We shall now consider more specifically the kinetic behaviour of reactions catalysed by enzymes. As we have previously remarked for the general case of catalysts, enzymes are not consumed during the course of the reactions that they catalyse and they do not alter the equilibrium constant.
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
We shall now consider more specifically the kinetic behaviour of reactions catalysed by enzymes. As we have previously remarked for the general case of catalysts, enzymes are not consumed during the course of the reactions that they catalyse and they do not alter the equilibrium constant.
Let us study the conversion of A into B:
whose constants k1 and k−1 are equal to 10−3 and 10−4 respectively. At equilibrium:
B is ten-fold more concentrated than A whether or not the enzyme is present. The enzyme accelerates the reaction by the same factor in one direction as it does in the reverse direction. The efficiency of enzymes is very variable; some are capable of accelerating a chemical reaction by a factor of 108 or even up to 1011.
Enzyme catalysis facilitates the conversion of a substrate S into a product P:
This conversion proceeds through the association of a substrate molecule to the enzyme, i.e. by the formation of at least one enzyme-substrate complex, ES. Substrate binding takes place in a precise location within the protein, at the active site of the enzyme. The first authors on the subject (see the introduction) considered that an enzyme-catalysed reaction comprised two steps, which are the reversible formation of a stereospecific enzyme-substrate complex, followed by the decomposition of this complex, the appearance of the reaction products and the regeneration of the active enzyme, according to the simple scheme:
This is a very short phase during which the first molecules of the ES complex are formed until the concentration of this intermediate complex reaches a constant value (steady state phase). The pre-steady phase only lasts a fraction of a second and is not detectable during kinetic experiments carried out under classic conditions. Study of this phase requires the use of fast techniques, which we shall look at later.
1.1.2 Steady State Phase
During the steady state phase the rate of appearance of the product P is constant. The Michaelis-Menten theory predicts that under these conditions the concentration of the enzyme-substrate complex, ES, is constant.
1.1.3 Phase of Inhibition by the Reaction Products
In this phase the concentrations of the reaction products are no longer negligible and, consequently, the reverse reaction tends to lower their concentrations. Of course, if the equilibrium constant is large, i.e. hugely in favour of the formation of P, the reverse reaction will be negligible.
1.1.4 Equilibrium Phase
During this phase equilibrium is reached. The quantities of S and P are constant. In these conditions:
The amount of P formed is equal to the amount of S converted.
1.2 Michaelis-Menten Theory
The Michaelis-Menten theory accounts for these phenomena in conditions where the enzyme concentration is very low compared to the substrate concentration. Consider the following simple reaction:
The respective concentrations of the species E, S, ES, and P are e, s, (ES) and p.
While the concentration of P is low relative to that at equilibrium, i.e. while in the initial conditions of the steady state phase, we may consider the rate of catalysis to be equal to the product of the concentration of the ES complex and the rate constant for its conversion:
Under conditions where the enzyme concentration is low compared to the concentration of substrate to be transformed, the concentration of free substrate in the initial phase of the reaction is: \({\rm{s}} = {\rm{s}}_0 \). The free-enzyme concentration is:
The maximum rate of the reaction Vm is reached when the active sites of the enzyme are saturated by the substrate, which is ensured when s is much larger than Km. In this case, the relationship s/(s + Km), which represents a function of saturation of the enzymeYs, tends towards 1 (Fig. 5.2 opposite). The maximum rate is Vm = k2e0. By substituting this value into equation [2], we obtain:
Thus, the fraction Ys of active sites occupied by the substrate is equal to v/Vm. The maximum rate Vm is therefore the maximum capacity of the enzyme to catalyse the reaction, i.e. the maximum quantity of substrate that can be converted per unit time. The ratio Vm/e0represents the molar activity or turnover number. Nevertheless, depending on the reaction and the substrate(s), it is possible that this value is never attained experimentally (due to Km being too high, poor substrate solubility etc.). The Michaelis constant, Km, represents in all cases the substrate concentration at which the rate is half maximal; indeed, for s = Km, v = Vm/2 (Fig. 5.2). However, the significance of these kinetic parameters depends essentially on the reaction scheme.
