
Abstract
The arms race between virus and host is a constant battle. APOBEC3 proteins are known to be potent innate cellular defenses against both endogenous retroelements and diverse retroviruses. However, retroviruses have developed their own methods to launch counter-strikes. Most primate lentiviruses encode a protein called the viral infectivity factor (Vif). Vif induces targeted destruction of APOBEC3 proteins by hijacking the cellular ubiquitin-proteasome pathway. Here we review the research that led up to the identification of A3G, the mechanisms by which APOBEC3 proteins can inhibit retroelements, and the counter-mechanisms that HIV-1 Vif has developed to evade its antiviral activities.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Human Immunodeficiency Virus
- Simian Immunodeficiency Virus
- Cytidine Deaminase
- Equine Infectious Anemia Virus
- Nuclear Magnetic Resonance Structure
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Great strides have been made in the research of the Human Immunodeficiency Virus (HIV) since it was first identified as the causative agent of Acquired Immunodeficiency Syndrome (AIDS). However, it was not until a few years ago that the function of the viral protein, viral infectivity factor (Vif), was revealed. Vif was found to overcome the host antiviral protein, APOBEC3G (A3G) (Sheehy et al. 2002). The A3G protein belongs to a family of human cytidine deaminases that can inhibit the replication of Vif-deficient viruses. A3G is packaged into newly formed virions of the defective virus, and upon infection of new cells, induces C→U mutations in the minus strand of the newly synthesized viral cDNA (Sheehy et al. 2002; Bieniasz 2004; Rose et al. 2004; Goff 2003; Navarro and Landau 2004; Turelli and Trono 2005; Zhang et al. 2003; Mangeat et al. 2003; Lecossier et al. 2003; Harris et al. 2002, 2003; Mariani et al. 2003; Chiu and Greene 2008; Bishop et al. 2008). In addition, A3G may also have other antiviral activities that have not yet been fully characterized (Newman et al. 2005). Vif, however, counteracts this potent antiviral element of our immune system by hijacking components of the cellular degradation pathway, and targeting A3G for proteasomal degradation (Yu et al. 2003). This prevents A3G from being packaged into budding viruses, and allows them to successfully infect new cells. In this review, we seek to analyze recent data in the field and address areas of research that still need to be tackled.
2 Non-Permissive Cells and the Identification of A3G
HIV-1 Vif was first identified in 1986 as small 192 residue protein, with a molecular weight of 23 kD. It was termed “sor” for short open reading frame protein and categorized as an accessory protein, since Vif deletion viruses were still able to replicate in certain cell lines (Kan et al. 1986; Lee et al. 1986; Sodroski et al. 1986; Strebel et al. 1987). These cell types were called permissive cells. However, it was found that in other cell types (non-permissive cells) virions produced in the absence of Vif were approximately 1,000 times less infectious than those produced from wild-type HIV-1 (Sheehy et al. 2002; Madani and Kabat 1998; Gabuzda et al. 1992; Simon et al. 1998). Permissive cell types included HeLa, HEK 293T, SupT1, and CEM-SS cell lines. On the other hand, primary human T-lymphocytes, macrophages, H9, and CEM cells were shown to be non-permissive, and thus incapable of producing infectious virions (Gabuzda et al. 1992; Simon et al. 1998; von Schwedler et al. 1993). Eventually, cell fusion experiments showed that this non-permissive phenotype was dominant. Permissive 293T cells were fused with the non-permissive HUT78 cells, resulting in virions that were less infectious than those produced from 293T cells alone (Madani and Kabat 1998; Simon et al. 1998). However, the particular restriction factor in these so-called non-permissive cells was not identified until 2002, when Sheehy et al. performed further experiments with two genetically related cell lines, the permissive CEM-SS line, and the parental non-permissive cells CEM T-cell line it was generated from (Sheehy et al. 2002). Comparison of cDNAs generated from these two cell lines consistently produced an approximately 1.5-kb cDNA segment that was expressed in all non-permissive cell lines tested, but not in permissive cell lines. This protein was identified as CEM15 or human cytidine deaminase apolipoprotein B mRNA-editing catalytic polypeptide-like 3G (APOBEC3G, A3G), a member of the APOBEC family of cytidine deaminases. Later studies showed that A3F (Bishop et al. 2004; Liddament et al. 2004; Wiegand et al. 2004; Zheng et al. 2004; Dang et al. 2006), A3B (Bishop et al. 2004; Dang et al. 2006; Yu et al. 2004a; Bogerd et al. 2006a, b) A3DE (Dang et al. 2006), and A3H (Dang et al. 2006; Tan et al. 2008; OhAinle et al. 2008), related deaminase proteins, are also capable of restricting Vif-deficient HIV-1.
3 The APOBEC Family of Cytidine Deaminases
The APOBEC family of cytidine deaminases is a large family of proteins with a conserved zinc-coordinating deaminase motif, H-X-E-(X)27–28-P-C-X2–4-C, that is capable of converting cytosine to uracil in RNA or DNA. This family includes APOBEC1, expressed mainly in the small intestine (Teng et al. 1993), and activation-induced cytidine deaminase (AID), a B cell-specific protein, both of which are found on chromosome 12. The family also includes APOBEC2 on chromosome 6, which is highly expressed in muscle tissue (Liao et al. 1999) and APOBEC4 on chromosome 1, reported to be expressed primarily in testes (Rogozin et al. 2005). APOBEC1 is responsible for the C-to-U editing of apolipoprotein B (apo-B) mRNA, and is catalyzed by a multiprotein complex that recognizes an 11-nucleotide sequence downstream of the editing site (Turelli and Trono 2005; Harris and Liddament 2004). AID on the other hand edits single-stranded DNA, and is required for somatic hypermutation (SHM), class switching recombination, and gene conversion of immunoglobulin genes (Turelli and Trono 2005; Harris and Liddament 2004). The cellular functions of the APOBEC2 and APOBEC4 proteins are poorly understood. Much is known on the other hand about the 7 members of the APOBEC3 family: A3A, A3B, A3C, A3DE, A3F, A3G, and A3H. These proteins are found on chromosome 22 (Jarmuz et al. 2002), and are thought to have been generated by multiple, and relatively recent, duplication events. In fact, rodents possess only one APOBEC3 gene which has been shown to be inessential for mouse development, fertility or survival (Mikl et al. 2005). It is thought that this single APOBEC3 gene expanded in primates 40–100 million years ago (Jarmuz et al. 2002). It is significant that several APOBEC3 genes appear to be affected by positive selection pressure, as evidenced by the accumulation of non-synonymous mutations (Sawyer et al. 2004; Zhang and Webb 2004; Conticello et al. 2005). One explanation for this high level of selection pressure is that these proteins evolved as a defense against endogenous retroelements, since the expansion of APOBEC3 proteins in primates appears to coincide with a sharp decline in retrotransposition rates in humans (Waterston et al. 2002). Indeed, it has been shown that several APOBEC3 members are capable of inhibiting a variety of endogenous retroelements as well as various retroviruses.
