26(1):145–150
Ceballos PI, Ruiz-Maldonado R, Mihm MC Jr (1995) Melanoma in children. N Engl J Med 332:656–662
Cerroni L, Barnhill R, Elder D et al (2010) Melanocytic tumors of uncertain malignant potential: results of a tutorial held at the XXIX symposium of the international society of dermatopathology in Graz, October 2008. Am J Surg Pathol 34:314–326
Chao MM, Schwartz JL, Wechsler DS, Thornburg CD, Griffith KA, Williams JA (2005) High-risk surgically resected pediatric melanoma and adjuvant interferon therapy. Pediatr Blood Cancer 44:441–448
Checketts SR, Hamilton TK, Baughman RD (2000) Congenital and childhood dermatofibrosarcoma protuberans: a case report and review of the literature. J Am Acad Dermatol 42:907–913
Chien CR, Chang YL, Lin DT, Yang RS (2007) Excellent survival of pediatric dermatofibrosarcoma protuberans in Taiwanese. Pediatr Surg Int 23:211–214
Coffin CM, Dehner LP (1993) Vascular tumors in children and adolescents: a clinicopathological study of 228 tumors in 222 patients. Pathol Annu 1:97–120
Conti EMS, Cercato MC, Gatta G et al (2001) Childhood melanoma in Europe since 1978: a population-based survival study. Eur J Cancer 37:780–784
Costa MJ, Weiss SW (1990) Angiomatoid malignant fibrous histiocytoma. A follow-up study of 108 cases with evaluation of possible histologic predictors of outcome. Am J Surg Pathol 14:1126–1132
Curtis RE, Rowlings PA, Deeg HJ et al (1997) Solid cancers after bone marrow transplantation. N Engl J Med 336:897–904
Danietti M, Ferrari A, Frigerio S et al (2009) Cutaneous melanoma on childhood and adolescence shows frequent loss of INK4A and gain of KIT. J Invest Dermatol 129(7):1759–1768
Deyrup AT, Lee VK, Hill CE et al (2006) Epstein-Barr virus-associated smooth muscle tumors are distinctive mesenchymal tumors reflecting multiple infection events: a clinicopathologic and molecular analysis of 29 tumors from 19 patients. Am J Surg Pathol 30:75–82
Easton DF, Eeles RA (2008) Genome-wide association studies in cancer. Hum Mol Genet 17(R2):R109–R115
Enzinger FM (1979) Angiomatoid malignant fibrous histiocytoma: a distinct fibrohistiocytic tumor of children and young adults simulating a vascular neoplasm. Cancer 44:2147–2157
Enzinger FM, Zhang RY (1988) Plexiform fibrohistiocytic tumor presenting in children and young adults. An analysis of 65 cases. Am J Surg Pathol 12:818–826
Euvrard S, Kanitakis J, Claudy A (2003) Skin cancers after organ transplantation. N Engl J Med 348:1681–1691
Fahrtash F, McCahon E, Arbuckle S (2010) Successful treatment of kaposiform hemangioendothelioma and tufted angioma with vincristine. J Pediatr Hematol Oncol 32:506–510
Fanburg-Smith JC, Miettinen M (1999) Angiomatoid “malignant” fibrous histiocytoma: a clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum Pathol 30:1336–1343
Fears TR, Bird CC, Guerry D et al (2002) Average midrange ultraviolet radiation flux and time outdoors predict melanoma risk. Cancer Res 62(14):3992–3996
Ferrari A, Casanova M, Meazza C et al (2001) Vascular tumors in pediatric age. The experience of the Italian and German Cooperative Group. Ital J Pediatr 27:774–778
Ferrari A, Casanova M, Bisogno G et al (2002) Malignant vascular tumors in children and adolescents: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Med Pediatr Oncol 39:109–114
Ferrari A, Bono A, Baldi M, Collini P, Casanova M, Pennacchioli E et al (2005) Does melanoma behave better in younger children than in adults? A retrospective study on 33 cases of childhood melanoma from a single institution. Pediatrics 115:649–654
