
Abstract
The HIV and SIV Nef accessory proteins are potent enhancers of viral persistence and accelerate progression to AIDS in HIV-1-infected patients and non-human primate models. Although relatively small (27–35 kD), Nef can interact with a multitude of cellular factors and induce complex changes in trafficking, signal transduction, and gene expression that together converge to promote viral replication and immune evasion. In particular, Nef recruits several immunologically relevant cellular receptors to the endocytic machinery to reduce the recognition and elimination of virally infected cells by the host immune system, while simultaneously interacting with various kinases to promote T cell activation and viral replication. This review provides an overview on selected Nef interactions with host cell proteins, and discusses their possible relevance for viral spread and pathogenicity.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Human Immunodeficiency Virus
- Simian Immunodeficiency Virus
- Sooty Mangabey
- Viral Immune Evasion
- Enhance Virion Infectivity
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The year 2008 has witnessed a sober reassessment of the state-of-the-art in AIDS (acquired immune deficiency syndrome) research since the discovery of the human immunodeficiency virus (HIV) 25 years ago. Despite many seminal advances in the field, HIV remains an elusive target for eradicating treatment or effective vaccination. Key to the elusive nature of the virus is its ability to evade host or treatment pressures through genetic hypervariability, its integration into the host cell genome, and its persistence in latent reservoirs (Stevenson 2003). In addition, the virus benefits from its ability to interact with components of the infected cell and subvert the cell trafficking, signal transduction, and transcriptional machineries to its advantage: to facilitate virus infection, increase the production of fully infectious progeny viruses, avoid recognition by the immune system, and establish latency.
The HIV and SIV Nef accessory proteins are particularly adept at interacting with their host cell and inducing complex changes that promote efficient virus spread and persistence. Although originally named on the mistaken understanding that it negatively regulates virus transcription (Nef is an acronym for negative factor), Nef acts as a potent viral enhancer of primate lentiviral persistence.
Particular interest in Nef lies in early observations that correlate its expression with progression to AIDS. Rhesus monkeys infected with simian immunodeficiency virus (SIV) carrying a large deletion in the nef gene showed low viral loads and did not progress to simian AIDS (Kestler et al. 1991). Similarly, defective nef genes have been detected in several long-term survivors of HIV-1 infection with normal CD4+ T cell counts and very low viral loads (Kirchhoff et al. 1995; Deacon et al. 1995; Mariani et al. 1996; Salvi et al. 1998). As a result, much effort has been undertaken to identify the mechanisms by which Nef promotes viral persistence and accelerates progression to AIDS and to define the molecular interactions with the host cell that are involved.
Nef is unique to primate lentiviruses and present in all HIV and SIV strains characterized to date. Nef proteins have a molecular weight ranging from 27 to about 35 kD and are encoded by sequences extending from the 3’ end of the viral envelope (env) into part of the 3’ long-terminal repeat (LTR). Although the amino acid sequence and length of HIV-1 Nef is variable, several distinct conserved functional domains have been identified (Geyer et al. 2001). HIV-1, HIV-2 and SIV Nef proteins frequently share only about 30% amino acid identity, but most structural properties and the majority of functions are well preserved (reviewed in Kirchhoff et al. 2008).
The best conserved feature of Nef is its N-terminal myristoylation signal, required for membrane binding and critical for most Nef activities. The flexible myristoylated N-terminal anchor is followed by a flexible loop containing a conserved acidic cluster involved in Nef effects on trafficking and a proline-rich region, conserved in HIV-1 but not in HIV-2 nor some SIVs. The latter is involved in SH3-domain binding of tyrosine kinases and Nef effects on signaling (Saksela et al. 1995; Renkema and Saksela 2000; Collette et al. 2000). Other important interfaces for interaction with cellular proteins are a highly ordered and well-conserved globular core domain and a flexible loop near the C-terminus of Nef, which contains a dileucine-based sorting motif that functions as an endocytosis signal (Craig et al. 1998). The remarkable ability of Nef to interact efficiently with multiple cellular partners may be due to the high degree of flexibility within its folded structure (Geyer and Peterlin 2001).
According to the current literature, Nef may interact with as many as 60 cellular factors and affect the function of more than 180 proteins. These are listed in the web site of the National Library of Medicine: http://www.ncbi.nlm.nih.gov/RefSeq/ HIVInteractions/nef.html. All these interactions and effects could potentially be advantageous for the virus. However, most of them remain to be confirmed in virally infected primary T cells or macrophages. Moreover, the underlying mechanisms and the role in viral persistence and progression to AIDS have only been examined for a small proportion of these Nef–host cell interactions.
The aim of this review is to expose some of the reported interactions of Nef with its host cell, and to discuss their possible implications for viral persistence and progression to AIDS. Although an attempt has been made to categorize Nef–host cell interactions into those that facilitate immune evasion and those that directly enhance viral spread, it should be noted that most Nef interactions with its host cell cooperate to ensure the production of progeny virions and to generate an environment that facilitates viral spread.
2 Interactions Facilitating Viral Immune Evasion
2.1 CD8+ T Cell Evasion: MHC-I Down-Modulation
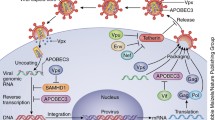
Like many other invading pathogens, HIV and SIV have developed multiple strategies to avoid elimination by the host immune system. One mechanism to reduce immune recognition of infected cells involves down-modulation of the major histocompatibility complex I (MHC-I) (Kerkau et al. 1989; Scheppler et al. 1989). The Nef protein is critical for this ability of HIV and SIV (Schwartz et al. 1996) and sufficient to protect infected primary T cells from killing by CD8+ cytotoxic T lymphocytes (CTL) (Collins et al. 1998). Although the exact mechanisms are still under investigation, consensus evidence indicates that Nef expression in infected cells leads to accumulation of MHC-I in the trans-golgi network (TGN) where it is redirected to TGN-associated endosomal compartments and to lysosomes for degradation (Roeth and Collins 2006). The literature supports two pathways for Nef-mediated down-modulation of MHC-I involving accelerated endocytosis of MHC-I from the plasma membrane (Greenberg et al. 1998a; Le Gall et al. 2000; Piguet et al. 2000) and disruption of normal anterograde transport of MHC-I from the Golgi to the cell surface (Le Gall et al. 2000; Swann et al. 2001; Kasper and Collins 2003; Roeth et al. 2004) (Fig. 1). The prevalence of one mechanism over the other may differ between T cell and non-T cell lines (Kasper and Collins 2003).

Schematic presentation of selected Nef functions and interactions in infected T cells. Nef interacts with HLA molecules, CD4, CD28, CXCR4, and CD3 to reduce their surface expression on infected CD4+ T cells, thereby reducing CTL lysis, suppressing cell migration, facilitating virus release and modulating signal transduction by the immunological synapse. Furthermore, Nef interacts with a variety of cellular kinases and other factors to modulate downstream signaling events. HIV-1 Nef promotes the activation of transcription factors, such as NF-AT, NF-κB, and AP-1, to induce the efficient transcription of the viral LTR promoter and of various cellular genes, e.g., those encoding for inflammatory cytokines, activation markers and death receptors. Furthermore, Nef down-modulates CD4 to promote virus release and to prevent superinfection and enhances virus replication and virion infectivity to directly promote virus spread. Please note that the presentation is preliminary and shows only a small proportion of all reported effects of Nef and that not all primate lentiviral Nefs perform all indicated functions
Nef interacts directly with the cytoplasmic tail of MHC-I, although this interaction is weak and may be transient or stabilized by other factors (Williams et al. 2002). An N-terminal α helical region, the polyproline repeat, and the acidic domain in HIV-1 Nef are involved in MHC-I down-modulation (Greenberg et al. 1998a; Mangasarian et al. 1999). Notably, however, some HIV-1 and SIV nef alleles lead to efficient MHC-I down-modulation although they contain alterations in these residues (Specht et al. 2008).
For rerouting of MHC-I to the lysosomes for degradation, Nef recruits the clathrin adaptor complex AP1 via its μ1 subunit and subsequently ß-COP, which are both implicated in endosomal trafficking and transport through the early secretory pathway, to the cytoplasmic tail of MHC-I (Roeth et al. 2004; Noviello et al. 2008; Schaefer et al. 2008). It is a matter of debate whether accelerated endocytosis of MHC-I from the cell surface requires an interaction between Nef and a coat protein called PACS-1 (phosphofurin acidic cluster sorting protein-1) (Piguet et al. 2000; Lubben et al. 2007; Blagoveshchenskaya et al. 2002). It has also been suggested that Nef-mediated endocytosis of MHC-I involves the ARF-6 (ADP-ribosylation factor 6) endocytic pathway (Blagoveshchenskaya et al. 2002), which is normally involved in the clathrin-independent trafficking of MHC-I between the plasma membrane and endosomal compartments, and PI3K (phosphoinositide 3-kinase) (Swann et al. 2001; Hung et al. 2007). However, the significance of these mechanisms requires further study (Larsen et al. 2004), and the exact mechanism of MHC-I down-modulation is still under investigation (Atkins et al. 2008; Schaefer et al. 2008).
It has been clearly shown that down-modulation of MHC-I by Nef contributes to the ability of SIV to avoid CTL responses in vivo. A point mutation of Y223F near the C-terminus of SIVmac Nef that selectively disrupts the effect on MHC-I reverted within 4 weeks after infection, shortly after the peak of the CTL response (Münch et al. 2001). Furthermore, macaques infected with SIVmac mutants containing difficult-to-revert Nef mutations specifically eliminating MHC-I down-modulation exhibited higher levels of CD8+ T cell responses and showed compensatory mutations in Nef that restored MHC-I down-modulation (Swigut et al. 2004). In support of a relevant role of Nef-mediated down-modulation of MHC-I for viral immune evasion and effective persistence in HIV-1-infected individuals, it has been shown that nef alleles from non-progressor perinatally infected children were less efficient in MHC-I down-modulation than those from rapid progressors (Casartelli et al. 2003). The efficiency of CTL responses in infected patients seems to exert a selective pressure on the ability of Nef to down-modulate MHC-I (Carl et al. 2001), and the ability of Nef to down-modulate MHC-I correlated positively with the breadth of the HIV-1-specific CTL response (Lewis et al. 2008). Finally, unusually strong CTL responses have been detected in individuals infected with Nef defective HIV-1 strains (Dyer et al. 1999). Thus, together with the high variability of HIV and SIV leading to the emergence of CTL escape variants and other factors, the ability of Nef to down-modulate MHC-I provides an explanation why CTL responses are usually unable to effectively control viral replication.