Although A3G was the first anti-HIV-1 deaminase identified, A3F, A3B, A3DE, and A3H can all inhibit Vif-deficient HIV-1. And although A3C has little effect on HIV-1, it is extremely effective against Vif-deficient simian immunodeficiency virus (SIV) (Yu et al. 2004a). In addition, several APOBEC3 proteins can also inhibit a variety of retroviruses such as mouse mammary tumor virus (MMTV), murine leukemia virus (MLV), human T-cell leukemia virus (HTLV), equine infectious anemia virus (EIAV), Rous sarcoma virus (RSV), Feline leukemia virus (FIV), as well as various foamy viruses (FV). Several APOBEC3 proteins can also inhibit the partial double-stranded DNA virus, Hepatitis B (HBV) ,as well as the single-stranded DNA virus, adeno-associated virus (AAV). And finally a recent study presents evidence that may extend the antiviral repertoire of APOBEC3 proteins to double-stranded DNA viruses, such as human papillomavirus (HPV). Several APOBEC3 proteins can also inhibit various retroelements, including both long terminal repeat (LTR) retrotransposons such as Ty1, IAP, MusD, and HERV-K, as well as non-LTR retroelements such as human LINE-1 and Alu. Table 1 offers a comprehensive list of the APOBEC family of proteins and their known effects on various microorganisms.
4 The Antiviral Mechanism of APOEBC3 Proteins: Deaminase Dependent
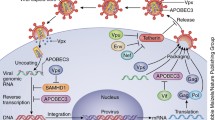
APOBEC3 proteins have been reported to have various inhibitory effects on multiple viruses, as mentioned above. For the purpose of simplicity, we will focus mainly on the antiviral effects of A3G on HIV-1, the most well-studied virus. Specifically, A3G has been reported to inhibit: (1) the accumulation of viral DNA (Mangeat et al. 2003; Mariani et al. 2003; Bishop et al. 2008; von Schwedler et al. 1993; Bishop et al. 2006; Holmes et al. 2007; Simon and Malim 1996); (2) the accumulation of two-LTR circle DNA (Luo et al. 2007; Anderson and Hope 2008); and (3) proviral DNA formation (Luo et al. 2007; Miyagi et al. 2007) (Fig. 1).

A3G in the cell. A3G can be packaged into newly budding virions, which when new infections take place in target cells (right), can edit newly reverse transcribed ssDNA. A3G may also have deaminase-independent functions in producer cells (left), such as inhibition of reverse transcription, strand transfer interactions, and DNA strand elongation
APOBEC3 proteins, like AID, prefer to edit single-stranded DNA. In particular, A3G edits the newly reverse transcribed minus-strand DNA of HIV-1, where it induces C-to-U mutations (Zhang et al. 2003; Mangeat et al. 2003; Lecossier et al. 2003; Harris et al. 2003; Mariani et al. 2003; Yu et al. 2004b; Suspene et al. 2004). Uracil-DNA glycosylases, such as UNG2 or SMUG1, may then generate abasic sites, which in turn may inhibit plus-strand DNA synthesis (Cai et al. 1993; Klarmann et al. 2003; Yang et al. 2007a), or even trigger DNA degradation by cellular endonucleases such as apurinic-apyrimidinic endonucleases (Harris et al. 2003; Yang et al. 2007a). Several laboratories have shown that host UNG-2 can be packaged into HIV-1 virions, via its interaction with other HIV-1 proteins, such as the viral protein R (Vpr) (Mansky et al. 2000) or integrase (IN) (Willetts et al. 1999; Priet et al. 2003). Alternatively, if the minus-strand uracil-containing viral DNA serves as a template for synthesizing the sense-strand DNA, the G-to-A-hypermutated viral DNA may generate premature stop codons or heavily mutated non-functional viral proteins.
Others, however, believe that UNG-2 is dispensable for the antiviral activity of A3G. One group used a bacteriophage protein, uracil DNA glycosylase inhibitor (UGI), in order to demonstrate that the levels of HIV-1 infectivity and A3G restriction were not affected, regardless of the presence or absence of UNG catalytic activity (Kaiser and Emerman 2006; Mbisa et al. 2007). Similar results were obtained with HBV and RSV systems (Nguyen et al. 2007; Langlois and Neuberger 2008). Nevertheless, it cannot be ruled out that Vpr may degrade UNG2 and SMUG1 (Schrofelbauer et al. 2005), thus overcoming the problem of abasic site generation. Alternatively, two other glycosylases, MBD4 and TDG, exist, both capable of acting on dsDNA (Barnes and Lindahl 2004). Neither of these proteins has been studied in the context of A3G antiviral activity. However, deamination may not be the only antiviral mechanism of APOBEC3 proteins.
5 The Antiviral Mechanism of APOEBC3 Proteins: Deaminase Independent
Several groups have shown that A3G and A3F deaminase mutants still retain some anti-HIV-1 activity (Newman et al. 2005; Bishop et al. 2006; Holmes et al. 2007). The first of these deaminase-independent mechanisms is inhibition of reverse transcription (Guo et al. 2006, 2007; Yang et al. 2007b). One group showed that much of the early inhibition of viral DNA production induced by A3G correlated with the inhibition of early minus-sense strong stop DNA, and the inability of tRNA Lys3 to prime reverse transcription (Guo et al. 2006). Later, the same group also used an in vitro system to show that A3G decreased the ability of tRNA Lys3 to bind to viral RNA and initiate reverse transcription (Guo et al. 2007). Similar results were obtained with A3F (Yang et al. 2007b).
The second mechanism described is the inhibition of strand transfer reactions (Mbisa et al. 2007; Li et al. 2007). Li et al. reported that A3G-induced inhibition of both minus- and plus-strand transfer in reverse transcription was responsible for the majority of the reduction in late DNA synthesis. Mbisa et al. showed that a deaminase-defective A3G resulted in defects not only in plus-strand DNA transfer and integration, but also in primer tRNA processing.
Finally, it was also shown that A3G can inhibit reverse transcriptase-catalyzed DNA elongations (Iwatani et al. 2007). Iwatani et al. used purified A3G, NC and RT in an established in vitro system to study reverse transcription. They found that A3G could inhibit all reverse transcriptase (RT)-catalyzed DNA elongation reactions, but did not have any effect on RNase H activity or NC's activities as a nucleic acid chaperone.
Interestingly, although A3G normally exerts its effects by inhibiting new infections after being packaged into newly budded viruses, it may also exert a post-entry block for HIV-1 in resting T cells (Chiu et al. 2005) and dendritic cells (Kreisberg et al. 2006). While activated CD4+ T lymphocytes are highly permissive to wild-type HIV-1 infection, resting PBMCs appear to be resistant to infection (Chiu et al. 2005). Chiu et al. showed that A3G in activated CD4+T cells exists in high-molecular mass ribonucleoprotein complexes (HMM), while A3G in resting CD4+ T cells is predominantly found in an enzymatically active low molecular mass (LMM) form. This LMM form of A3G is thought to cause a post-entry block early in the HIV-1 replication cycle, by impairing reverse transcription (Chiu et al. 2005). These results were reproducible in both naive and memory T cells (Kreisberg et al. 2006), as well as macrophages and dendritic cells (Stopak et al. 2007). Activation of these resting cells with various mitogens and cytokines caused a shift in A3G from LMM to HMM complexes, which also correlated with increased susceptibility to HIV-1 infection (Chiu et al. 2005; Stopak et al. 2007). That this post-entry block is caused by A3G is further supported by data showing that siRNA directed against A3G in unstimulated CD4+ T cells increased cell permissivity to HIV-1 (Chiu et al. 2005).