Fiore M, Miceli R, Mussi C et al (2005) Dermatofibrosarcoma Protuberans Treated at a Single Institution: a surgical disease with a high cure rate. J Clin Oncol 23:7669–7675
Flaherty KT, Puzanov I, Kim KB et al (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363:809–819
Friedman DL, Whitton J, Leisenring W et al (2010) Subsequent neoplasms in 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst 102:1083–1095
Gerami P, Jewell SS, Morrison LE et al (2009) Fluorescence in situ hybridization (FISH) as an ancillary diagnostic tool in the diagnosis of melanoma. Am J Surg Pathol 33:1146–1156
Gibbs P, Moore A, Robinson W et al (2000) Pediatric melanoma: are recent advances in the management of adult melanoma relevant to the pediatric population. J Pediatr Hematol Oncol 22:428–432
Giblin AV, Clover AJ, Athanassopoulos A, Budny PG (2007) Pyogenic granuloma – the quest for optimum treatment: audit of treatment of 408 cases. J Plast Reconstr Aesthet Surg 60:1030–1035
Gilchrest BA, Eller MS, Geller AC, Yaar M (1999) The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med 340(17):1341–1348
Gill M, Renwick N, Silvers DN, Celebi JT (2004) Lack of BRAF mutations in Spitz nevi. J Invest Dermatol 122:1325–1326
Godfrey JC, Vaughan MC, Williams JV (2003) Successful treatment of bowenoid papulosis in a 9-year-old girl with vertically acquired human immunodeficiency virus. Pediatrics 112:e73–e76
Gooskens SL, Oranje AP, van Adrichem LN et al (2010) Imatinib mesylate for children with dermatofibrosarcoma protuberans (DFSP). Pediatr Blood Cancer 55:369–373
Goto M, Miller RW, Ishikawa Y, Sugano H (1996) Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5:239–246
Haggstrom AN, Drolet BA, Baselga E et al (2006) Prospective study of infantile hemangiomas: clinical characteristics predicting complications and treatment. Pediatrics 118(3):882–887
Hale EK, Stein J, Ben-Porat L et al (2005) Association of melanoma and neurocutaneous melanocytosis with large congenital melanocytic naevi – results from the NYU-LCMN registry. Br J Dermatol 152:512–517
Hamre MR, Chuba P, Bakhshi S et al (2002) Cutaneous melanoma in childhood and adolescence. Pediatr Hematol Oncol 19:309–317
Handfield-Jones SE, Smith NP (1996) Malignant melanoma in childhood. Br J Dermatol 134:607–616
Hayward NK (2003) Genetics of melanoma predisposition. Oncogene 22:3053–3062
Hodi FS, O’Day SJ, McDermott DF et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723
Horn HM, Tidman MJ (2002) The clinical spectrum of dystrophic epidermolysis bullosa. Br J Dermatol 146:267–274
Isowa N, Hasegawa S, Mino M et al (2002) Mediastinal epithelioid hemangioendothelioma resected by Hemi-Plastron Window technique. Ann Thorac Surg 74:567–569
Jafarian F, McCuaig C, Kokta V, Laberge L, Ben Nejma B (2008) Dermatofibrosarcoma protuberans in childhood and adolescence: report of eight patients. Pediatr Dermatol 25:317–325
Jensen P, Hansen S, Moller B et al (1999) Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol 40:177–186
Jha P, Moosavi C, Fanburg-Smith JC (2007) Giant cell fibroblastoma: an update and addition of 86 new cases from the Armed Forces Institute of Pathology, in honor of Dr. Franz M. Enzinger. Ann Diagn Pathol 11:81–88
Karlsson P, Boeryd B, Sander B et al (1998) Increasing incidence of cutaneous malignant melanoma in children and adolescents 12–19 years of age in Sweden 1973–1992. Acta Derm Venereol 78:289–292