2.2 NK Cell Evasion: Selective Down-Modulation of HLA-A and –B
Regulation of cell surface expression of MHC-I is a mechanism used by a number of viruses to evade recognition by CTL (for example herpes viruses, papillomaviruses, HIV). As a counteractive measure, natural killer (NK) cells can gage the level of MHC-I expressed on cell surfaces and preferentially lyse cells that lack MHC-I. Under normal conditions, NK cell cytotoxicity is blocked by specific recognition of MHC-I molecules by inhibitory NK cells receptors (iNKRs). Reduced expression of MHC-I molecules on the surface of infected cells results in reduced engagement of iNKRs and triggers NK-cell-mediated cytolysis. Thus, down-modulation of MHC-I by HIV-1 Nef (among other viral proteins) should expose infected cells to lysis by NK cells. However, evidence shows that HIV-1 Nef down-modulates class I MHC proteins selectively, reducing surface expression of HLA-A and -B, which are recognized by the majority of CTL, but not of HLA-C and -E, which are recognized by iNKRs (LeGall et al. 1998; Cohen et al. 1999). Selective down-modulation of class I proteins by Nef is accounted for by differences in the cytoplasmic tail of MHC-I molecules, and residues Y320, A323, and/or D327 important for Nef-dependent down-modulation of HLA-A and -B are mutated in HLA-C and -E (LeGall et al. 1998; Cohen et al. 1999). The ability of Nef to selectively down-modulate HLA-A and -B reduces the ability of NK cells to kill HIV-infected cells despite reduced MHC-I surface molecules (Bonaparte and Barker 2003) and is a well conserved property among primate lentiviruses (Specht et al. 2008).
Nevertheless, studies have demonstrated that 30% or more of NK cells present in the peripheral blood do not express any receptors able to bind to HLA-C or -E (Bonaparte and Barker 2004; Mavilio et al. 2003), and that these NK cells have a greater ability to kill CD4+ T cells infected with HIV in which HLA-A and -B are decreased (Bonaparte and Barker 2004). It is therefore very likely that HIV-mediated perturbation of NK cell function (through aberrant expression and function of inhibitory receptors, defective cytokine production, and preferential expansion of NK cell subsets) also plays a role in the decreased ability of NK cells to kill HIV-infected cells. A recent work showed that Nef-pulsed dendritic cells (DCs) modulate NK cell effector function, inhibiting cytotoxic NK cell function while stimulating the pro-inflammatory cytokine-producing NK fraction (Quaranta et al. 2007).
Interestingly, although HLA-C presents self-peptides to NK cells to inhibit cell killing, it also has the ability to present viral peptides to CTL and thus restrict HIV-1 infection (Goulder et al. 1997; Adnan et al. 2006). Moreover, a single nucleotide polymorphism (SNP) upstream of the HLA-C locus associates with increased HLA-C expression and lower viral load at set-point (Fellay et al. 2007). Therefore, the resistance of HLA-C to Nef-mediated down-modulation could offer a promising opportunity for vaccine developments targeting HLA-C-restricted CTL responses.
2.3 Restricting MHC-II Antigen Presentation: Ii up-Modulation
MHC class II molecules, expressed chiefly on B cells, macrophages, and DCs, are specialized in exogenous antigen presentation to CD4+ T cells and are synthesized in the endoplasmic reticulum (ER) together with the invariant chain (Ii or CD74). Ii caps the MHC-II peptide binding site during its transport to endosomal compartments, where acidic pH leads to proteolytic cleavage of Ii thus allowing loading of appropriate peptides on the MHC-II groove and subsequent transport of mature MHC-II-antigen complexes to the cell surface (reviewed by Rocha and Neefjes 2008). Interestingly, a fraction of MHC II-Ii complexes reaches endosomes not directly from the ER but rather by rapid internalization from the cell surface (Roche et al. 1993). It seems that this MHC-II-Ii fraction shuttles between the endosomes and plasma membrane before peptide loading in endosomes, undergoing repeated cycles of surface delivery and rapid internalization (Lindner 2002) via adaptor protein 2 (AP-2)-dependent endocytosis (Dugast et al. 2005).
Expression of HIV-1 Nef may perturb MHC-II-restricted antigen presentation by up-modulation of Ii cell surface expression (Stumptner et al. 2001; Stumptner et al. 2003). Indeed, stable expression of Ii hampers peptide presentation (Roche et al. 1992; Bertolino and Rabourdin 1996), and Nef-mediated up-modulation of surface Ii might therefore contribute to impaired helper T cell responses observed in AIDS patients (Norris and Rosenberg 2002). Nef interacts directly with both AP-2 and Ii in a dileucine-dependent manner (Toussaint et al. 2008), and it has been suggested that Nef may up-modulate Ii because both compete for AP-2 binding (Mitchell et al. 2008). More recently, however, it has been suggested that Ii up-modulation by Nef is due to impaired AP-2-mediated endocytosis rather than direct competition for AP-2 (Toussaint et al. 2008). Efficient Nef-mediated up-modulation of surface Ii is a well conserved property of primate lentiviruses (Schindler et al. 2003) and can be observed at very low levels of Nef expression (Stumptner et al. 2001). Significant Nef-mediated up-regulation of Ii was also observed in HIV-1-infected macrophages (Schindler et al. 2007a). Further studies are required to obtain definitive proof but our current knowledge suggests that the effect of Nef on Ii represents an important viral immune evasion mechanism in vivo in HIV-1-infected individuals.
2.4 Modulation of Signaling from the Cell Surface
2.4.1 Down-Modulation of TCR-CD3
The T cell receptor (TCR) is a heterodimeric protein consisting of α and β chains. Its function as a receptor is entirely dependent on its association with the CD3 complex, comprised of four transmembrane protein chains: γ, δ, ε, and ζ (zeta), which mediates signal transduction after antigen recognition. All four CD3 chains have intracellular immunoreceptor tyrosine-based activation motifs (ITAM) that become phosphorylated upon receptor ligation, enabling their interaction with cytoplasmic signaling proteins.
Early work showed that the central region of SIVmac239, SIVsmm, and HIV-2 Nef can directly associate with the TCR zeta chain and that this interaction correlates with their ability to down-modulate CD3 from the cell surface (Bell et al. 1998; Howe et al. 1998; Schaefer et al. 2002). Although Nef’s endocytic motifs do not appear to be involved, TCR-CD3 endocytosis occurs via the AP-2 clathrin adaptor pathway (Schaefer et al. 2002; Swigut et al. 2003). HIV-1 Nef, on the other hand, is unable to induce CD3 down-modulation. One study reported that HIV-1 Nef maintains some ability to interact with TCRζ (Xu et al. 1999), whereas several others found that only SIV and HIV-2, but not HIV-1, Nef proteins show this association (Bell et al. 1998; Howe et al. 1998; Schaefer et al. 2002).
To date, SIVs have been detected in about 40 African non-human primate species (reviewed in Pandrea et al. 2008), and phylogenetic studies indicate that SIVs from two of these species, chimpanzees and sooty mangabeys, have been transmitted to humans and gave rise to HIV-1 and HIV-2, respectively (Hahn et al. 2000). A recent study of a wide range of primate lentiviral Nef proteins revealed that the ability of SIVmac239, SIVsmm, and HIV-2 Nefs to down-modulate CD3 is shared by the great majority of primate lentiviruses, and blocks the responsiveness of infected T cells to activation (Schindler et al. 2006). Only Nef proteins from HIV-1 and its SIV counter parts failed to down-modulate TCR-CD3 and to suppress T cell activation and programmed cell death. The structural basis for this fundamental difference in the ability of primate lentiviral Nef proteins to modulate CD3 remains elusive since the core region of Nef that is critical for the interaction with the TCR zeta chain (Schaefer et al. 2002; Swigut et al. 2003) is generally well conserved.
While HIV-1 infection in humans is typically associated with high levels of immune activation and progressive CD4+ T cell depletion, natural SIV infections, such as by SIVsmm in sooty mangabeys and by SIVagm in African green monkeys, do not bring about generalized chronic immune activation or disease. The effect of HIV-1 Nef versus SIVsmm/SIVagm Nef on CD3 surface expression and T cell activation suggests a protective role for SIV Nef against immune activation (Schindler et al. 2006), and TCR-CD3 down-modulation may help the natural hosts of SIV to maintain stable numbers of CD4+ T cells (Schindler et al. 2008). It is interesting, however, that even profound depletion of CD4+ T cells is not associated with disease in naturally infected sooty mangabeys, and it has been proposed that attenuated immune activation following acute viral infection protects these animals from progressing to AIDS (Gordon et al. 2007; Milush et al. 2007).
2.4.2 Down-Modulation of the CD4 Receptor
CD4 assists signal transduction and T cell activation after TCR ligation. It is involved in clustering at the immunological synapse and binding to MHC-II on antigen presenting cells (APCs). CD4 is also the primary receptor for HIV and SIV entry. In support of a critical role for virus replication, three HIV-1 encoded proteins, Vpu, Env, and Nef, ensure that CD4 molecules are kept away from the cell surface upon HIV-1 infection.
It has long been known that Nef down-modulates CD4 (Garcia and Miller 1991) and the underlying mechanisms have been extensively investigated (reviewed by Lama 2003; Roeth and Collins 2006). Notably, Nef uses distinct surfaces to down-regulate MHC-I, CD3 and CD4 from the cell surface. Nef-mediated down-modulation of CD4 involves a dileucine motif in the membrane-proximal cytoplasmic domain of CD4 (Aiken et al. 1994) and an intact dileucine motif, two diacidic motifs, and a hydrophobic pocket in Nef (Mangasarian et al. 1999; Bresnahan et al. 1998; Craig et al. 1998; Piguet et al. 1998; Lindwasser et al. 2008).
Expression of Nef leads to the endocytosis of surface CD4 by recruitment of the AP-2 clathrin adaptor complex, which directs the receptor to lysosomes for degradation (Aiken et al. 1994; Piguet et al. 1998; Greenberg et al. 1998b; Bresnahan et al. 1998; Craig et al. 1998). Recent findings suggest that ubiquitination of Nef may play a role in CD4 down-regulation (Jin et al. 2008). In contrast to Nef, Vpu and Env are expressed during the late stage of the viral life cycle and interfere with the transport of newly synthesized CD4 to the cell membrane rather than down-modulating CD4 molecules already present at the cell surface (Malim and Emerman 2008). However, intracellular retention mechanisms may also contribute to Nef-mediated down-modulation of CD4 (Rose et al. 2005). In either case, down-modulation of CD4 by Nef is highly effective and most likely important for efficient viral persistence in vivo (Brenner et al. 2006).
The role of CD4 in mediating T cell activation following receptor ligation suggests that down-modulation of CD4 by Nef may be advantageous for the virus because it limits T cell activation. However, since essentially all primate lentiviral Nef proteins down-modulate CD4 with high efficiency, while having very different effects on T cell responsiveness to activation (Schindler et al. 2006), it is likely that other consequences of low CD4 cell surface levels discussed Sect. 3.2.3 are of higher physiological relevance.