In addition, sequencing of reverse transcripts from infected cells showed only low levels of G-to-A hypermutation, suggesting that this early restriction is deaminase-independent. Nonetheless, this restriction is not absolute, as newly synthesized viral DNA was detected in resting CD4+ T cells after a 24- to 48-h delay (Chiu et al. 2005). Inactivated T cells was also reported by others to be permissive to HIV-1 infection in some cases (Stevenson et al. 1990; Watson and Wilburn 1992).
In addition, several APOBEC3 proteins appear to inhibit HBV, AAV, IAP, MusD, and LINE-1 in a deaminase-independent manner, which has yet to be described (Bogerd et al. 2006a; Chen et al. 2006; Stenglein and Harris 2006; Muckenfuss et al. 2006), supporting the idea of alternative, and deaminase-independent inhibition mechanisms.
However, there is still no clear consensus on these deaminase-independent activities, with some arguing that these effects may be just a result of protein overexpression to levels far above normal physiological concentrations. One group showed that active site mutants of A3G had no antiviral activity when expressed at levels similar to those observed in primary human T cells (Holmes et al. 2007; Miyagi et al. 2007; Mbisa et al. 2007; Schumacher et al. 2008). Another showed that the catalytic site of A3G and DNA cytosine deamination is important in inhibiting Ty1, MusD, and HIV-1 when expressed at near-physiologic levels (Schumacher et al. 2008). Nevertheless, this does not rule out the deaminase-independent mechanisms described above, as the active site mutants tested may also block important protein functions such as protein–protein or protein–DNA/RNA interactions.
6 A3G Structural Features
To better understand the structural features of APOBEC3 proteins that are important for relationships with viral proteins such as HIV-1 Vif and NC, and in order to inhibit these interactions and thus achieve our ultimate goal of preventing disease, it is essential to have high resolution structures of these proteins.
Three recent reports on the structure of the C-terminal cytidine deaminase domain (CDD) have highlighted several of these structural features. The first structure was resolved by nuclear magnetic resonance (NMR) (Chen et al. 2008). The second more recent structure was resolved by x-ray crystallography (Holden et al. 2008). The second report confirmed many of the structural features and characteristics first reported by Chen et al. but also reported several important differences. While the crystal structure had a five β-sheet core structure surrounded by six α-helices, the NMR structure was reported to have the same core β-structure, but was surrounded by only five α-helices. Furthermore, the A3G crystal structure was reported to have a long and well-defined β2 strand, while the same stretch of amino acids in the NMR structure was short, and interrupted by a 6-residue bulge. In addition, the two active center loops located near the active site were found at different positions. However, the most important difference was in the proposed nucleic acid substrate-binding area. While the crystal structure had a deep, well-defined groove that ran alongside the AC loops and an active site where the cytosine substrate is thought to be deaminated, the NMR structure did not have this groove. Instead, the authors noticed several positively charged residues surrounding the active site region. Several of these amino acids overlap with residues found in the crystal structure groove. These structural differences could be attributed to several mutations made in the A3G construct that was used to obtain the NMR structure. However, these differences may also be a result of the different methodologies used for isolating the protein and/or structural analysis.
Finally, the most recent report (Furukawa et al. 2009), also presents the structure of the wild-type CDD of A3G, resolved in solution by NMR. While this latest structure shares some similarities with the crystal structure (such as the presence of a sixth alpha helix), it also has some features in common with Chen et al.’s NMR structure (such as the second interrupted beta strand). The convergence for the two active center loops near the active site was poor and these elements are not well defined in this structure. In addition, the authors presenting this third structure identified yet another position for the binding of the ssDNA substrate. They propose a model where the ssDNA is positioned along the two alpha helices α1 and α2. Clearly, additional experimental studies, in particular mutational assays will be necessary to determine which, if any, of these three models is correct.
Future studies in this field will most assuredly include attempts to crystallize the N-terminal deaminase domain of A3G, as well as the entire molecule. Furthermore, molecular modeling studies of other deaminase proteins based on this structure may shed light on their different substrate-binding and antiviral specificities.
7 Packaging of APOBEC3 Molecules
In order for APOBEC3 molecules to be effective in inhibiting HIV-1, they need to be effectively packaged into newly budded virions. Studies of A3G packaging requirements revealed that A3G binds to the HIV-1 Gag protein, and in particular to the nucleocapsid (NC) region (Cen et al. 2004; Khan et al. 2005; Schafer et al. 2004; Alce and Popik 2004; Zennou et al. 2004; Luo et al. 2004). Several groups have reported that HIV-1 Gag is necessary and sufficient for A3G packaging; however, these experiments were performed with virus-like particles rather than whole virus, which may have different requirements (Cen et al. 2004; Schafer et al. 2004; Alce and Popik 2004; Svarovskaia et al. 2004; Douaisi et al. 2004). Most groups, however, agree that the efficiency of A3G packaging is significantly enhanced by RNA interactions (Svarovskaia et al. 2004). Although A3G appears to edit only ssDNA, it is nevertheless capable of strongly binding to RNA (Jarmuz et al. 2002; Yu et al. 2004b; Khan et al. 2005; Iwatani et al. 2006; Khan et al. 2007; Wang et al. 2007a; Tian et al. 2007). The NC region of Gag is also important for this interaction with RNA, and, together with A3G, may form a protein:RNA complex (Iwatani et al. 2006; Burnett and Spearman 2007).
In the case of A3G, it is the N terminal cytidine deaminase that is thought to be responsible for the nucleic acid binding properties that contribute to packaging, while the C terminal domain is generally thought to be responsible for deamination (Newman et al. 2005; Iwatani et al. 2006; Navarro et al. 2005; Hache et al. 2005). Several groups have identified specific residues in the N-terminal region of A3G that were shown to affect packaging, in particular, W127 (Huthoff and Malim 2007), which was also shown to be important for RNA-binding.
Although many agree that an RNA interaction is important for efficient packaging, the nature of this RNA is still highly debated. One proposed candidate is the HIV-1 viral genomic RNA, where studies showed that, although A3G could be successfully packaged in the absence of viral RNA, these A3G molecules were not associated with the viral cores of the virions (Khan et al. 2005,2007). A3G molecules were able to be re-associated with the viral cores by the addition of viral RNA in trans. Another RNA candidate is a cellular RNA, and a component of signal recognition particles (SRP), 7SL RNA (Wang et al. 2007a). Indeed, 7SL RNA has been reported to be both abundantly and selectively packaged in HIV-1 virions (Khan et al. 2007; Wang et al. 2007a; Onafuwa-Nuga et al. 2006). Both A3G and NC mutants that were known to have packaging defects, also showed a reduced ability to bind to and package 7SL RNA (Wang et al. 2007a; Bach et al. 2008). Experiments involving overexpression of SRP19, a known 7SL-binding protein, in increasing concentrations in order to reduce the level of free 7SL RNA found in the cell were performed. This overexpression was associated with reduced levels of A3G packaging (Wang et al. 2007a), further supporting the idea that 7SL RNA contributes to the A3G packaging process. Others, however, believe that, although 7SL RNA can bind both A3G and NC, it is not an essential factor for the A3G packaging process (Bach et al. 2008). And although regions in both NC and A3G have been shown to be important for A3G packaging, it is not known whether this interaction is direct or if their interaction could be mediated by other molecules, such as 7SL RNA. This field is still far from a consensus, and more research is needed in order to resolve these discrepancies.