Enzyme reactions are, in fact, more complex than the original authors on the subject were able to foresee. Generally, they comprise more than one substrate and involve more than one intermediate complex. We shall tackle the study of kinetic schemes of increasing complexity and for each, the significance of the kinetic parameters will be discussed.
2 Enzymatic Reactions with a Single Substrate and a Single Intermediate Complex
Most enzymatic reactions involve at least two substrates. However, there are borderline cases in which the concentration of one of the substrates is in excess and the system behaves mechanistically like a single-substrate enzymatic reaction. Let us take the example of enzymatic hydrolysis reactions in aqueous solution. Water, the second substrate, is in vast excess and plays no part in the reaction kinetics.
A great number of concepts that remain valid today were established after studying enzymatic reactions with a single substrate and a single intermediate complex. This is why this topic is particularly well developed.
2.1 Reversibility of Enzymatic Reactions
Let us consider the simplest borderline case involving a single substrate and a single intermediate complex:
Enzymatic reactions, like all chemical reactions, are reversible. Nevertheless, the equilibrium can be strongly shifted in favour of the formation or, conversely, the degradation of a given metabolite, such that the reaction is practically irreversible. In certain cases, the reaction products undergo transformations (ionisations for example) that render the reaction quasi-irreversible. Thus, in peptide bond hydrolysis reactions at a pH conducive to protease action:
We can show that a relationship exists between the equilibrium constant and the parameters of both the forward reaction \(\left( {{\rm{S}} \to {\rm{P}}} \right)\) and the reverse reaction \(\left( {{\rm{P}} \to {\rm{S}}} \right)\). Indeed, the kinetic parameters of the forward reaction are written:
These conditions are practically attained when the equilibrium strongly favours the formation of P, or in the initial reaction conditions when the concentration of P is zero or practically zero, or even when P undergoes subsequent conversions precluding the reverse reaction, as described above.
This scheme, a highly simplified case, was historically the first scheme proposed (V. Henri, L. Michaelis and M. Menten), and which J.B.S. Haldane resolved in 1925 for the general case that assumes a steady state. Indeed, the general equation for enzymatic reactions, i.e. the Michaelis-Menten equation described above, rests on two assumptions:
a low enzyme concentration,
the presence of a steady state.
When the enzyme concentration becomes too high, (ES) is no longer negligible relative to s. There is no longer a first-order reaction with respect to the enzyme, but a more complex form that leads to a second-degree equation with respect to the enzyme, as analysed by Strauss and Goldstein in 1943. The rate equation can no longer be put in a linear form. The simplest procedure, whenever possible in in vitro studies, is to work with an enzyme concentration that ensures linearity between the reaction rate and this concentration. Again, it is worthwhile checking this (see later). In reactions that take place in a cellular environment (see Part VI), however, frequently the enzyme concentration becomes high relative to the substrate concentration.
The Michaelis equation assumes a steady state, but the steady state is not reached immediately; everything depends on the time-scale of the reaction. If it is possible to work in a zone of small time constants, we can follow the establishment of a steady state system. This topic will be explored in the following sections.
2.2.1 Kinetics in the Pre-Steady State
The study of reaction kinetics in the pre-steady state involves estimating the reaction product during the very short time preceding the steady state, which is characterised by a constant concentration of the ES complex. There is an initial acceleration that lasts only a fraction of a second before the system reaches a steady state.
When the appearance of the reaction product is measured using a rapid kinetics device, we obtain a curve that has the form indicated in Fig. 5.3 below.
Fig. 5.3
Appearance of the reaction product over time
The initial curvature represents the pre-steady state; the linear part that follows corresponds to arrival at the steady state
The solution to this equation is valid for both the pre-steady state and the steady state in the first period of the reaction as long as the concentration of s is little different from s0, the initial substrate concentration:
This simplified equation is only valid for the initial part of the acceleration period. Thus, by measuring p in this initial period, it is possible to obtain k1k2e0. As k2e0 is determined by measuring Vm at the steady state by the previously described methods, we know the value of k2 and consequently we can calculate k1. Having the value of Km by the same study at the steady state, we can also calculate k−1 and afterwards the true dissociation constant for the enzyme-substrate complex.