2.4.3 Down-Modulation of Co-Stimulatory Molecules
The complete activation of a T cell following antigen-MHC recognition requires a second co-stimulatory signal, provided for predominantly by the CD28 receptor on T cells and the B7 (CD80/CD86) ligand on APCs. Absence of this co-stimulatory signal results in the suppression of the immune response, and induces antigen-specific tolerance and T cell anergy.
In T cells, the Nef proteins of SIVmac239, SIVsmm and HIV-2, and to a lesser extent of HIV-1, down-modulate surface CD28 expression by binding to the cytoplasmic domain of CD28 and accelerating its endocytosis via the AP2 clathrin adaptor (Swigut et al. 2001; Bell et al. 2001; Münch et al. 2005). Thus, the mechanism is similar to CD4 and CD3 down-modulation, although the ability of Nef to down-regulate CD28 can be genetically separated from these functions (Swigut et al. 2001).
In APCs, the HIV-1 Nef protein has been shown to redirect the co-stimulatory molecules CD80 and CD86 away from the surface by binding to their cytoplasmic tails and rerouting them to the Golgi apparatus by a clathrin- and dynamin-independent actin-based endocytic pathway that seems to involve the activation of c-src and Rac (Chaudhry et al. 2005, 2007, 2008). By down-modulating co-stimulatory molecules, as well as other surface receptors, HIV-1 Nef manipulates the functional interaction between T cells and APCs (discussed further in Sect. 2.4.5) to impede the mounting of an effective immune response against the virus.
2.4.4 CXCR4 Down-Modulation
In addition to CD4, chemokine receptors are also essential as co-receptors for HIV-1 entry into target cells (Deng et al. 1996; Feng et al. 1996). Transmitted HIV-1 strains, and those that persist during chronic infection, generally use the CCR5 co-receptor, while HIV-1 variants that utilize CXCR4 or are dual tropic are observed in about 50% of all AIDS patients. HIV-2, on the other hand, is more promiscuous in its use of chemokine co-receptors, and most SIVs use only CCR5, but not CXCR4, as entry co-factors (Marx and Chen 1998).
Some HIV-2 and SIV Nef proteins effectively down-modulate CXCR4 to inhibit T cell migration to the CXCR4 natural ligand, the chemokine stromal-derived factor 1 (SDF-1), whereas HIV-1 Nefs display only weak activity (Choe et al. 2002; Hrecka et al. 2005; Venzke et al. 2006; Wildum et al. 2006). Similar to its effects on CD4, CD28, and CD3, Nef seems to down-modulate CXCR4 by recruiting it to sites of the AP-2 clathrin-adaptor-dependent endocytosis (Hrecka et al. 2005). Notably, HIV-1 and SIVmac239 Nefs also inhibit chemotaxis by binding to the guanine exchange factor DOCK2-ELMO1, a key activator of the Rho GTPase Rac in antigen- and chemokine-initiated signaling pathways (Janardhan et al. 2004; Hrecka et al. 2005).
The chemotaxis of T cells mediated by SDF-1 through CXCR4 is essential for trafficking of T cells during development and the initiation of immune responses, with recruitment of lymphocytes to lymphoid tissues. CXCR4 down-modulation also reduces superinfection of infected cells (Venzke et al. 2006; Wildum et al. 2006). However, this effect is obviously of limited relevance since the primate lentiviruses that down-modulate CXCR4 with the highest efficiency do not utilize it as an entry cofactor. Nef-mediated impairment of T cell chemotaxis, with or without affecting CXCR4 surface expression levels, likely reduces contact with APCs and contributes to immune evasion.
2.4.5 Effects on the Immunological Synapse
As outlined above, Nef most likely forms trimeric complexes with AP-2 and various receptors, i.e., CD3, CD4, CD28, and CXCR4, to target them for endocytosis via AP-2 clathrin adaptors and subsequent degradation in lysosomes. Notably, Nef can use distinct surfaces to interact with AP-2 and to recruit different receptors. As a consequence, all these Nef activities are genetically separable (Akari et al. 2000; Craig et al. 1998; Iafrate et al. 1997; Swigut et al. 2000, 2001), and can (at least in part) be independently adapted to the specific host environment to optimize viral spread (Carl et al. 2001; Patel et al. 2002). Another interesting aspect is that some conserved functions are mediated by different domains in Nef proteins derived from various lineages of HIV and SIV (Swigut et al. 2000; Bresnahan et al. 1999; Hua and Cullen 1997; Lock et al. 1999) suggesting that they evolved independently during primate lentiviral evolution.
Many of the receptors modulated by Nef are involved in the formation of the immunological synapse (Fig. 1), which requires the interaction of the TCR-CD3 complex on T cells with the Ag/MHC-II complex expressed by APCs and of co-stimulatory and adhesion molecules on both cells. As described above, HIV-1 Nefs interfere with this process by modulating MHC-II, Ii, CD4, and, to some extent, CD28. It has been suggested that the ability of HIV-1 Nef to deregulate the function of the immunological synapse may reduce T cell activation and help to prevent damaging high levels of immune activation (Fackler et al. 2007). It is obvious, however, that HIV-1 Nef does a poor job in protecting the infected host because HIV-1 infection is almost invariably associated with high levels of chronic immune activation and progression to AIDS in infected humans. In comparison, HIV-2 and most SIV Nefs impair synapse formation more severely because they also down-regulate TCR-CD3 and are usually more effective in down-regulating CD28 and CXCR4 (Hrecka et al. 2005; Schindler et al. 2006). Cell cultures infected with viruses expressing HIV-1 Nefs are characterized by high levels of activation and apoptosis, whereas PBMCs expressing HIV-2, SIVsmm, or SIVagm Nefs show low levels of activation and cell death. The distinct characteristics of these in vitro cultures are similar to the documented different characteristics of HIV-1, HIV-2, and SIVsmm or SIVagm infection in vivo (Pandrea et al. 2008). Altogether, these findings suggest that HIV-1 Nefs dysregulate the functional interaction of infected T cells with APCs, whereas those of most other primate lentiviruses may disrupt it entirely. The possible importance of this differential ability of primate lentiviruses to disrupt the immunological synapse has been addressed in a recent review (Kirchhoff et al. 2008).
3 Interactions Supporting SIV and HIV Replication
Besides performing multiple functions that facilitate viral immune evasion, Nef also modulates the activation status of the infected T cells and enhances the infectivity of progeny virions to promote viral replication (Fig. 1). As described in the previous chapters, Nef down-modulates several receptors to dysregulate or disrupt antigen-specific signaling by the TCR-CD3 complex. However, HIV-1 Nef proteins also interact with numerous cellular factors to increase the responsiveness of virally-infected T cells to stimulation. The induction of downstream signaling pathways leads to the activation of transcription factors that increase the expression of the HIV-1 provirus as well as of many cellular genes. In addition, HIV and SIV Nefs also act at the latest stage of the virus life cycle by enhancing the infectivity of progeny virions. The effect of Nef on viral replication is most pronounced in primary T cell cultures, particularly if these are infected prior to stimulation (Miller et al. 1994; Chowers et al. 1994; Schwartz et al. 1995) and is most likely dependent on a variety of Nef effects, such as modulation of signal transduction pathway, induction of transcription factors and cellular activation, CD4 down-modulation, and enhancement of virion infectivity.
3.1 Enhancement of Virus Production
3.1.1 Subversion of T Cell Signaling Pathways
Although HIV-1 Nef down-modulates CD4 and (to some extent) CD28, it enhances the responsiveness of T cells to stimulation (Skowronski et al. 1993; Schrager and Marsh 1999; Fenard et al. 2005; Fortin et al. 2004; Wang et al. 2000) and interacts with a large number of cellular signaling proteins (reviewed in Greenway et al. 2003; Renkema and Saksela 2000). While the physiological relevance of these interactions is often poorly understood, they may lead to the induction of various signaling pathways and the activation of transcription factors (Maninnen et al. 2000; Wang et al. 2000; Fortin et al. 2004) that enhance viral and cellular gene expression (Arendt and Littman 2001; Simmons et al. 2001). Thus, HIV-1 Nef may partly uncouple T cell activation from the “normal” antigen-dependent interaction with APCs to increase virus production (Fig. 1). Notably, Nef can manipulate the infected cells very rapidly, as it is abundantly produced early during the viral life cycle. Moreover, it has been shown that selective transcription of nef and tat in quiescent cells can increase T cell activation and viral replication even prior to viral integration into the host cell genome (Wu and Marsh 2001).
It is well established that the PxxP motif in HIV-1 Nef interacts with the SH3 domains of a number of tyrosine kinases, including Lck, Fyn, Hck, Lyn, and c-Src itself (Saksela et al. 1995; Greenway et al. 1996; Collette et al. 1996; Baur et al. 1997). Lck and Fyn are the first recruited kinases upon T cell receptor ligation and Lck also mediates signaling from CD4, CD8, and IL-2 receptors. In yeast, Nef activated the kinase activities of Hck, Lyn, and c-Src but not of Fyn, Lck, or Yes (Trible et al. 2006). It is conceivable that the well-conserved albeit frequently low-affinity interactions of HIV-1 Nef with various Src kinases contribute to its effect on signal transduction and T cell activation (Renkema and Saksela 2000). It has also been reported that Nef interacts with the SH3 domain of VAV, a Rac1 guanine nucleotide exchange factor (GEF), possibly to trigger the JNK/SAPK signaling cascade and to induce rearrangements of the cytoskeleton (Fackler et al. 1999). However, another study using a comprehensive proteomics approach to directly identify Nef interacting proteins did not detect several of the proteins mentioned above including VAV, but suggests that binding of Nef to the DOCK2–ELMO1 complex, a key activator of Rac, plays a major role in its ability to inhibit chemotaxis and to promote T cell activation (Janardhan et al. 2004).
Nef can be phosphorylated on serine residues and associates with a number of serine and threonine kinases (Renkema and Saksela 2000). Particularly, the interaction of Nef with the p21-activated serine–threonine kinase 2 (PAK-2) has been extensively investigated (Sawai et al. 1994; Lu et al. 1996). PAK-2 is usually activated by Rac1 and Cdc42 and involved in the regulation of several cellular processes, such as cytoskeleton rearrangement, cell morphology, motility, apoptosis, and gene transcription (Daniels and Bokoch 1999; Chu et al. 2004). It has been proposed that the interaction of PAK-2 with Nef plays a role in T cell activation, viral replication, apoptosis, and progression to AIDS (Lu et al. 1996; Wiskerchen and Cheng-Mayer 1996; Fackler et al. 2000; Linneman et al. 2002; Chu et al. 2004). However, data obtained using Nef mutants selectively impaired in this interaction (Agopian et al. 2006) failed to detect definitive effects on T cell activation, viral replication, and apoptosis (Schindler et al. 2007b). Further effects of HIV-1 Nef involve the modulation of additional effectors and signaling pathways such as protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) families (Lu et al. 1996; Smith et al. 1996; Greenway et al. 1996; Yang and Gabuzda 1999; Wolf et al. 2008), interactions with the Ras-Raf-MAP kinase pathway (Maninnen et al. 2000; Hodge et al. 1998), and modulation of calcium signaling (Skowronski et al. 1993; Baur et al. 1994; Maninnen and Saksela 2002).