In addition to the mechanism of packaging, the location of packaging is also an important factor for antiviral activity. A3A, another member of the APOBEC3 family, has a potent inhibitory effect on several retroelements (Bogerd et al. 2006b; Chen et al. 2006), but has no effect on HIV-1 (Bishop et al. 2004). However, when A3A was targeted to the viral core, by creating a Vpr-A3A fusion protein, this cytidine deaminase was able to restrict both HIV-1 and SIV in a Vif-independent manner (Aguiar et al. 2008).
However, not all APOBEC3 proteins are packaged by the same mechanism. APOBEC3C, which efficiently restricts SIV but not HIV (Yu et al. 2004a), can nevertheless be packaged into both. A3C also interacts with HIV-1 Gag protein, but unlike A3G, A3C is packaged through a RNA-dependent and NC-independent fashion (Wang et al. 2008). Thus, it appears that individual APOBEC3 proteins have evolved to use different mechanisms for targeting retroviruses, possibly to broaden the range of viruses targeted.
8 Vif Targets APOBEC3 Proteins for Proteasome-Mediated Degradation
Although APOBEC3 proteins may be a formidable weapon in the cell’s antiviral arsenal, several retroviruses have developed their own counter measures that inhibit HIV. The SIV and HIV-1 lentiviruses encode the Vif protein, which can induce the polyubiquitination and degradation of multiple APOBEC3 molecules (Yu et al. 2003; Mehle et al. 2004; Stopak et al. 2003; Marin et al. 2003; Conticello et al. 2003; Sheehy et al. 2003; Liu et al. 2004, 2005). Vif proteins assemble with Cul5, ElonginB, ElonginC, and Rbx1 proteins to form an E3 ubiquitin ligase (Yu et al. 2003; Mehle et al. 2004; Liu et al. 2005; Yu et al. 2004c; Luo et al. 2005; Kobayashi et al. 2005) (Fig. 2).

The HIV-1 Vif-Cul5 E3 ubiquitin ligase complex; a model of Vif-induced APOBEC3 degradation. Vif binds ElonginC through a viral specific BC-box; ElonginC also interacts with ElonginB. The Vif-ElonginC-ElonginB complex also binds to Cul5, forming the Cul5-EloB/C-Vif E3 ligase. Cul5 also binds Rbx which recruits the E2-ubiquitin conjugating enzyme. Ubiquitin activated by E1 enzyme is transiently transferred to the E2 enzyme, then to the APOBEC3 target molecule. Polyubiquitination of APOBEC3 molecules takes place, followed by degradation by the 26S proteasome
E3 ubiquitin ligases are critical for regulating cellular processes such as mitosis and the cell cycle through targeted protein degradation (Pickart 2004). It is members of the E3 ubiquitin ligase family, such as the cullin-based E3 ligases that mediate protein degradation specificity. These cullins can then form a scaffold on which other E3 ligase protein components assemble and convey the substrate to the E2 ubiquitin-conjugating enzyme. The Vif binding cullin, Cul5, is commonly associated with the ElonginC and ElonginB adaptor molecules. ElonginC recognizes substrate receptor proteins containing a BC-box. The SLQxLA motif in Vif is highly conserved in lentiviral Vifs, and forms a virus-specific BC-box motif that mediates the interaction with ElonginC, which in turn interacts with both ElonginB and Cul5 (Figs. 2 and 3). Vif in turn binds to Cul5 through two other highly conserved sites, the Hx5Cx17-18Cx3-5H motif (Luo et al. 2005; Xiao et al. 2007; Mehle et al. 2006) and a LPx4L motif downstream (Stanley et al. 2008) (Fig. 3). The first motif is responsible for binding zinc, which stabilizes the molecule, and the second is a highly conserved hydrophobic interface that mediates Cul5 selection.

APOBEC3 and Vif interaction domains. Various domains for interaction of Vif and APOBEC3 with each other as well as other proteins have been mapped. A3F and A3G have different binding requirements on Vif
In terms of Vif binding to APOBEC3 proteins, several regions have been described in both A3G and A3F, and will be further discussed below. It is also of interest that A3C and A3DE, two APOBEC3 proteins with weaker effects on HIV-1, are also ubiquitinated and degraded by Vif (Dang et al. 2006; Zhang et al. 2008a). And although A3B may also inhibit HIV-1, it is not degraded by Vif, possibly because of its low expression level in T cells.
9 Vif May also Inhibit A3G by Degradation-Independent Mechanisms
Several lines of evidence point to the existence of degradation-independent mechanisms of A3G inhibition in Vif. Recently, a degradation-resistant variant of A3G was identified (Opi et al. 2007). An A3G mutant with a single point mutation at position 97 was found to be defective in multimerization, and at the same time to be impervious to Vif-induced proteasomal degradation, although still capable of binding to Vif (Opi et al. 2007). Surprisingly, although this A3G mutant was not degraded, Vif was still able to prevent its encapsidation into HIV-1 virions, as well as inhibit its antiviral activity (Opi et al. 2007). In fact, one report suggests that Vif’s abilities to establish the production of infectious virus and to degrade APOBEC3 proteins are completely separable functions (Kao et al. 2007). In this study, several different forms of tagged HIV-1Vif were studied. It was found that although Vif expressed from a proviral vector was less efficient at degrading A3G than Vif expressed from codon-optimized vectors, it still was able to efficiently inhibit A3G activity (Kao et al. 2007). In addition, although a YFP-tagged form of Vif was able to efficiently degrade A3G, it was unable to restore viral infectivity (Kao et al. 2007). Further support for this theory comes from Vif mutational studies. Although a point mutation of a serine at position 144 in Vif resulted in reduced levels of viral infectivity, this mutant was found to be able to degrade A36 effectively and efficiently (Mehle et al. 2004). Moreover, HIV-1 is capable of inhibiting the deamination activity of both A3G (Santa-Marta et al. 2004) and AID (Santa-Marta et al. 2007) in bacterial E. coli systems, which do not contain any proteasomal-degradation machinery.
The details and mechanism of this degradation-independent inhibition are not yet known. Two theories that have been proposed are that: (1) Vif competitively binds to a common Vif/A3G binding element that interferes with A3G packaging (Goila-Gaur et al. 2008a); or (2) Vif promotes the shift of A3G from LMM to HMM states (Goila-Gaur et al. 2008a, 2008b). A recent paper also notes that although newly synthesized A3G and stable pre-existing A3G are packaged with the same efficiency into virions, HIV-1 will preferentially degrade newly synthesized A3G (Goila-Gaur et al. 2008b). It is thus possible that Vif may have to use alternative methods to inhibit the action of pre-existing A3G in the cell. Again, this is another area of the Vif:APOBEC field that is ripe for further exploration.
10 Specificity of the Vif:APOBEC Interaction
As discussed above, several important structural elements exist in Vif that allow interactions with various molecules. In addition to the structural elements of Vif necessary for interaction with APOBEC3 proteins, there also exist certain domains on APOBEC3 proteins, important for interactions with Vif. For example, it is known that it is the N-terminal cytidine deaminase domain of A3G which interacts with Vif (Conticello et al. 2003) (Fig. 3). In contrast, it was noticed that adding tags to the C-terminal of A3F induced resistance to degradation by Vif, which was later shown to be due to reduced interaction with Vif (Tian et al. 2006). In fact, the C-terminal of A3F alone is sufficient for interaction with, and degradation by, Vif (Zhang et al. 2008b). Recently published data further specify the region of A3F necessary for interaction with Vif, and map it to an area between amino acids 283–300 (Russell et al. 2008). Furthermore, the authors also show that this stretch of amino acids was sufficient for both interaction with, and degradation by the Vif protein. Therefore, although interaction with Vif maps to the N-terminal of A3G, it appears to be the C-terminal domain that is important for this interaction in A3F (Fig. 3).