The complex equation obtained by integration can also be used in a different way. If we plot p as a function of time, we obtain a curve whose initial curvature is due to the exponential term. As t increases, the exponential term can be ignored since the steady state is reached. The straight line obtained arises from:
And for p = 0, i.e. where the linear part of the curve intersects the horizontal axis (see Fig. 5.3), then t = 1/k1s0.
Therefore, the value of t gives k1 directly. The validity of these expressions naturally implies the existence of a single intermediate complex. If several intermediate complexes are formed, these expressions become more complicated and depend on the rate-limiting steps.
2.2.2 Reaching the Steady State
Let us look again at the expression for p which comprises a first Michaelian term (I) followed by an exponential term (II). When time becomes very large, the exponential term cancels out. We rediscover the Michaelis equation by deriving dp/dt.
Figure 5.4 represents the changes in s, p and (ES) at different phases of the reaction. It shows clearly that a linear change in p over time is only obtained when (ES) is constant, i.e. when the steady state is reached.
Under conditions approximating the steady state with the simplified scheme that we have already considered, the kinetic parameters have the following significance: the maximum rate, Vm, is equal to k2e0and the Michaelis constant, Km, is equal to (k–1+ k2)/k1. It is a complex constant that does not reflect the inverse of the enzyme′s affinity for the substrate, but depends on all the rate constants.
2.2.3 Approximation to a Quasi-Equilibrium
Whereas the steady state approximation involves no assumption as to the respective values of the specific rate constants, the approximation to a quasi-equilibrium is based on such an assumption. It assumes that k2 « k−1, i.e. that the equilibrium between E, S and ES is attained rapidly and that the chemical reaction is the limiting step. In these conditions, we always have:
Under these conditions, the experimental Michaelis constant, Km, is equal to Ks, i.e. to the ratiok−1/k1, the dissociation constant of the enzyme-substrate complex. In this extreme case, it represents the inverse of the enzyme’s affinity for its substrate. In other words, Km for a given substrate can be likened to a constant of the substrate’s dissociation from the ES complex. This approximation is only valid if the catalytic constant, k2, is much smaller than k−1, which is justified for certain enzymatic reactions, but it is always necessary to show it. Never must Kmimmediately be likened to a dissociation constant.
2.2.4 Order of Enzymatic Reactions
As for chemical reactions (see Chap. 4) we must consider, on the one hand, the order with respect to time, and on the other, the order with respect to concentration or the initial order.
2.2.4.1 Order with Respect to Time
It is possible to follow the kinetics of an enzymatic reaction until the amount of substrate is exhausted. Only the substrate is consumed, the enzyme concentration is the same at the end as at the start of the reaction. The kinetics are intermediate between zero and first order. Indeed, starting with an initial substrate concentration s0, with s being the concentration at time t, we may write:
Conversely, if s0 is large compared to Km and the product concentrations, i.e. at the beginning of the reaction when the substrate concentration is saturating (s » Km), we have:
This corresponds to the portion of the curve that is zero-order and observed at the start of the reaction when the substrate concentration is sufficiently high.
2.2.4.2 Order with Respect to Concentration or Initial Order
As for chemical reactions, in order to determine the reaction order, we must vary the initial substrate or enzyme concentration and then determine the initial reaction rate at each concentration.
2.2.4.2.1 Study of the Changes in the Reaction Rate as a Function of the Enzyme Concentration
One of the experimenter’s primary objectives is to determine the change in the initial reaction rate as a function of the enzyme concentration. The curve obtained generally contains a linear part, before curving to reach a plateau (Fig. 5.5).