The biological consequences of most of these Nef interactions are still poorly understood. Taken together, however, our current knowledge suggests that HIV-1 Nef recruits various signaling proteins to the inner cell membrane and lipid rafts and, by modulating their catalytic activity and bringing them into close proximity, primes T cells for activation. In fact, it has been shown Nef alone can trigger T cell activation signaling pathways, inducing a transcriptional program that is highly similar to that of anti-CD3 T cell activation at least in Jurkat T cells (Simmons et al. 2001). In primary HIV-1 infected T cells, however, expression of Nef is usually not sufficient to induce T cell activation and viral replication but rather increases their responsiveness to stimulation (Djordjevic et al. 2004; Fenard et al. 2005; Fortin et al. 2004; Wang et al. 2000). The exceptions are SIV nef alleles that contain an additional SH2 domain and are highly active in enhancing T cell activation and associated with an acute lethal disease in infected rhesus monkeys (Du et al. 1995). Typically, Nef expression in HIV-1-infected T cells increases the induction of various transcription factors, such as NFAT, NFκB, and AP1 Activation Protein 1 upon stimulation with various agents (Wang et al. 2000; Manninen et al. 2000; Manninen et al. 2001; Fortin et al. 2004). Since HIV-1 requires T cell activation for efficient replication, Nef’s subversion of T cell signaling pathways increases the transcriptional activity of the viral LTR promoter to promote efficient viral replication (Fig. 1).
Although the polyproline motif mediating interactions between Nef and Src kinases is highly conserved between HIV-1 isolates, three amino acid substitutions in the HIV-2 and SIVmac Nef proteins result in the targeting of different Src kinases (Collette et al. 2000; Karn et al. 1998). To date, relatively little is known about the interaction of SIV and HIV-2 Nefs with cellular kinases and their effects on signal transduction pathways, T cell activation, and virus replication. Further studies seem warranted since differences in the ability of Nef to modulate cellular signaling pathways may affect the levels of immune activation and hence the development of immunodeficiency. The observation that an additional SH2 domain in Nef is associated with acute disease in SIV-infected rhesus macaques (Du et al. 1995) represents a particular striking example for the importance of cellular activation in the clinical outcome of primate lentiviral infections.
3.1.2 Activation of Viral and Cellular Transcription
By modulating the signal transduction machinery Nef augments the expression of its own genome and of a large number of cellular genes (reviewed by Arendt and Littman 2001). Gene expression profiling studies showed that Nef induces a transcriptional program in Jurkat T cells that is highly similar (but not identical) to that of anti-CD3 T cell activation and partly dependent on ZAP-70 and the zeta chain of the TCR (Simmons et al. 2001). In particular, Nef induces transcription factors that transactivate the HIV-1 LTR promoter [NFAT, NFκB, IRF-1/2 (interferon regulatory factor), c-fos, Jun-D] and several cellular co-factors of viral replication, such as the Tat co-factor CDK9, the transcription elongation factor Tat-SF1, the transactivator NFIB-2, and the spliceosome component U1 snRNP (small nuclear ribonucleic protein) (Simmons et al. 2001). Nef also induces the expression of cytokines and chemokines which are thought to favor viral replication (IL-2 IL-4, TGFβ, MIP1α, MIP1β) (Arendt and Littman 2001). At least in HeLa cells, many effects on cellular gene expression were dependent on an intact PxxP motif in HIV-1 Nef (Shaheduzzaman et al. 2002). Relatively little is known about the effect of other primate lentiviral Nef proteins on the transcription of cellular genes, but one study reported that SIVmac Nef down-modulates genes associated with antigen presentation and induces the transcription of genes involved in cell survival and in the synthesis of membrane glycolipids and phospholipids (Ndolo et al. 2006). In contrast to the HIV-1 Nef (Simmons et al. 2001), that of SIVmac did not significantly modulate genes involved in T cell activation (Ndolo et al. 2006). Although all these findings need to be confirmed in virally infected primary T cells, they indicate that Nef exerts profound effects on the transcriptional response of infected T cells to favor viral replication. Notably, HIV-1 Nef also induces the transcription of the genes encoding T cell activation markers and of death receptors, such as the programmed death receptor 1 (PD-1) and the Fas ligand (Xu et al. 1999; Muthumani et al. 2008). These effects of Nef may play a role in the pathogenesis of AIDS since they may cause the death or dysfunction of uninfected bystander CD8+ T cells.
3.1.3 CD4 Down-Modulation
As discussed in Sect. 2.4.2, down-modulation of CD4 is one of the hallmarks of primate lentiviral infections and the best characterized Nef function. However, it is still not well understood which of its multiple consequences is most critical for efficient viral spread in vitro and in vivo (reviewed by Lama 2003; Kirchhoff et al. 2008). It has been reported that cell surface CD4 interferes with viral particle release by interacting with the HIV-1 envelope protein present on budding virions, and that Nef-mediated down-modulation of surface CD4 relieves progeny virions from this block (Arganaraz et al. 2003; Cortes et al. 2002; Lama et al. 1999; Ross et al. 1999). Furthermore, Nef-mediated down-modulation of CD4 may increase the incorporation of functional Env glycoproteins into progeny virus (Lama et al. 1999; Argañaraz et al. 2003). In support of a relevant role in virus replication, it has been shown that the potency of CD4 down-modulation correlates with the efficiency of viral replication in primary lymphocyte cultures in human lymphoid tissue ex vivo (Glushakova et al. 2001; Lundquist et al. 2002). Moreover, down-modulation by HIV-1 of its own entry receptor reduces superinfection (Benson et al. 1993; Wildum et al. 2006), which would otherwise likely lead to cell death before virus production is accomplished. However, effective down-modulation of CD4 is not sufficient for effective replication in primary T cells (Saksela et al. 1995). To our current knowledge, at least three HIV-1 Nef activities, i.e., CD4 down-modulation, T cell activation, and enhancement of virion infection, contribute to efficient viral replication. Some seeming discrepancies may be due to the fact that the relative importance of these Nef functions for viral replication varies depending on the activation status and CD4 expression level of the target cells. CD4 down-modulation is most likely particularly important for the efficient release of fully infectious progeny virions from primary T cells since they usually express high levels of this receptor (Pham et al. 2004).
3.1.4 Enhancement of Viral Transfer from DCs to T Cells
Although the main target cells for HIV and SIV are CD4+ T cells and macrophages, it has been proposed that immature DCs present in the mucosa can capture virus particles and, following maturation and migration towards secondary lymphoid organs, transmit the virus to T cells (Granelli-Piperno et al. 1999). Although DCs are poorly infected by HIV-1, they can capture HIV-1 virions using the lectin DC-SIGN and maintain them in an infectious state for several days before transmitting them to lymphocytes (Geijtenbeek et al. 2000).
HIV-1 Nef is required for optimal virus production in DC-T cell co-cultures (Petit et al. 2001) and is thought to be implicated in transmission of virus to T cells (Sol-Foulon et al. 2002). Moreover, increased CD4 surface expression in APCs impairs DC-SIGN-mediated transmission by increasing internalization of particles through productive infection, and Nef-mediated CD4 down-modulation in DCs correlates with enhanced viral transmission to T cells (Wang et al. 2007). Together, these studies provide possible mechanisms by which Nef expression converts DCs into more efficient HIV-1 transmitters to T cells.
3.2 Enhancement of Virion Infectivity
Nef also enhances virion infectivity in a CD4-independent manner. Early studies suggested that Nef acts at an early postentry step because viral particles produced in the absence of Nef have an impaired ability to undergo reverse transcription (Aiken and Trono 1995; Chowers et al. 1995; Miller et al. 1995; Schwartz et al. 1995). This Nef-mediated enhancement of infectivity may depend on the route of entry since it is lost with HIV-1 virions pseudotyped with the vesicular stomatits virus glycoprotein (VSV-G) which targets virions for entry by endocytosis rather than surface fusion (Aiken 1997; Chazal et al. 2001). However, recent data suggest that entry via low pH-dependent Envs does not always bypass the requirement for Nef (Pizzato et al. 2008). Nef does not affect the efficiency of virion fusion with target cells (Tobiume et al. 2003; Cavrois et al. 2004). Instead, it may promote cytoplasmic delivery of HIV-1 virions (Schaeffer et al. 2001), to the detriment of endocytosis, which may lead to non-productive infection (Maréchal et al. 1998). Other reports suggested that Nef enables HIV-1 complexes to cross the cortical actin network underlying the plasma membrane (Campbell et al. 2004) and may reduce the susceptibility of incoming virions to proteasomal degradation in the target cells (Qi and Aiken 2007). Altogether, the results suggest that Nef may help the virus to penetrate the cortical actin barrier and that this function becomes dispensable if entry is mediated by endocytosis (Campbell et al. 2004).
The enhancement of virion infectivity is dependent on the expression of Nef in the virus producer cells, suggesting that Nef alters the molecular composition or properties of the progeny virions. Small quantities of Nef are incorporated into the viral particles and cleaved by the viral protease (Welker et al. 1996; Pandori et al. 1996). However, virion incorporation and cleavage of Nef does not correlate with its ability to enhance virion infectivity (Chen et al. 1998; Miller et al. 1997). Recently, it has been shown that Nef interacts with the GTPase Dynamin-2 (Dyn-2), an essential regulator of clathrin-mediated endocytosis, and that the infectivity enhancement of Nef is dependent on both Nef interaction with Dyn-2 and clathrin-coated pit formation (Pizzato et al. 2007). Nef itself has also been shown to increase clathrin-coated pit formation in an in vitro model (Foti et al. 1997), suggesting that Nef’s ability to enhance virion infectivity may be linked to its ability to enhance clathrin-dependent endocytosis. In support of this possibility it has been shown that the dileucine motif in Nef, which acts as an endocytosis signal, is required for optimal viral infectivity (Craig et al. 1998; Madrid et al. 2005).
The capability of Nef to enhance virion infectivity is highly conserved between different primate lentiviral lineages (Münch et al. 2007), and some evidence suggests that it is relevant for viral spread in vivo (Brenner et al. 2007). However, it is noteworthy that the effect of Nef on virion infectivity does not correlate with its ability to promote viral replication in primary T cells or ex vivo infected human lymphoid tissues (Glushakova et al. 2001; Lundquist et al. 2002) and has mainly been examined using HeLa-derived indicator cell lines. Thus, it will be important to further investigate how Nef modifies progeny virions and enhances virion infectivity in primary producer and target cells.