Studies to map the specific regions in Vif that are important for A3G and A3F binding revealed that various amino-terminal domains of HIV-1 Vif molecules are involved in distinct substrate APOBEC3 recognitions (Marin et al. 2003; Goila-Gaur et al. 2008b; Tian et al. 2006; Simon et al. 2005; Schrofelbauer et al. 2006; Russell and Pathak 2007; Mehle et al. 2007; He et al. 2008). A region in HIV-1 Vif, spanning a stretch of amino acids from 22 to 44, was found to be important for the suppression of A3G but not A3F (Simon et al. 2005; Russell and Pathak 2007; Mehle et al. 2007). This region is known as the G-box (Fig. 3). On the other hand, several amino acids from 11 to 17 and 74 to 79 of HIV-1 Vif were found to be important for the suppression of A3F but not A3G (Tian et al. 2006; Simon et al. 2005; Schrofelbauer et al. 2006; Russell and Pathak 2007; He et al. 2008) (Fig. 3). Yet another region, from amino acids 52 to 72, was found to be important for both A3G and A3F suppression (He et al. 2008) (Fig. 3). In addition to A3G and A3F, other APOBEC3 family members, such as A3A, A3B, A3C, and A3DE, can also bind to HIV-1 Vif (Dang et al. 2006; Zhang et al. 2008a; Marin et al. 2008). And although A3A and A3B are not thought to be normally degraded by HIV-1 Vif even though they are re-localized by it, certain Vif variants from different HIV-1 strains were capable of degrading them (Marin et al. 2008). However, little is known about how these proteins are recognized by HIV-1 Vifs of different HIV-1 strains, and further exploration is needed.
In addition to the many Vif regions that determine specificity of binding to various APOBEC3 and cellular proteins, various Vifs can also exert species-specific selectivity on the proteins they degrade. For example, HIV-1 Vif can effectively inhibit human A3G, and African green monkey SIV (SIVAgm) Vif is able to inhibit Agm A3G. However, HIV-1 Vif is unable to recognize and inhibit simian (Agm or rhesus macaque) A3G. Vice versa, SIVAgm Vif cannot inhibit human or macaque A3G. On the other hand, the Vif protein from macaque-specific SIV (SIVMac) can counteract all three A3Gs from humans, Agms, and rhesus macaques (Mariani et al. 2003). It was shown that this species specificity was conferred by a single amino acid, an aspartate at position 128 in A3G. Several independent studies found that this single amino acid residue at position 128 in human A3G was responsible for the species specificity. Changing this residue to its equivalent in Agms (D128K) was enough to reverse the specificity of HIV-1 and SIVAgm Vifs for the binding and degradation of their respective human and Agm A3Gs (Bogerd et al. 2004; Schrofelbauer et al. 2004; Mangeat et al. 2004; Xu et al. 2004). In contrast, mutations at the equivalent position in A3F, at amino acid 127, had no influence on Vif suppression (Liu et al. 2005).
Finally, it is of interest that phosphorylation of both Vif and A3G can play a role in modulating these interactions. Vif can be phosphorylated at several serine and threonine residues: T96, S144, and T188 (Yang and Gabuzda 1998; Yang et al. 1996). Mutation of these conserved residues does not affect Vif-A3G binding, or A3G degradation (Mehle et al. 2004); however, mutations preventing phosphorylation of S144 in the Vif BC-box significantly decreased Vif function by inhibiting the Vif-ElonginC interaction (Mehle et al. 2004), and thus hindering the process of proteasomal degradation. In addition, a recent report has proposed that A3G is also capable of being phosphorylated (Shirakawa et al. 2008).
Shirakawa et al. showed that protein kinase A (PKA) can bind to and phosphorylate A3G at a threonine at position 32. Phosphorylation of A3G at this position appears to reduce Vif–A3G interaction, thus reducing levels of A3G ubiquitination and degradation. Computer modeling and mutagenesis studies were also used to study an interaction between two amino acids in A3G (T32 and R24) that are proposed to be important for binding to and subsequent degradation by Vif. Thus, phosphorylation may be yet another mechanism whereby protein interaction between Vif, A3G, and proteasomal degradation machinery can be regulated.
11 Vif and Cell Cycle Inhibition
In addition to the antiviral effects Vif exerts through degradation of APOBEC3 proteins, several reports describe a Vif-induced cell cycle delay (Sakai et al. 2006; Wang et al. 2007b; Dehart et al. 2007). Vpr is normally thought to be the HIV-1 protein responsible for the G 2 cell cycle arrest that has been observed in CD4+ T cells (He et al. 1995; Jowett et al. 1995; Stewart et al. 1997). Unexpectedly, though, studies in HIV-1-infected cells in the absence of Vpr showed an accumulation of cells at the G 2 phase, and further studies in the presence or absence of Vif confirmed that this phenotype was Vif dependent (Sakai et al. 2006). Additional experiments showed that expression of the Vif protein alone in the absence of HIV-1 infection was enough to increase the ratio of G 2 :G 1 cells (Wang et al. 2007b). Moreover, another recent report showed that the G 2 arrest phenomenon was actually a cell cycle delay, and that Vif uses the same machinery to induce cell cycle delays that it does for APOBEC3 degradation (DeHart et al. 2008). DeHart et al. showed that a Cul5 E3 ligase was required for this phenotype, regardless of the presence or absence of APOBEC3 proteins. However, although the presence of Vif resulted in an accumulation of cells in the G 2 phase, when followed over a period of several days, it was shown that Vif did not inhibit cell division or reduce the number of dividing cells (DeHart et al. 2008). The degraded substrate responsible for the G2 2 disruption has not yet been identified.
12 Other Roles for Vif
In addition to APOBEC3 and proteasomal-degradation machinery proteins, several laboratories have reported that Vif also binds to many other cellular proteins. Ku70, a cellular protein involved in DNA double-strand break repair, and a component of HIV-1 pre-integration complexes (Li et al. 2001), was shown to bind to HIV-1 Vif in a yeast two-hybrid screen of a human lymphocyte cDNA library (Madani et al. 2002). Ku70, together with another protein, Ku80, form a complex (Ku70/80) that can function as a single-stranded DNA-dependent helicase. Therefore, Vif may recruit Ku70 early in the HIV-1 replication cycle to aid in the integration process of the HIV-1 PIC.
In another experiment, a glutathione S-transferase (GST) pull-down assay used to identify Vif-binding partners, showed that Vif bound specifically to the SH3 domain of Hck, a Src family tyrosine kinase (Hassaine et al. 2001). This group further showed that Hck inhibited both the production and the infectivity of HIV-1 viruses in the absence of Vif, and that expression of Hck in Jurkat cells rendered these cells less permissive to infection by Vif deletion viruses. Hck is present in monocytes but not in primary restrictive T cells (Hassaine et al. 2001). Interestingly, Hck also binds to the HIV-1 Nef protein (Saksela et al. 1995), and dominant negative forms of Hck have been shown to block HIV-1 Nef-induced MHC class I downregulation (Chang et al. 2001).