Kassi K, Kouame K, Allen W et al (2010) Squamous cell carcinoma secondary to Buruli ulcer: a clinical case report in a young girl. Bacteriol Virusol Parazitol Epidemiol 55:25–28
Kaste SC, Pappo AS, Jenkins JJ III, Pratt CB (1996) Malignant melanoma in children: imaging spectrum. Pediatr Radiol 26:800–805
Kehagias DT, Moulopoulos LA, Antoniou A, Psychogios V, Vourtsi A, Vlahos LJ (2000) Hepatic epithelioid hemangioendothelioma: MR imaging findings. Hepatogastroenterology 47:1711–1713
Kelleher MB, Iwatsuki S, Sheahan DG (1989) Epithelioid hemangioendothelioma of liver. Clinicopathological correlation of 10 cases treated by orthotopic liver transplantation. Am J Surg Pathol 13(12):999–1008
Kirkwood JM, Manola J, Ibrahim J, Sondak V, Ernstoff MS, Rao U (2004) A pooled analysis of eastern cooperative oncology group and intergroup trials of adjuvant high-dose interferon for melanoma. Clin Cancer Res 10:1670–1677
Kleinerman RA, Tucker MA, Tarone RE et al (2005) Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol 23:2272–2279
Kraemer KH, Lee MM, Scotto J (1987) Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 123:241–250
Kraemer KH, DiGiovanna JJ, Moshell AN, Tarone RE, Peck GL (1988) Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N Engl J Med 318:1633–1637
Kraemer KH, Levy DD, Parris CN et al (1994) Xeroderma pigmentosum and related disorders: examining the linkage between defective DNA repair and cancer. J Invest Dermatol 103:96S–101S
Kumar P, Ladas GP, Judson I, Nicholson AG (2003) Epithelioid hemangioendothelioma and other vascular mediastinal tumors: a role for alpha-2a interferon? Ann Thorac Surg 76(2):653
Lange JR, Palis BE, Chang DC, Soong SJ, Balch CM (2007) Melanoma in children and teenagers: an analysis of patients from the National Cancer Data Base. J Clin Oncol 25:1363–1368
Lazovich D, Vogel RI, Berwick M, Weinstock MA, Anderson KE, Warshaw EM (2010) Indoor tanning and risk of melanoma: a case-control study in a highly exposed population. Cancer Epidemiol Biomarkers Prev 19:1557–1568
Léauté-Labrèze C, Dumas de la Roque E, Hubiche T et al (2008) Propranolol for severe hemangiomas of infancy. N Engl J Med 358:2649–2651. doi:24
Leman JA, Evans A, Mooi W, Mackie RM (2005) Outcomes and pathological review of a cohort of children with melanoma. Br J Dermatol 152(6):1321–1323
Lohmann CM, Coit DG, Brady MS, Berwick M, Busam KJ (2002) Sentinel lymph node biopsy in patients with diagnostically controversial spitzoid melanocytic tumors. Am J Surg Pathol 26:47–55
Loria D, Matos E (2001) Risk factors for cutaneous melanoma: a case-control study in Argentina. Int J Dermatol 40(2):108–114
Luzar B, Calonje E (2010) Cutaneous fibrohistiocytic tumours – an update. Histopathology 56:148–165
Lyburn ID, Torreggiani WC, Harris AC et al (2003) Hepatic epithelioid hemangioendothelioma: sonographic, CT, and MR imaging appearances. AJR 180:1359–1364
Madariaga JR, Marino IR, Karavias DD et al (1995) Long-term results after liver transplantation for primary hepatic epithelioid hemangioendothelioma. Ann Surg Oncol 2(6):483–487
Makkar HS, Frieden IJ (2004) Neurocutaneous melanosis. Semin Cutan Med Surg 23:138–144
Marsciani A, Pericoli R, Alaggio R et al (2010) Massive response of severe infantile hepatic hemangioma to propanolol. Pediatr Blood Cancer 54(1):176
Matsumoto K, Nakanishi H, Seike T, Koizumi Y, Mihara K, Kubo Y (2001) Treatment of pyogenic granuloma with a sclerosing agent. Dermatol Surg 27:521–523
Mazereeuw-Hautier J, Hoeger PH, Benlahrech S et al (2010) Efficacy of propranolol in hepatic infantile hemangiomas with diffuse neonatal hemangiomatosis. J Pediatr 157(2):340–342