3.3 Induction of Soluble Factors Facilitating Virus Spread
Nef not only manipulates the infected host cells but may also induce changes in the cellular environment to render it more conducive to viral spread. Specifically, it has been shown that Nef expression in HIV-1-infected macrophages induces the secretion of the chemokines MIP-1alpha and MIP-1beta (Swingler et al. 1999). These chemokines can promote the chemotaxis of resting T-lymphocytes, thus recruiting them to sites of HIV-1 virion release from infected macrophages. Furthermore, it has been suggested that Nef expression in macrophages leads to the production of the soluble factors sCD23 and sICAM-1, which stimulate the production of accessory surface molecules on neighboring B cells (Swingler et al. 2003), which in turn can render resting T cells permissive for productive HIV-1 infection. These results indicate that HIV and SIV Nef proteins have evolved highly sophisticated ways to manipulate the cross-talk between different cell types, and that some effects that may be highly important for viral spread in vivo can be missed in standard cell cultures.
4 Effects of Nef on Programmed Cell Death
One can conceive how it might be advantageous for primate lentiviruses to inhibit apoptosis of the infected host cell to prolong the time of virus production. No less than five viral proteins, Tat, Nef, Vpr, Vpu, and Env, were reported to modulate the programmed cell death of virally infected T cells (Gougeon 2003; Fackler and Baur 2002). As discussed in Chap. 2, most SIV and HIV-2, but not HIV-1, Nefs inhibit apoptosis by blocking the responsiveness of infected T cells to activation (Schindler et al. 2006). Furthermore, Nef induces the expression of the Fas ligand (CD95L), possibly to induce the apoptosis of “attacking” bystander CD8+ cytotoxic T-cells (Xu et al. 1999; Muthumani et al. 2005). It has also been suggested that Nef directly represses pro-apoptotic signaling in infected cells by inhibiting apoptosis signal-regulating kinase 1 (ASK1) (Geleziunas et al. 2001) and by the inactivation of the pro-apoptotic Bad-2 protein through phosphorylation (Wolf et al. 2001). However, it is controversial whether Nef inhibits or enhances apoptosis. In support of a pro-apoptotic role, it has been described that Nef sensitizes CD4+ T cells to apoptosis by up-regulating CD95 and CD95L (Zauli et al. 1999) and by reducing the expression of the anti-apoptotic proteins Bcl-2 and Bcl-XL (Rasola et al. 2001). Experiments using HIV-1 constructs coexpressing Nef and eGFP from single bicistronic RNAs that allowed to directly correlate the levels of cell death and Nef expression failed to detect a significant effect of Nef on apoptosis in HIV-1-infected primary T cells (Schindler et al. 2005). This suggests that Nef affects the survival of the infected cells mainly indirectly, e.g., by reducing CTL lysis and suppressing T cell activation (in the case of HIV-2 and most SIVs), rather than by direct effects on apoptosis.
5 Interactions of Exogenous Nef with Host Cells
In addition to its expression in virally infected cells, Nef is also present in the extracellular environment and can reach concentrations of up to 10 ng/ml in the sera of HIV-infected individuals (Fujii et al. 1996). Extracellular Nef may activate various transcription factors in monocytes and macrophages (Alessandrini et al. 2000; Mangino et al. 2007; Olivetta et al. 2003; Varin et al. 2003) and induce apoptosis (Fujii et al. 1996; Okada et al. 1997; Huang et al. 2004). It has been proposed that soluble Nef is internalized and blocks immunoglobulin class switch DNA recombination in B cells by perturbing CD40 ligand activation of B cells by T cells (Qiao et al. 2006). Recently, soluble Nef has also been shown to interact with CD34+ hematopoietic stem cells and to inhibit their clonogenic potential through induction of PPARγ and down-modulation of STAT5A and 5B expression (Prost et al. 2008), suggesting that it may contribute to the hematopoietic abnormalities observed in HIV-infected patients (Marandin et al. 1996; Sloand et al. 1997). These studies suggest that extracellular Nef may play a relevant role in the pathogenesis of AIDS. However, it is currently largely unknown how effectively Nef is released and how it can interact or be taken up by uninfected bystander cells. Furthermore, the significance of some of these findings is difficult to assess since they were obtained using rather artificial experimental conditions, such as high levels of Nef produced by bacteria or insect cells.
6 Conclusion
HIV and SIV Nef proteins perform a large number of interactions and functions that help the virus to persist in the infected host by facilitating immune evasion and by increasing virus spread. Although (or perhaps because) a myriad of Nef effects has been reported, we are still far away from a comprehensive understanding of the role of this “all-rounder” protein in viral persistence and pathogenesis. However, some principles of Nef function have become clear: (1) Nef interacts with the cytoplasmic domains of various cellular receptors to recruit them to the endocytic machinery, or to reroute them to endosomes, for ultimate degradation in lysosomes. The reduced surface expression of MHC-I prevents CTL lysis, that of CXCR4 impairs cellular migration, and that of several others, such as CD4, CD28, and CD3, interferes with TCR signaling. Together, these effects of Nef help the virus to evade the host immune system by making infected cells less “visible” to the immune system and prolonging their survival time and by preventing or deregulating the crosstalk between infected T cells and APCs. (2) Simultaneously, Nef interacts with various cellular factors involved in signaling, trafficking, cell activation and migration to alter the responsiveness of virally infected cells to stimulation and to facilitate the transcription of the viral genome and various cellular genes. The preliminary evidence suggests that HIV and SIV have evolved highly elaborate mechanisms to favor the expression of cellular genes that promote viral spread at the cost of antiviral cellular factors. (3) By reducing the surface levels of CD4 and by another mechanism that is currently incompletely understood, Nef facilitates viral release from primary T cells and enhances the infectivity of progeny virions. (4) Nef may also facilitate viral immune evasion by up-modulating several death receptors and inducing the secretion of cytokines that affect the survival and function of uninfected bystander CD8 T cells. (5) Finally, by inducing the release of cellular factors, Nef may recruit T cells to the sites of infection and render them susceptible to infection. Thus, although Nef is commonly considered as an early viral gene product, it acts essentially at every stage of the virus life cycle and may even modify the microenvironment of the infected cells to facilitate viral spread. The combination of these Nef interactions and functions allows HIV and SIV to persist efficiently at high levels in their respective hosts. Most likely, the great majority of primate lentiviral Nefs also limits the detrimental effects associated with these high viral loads by efficiently suppressing the responsiveness of virally-infected T cells to activation. Exceptions are nef alleles from vpu-containing viruses, i.e., HIV-1 and its simian precursors, which increase rather than block the responsiveness of infected T cells to stimulation. These differences in Nef function could contribute to the differential levels of immune activation associated with pathogenic and nonpathogenic primate lentiviral infections and hence play a relevant role in the clinical outcome of infection (Kirchhoff, 2009). Understanding the sophisticated mechanisms that primate lentiviruses have evolved to evade the host immune response and to manipulate cells to their advantage may help us to develop novel preventive and therapeutic strategies.
Note Some SIV Nef Proteins Are Tetherin Antagonists: Very recent data demonstrate that the Nef proteins of some SIVs antagonize the recently identified interferon-inducible host-cell factor Tetherin, also known as BST2, CD317 or HM1.24 (Jia et al., 2009; Zhang et al., 2009). This further adds to the list of Nef functions that facilitate virus spread. Furthermore, these studies identify the second function (beside the suppression of CD4 cell surface expression) that Nef shares with the small accessory viral protein U (Vpu), which is used by HIV-1 to counteract tetherin (Neil et al., 2008).
References
Adnan S, Balamurugan A, Trocha A et al (2006) Nef interference with HIV-1-specific CTL antiviral activity is epitope specific. Blood 108:3414–3419
Agopian K, Wei BL, Garcia JV et al (2006) A hydrophobic binding surface on the human immunodeficiency virus type 1 Nef core is critical for association with p21-activated kinase 2. J Virol 80:3050–3061
Aiken C, Trono D (1995) Nef stimulates human immunodeficiency virus type 1 proviral DNA synthesis. J Virol 69:5048–5056
Aiken C, Konner J, Landau NR et al (1994) Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 76:853–864
Akari H, Arold S, Fukumori T et al (2000) Nef-induced major histocompatibility complex class I down-regulation is functionally dissociated from its virion incorporation, enhancement of viral infectivity, and CD4 down-regulation. J Virol 74:2907–2912
Alessandrini L, Santarcangelo AC, Olivetta E et al (2000) T-tropic human immunodeficiency virus (HIV) type 1 Nef protein enters human monocyte-macrophages and induces resistance to HIV replication: a possible mechanism of HIV T-tropic emergence in AIDS. J Gen Virol 81:2905–2917
Arendt CW, Littman DR (2001) HIV: master of the host cell. Genome Biol 2:Reviews1030
Arganaraz ER, Schindler M, Kirchhoff F et al (2003) Enhanced CD4 down-modulation by late stage HIV-1 nef alleles is associated with increased Env incorporation and viral replication. J Biol Chem 278:33912–33919
Atkins KM, Thomas L, Youker RT et al (2008) HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation: analysis using short interfering RNA and knock-out mice. J Biol Chem 283:11772–11784
Baur AS, Sass G, Laffert B et al (1997) The N-terminus of Nef from HIV-1/SIV associates with a protein complex containing Lck and a serine kinase. Immunity 6:283–291
Baur AS, Sawai ET, Dazin P et al (1994) HIV-1 Nef leads to inhibition or activation of T cells depending on its intracellular localization. Immunity 1:373–384
Bell I, Schaefer TM, Trible RP et al (2001) Down-modulation of the costimulatory molecule, CD28, is a conserved activity of multiple SIV Nefs and is dependent on histidine 196 of Nef. Virology 283:148–158