SP140, the nuclear speckle factor, is yet another cellular protein that has been reported to bind to Vif (Madani et al. 2002). SP140 was found to be expressed in non-permissive cells, but absent in permissive cells; however, expressing SP140 in permissive cells did not render them non-permissive. Nonetheless, expression of Vif in cells caused the dispersal of SP140 from nuclear speckles, or its retention in the cytoplasm (Madani et al. 2002). These nuclear speckles are known as PML bodies and have been reported to have functions in transcription, DNA repair, viral defense, cell stress, cell cycle regulation, proteolysis, and apoptosis (Bernardi and Pandolfi 2007).
Other proteins reported to associate with Vif include: SSAT, the spermine/spermidine N1-acetyl-transferase (Lee et al. 1999), a protein involved in polyamine metabolism, the regulation of which could affect viral RNA; cyclophilin A, a member of the peptidyl-prolyl cis-trans isomerase (PPIase) family, which has been shown to interact with many HIV-1 proteins including p55 gag, Vpr, and capsid protein. Cyclophilin A has been shown to be necessary for the formation of infectious HIV virions (Billich et al. 1995); and NVBP, a novel Vif-binding protein (Lee et al. 1999).
Although the interaction of Vif with most of these proteins was reported before Vif was known to degrade A3G, it may be worth re-examining the roles of these proteins in relation to Vif, particularly in light of the recently reported cell cycle disruptions, and the degradation-independent inhibition of A3G induced by Vif.
13 Conclusions
Although the HIV field, and in particular the APOBEC/Vif area, is a rapidly advancing one that aggressively pursues new discoveries, several important questions still remain. It is unclear what (if any) other cellular and antiviral functions APOBEC3 proteins may possess. Similarly, Vif has shown signs of being involved in processes other than APOBEC3 degradation, and these need to be further investigated. The regions and particular amino acids of APOBEC3 and Vif involved in their interaction still need to be fully mapped. In addition, high resolution structures for both Vif and full-length A3G still do not exist. Such structures and further mapping of protein interactions are essential for initiating new drug design studies.
References
Aguiar RS et al (2008) Vpr.A3A chimera inhibits HIV replication. J Biol Chem 283(5):2518–2525
Alce TM, Popik W (2004) APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J Biol Chem 279(33):34083–34086
Anderson JL, Hope TJ (2008) APOBEC3G restricts early HIV-1 replication in the cytoplasm of target cells. Virology 375(1):1–12
Bach D et al (2008) Characterization of APOBEC3G binding to 7SL RNA. Retrovirology 5:54
Barnes DE, Lindahl T (2004) Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet 38:445–476
Baumert TF et al (2007) Hepatitis B virus DNA is subject to extensive editing by the human deaminase APOBEC3C. Hepatology 46(3):683–689
Bernardi R, Pandolfi PP (2007) Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8(12):1006–1016
Bieniasz PD (2004) Intrinsic immunity: a front-line defense against viral attack. Nat Immunol 5(11):1109–1115
Billich A et al (1995) Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus (HIV) type 1: interference with HIV protein-cyclophilin A interactions. J Virol 69(4):2451–2461
Bishop KN et al (2004) Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol 14(15):1392–1396
Bishop KN, Holmes RK, Malim MH (2006) Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol 80(17):8450–8458
Bishop KN et al (2008) APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog 4(12):e1000231
Bogerd HP et al (2004) A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc Natl Acad Sci USA 101(11):3770–3774
Bogerd HP et al (2006a) Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci USA 103(23):8780–8785
Bogerd HP et al (2006b) APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res 34(1):89–95
Bonvin M et al (2006) Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 43(6):1364–1374
Burnett A, Spearman P (2007) APOBEC3G multimers are recruited to the plasma membrane for packaging into human immunodeficiency virus type 1 virus-like particles in an RNA-dependent process requiring the NC basic linker. J Virol 81(10):5000–5013
Cai H et al (1993) Kinetics of deoxyribonucleotide insertion and extension at abasic template lesions in different sequence contexts using HIV-1 reverse transcriptase. J Biol Chem 268(231):23567–23572
Cen S et al (2004) The interaction between HIV-1 Gag and APOBEC3G. J Biol Chem 279(32):33177–33184
Chang AH, O'Shaughnessy MV, Jirik FR (2001) Hck SH3 domain-dependent abrogation of Nef-induced class 1 MHC down-regulation. Eur J Immunol 31(8):2382–2387
Chen H et al (2006) APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol 16(5):480–485
Chen KM et al (2008) Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 452(7183):116–119
Chiu YL, Greene WC (2008) The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol 26:317–353
Chiu YL et al (2005) Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature 435(7038):108–114
Chiu YL et al (2006) High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc Natl Acad Sci USA 103(42):15588–15593
Conticello SG, Harris RS, Neuberger MS (2003) The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol 13(22):2009–2013
Conticello SG et al (2005) Evolution of the AID/APOBEC family of polynucleotide (deoxy) cytidine deaminases. Mol Biol Evol 22(2):367–377
Dang Y et al (2006) Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol 80(21):10522–10533
Dehart JL et al (2007) HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol J 4(1):57
DeHart JL et al (2008) Human immunodeficiency virus type 1 Vif induces cell cycle delay via recruitment of the same E3 ubiquitin ligase complex that targets APOBEC3 proteins for degradation. J Virol 82(18):9265–9272
Derse D et al (2007) Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc Natl Acad Sci USA 104(8):2915–2920
Douaisi M et al (2004) HIV-1 and MLV Gag proteins are sufficient to recruit APOBEC3G into virus-like particles. Biochem Biophys Res Commun 321(3):566–573
Dutko JA et al (2005) Inhibition of a yeast LTR retrotransposon by human APOBEC3 cytidine deaminases. Curr Biol 15(7):661–666
Esnault C et al (2006) Dual inhibitory effects of APOBEC family proteins on retrotransposition of mammalian endogenous retroviruses. Nucleic Acids Res 34(5):1522–1531
Furukawa A et al (2009) Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. Embo J 28(4):440–451
Gabuzda DH et al (1992) Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J Virol 66(11):6489–6495
Goff SP (2003) Death by deamination: a novel host restriction system for HIV-1. Cell 114(3):281–283
Goila-Gaur R, Strebel K (2008) HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 5:51
Goila-Gaur R et al (2008) HIV-1 Vif promotes the formation of high molecular mass APOBEC3G complexes. Virology 372(1):136–146
Guo F et al (2006) Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J Virol 80(23):11710–11722
Guo F, et al. (2007) The interaction of APOBEC3G with HIV-1 nucleocapsid inhibits tRNALys3 annealing to viral RNA. J Virol doi:10.1128/JVI.00162-07
Hache G, Liddament MT, Harris RS (2005) The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J Biol Chem 280(12):10920–10924
Harris RS, Liddament MT (2004) Retroviral restriction by APOBEC proteins. Nat Rev Immunol 4(11):868–877
Harris RS, Petersen-Mahrt SK, Neuberger MS (2002) RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell 10(5):1247–1253
Harris RS et al (2003) DNA deamination mediates innate immunity to retroviral infection. Cell 113(6):803–809
Hassaine G et al (2001) The tyrosine kinase Hck is an inhibitor of HIV-1 replication counteracted by the viral vif protein. J Biol Chem 276(20):16885–16893
He J et al (1995) Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol 69(11):6705–6711
He Z et al (2008) Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J Mol Biol 381(4):1000–1011