McArthur G (2007) Dermatofibrosarcoma protuberans: recent clinical progress. Ann Surg Oncol 14:2876–2886
McKee PH, Fletcher CD (1991) Dermatofibrosarcoma protuberans presenting in infancy and childhood. J Cutan Pathol 18:241–246
Melnik MK, Urdaneta LF, Al-Jurf AS, Foucar E, Jochimsen PR, Soper RT (1986) Malignant melanoma in childhood and adolescence. Am Surg 52:142–147
Mentzel T, Beham A, Calonje E et al (1997) Epithelioid hemangioendothelioma of skin and soft tissues: clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol 21:363
Mentzel T, Beham A, Katenkamp D, Dei Tos AP, Fletcher CD (1998) Fibrosarcomatous (“high-grade”) dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol 22:576–587
Milton GW, Shaw HM, Thompson JF, McCarthy WH (1997) Cutaneous melanoma in childhood: incidence and prognosis. Australas J Dermatol 38:44–48
Moehrle M, Breuninger H, Rocken M (2007) A confusing world: what to call histology of three-dimensional tumour margins? J Eur Acad Dermatol Venereol 21:591–595
Mones JM, Ackerman AB (2003) Melanomas in prepubescent children. Review comprehensively, critique historically, criteria diagnostically, and course biologically. Am J Dermatopathol 25:223–238
Mones JM, Ackerman AB (2004) “Atypical” Spitz’s nevus, “malignant” Spitz’s nevus, and “metastasizing” Spitz’s nevus: a critique in historical perspective of three concepts flawed fatally. Am J Dermatopathol 26:310–333
Moosavi C, Jha P, Fanburg-Smith JC (2007) An update on plexiform fibrohistiocytic tumor and addition of 66 new cases from the Armed Forces Institute of Pathology, in honor of Franz M. Enzinger, MD. Ann Diagn Pathol 11:313–319
Morton DL, Thompson JF, Cochran AJ et al (2006) Sentinel-node biopsy or nodal observation in melanoma. N Engl J Med 355:1307–1317
Muftuoglu M, Oshima J, von Kobbe C, Cheng WH, Leistritz DF, Bohr VA (2008) The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet 124:369–377
Navid F, Furman WL, Fleming M et al (2005) The feasibility of adjuvant interferon alpha-2b in children with high-risk melanoma. Cancer 103:780–787
Pappo AS, Rao BN, Cain A, Bodner S, Pratt CB (1997) Dermatofibrosarcoma protuberans: the pediatric experience at St. Jude Children’s Research Hospital. Pediatr Hematol Oncol 14:563–568
Paradela S, Fonseca E, Pita-Fernandez S et al (2010) Prognostic factors for melanoma in children and adolescents: a clinicopathologic, single-center study of 137 patients. Cancer 116:4334–4344
Pastore G, De Salvo GL, Bisogno G et al (2009) Evaluating the access to pediatric cancer care centres of children and adolescents with rare tumors in Italy: the TREP project. Pediatr Blood Cancer 53:152–155
Pearce MS, Parker L, Cotterill SJ et al (2003) Skin cancer in children and young adults: 28 years’ experience from the Northern Region Young Person’s Malignant Disease Registry, UK. Melanoma Res 13(4):421–426
Penel N, Bui B, Bay JO et al (2008) Phase II trial of weekly paclitaxel for unresectable angiosarcoma: the ANGIOTAX study. J Clin Oncol 26:5269–5274
Penel N, Marreaud S, Robin YM, Hohenberger P (2010) Angiosarcoma: state of the art and perspectives. Crit Rev Oncol Hematol. Nov. 3 (Epub ahead of print)
Penn I (1994) De novo malignancy in pediatric organ transplant recipients. J Pediatr Surg 29:221–228
Price VE, Fletcher JA, Zielenska M et al (2005) Imatinib mesylate: an attractive alternative in young children with large, surgically challenging dermatofibrosarcoma protuberans. Pediatr Blood Cancer 44:511–515