Bell I, Ashman C, Maughan J et al (1998) Association of simian immunodeficiency virus Nef with the T-cell receptor (TCR) zeta chain leads to TCR down-modulation. J Gen Virol 79:2717–2727
Benson RE, Sanfridson A, Ottinger JS et al (1993) Downregulation of cell-surface CD4 expression by simian immunodeficiency virus Nef prevents viral super infection. J Exp Med 177:1561–1566
Bertolino P, Rabourdin-Combe C (1996) The MHC class II-associated invariant chain: a molecule with multiple roles in MHC class II biosynthesis and antigen presentation to CD4+ T cells. Crit Rev Immunol 16:359–379
Blagoveshchenskaya AD, Thomas L, Feliciangeli SF et al (2002) HIV-1 Nef downregulates MHC-I by a PACS-1- and PI3K-regulated ARF6 endocytic pathway. Cell 111:853–866
Bonaparte MI, Barker E (2003) Inability of natural killer cells to destroy autologous HIV-infected T lymphocytes. AIDS 17:487–494
Bonaparte MI, Barker E (2004) Killing of human immunodeficiency virus-infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood 104:2087–2094
Brenchley JM, Price DA, Schacker TW et al (2006) Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12:1365–1371
Brenner M, Munch J, Schindler M et al (2006) Importance of the N-distal AP-2 binding element in Nef for simian immunodeficiency virus replication and pathogenicity in rhesus macaques. J Virol 80:4469–4481
Bresnahan PA, Yonemoto W, Greene WC (1999) Cutting edge: SIV Nef protein utilizes both leucine- and tyrosine-based protein sorting pathways for down-regulation of CD4. J Immunol 163:2977–2981
Campbell EM, Nunez R, Hope TJ (2004) Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J Virol 78:5745–5755
Carl S, Greenough TC, Krumbiegel M et al (2001) Modulation of different human immunodeficiency virus type 1 Nef functions during progression to AIDS. J Virol 75:3657–3665
Casartelli N, Di MG, Potesta M et al (2003) CD4 and major histocompatibility complex class I downregulation by the human immunodeficiency virus type 1 nef protein in pediatric AIDS progression. J Virol 77:11536–11545
Cavrois M, Neidleman J, Yonemoto W et al (2004) HIV-1 virion fusion assay: uncoating not required and no effect of Nef on fusion. Virology 328:36–44
Chaudhry A, Das SR, Jameel S et al (2008) HIV-1 Nef induces a Rab11-dependent routing of endocytosed immune costimulatory proteins CD80 and CD86 to the Golgi. Traffic 9:1925–1935
Chaudhry A, Das SR, Jameel S et al (2007) A two-pronged mechanism for HIV-1 Nef-mediated endocytosis of immune costimulatory molecules CD80 and CD86. Cell Host Microbe 1:37–49
Chaudhry A, Das SR, Hussain A et al (2005) The Nef protein of HIV-1 induces loss of cell surface costimulatory molecules CD80 and CD86 in APCs. J Immunol 175:4566–4574
Chazal N, Singer G, Aiken C et al (2001) Human immunodeficiency virus type 1 particles pseudotyped with envelope proteins that fuse at low pH no longer require Nef for optimal infectivity. J Virol 75:4014–4018
Chen YL, Trono D, Camaur D (1998) The proteolytic cleavage of human immunodeficiency virus type 1 Nef does not correlate with its ability to stimulate virion infectivity. J Virol 72:3178–3184
Choe EY, Schoenberger ES, Groopman JE et al (2002) HIV Nef inhibits T cell migration. J Biol Chem 277:46079–46084
Chowers MY, Pandori MW, Spina CA et al (1995) The growth advantage conferred by HIV-1 nef is determined at the level of viral DNA formation and is independent of CD4 downregulation. Virology 212:451–457
Chowers MY, Spina CA, Kwoh TJ et al (1994) Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J Virol 68:2906–2914
Chu PC, Wu J, Liao XC et al (2004) A novel role for p21-activated protein kinase 2 in T cell activation. J Immunol 172:7324–7334
Cohen GB, Gandhi RT, Davis DM et al (1999) The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671
Collette Y, Dutartre H, Benziane A et al (1996) Physical and functional interaction of Nef with Lck. HIV-1 Nef-induced T-cell signaling defects. J Biol Chem 271:6333–6341
Collins KL, Chen BK, Kalams SA et al (1998) HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391:397–401
Cortes MJ, Wong-Staal F, Lama J (2002) Cell surface CD4 interferes with the infectivity of HIV-1 particles released from T cells. J Biol Chem 277:1770–1779
Craig HM, Pandori MW, Guatelli JC (1998) Interaction of HIV-1 Nef with the cellular dileucine-based sorting pathway is required for CD4 down-regulation and optimal viral infectivity. Proc Natl Acad Sci U S A 95:11229–11234
Daniels RH, Bokoch GM (1999) p21-activated protein kinase: a crucial component of morphological signaling? Trends Biochem Sci 24:350–355
Deacon NJ, Tsykin A, Solomon A et al (1995) Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270:988–991
Deng H, Liu R, Ellmeier W et al (1996) Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661–666
Djordjevic JT, Schibeci SD, Stewart GJ et al (2004) HIV type 1 Nef increases the association of T cell receptor (TCR)-signaling molecules with T cell rafts and promotes activation-induced raft fusion. AIDS Res Hum Retroviruses 20:547–555
Du Z, Lang SM, Sasseville VG et al (1995) Identification of a nef allele that causes lymphocyte activation and acute disease in macaque monkeys. Cell 82:665–674
Dugast M, Toussaint H, Dousset C et al (2005) AP2 clathrin adaptor complex, but not AP1, controls the access of the major histocompatibility complex (MHC) class II to endosomes. J Biol Chem 280:19656–19664
Dyer WB, Ogg GS, Demoitie MA et al (1999) Strong human immunodeficiency virus (HIV)-specific cytotoxic T-lymphocyte activity in Sydney Blood Bank Cohort patients infected with nef-defective HIV type 1. J Virol 73:436–443
Fackler OT, Alcover A, Schwartz O (2007) Modulation of the immunological synapse: a key to HIV-1 pathogenesis? Nat Rev Immunol 7:310–317
Fackler OT, Baur AS (2002) Live and let die: Nef functions beyond HIV replication. Immunity 16:493–497
Fackler OT, Lu X, Frost JA et al (2000) p21-activated kinase 1 plays a critical role in cellular activation by Nef. Mol Cell Biol 20:2619–2627
Fackler OT, Luo W, Geyer M et al (1999) Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol Cell 3:729–739
Fellay J, Shianna KV, Ge D et al (2007) A whole-genome association study of major determinants for host control of HIV-1. Science 317:944–947
Fenard D, Yonemoto W, de Noronha C et al (2005) Nef is physically recruited into the immunological synapse and potentiates T cell activation early after TCR engagement. J Immunol 175:6050–6057
Feng Y, Broder CC, Kennedy PE et al (1996) HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872–877
Fortin JF, Barat C, Beausejour Y et al (2004) Hyper-responsiveness to stimulation of human immunodeficiency virus-infected CD4+ T cells requires Nef and Tat virus gene products and results from higher NFAT, NF-kappaB, and AP-1 induction. J Biol Chem 279:39520–39531
Foti M, Mangasarian A, Piguet V et al (1997) Nef-mediated clathrin-coated pit formation. J Cell Biol 139:37–47
Fujii Y, Otake K, Tashiro M et al (1996) Soluble Nef antigen of HIV-1 is cytotoxic for human CD4+ T cells. FEBS Lett 393:93–96
Garcia JV, Miller AD (1991) Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350:508–511
Geijtenbeek TB, Kwon DS, Torensma R et al (2000) DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100:587–597
Geleziunas R, Xu W, Takeda K et al (2001) HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 410:834–838
Geyer M, Fackler OT, Peterlin BM (2001) Structure–function relationships in HIV-1 Nef. EMBO Rep 2:580–585
Geyer M, Peterlin BM (2001) Domain assembly, surface accessibility and sequence conservation in full length HIV-1 Nef. FEBS Lett 496:91–95
Glushakova S, Munch J, Carl S et al (2001) CD4 down-modulation by human immunodeficiency virus type 1 Nef correlates with the efficiency of viral replication and with CD4(+) T-cell depletion in human lymphoid tissue ex vivo. J Virol 75:10113–10117
Gordon S, Klatt NR, Bosinger SE et al (2007) Severe depletion of mucosal CD4+ T cells in AIDS-free SIV-infected sooty mangabeys. J Immunol 179:3026–3034
Gougeon ML (2003) Apoptosis as an HIV strategy to escape immune attack. Nat Rev Immunol 3:392–404
Goulder PJ, Bunce M, Luzzi G et al (1997) Potential underestimation of HLA-C-restricted cytotoxic T-lymphocyte responses. AIDS 11:1884–1886
Granelli-Piperno A, Finkel V, Delgado E et al (1999) Virus replication begins in dendritic cells during the transmission of HIV-1 from mature dendritic cells to T cells. Curr Biol 9:21–29
Greenberg ME, Iafrate AJ, Skowronski J (1998a) The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J 17:2777–2789
Greenberg M, DeTulleo L, Rapoport I et al (1998b) A dileucine motif in HIV-1 Nef is essential for sorting into clathrin-coated pits and for downregulation of CD4. Curr Biol 8:1239–1242
Greenway AL, Holloway G, McPhee DA et al (2003) HIV-1 Nef control of cell signalling molecules: multiple strategies to promote virus replication. J Biosci 28:323–335
Greenway A, Azad A, Mills J et al (1996) Human immunodeficiency virus type 1 Nef binds directly to Lck and mitogen-activated protein kinase, inhibiting kinase activity. J Virol 70:6701–6708
Hahn BH, Shaw GM, De Cock KM et al (2000) AIDS as a zoonosis: scientific and public health implications. Science 287:607–614
Hodge DR, Dunn KJ, Pei GK et al (1998) Binding of c-Raf1 kinase to a conserved acidic sequence within the carboxyl-terminal region of the HIV-1 Nef protein. J Biol Chem 273:15727–15733
Howe AY, Jung JU, Desrosiers RC (1998) Zeta chain of the T-cell receptor interacts with nef of simian immunodeficiency virus and human immunodeficiency virus type 2. J Virol 72:9827–9834
Hrecka K, Swigut T, Schindler M et al (2005) Nef proteins from diverse groups of primate lentiviruses downmodulate CXCR4 to inhibit migration to the chemokine stromal derived factor 1. J Virol 79:10650–10659
Hua J, Cullen BR (1997) Human immunodeficiency virus types 1 and 2 and simian immunodeficiency virus Nef use distinct but overlapping target sites for downregulation of cell surface CD4. J Virol 71:6742–6748
Hung CH, Thomas L, Ruby CE et al (2007) HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe 1:121–133
Iafrate AJ, Bronson S, Skowronski J (1997) Separable functions of Nef disrupt two aspects of T cell receptor machinery: CD4 expression and CD3 signaling. EMBO J 16:673–684
Janardhan A, Swigut T, Hill B et al (2004) HIV-1 Nef binds the DOCK2-ELMO1 complex to activate rac and inhibit lymphocyte chemotaxis. PLoS Biol 2:E6
Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, Johnson WE, Westmoreland S, Evans DT (2009) Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog May; 5(5):e1000429
Jin YJ, Cai CY, Zhang X et al (2008) Lysine 144, a ubiquitin attachment site in HIV-1 Nef, is required for Nef-mediated CD4 down-regulation. J Immunol 180:7878–7886