Holden LG et al (2008) Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature 456(7218):121–124
Holmes RK et al (2007) APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem 4:2587–2595
Huthoff H, Malim MH (2007) Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and virion encapsidation. J Virol 81(8):3807–3815
Iwatani Y et al (2006) Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J Virol 80(12):5992–6002
Iwatani Y et al (2007) Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res 35(21):7096–7108
Jarmuz A et al (2002) An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79(3):285–296
Jost S et al (2007) Induction of antiviral cytidine deaminases does not explain the inhibition of hepatitis B virus replication by interferons. J Virol 81(19):10588–10596
Jowett JB et al (1995) The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J Virol 69(10):6304–6313
Kaiser SM, Emerman M (2006) Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J Virol 80(2):875–882
Kan NC et al (1986) Identification of HTLV-III/LAV sor gene product and detection of antibodies in human sera. Science 231(4745):1553–1555
Kao S et al (2007) Production of infectious virus and degradation of APOBEC3G are separable functional properties of human immunodeficiency virus type 1 Vif. Virology 369(2):329–339
Khan MA et al (2005) Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J Virol 79(9):5870–5874
Khan MA et al (2007) Analysis of the contribution of cellular and viral RNA to the packaging of APOBEC3G into HIV-1 virions. Retrovirology 4:48
Klarmann GJ et al (2003) Incorporation of uracil into minus strand DNA affects the specificity of plus strand synthesis initiation during lentiviral reverse transcription. J Biol Chem 278(10):7902–7909
Kobayashi M et al (2004) APOBEC3G targets specific virus species. J Virol 78(15):8238–8244
Kobayashi M et al (2005) Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem 280(19):18573–18578
Kreisberg JF, Yonemoto W, Greene WC (2006) Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J Exp Med 203(4):865–870
Langlois MA, Neuberger MS (2008) Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J Virol 82(9):4660–4664
Lecossier D et al (2003) Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 300(5622):1112
Lee YN, Bieniasz PD (2007) Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog 3(1):e10
Lee TH et al (1986) A new HTLV-III/LAV protein encoded by a gene found in cytopathic retroviruses. Science 231(4745):1546–1549
Lee B et al (1999) Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci USA 96(9):5215–5220
Lee YN, Malim MH, Bieniasz PD (2008) Hypermutation of an ancient human retrovirus by APOBEC3G. J Virol 82(17):8762–8770
Li L et al (2001) Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. Embo J 20(12):3272–3281
Li XY et al (2007) APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem 282(44):32065–32074
Liao W et al (1999) APOBEC-2, a cardiac- and skeletal muscle-specific member of the cytidine deaminase supergene family. Biochem Biophys Res Commun 260(2):398–404
Liddament MT et al (2004) APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol 14(15):1385–1391
Liu B et al (2004) Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J Virol 78(4):2072–2081
Liu B et al (2005) Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J Virol 79(15):9579–9587
Luo K et al (2004) Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J Virol 78(21):11841–11852
Luo K et al (2005) Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5–E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci USA 102(32):11444–11449
Luo K et al (2007) Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J Virol 81(13):7238–7248
Madani N, Kabat D (1998) An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. J Virol 72(12):10251–10255
Madani N et al (2002) Implication of the lymphocyte-specific nuclear body protein Sp140 in an innate response to human immunodeficiency virus type 1. J Virol 76(21):11133–11138
Mahieux R et al (2005) Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J Gen Virol 86(Pt 9):2489–2494
Mangeat B et al (2003) Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424(6944):99–103
Mangeat B et al (2004) A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J Biol Chem 279(15):14481–14483
Mansky LM et al (2000) The interaction of vpr with uracil DNA glycosylase modulates the human immunodeficiency virus type 1 In vivo mutation rate. J Virol 74(15):7039–7047
Mariani R et al (2003) Species-Specific Exclusion of APOBEC3G from HIV-1 Virions by Vif. Cell 114(1):21–31
Marin M et al (2003) HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med 9(11):1398–1403
Marin M et al (2008) Human immunodeficiency virus type 1 Vif functionally interacts with diverse APOBEC3 cytidine deaminases and moves with them between cytoplasmic sites of mRNA metabolism. J Virol 82(2):987–998
Mbisa JL et al (2007) Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol 81(13):7099–7110
Mehle A et al (2004) Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev 18(23):2861–2866
Mehle A et al (2006) A zinc-binding region in Vif binds Cul5 and determines cullin selection. J Biol Chem 281(25):17259–17265
Mehle A et al (2007) Identification of an APOBEC3G binding site in human immunodeficiency virus type 1 Vif and inhibitors of Vif-APOBEC3G binding. J Virol 81(23):13235–13241
Mikl MC et al (2005) Mice deficient in APOBEC2 and APOBEC3. Mol Cell Biol 25(16):7270–7277
Miyagi E et al (2007) Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J Virol 81(24):13346–13353
Muckenfuss H et al (2006) APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem 281(31):22161–22172
Navarro F, Landau NR (2004) Recent insights into HIV-1 Vif. Curr Opin Immunol 16(4):477–482
Navarro F et al (2005) Complementary function of the two catalytic domains of APOBEC3G. Virology 333(2):374–386
Newman EN et al (2005) Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol 15(2):166–170
Nguyen DH, Gummuluru S, Hu J (2007) Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol 81(9):4465–4472
Niewiadomska AM et al (2007) Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J Virol 81(17):9577–9583
Noguchi C et al (2005) G to A hypermutation of hepatitis B virus. Hepatology 41(3):626–33
OhAinle M et al (2008) Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4(3):249–259
Ohsugi T, Koito A (2007) Human T cell leukemia virus type I is resistant to the antiviral effects of APOBEC3. J Virol Methods 139(1):93–96
Onafuwa-Nuga AA, Telesnitsky A, King SR (2006) 7SL RNA, but not the 54-kd signal recognition particle protein, is an abundant component of both infectious HIV-1 and minimal virus-like particles. Rna 12(4):542–546
Opi S et al (2007) Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J Virol 81(15):8236–8246
Pickart CM (2004) Back to the future with ubiquitin. Cell 116(2):181–190
Priet S et al (2003) Differential incorporation of uracil DNA glycosylase UNG2 into HIV-1, HIV-2, and SIV(MAC) viral particles. Virology 307(2):283–289
Rogozin IB et al (2005) APOBEC4, a new member of the AID/APOBEC family of polynucleotide (deoxy) cytidine deaminases predicted by computational analysis. Cell Cycle 4(9):1281–1285
Rose KM et al (2004) Transcriptional regulation of APOBEC3G, a cytidine deaminase that hypermutates human immunodeficiency virus. J Biol Chem 279(40):41744–41749
Rosler C et al (2005) APOBEC-mediated interference with hepadnavirus production. Hepatology 42(2):301–309
Russell RA, Pathak VK (2007) Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol 81(15):8201–8210
Russell RA et al (2008) Distinct domains within apobec3g and apobec3f interact with separate regions of hiv-1 Vif. J Virol 83(4):1992–2003
Sakai K, Dimas J, Lenardo MJ (2006) The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc Natl Acad Sci USA 103(9):3369–3374
Saksela K, Cheng G, Baltimore D (1995) Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. Embo J 14(3):484–491
Santa-Marta M et al (2004) HIV-1 Vif can directly inhibit APOBEC3G-mediated cytidine deamination by using a single amino acid interaction and without protein degradation. J Biol Chem 280(10):8765–8775