Pryor SG, Lewis JE, Weaver AL, Orvidas LJ (2005) Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg 132:938–942
Rao BN, Hayes FA, Pratt CB et al (1990) Malignant melanoma in children: its management and prognosis. J Pediatr Surg 25:198–203
Ribeiro RC, Rill D, Roberson PK et al (1993) Continuous infusion of interleukin-2 in children with refractory malignancies. Cancer 72:623–628
Ries L SM, Gurney J, Tamra T, Young J, Bunin G (1999) Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute SEER Program NIH Pub 99-4649, Bethesda
Rizzo C, Brightman L, Chapas A et al (2009) Outcomes of childhood hemangiomas treated with the pulsed dye laser with dynamic cooling: a retrospective chart analysis. Dermatol Surg 35:1947–1954
Roaten JB, Partrick DA, Bensard D et al (2005) Survival in sentinel lymph node-positive pediatric melanoma. J Pediatr Surg 40(6):988–992
Roudier-Pujol C, Enjolras O, Lacronique J et al (1994) Multifocal epithelioid hemangioendothelioma with partial remission after interferon alfa-2a treatment. Ann Dermatol Venereol 121(12):898–904
Rubin BP, Schuetze SM, Eary JF et al (2002) Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans. J Clin Oncol 20:3586–3591
Rutkowski P, Van Glabbeke M, Rankin CJ et al (2010) Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol 28:1772–1779
Saenz NC, Saenz-Badillos J, Busam K et al (1999) Childhood melanoma survival. Cancer 85:750–754
Sahin G, Palanduz A, Aydogan G et al (2010) Classic Kaposi sarcoma in 3 unrelated Turkish children born to consanguineous kindreds. Pediatrics 125:e704–e708
Salomao DR, Nascimento AG (1997) Plexiform fibrohistiocytic tumor with systemic metastases: a case report. Am J Surg Pathol 21:469–476
Sander B, Karlsson P, Rosdahl I et al (1999) Cutaneous malignant melanoma in Swedish children and teenagers 1973-1992: a clinico-pathological study of 130 cases. Int J Cancer 80:646–651
Schmid-Wendtner MH, Berking C, Baumert J et al (2002) Cutaneous melanoma in childhood and adolescence: an analysis of 36 patients. J Am Acad Dermatol 46:874–879
Scoazec JY, Lamy P, Degott C et al (1988) Epithelioid hemangioendothelioma of the liver. Diagnostic features and role of liver transplantation. Gastroenterology 94(6):1447–1453
Shah NC, Gerstle JT, Stuart M, Winter C, Pappo A (2006) Use of sentinel lymph node biopsy and high-dose interferon in pediatric patients with high-risk melanoma: the Hospital for Sick Children experience. J Pediatr Hematol Oncol 28:496–500
Sondak VK, Cimmino VM, Lowe LM, Dubay DA, Johnson TM (1999) Dermatofibrosarcoma protuberans: what is the best surgical approach? Surg Oncol 8:183–189
Spatz A, Ruiter D, Hardmeier T et al (1996) Melanoma in childhood: an EORTC-MCG multicenter study on the clinicopathological aspects. Int J Cancer 4:317–324
Spitz S (1948) Melanomas of childhood. Am J Pathol 24:591–609
Strouse JJ, Fears TR, Tucker MA, Wayne AS (2005) Pediatric melanoma: risk factor and survival analysis of the surveillance, epidemiology and end results database. J Clin Oncol 23:4735–4741
Su LD, Fullen DR, Sondak VK, Johnson TM, Lowe L (2003) Sentinel lymph node biopsy for patients with problematic spitzoid melanocytic lesions: a report on 18 patients. Cancer 97:499–507
Sybert V (1991) Six children with malignant melanoma. J Am Acad Dermatol 24:666–667
Taher A, Pushpanathan C (2007) Plexiform fibrohistiocytic tumor: a brief review. Arch Pathol Lab Med 131:1135–1138
Tate PS, Ronan SG, Feucht KA et al (1993) Melanoma in childhood and adolescence: clinical and pathological features of 48 cases. J Pediatr Surg 28:217–222