Kärkkäinen S, Hiipakka M, Wang JH et al (2006) Identification of preferred protein interactions by phage-display of the human Src homology-3 proteome. EMBO Rep 7:186–191
Karn T, Hock B, Holtrich U et al (1998) Nef proteins of distinct HIV-1 or -2 isolates differ in their binding properties for HCK: isolation of a novel Nef binding factor with characteristics of an adaptor protein. Virology 246:45–52
Kasper MR, Collins KL (2003) Nef-mediated disruption of HLA-A2 transport to the cell surface in T cells. J Virol 77:3041–3049
Kerkau T, Schmitt-Landgraf R, Schimpl A et al (1989) Downregulation of HLA class I antigens in HIV-1-infected cells. AIDS Res Hum Retroviruses 5:613–620
Kestler HW, Ringler DJ, Mori K et al (1991) Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65:651–662
Kirchhoff F (2009) Is the high virulence of HIV-1 an unfortunate coincidence of primate lentiviral evolution? Nat Rev Microbiol 7: 467–476
Kirchhoff F, Schindler M, Specht A et al (2008) Role of Nef in primate lentiviral immunopathogenesis. Cell Mol Life Sci 65:2621–2636
Kirchhoff F, Greenough TC, Brettler DB et al (1995) Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med 332:228–232
Lama J (2003) The physiological relevance of CD4 receptor down-modulation during HIV infection. Curr HIV Res 1:167–184
Lama J, Mangasarian A, Trono D (1999) Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr Biol 9:622–631
Larsen JE, Massol RH, Nieland TJ et al (2004) HIV Nef-mediated major histocompatibility complex class I down-modulation is independent of Arf6 activity. Mol Biol Cell 15:323–331
Le Gall S, Buseyne F, Trocha A et al (2000) Distinct trafficking pathways mediate Nef-induced and clathrin-dependent major histocompatibility complex class I down-regulation. J Virol 74:9256–9266
Le Gall S, Erdtmann L, Benichou S et al (1998) Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 8:483–495
Lewis MJ, Balamurugan A, Ohno A et al (2008) Functional adaptation of Nef to the immune milieu of HIV-1 infection in vivo. J Immunol 180:4075–4081
Lindner R (2002) Transient surface delivery of invariant chain-MHC II complexes via endosomes: a quantitative study. Traffic 3:133–146
Lindwasser OW, Smith WJ, Chaudhuri R et al (2008) A diacidic motif in human immunodeficiency virus type 1 Nef is a novel determinant of binding to AP-2. J Virol 82:1166–1174
Linnemann T, Zheng YH, Mandic R et al (2002) Interaction between Nef and phosphatidylinositol-3-kinase leads to activation of p21-activated kinase and increased production of HIV. Virology 294:246–255
Lock M, Greenberg ME, Iafrate AJ et al (1999) Two elements target SIV Nef to the AP-2 clathrin adaptor complex, but only one is required for the induction of CD4 endocytosis. EMBO J 18:2722–2733
Lu X, Wu X, Plemenitas A et al (1996) CDC42 and Rac1 are implicated in the activation of the Nef-associated kinase and replication of HIV-1. Curr Biol 6:1677–1684
Lubben NB, Sahlender DA, Motley AM et al (2007) HIV-1 Nef-induced down-regulation of MHC class I requires AP-1 and clathrin but not PACS-1 and is impeded by AP-2. Mol Biol Cell 18:3351–3365
Lundquist CA, Tobiume M, Zhou J et al (2002) Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. J Virol 76:4625–4633
Madrid R, Janvier K, Hitchin D et al (2005) Nef-induced alteration of the early/recycling endosomal compartment correlates with enhancement of HIV-1 infectivity. J Biol Chem 280:5032–5044
Malim MH, Emerman M (2008) HIV-1 accessory proteins–ensuring viral survival in a hostile environment. Cell Host Microbe 3:388–398
Mangasarian A, Piguet V, Wang JK et al (1999) Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal alpha helix and proline repeat of Nef selectively regulate MHC-I trafficking. J Virol 73:1964–1973
Mangino G, Percario ZA, Fiorucci G et al (2007) In vitro treatment of human monocytes/macrophages with myristoylated recombinant Nef of human immunodeficiency virus type 1 leads to the activation of mitogen-activated protein kinases, IkappaB kinases, and interferon regulatory factor 3 and to the release of beta interferon. J Virol 81:2777–2791
Manninen A, Huotari P, Hiipakka M et al (2001) Activation of NFAT-dependent gene expression by Nef: conservation among divergent Nef alleles, dependence on SH3 binding and membrane association, and cooperation with protein kinase C-theta. J Virol 75:3034–3037
Manninen A, Renkema GH, Saksela K (2000) Synergistic activation of NFAT by HIV-1 nef and the Ras/MAPK pathway. J Biol Chem 275:16513–16517
Marandin A, Katz A, Oksenhendler E et al (1996) Loss of primitive hematopoietic progenitors in patients with human immunodeficiency virus infection. Blood 88:4568–4578
Marechal V, Clavel F, Heard JM et al (1998) Cytosolic Gag p24 as an index of productive entry of human immunodeficiency virus type 1. J Virol 72:2208–2212
Mariani R, Kirchhoff F, Greenough TC et al (1996) High frequency of defective nef alleles in a long-term survivor with nonprogressive human immunodeficiency virus type 1 infection. J Virol 70:7752–7764
Marx PA, Chen Z (1998) The function of simian chemokine receptors in the replication of SIV. Semin Immunol 10:215–223
Mavilio D, Benjamin J, Daucher M et al (2003) Natural killer cells in HIV-1 infection: dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. Proc Natl Acad Sci U S A 100:15011–15016
Miller MD, Warmerdam MT, Ferrell SS et al (1997) Intravirion generation of the C-terminal core domain of HIV-1 Nef by the HIV-1 protease is insufficient to enhance viral infectivity. Virology 234:215–225
Miller MD, Warmerdam MT, Page KA et al (1995) Expression of the human immunodeficiency virus type 1 (HIV-1) nef gene during HIV-1 production increases progeny particle infectivity independently of gp160 or viral entry. J Virol 69:579–584
Miller MD, Warmerdam MT, Gaston I et al (1994) The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J Exp Med 179:101–113
Milush JM, Reeves JD, Gordon SN et al (2007) Virally induced CD4+ T cell depletion is not sufficient to induce AIDS in a natural host. J Immunol 179:3047–3056
Mitchell RS, Chaudhuri R, Lindwasser OW et al (2008) Competition model for upregulation of the major histocompatibility complex class II-associated invariant chain by human immunodeficiency virus type 1 Nef. J Virol 82:7758–7767
Munch J, Rajan D, Schindler M et al (2007) Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J Virol 81:13852–13864
Munch J, Stolte N, Fuchs D et al (2001) Efficient class I major histocompatibility complex down-regulation by simian immunodeficiency virus Nef is associated with a strong selective advantage in infected rhesus macaques. J Virol 75:10532–10536
Muthumani K, Choo AY, Hwang DS et al (2005) HIV-1 Nef-induced FasL induction and bystander killing requires p38 MAPK activation. Blood 106:2059–2068
Ndolo T, George M, Nguyen H et al (2006) Expression of simian immunodeficiency virus Nef protein in CD4+ T cells leads to a molecular profile of viral persistence and immune evasion. Virology 353:374–387
Neil SJ, Zang T, Bieniasz PD (2008). Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008 Jan 24; 451(7177):425–430
Norris PJ, Rosenberg ES (2002) CD4(+) T helper cells and the role they play in viral control. J Mol Med 80:397–405
Noviello CM, Benichou S, Guatelli JC (2008) Cooperative binding of the class I major histocompatibility complex cytoplasmic domain and human immunodeficiency virus type 1 Nef to the endosomal AP-1 complex via its mu subunit. J Virol 82:1249–1258
Okada H, Takei R, Tashiro M (1997) HIV-1 Nef protein-induced apoptotic cytolysis of a broad spectrum of uninfected human blood cells independently of CD95(Fas). FEBS Lett 414:603–606
Olivetta E, Percario Z, Fiorucci G et al (2003) HIV-1 Nef induces the release of inflammatory factors from human monocyte/macrophages: involvement of Nef endocytotic signals and NF-kappa B activation. J Immunol 170:1716–1727
Pandori MW, Fitch NJ, Craig HM et al (1996) Producer-cell modification of human immunodeficiency virus type 1: Nef is a virion protein. J Virol 70:4283–4290
Pandrea I, Sodora DL, Silvestri G et al (2008) Into the wild: simian immunodeficiency virus (SIV) infection in natural hosts. Trends Immunol 29:419–428
Patel PG, Yu Kimata MT, Biggins JE et al (2002) Highly pathogenic simian immunodeficiency virus mne variants that emerge during the course of infection evolve enhanced infectivity and the ability to downregulate CD4 but not class I major histocompatibility complex antigens. J Virol 76:6425–6434
Petit C, Buseyne F, Boccaccio C et al (2001) Nef is required for efficient HIV-1 replication in cocultures of dendritic cells and lymphocytes. Virology 286:225–236
Pham HM, Arganaraz ER, Groschel B et al (2004) Lentiviral vectors interfering with virus-induced CD4 down-modulation potently block human immunodeficiency virus type 1 replication in primary lymphocytes. J Virol 78:13072–13081
Piguet V, Wan L, Borel C et al (2000) HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat Cell Biol 2:163–167
Piguet V, Chen YL, Mangasarian A et al (1998) Mechanism of Nef-induced CD4 endocytosis: Nef connects CD4 with the mu chain of adaptor complexes. EMBO J 17:2472–2481
Pizzato M, Popova E, Gottlinger HG (2008) Nef can enhance the infectivity of receptor-pseudotyped human immunodeficiency virus type 1 particles. J Virol 82(21):10811–10810
Pizzato M, Helander A, Popova E et al (2007) Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc Natl Acad Sci U S A 104:6812–6817
Prost S, Le DM, Auge S et al (2008) Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARgamma/STAT5 signaling pathway in macaques. J Clin Invest 118:1765–1775
Qi M, Aiken C (2007) Selective restriction of Nef-defective human immunodeficiency virus type 1 by a proteasome-dependent mechanism. J Virol 81:1534–1536
Qiao X, He B, Chiu A et al (2006) Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in bystander B cells. Nat Immunol 7:302–310
Quaranta MG, Napolitano A, Sanchez M et al (2007) HIV-1 Nef impairs the dynamic of DC/NK crosstalk: different outcome of CD56(dim) and CD56(bright) NK cell subsets. FASEB J 21:2323–2334
Rasola A, Gramaglia D, Boccaccio C et al (2001) Apoptosis enhancement by the HIV-1 Nef protein. J Immunol 166:81–88
Rauch S, Pulkkinen K, Saksela K, Fackler OT (2008) Human immunodeficiency virus type 1 Nef recruits the guanine exchange factor Vav1 via an unexpected interface into plasma membrane microdomains for association with p21-activated kinase 2 activity. J Virol 82:2918–2929
Renkema GH, Saksela K (2000) Interactions of HIV-1 NEF with cellular signal transducing proteins. Front Biosci 5:D268–D283
Rocha N, Neefjes J (2008) MHC class II molecules on the move for successful antigen presentation. EMBO J 27:1–5