Santa-Marta M et al (2007) HIV-1 Vif protein blocks the cytidine deaminase activity of B-cell specific AID in E. coli by a similar mechanism of action. Mol Immunol 44(4):583–590
Sasada A et al (2005) APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 2(1):32
Sawyer SL, Emerman M, Malik HS (2004) Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol 2(9):E275
Schafer A, Bogerd HP, Cullen BR (2004) Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology 328(2):163–168
Schrofelbauer B, Chen D, Landau NR (2004) A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc Natl Acad Sci USA 101(11):3927–3932
Schrofelbauer B et al (2005) Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol 79(17):10978–10987
Schrofelbauer B et al (2006) Mutational alteration of human immunodeficiency virus type 1 Vif allows for functional interaction with nonhuman primate APOBEC3G. J Virol 80(12):5984–5991
Schumacher AJ, Nissley DV, Harris RS (2005) APOBEC3G hypermutates genomic DNA and inhibits Ty1 retrotransposition in yeast. Proc Natl Acad Sci USA 102(28):9854–9859
Schumacher AJ et al (2008) The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J Virol 82(6):2652–2660
Sheehy AM et al (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418(6898):646–650
Sheehy AM, Gaddis NC, Malim MH (2003) The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 9(11):1404–1407
Shirakawa K et al (2008) Phosphorylation of APOBEC3G by protein kinase A regulates its interaction with HIV-1 Vif. Nat Struct Mol Biol 15(11):1184–1191
Simon JH, Malim MH (1996) The human immunodeficiency virus type 1 Vif protein modulates the postpenetration stability of viral nucleoprotein complexes. J Virol 70(8):5297–5305
Simon JH et al (1998) Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat Med 4(12):1397–1400
Simon V et al (2005) Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog 1(1):e6
Sodroski J et al (1986) Replicative and cytopathic potential of HTLV-III/LAV with sor gene deletions. Science 231(4745):1549–1553
Stanley BJ et al (2008) Structural insight into the human immunodeficiency virus Vif SOCS box and its role in human E3 ubiquitin ligase assembly. J Virol 82(17):8656–8663
Stenglein MD, Harris RS (2006) APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem 281(25):16837–16841
Stevenson M et al (1990) HIV-1 replication is controlled at the level of T cell activation and proviral integration. Embo J 9(5):1551–1560
Stewart SA et al (1997) Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J Virol 71(7):5579–5592
Stopak K et al (2003) HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell 12(3):591–601
Stopak KS et al (2007) Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J Biol Chem 282(6):3539–3546
Strebel K et al (1987) The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature 328(6132):728–730
Suspene R et al (2004) APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res 32(8):2421–2429
Suspene R et al (2005) Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci USA 102(23):8321–8326
Svarovskaia ES et al (2004) Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J Biol Chem 279(34):35822–35828
Tan L et al (2008) Sole copy of Z2-type human cytidine deaminase APOBEC3H has inhibitory activity against retrotransposons and HIV-1. Faseb J 23(1):279–287
Tanaka Y et al (2006) Anti-viral protein APOBEC3G is induced by interferon-alpha stimulation in human hepatocytes. Biochem Biophys Res Commun 341(2):314–319
Teng B, Burant CF, Davidson NO (1993) Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science 260(5115):1816–1819
Tian C et al (2006) Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J Virol 80(6):3112–3115
Tian C et al (2007) Virion packaging determinants and reverse transcription of SRP RNA in HIV-1 particles. Nucleic Acids Res 35(21):7288–7302
Turelli P, Trono D (2005) Editing at the crossroad of innate and adaptive immunity. Science 307(5712):1061–1065
Turelli P et al (2004a) Inhibition of hepatitis B virus replication by APOBEC3G. Science 303(5665):1829
Turelli P, Vianin S, Trono D (2004b) The innate antiretroviral factor APOBEC3G does not affect human LINE-1 retrotransposition in a cell culture assay. J Biol Chem 279(42):43371–43373
Vartanian JP et al (2008) Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 320(5873):230–233
von Schwedler U et al (1993) Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J Virol 67(8):4945–4955
Wang T et al (2007a) 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J Virol 81(23):13112–13124
Wang J et al (2007b) The Vif accessory protein alters the cell cycle of human immunodeficiency virus type 1 infected cells. Virology 359(2):243–252
Wang T et al (2008) Distinct viral determinants for the packaging of human cytidine deaminases APOBEC3G and APOBEC3C. Virology 377(1):71–79
Waterston RH et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420(6915):520–562
Watson AJ, Wilburn LM (1992) Inhibition of HIV infection of resting peripheral blood lymphocytes by nucleosides. AIDS Res Hum Retroviruses 8(7):1221–1227
Wiegand HL, Cullen BR (2007) Inhibition of alpharetrovirus replication by a range of human APOBEC3 proteins. J Virol 81(24):13694–13699
Wiegand HL et al (2004) A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. Embo J 23(12):2451–2458
Willetts KE et al (1999) DNA repair enzyme uracil DNA glycosylase is specifically incorporated into human immunodeficiency virus type 1 viral particles through a Vpr-independent mechanism. J Virol 73(2):1682–1688
Xiao Z et al (2007) Characterization of a novel cullin5 binding domain in HIV-1 Vif. J Mol Biol 373(3):541–550
Xu H et al (2004) A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci USA 101(15):5652–5657
Yang X, Gabuzda D (1998) Mitogen-activated protein kinase phosphorylates and regulates the HIV-1 Vif protein. J Biol Chem 273(45):29879–29887
Yang X, Goncalves J, Gabuzda D (1996) Phosphorylation of Vif and its role in HIV-1 replication. J Biol Chem 271(17):10121–10129
Yang B et al (2007a) Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J Biol Chem 282(16):11667–11675
Yang Y et al (2007b) Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology 365(1):92–100
Yu X et al (2003) Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302(5647):1056–1060
Yu Q et al (2004a) APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem 279(51):53379–53386
Yu Q et al (2004b) Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol 11(5):435–442
Yu Y et al (2004c) Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev 18(23):2867–2872
Zennou V et al (2004) APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J Virol 78(21):12058–12061
Zhang J, Webb DM (2004) Rapid evolution of primate antiviral enzyme APOBEC3G. Hum Mol Genet 13(16):1785–1791
Zhang H et al (2003) The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424(6944):94–98
Zhang W et al (2008a) Conserved and non-conserved features of HIV-1 and SIVagm Vif mediated suppression of APOBEC3 cytidine deaminases. Cell Microbiol 10(8):1662–1675
Zhang W et al (2008b) Distinct determinants in HIV-1 Vif and human APOBEC3 proteins are required for the suppression of diverse host anti-viral proteins. PLoS ONE 3(12):e3963
Zhang W et al (2008c) Cytidine deaminase APOBEC3B interacts with heterogeneous nuclear ribonucleoprotein K and suppresses hepatitis B virus expression. Cell Microbiol 10(1):112–121
Zheng YH et al (2004) Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol 78(11):6073–6076
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Niewiadomska, A.M., Yu, XF. (2009). Host Restriction of HIV-1 by APOBEC3 and Viral Evasion Through Vif. In: Spearman, P., Freed, E. (eds) HIV Interactions with Host Cell Proteins. Current Topics in Microbiology and Immunology, vol 339. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-02175-6_1
Download citation
DOI: https://doi.org/10.1007/978-3-642-02175-6_1
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-02174-9
Online ISBN: 978-3-642-02175-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)