Tay YK, Weston WL, Morelli JG (1997) Treatment of pyogenic granuloma in children with the flashlamp-pumped pulsed dye laser. Pediatrics 99:368–370
Terrier-Lacombe MJ, Guillou L, Maire G et al (2003) Dermatofibrosarcoma protuberans, giant cell fibroblastoma, and hybrid lesions in children: clinicopathologic comparative analysis of 28 cases with molecular data – a study from the French Federation of Cancer Centers Sarcoma Group. Am J Surg Pathol 27:27–39
Terui K, Nakatani Y, Kambe M, Fukunaga M, Hishiki T, Saito T, Sato Y, Takenouchi A, Saito E, Ono S, Yoshida H (2010) Kaposiform hemangioendothelioma of the choledochus. J Pediatr Surg 45:1887–1889
Thway K (2008) Angiomatoid fibrous histiocytoma: a review with recent genetic findings. Arch Pathol Lab Med 132:273–277
Toyoda H, Ido M, Nakanishi K et al (2010) Multiple cutaneous squamous cell carcinomas in a patient with interferon gamma receptor 2 (IFN gamma R2) deficiency. J Med Genet 47:631–634
Tulpule A, Yung RC, Wernz J, Espina BM, Myers A, Scadden DT, Cabriales S, Ilaw M, Boswell W, Gill PS (1998) Phase II trial of liposomal daunorubicin in the treatment of AIDS-related pulmonary Kaposi’s sarcoma. J Clin Oncol 16:3369–3374
Uribe P, Wistuba II, Solar A et al (2005) Comparative analysis of loss of heterozygosity and microsatellite instability in adult and pediatric melanoma. Am J Dermatopathol 27:279–285
Wechsler J, Bastuji-Garin S, Spatz A et al (2002) Reliability of the histopathologic diagnosis of malignant melanoma in childhood. Arch Dermatol 138(5):625–628
Weiss SW, Enzinger FM (1982) Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer 50:970–981
Whiteman D, Valery P, McWhirter W, Green A (1995) Incidence of cutaneous childhood melanoma in Queensland, Australia. Int J Cancer 63:765–768
Whiteman DC, Milligan A, Welch J et al (1997) Germline CDKN2A mutations in childhood melanoma. J Natl Cancer Inst 89:1460
Wilson MW, Hoehn ME, Haik BG, Rieman M, Reiss U (2007) Low-dose cyclophosphamide and interferon alfa 2a for the treatment of capillary hemangioma of the orbit. Ophthalmology 114(5):1007–1011
Wollina U, Buslau M, Weyers W (2002) Squamous cell carcinoma in pansclerotic morphea of childhood. Pediatr Dermatol 19:151–154
Youl P, Aitken J, Hayward N et al (2002) Melanoma in adolescents: a case-control study of risk factors in Queensland, Australia. Int J Cancer 98:92–98
Yuksekkaya HA, Arikan C, Yazici A et al (2009) Successful treatment of a child having generalized Kaposi’s sarcoma after living donor liver transplantation with conversion to sirolimus. Pediatr Transplant 13:375–378
Ziegler JL, Katongole-Mbidde E (1996) Kaposi’s sarcoma in childhood: an analysis of 100 cases from Uganda and relationship to HIV infection. Int J Cancer 65:200–203
Zuckerman R, Maier JP, Guiney WB Jr et al (2001) Pediatric melanoma: confirming the diagnosis with sentinel node biopsy. Ann Plast Surg 46:394–399
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Pappo, A.S., Eigentler, T.K., Garbe, C., Collini, P., Ferrari, A., Rössler, J. (2012). Rare Tumors of the Skin and Subcutaneous Tissues. In: Schneider, D., Brecht, I., Olson, T., Ferrari, A. (eds) Rare Tumors In Children and Adolescents. Pediatric Oncology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-04197-6_43
Download citation
DOI: https://doi.org/10.1007/978-3-642-04197-6_43
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-04196-9
Online ISBN: 978-3-642-04197-6
eBook Packages: MedicineMedicine (R0)