Roche PA, Teletski CL, Stang E et al (1993) Cell surface HLA-DR-invariant chain complexes are targeted to endosomes by rapid internalization. Proc Natl Acad Sci U S A 90:8581–8585
Roche PA, Teletski CL, Karp DR et al (1992) Stable surface expression of invariant chain prevents peptide presentation by HLA-DR. EMBO J 11:2841–2847
Roeth JF, Collins KL (2006) Human immunodeficiency virus type 1 Nef: adapting to intracellular trafficking pathways. Microbiol Mol Biol Rev 70:548–563
Roeth JF, Williams M, Kasper MR et al (2004) HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J Cell Biol 167:903–913
Rose JJ, Janvier K, Chandrasekhar S et al (2005) CD4 down-regulation by HIV-1 and simian immunodeficiency virus (SIV) Nef proteins involves both internalization and intracellular retention mechanisms. J Biol Chem 280:7413–7426
Ross TM, Oran AE, Cullen BR (1999) Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr Biol 9:613–621
Saksela K, Cheng G, Baltimore D (1995) Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef + viruses but not for down-regulation of CD4. EMBO J 14:484–491
Salvi R, Garbuglia AR, Di CA et al (1998) Grossly defective nef gene sequences in a human immunodeficiency virus type 1-seropositive long-term nonprogressor. J Virol 72:3646–3657
Sawai ET, Baur A, Struble H et al (1994) Human immunodeficiency virus type 1 Nef associates with a cellular serine kinase in T lymphocytes. Proc Natl Acad Sci U S A 91:1539–1543
Schaefer MR, Wonderlich ER, Roeth JF et al (2008) HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog 4:e1000131
Schaefer TM, Bell I, Pfeifer ME et al (2002) The conserved process of TCR/CD3 complex down-modulation by SIV Nef is mediated by the central core, not endocytic motifs. Virology 302:106–122
Schaeffer E, Geleziunas R, Greene WC (2001) Human immunodeficiency virus type 1 Nef functions at the level of virus entry by enhancing cytoplasmic delivery of virions. J Virol 75:2993–3000
Scheppler JA, Nicholson JK, Swan DC et al (1989) Down-modulation of MHC-I in a CD4+ T cell line, CEM-E5, after HIV-1 infection. J Immunol 143:2858–2866
Schindler M, Schmokel J, Specht A et al (2008) Inefficient Nef-mediated downmodulation of CD3 and MHC-I correlates with loss of CD4 + T cells in natural SIV infection. PLoS Pathog 4:e1000107
Schindler M, Wildum S, Casartelli N et al (2007a) Nef alleles from children with non-progressive HIV-1 infection modulate MHC-II expression more efficiently than those from rapid progressors. AIDS 21:1103–1107
Schindler M, Rajan D, Specht A et al (2007b) Association of Nef with p21-activated kinase 2 is dispensable for efficient human immunodeficiency virus type 1 replication and cytopathicity in ex vivo-infected human lymphoid tissue. J Virol 81:13005–13014
Schindler M, Munch J, Kutsch O et al (2006) Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell 125:1055–1067
Schindler M, Munch J, Kirchhoff F (2005) Human immunodeficiency virus type 1 inhibits DNA damage-triggered apoptosis by a Nef-independent mechanism. J Virol 79:5489–5498
Schindler M, Wurfl S, Benaroch P et al (2003) Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J Virol 77:10548–10556
Schrager JA, Marsh JW (1999) HIV-1 Nef increases T cell activation in a stimulus-dependent manner. Proc Natl Acad Sci U S A 96:8167–8172
Schwartz O, Marechal V, Le Gall S et al (1996) Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med 2:338–342
Schwartz O, Marechal V, Danos O et al (1995) Human immunodeficiency virus type 1 Nef increases the efficiency of reverse transcription in the infected cell. J Virol 69:4053–4059
Shaheduzzaman S, Krishnan V, Petrovic A et al (2002) Effects of HIV-1 Nef on cellular gene expression profiles. J Biomed Sci 9:82–96
Simmons A, Aluvihare V, McMichael A (2001) Nef triggers a transcriptional program in T cells imitating single-signal T cell activation and inducing HIV virulence mediators. Immunity 14:763–777
Skowronski J, Parks D, Mariani R (1993) Altered T cell activation and development in transgenic mice expressing the HIV-1 nef gene. EMBO J 12:703–713
Sloand EM, Young NS, Sato T et al (1997) Secondary colony formation after long-term bone marrow culture using peripheral blood and bone marrow of HIV-infected patients. AIDS 11:1547–1553
Smith BL, Krushelnycky BW, Mochly-Rosen D et al (1996) The HIV nef protein associates with protein kinase C theta. J Biol Chem 271:16753–16757
Sol-Foulon N, Moris A, Nobile C et al (2002) HIV-1 Nef-induced upregulation of DC-SIGN in dendritic cells promotes lymphocyte clustering and viral spread. Immunity 16:145–155
Specht A, DeGottardi MQ, Schindler M et al (2008) Selective downmodulation of HLA-A and -B by Nef alleles from different groups of primate lentiviruses. Virology 373:229–237
Stevenson M (2003) HIV-1 pathogenesis. Nat Med 9:853–860
Stumptner-Cuvelette P, Jouve M, Helft J et al (2003) Human immunodeficiency virus-1 Nef expression induces intracellular accumulation of multivesicular bodies and major histocompatibility complex class II complexes: potential role of phosphatidylinositol 3-kinase. Mol Biol Cell 14:4857–4870
Stumptner-Cuvelette P, Morchoisne S, Dugast M et al (2001) HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc Natl Acad Sci U S A 98:12144–12149
Swann SA, Williams M, Story CM et al (2001) HIV-1 Nef blocks transport of MHC class I molecules to the cell surface via a PI 3-kinase-dependent pathway. Virology 282:267–277
Swigut T, Alexander L, Morgan J et al (2004) Impact of Nef-mediated downregulation of major histocompatibility complex class I on immune response to simian immunodeficiency virus. J Virol 78:13335–13344
Swigut T, Greenberg M, Skowronski J (2003) Cooperative interactions of simian immunodeficiency virus Nef, AP-2, and CD3-zeta mediate the selective induction of T-cell receptor-CD3 endocytosis. J Virol 77:8116–8126
Swigut T, Shohdy N, Skowronski J (2001) Mechanism for down-regulation of CD28 by Nef. EMBO J 20:1593–1604
Swigut T, Iafrate AJ, Muench J et al (2000) Simian and human immunodeficiency virus Nef proteins use different surfaces to downregulate class I major histocompatibility complex antigen expression. J Virol 74:5691–5701
Swingler S, Brichacek B, Jacque JM et al (2003) HIV-1 Nef intersects the macrophage CD40L signalling pathway to promote resting-cell infection. Nature 424:213–219
Swingler S, Mann A, Jacque J et al (1999) HIV-1 Nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat Med 5:997–103
Swingler S, Zhou J, Swingler C et al (2008) Evidence for a pathogenic determinant in HIV-1 Nef involved in B cell dysfunction in HIV/AIDS. Cell Host Microbe 17:63–76
Tobiume M, Lineberger JE, Lundquist CA et al (2003) Nef does not affect the efficiency of human immunodeficiency virus type 1 fusion with target cells. J Virol 77:10645–10650
Toussaint H, Gobert FX, Schindler M et al (2008) Human immunodeficiency virus type 1 nef expression prevents AP-2-mediated internalization of the major histocompatibility complex class II-associated invariant chain. J Virol 82:8373–8382
Trible RP, Emert-Sedlak L, Smithgall TE (2006) HIV-1 Nef selectively activates Src family kinases Hck, Lyn, and c-Src through direct SH3 domain interaction. J Biol Chem 281:27029–27038
Varin A, Manna SK, Quivy V et al (2003) Exogenous Nef protein activates NF-kappa B, AP-1, and c-Jun N-terminal kinase and stimulates HIV transcription in promonocytic cells. Role in AIDS pathogenesis. J Biol Chem 278:2219–2227
Venzke S, Michel N, Allespach I et al (2006) Expression of Nef downregulates CXCR4, the major coreceptor of human immunodeficiency virus, from the surfaces of target cells and thereby enhances resistance to superinfection. J Virol 80:11141–11152
Wang JH, Janas AM, Olson WJ et al (2007) CD4 coexpression regulates DC-SIGN-mediated transmission of human immunodeficiency virus type 1. J Virol 81:2497–2507
Wang JK, Kiyokawa E, Verdin E et al (2000) The Nef protein of HIV-1 associates with rafts and primes T cells for activation. Proc Natl Acad Sci U S A 97:394–399
Welker R, Kottler H, Kalbitzer HR et al (1996) Human immunodeficiency virus type 1 Nef protein is incorporated into virus particles and specifically cleaved by the viral proteinase. Virology 219:228–236
Wildum S, Schindler M, Munch J et al (2006) Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J Virol 80:8047–8059
Williams M, Roeth JF, Kasper MR et al (2002) Direct binding of human immunodeficiency virus type 1 Nef to the major histocompatibility complex class I (MHC-I) cytoplasmic tail disrupts MHC-I trafficking. J Virol 76:12173–12184
Wiskerchen M, Cheng-Mayer C (1996) HIV-1 Nef association with cellular serine kinase correlates with enhanced virion infectivity and efficient proviral DNA synthesis. Virology 224:292–301
Wolf D, Witte V, Laffert B et al (2001) HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat Med 7:1217–1224
Wu Y, Marsh JW (2001) Selective transcription and modulation of resting T cell activity by preintegrated HIV DNA. Science 293:1503–1506
Xu XN, Laffert B, Screaton GR et al (1999) Induction of Fas ligand expression by HIV involves the interaction of Nef with the T cell receptor zeta chain. J Exp Med 189:1489–1496
Yang X, Gabuzda D (1999) Regulation of human immunodeficiency virus type 1 infectivity by the ERK mitogen-activated protein kinase signaling pathway. J Virol 73:3460–3466
Zauli G, Gibellini D, Secchiero P et al (1999) Human immunodeficiency virus type 1 Nef protein sensitizes CD4(+) T lymphoid cells to apoptosis via functional upregulation of the CD95/CD95 ligand pathway. Blood 93:1000–1010
Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, Munch J, Kirchhoff F, Bieniasz PD, Hatziioannou T (2009) Nef Proteins from Simian Immunodeficiency Viruses Are Tetherin Antagonists. Cell Host Microbe 2009 Jun 3. [Epub ahead of print]
Acknowledgments
We thank all current and former members of the Kirchhoff laboratory and our many excellent collaborators, especially Beatrice Hahn, Leonid Margolis, Jean-Chartes Grivel, David Evans, Guido Silvestri, Jacek Skowronski and Christiane Stahl-Hennig, for support, helpful discussion and sharing reagents and knowledge. We apologize to those colleagues whose studies on Nef function could not be mentioned due to space limitations.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Arhel, N.J., Kirchhoff, F. (2009). Implications of Nef: Host Cell Interactions in Viral Persistence and Progression to AIDS. In: Spearman, P., Freed, E. (eds) HIV Interactions with Host Cell Proteins. Current Topics in Microbiology and Immunology, vol 339. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-02175-6_8
Download citation
DOI: https://doi.org/10.1007/978-3-642-02175-6_8
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-02174-9
Online ISBN: 978-3-642-02175-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)