Fig. 5.5
Change in an enzymatic reaction as a function of the enzyme concentration
The simplest procedure whenever possible is to work in a range of enzyme concentrations that ensures proportionality between the reaction rate and the enzyme concentration. In this range, the reaction is first order with respect to the concentration. It is important here to emphasise that care must be taken in determining the initial rate. Indeed, some enzymatic reactions cannot be followed by continuous-flow methods (see Chap.6), and so only one method, “point by point”, is at our disposal. This involves taking samples and then stopping the reaction after a certain time. It is advisable, therefore, to choose carefully the time period otherwise the initial rate risks being underestimated. Thus, if an experimenter titrates, relative to a control solution, an enzyme solution of unknown concentration for too long a time interval that no longer respects the conditions of linearity, an error DP will be made (Figs. 5.6 and 5.7a). It is essential, therefore, at least in an initial series of trials to obtain several points in order to check at what time there is a deviation from the initial rate (Fig. 5.6).
Fig. 5.6
Error in the determination of the rate by measuring p at time t1
(a) kinetics of the appearance of P over time for different enzyme concentrations: E1, E2and E3. Evaluation of the rate at times t1and t2(b) change in the reaction rate as a function of the enzyme concentration at times t1and t2, taken from the data in the preceding curve
Indeed, when no longer working under the initial-rate conditions, the Michaelis equation is no longer satisfied and the change in reaction rate with respect to the enzyme concentration is no longer linear (Fig. 5.7a and b). The rate expression given previously is only applicable in these conditions. For the same reasons, it is wise to ensure that the pH of the reaction medium stays constant for the duration of the measurements (see Chap.9).
2.2.4.2.2 Study of the Changes in Reaction Rate as a Function of the Substrate Concentration
For a given enzyme concentration, changes in the reaction rate as a function of the initial substrate concentration follow the hyperbolic law given by the Michaelis equation:
This relation also expresses a kinetic intermediate between zero and first order. Under conditions of extreme substrate concentrations, there is a tendency to lean either towards zero-order kinetics or towards first-order kinetics.
When s0 is large relative to Km (s0 » Km), this expression simplifies to:
The rate tends towards the maximal rate and Ys becomes practically equal to 1. Therefore, the reaction rate becomes practically independent of the substrate concentration. These conditions are important from an experimental point of view.
The reaction rate is proportional to the ratio k2e0/Km, which is the first-order rate constant for the reaction. The rate varies linearly as a function of s0. Now, we can write:
The limit of the constant k2/Km is therefore determined by k1, the rate constant for the formation of the ES complex. This rate is limited by molecular diffusion. It cannot be faster than the speed with which the molecules of E and S meet, which is controlled by diffusion. This is clearly a physical barrier that cannot be exceeded. At physiological temperatures, the magnitude of diffusion is between 108 and 109mol−1 . s−1, implying that:
Certain enzymes such as catalase or carbonic anhydrase have kcat/Km values that are this order of magnitude. These enzymes have attained catalytic perfection and hence their activity rates are only limited by diffusion (see Chap.11). Biological systems have found a way round this physical limit by creating multi-enzyme complexes in which the reaction product is transferred directly to the catalytic site of the enzyme catalysing the subsequent reaction. In Part V, we shall see examples of these multi-enzyme complexes, which catalyse several successive reactions on the same metabolic pathway.
In summary, under extreme conditions when the substrate concentration is low compared to the Michaelis constant, the kinetics are first order; conversely, if s is large relative to Km, the kinetics are practically zero order. The reaction rate tends towards a maximum value Vm = k2e0.
2.3 Methods to Determine Kinetic Parameters
In order to obtain with satisfactory precision the experimental parameters Km and Vm, it is important to determine the initial reaction rate for a sufficient number of substrate concentrations situated either side of the Km value. Several linear graphical plots are available for determining these parameters.
2.3.1 Semi-Logarithmic Plot
The first graphical plot used by Michaelis involved plotting the reaction rate as a function of the logarithm of the substrate concentration, for a given enzyme concentration (Fig. 5.8). The inflexion point corresponds to logKm on the horizontal axis. This method is very imprecise as a result of the difficulty in reliably determining Vm.
Fig. 5.8
Determination of the Michaelis constant and Vmby the semi-logarithmic plot
The slope of the straight line obtained by plotting 1/v versus 1/s is equal to Km/Vm and the vertical-axis intercept, 1/Vm. The intercept on the horizontal axis gives the value −1/Km (Fig. 5.10).
The plot of s/v as a function of s leads to a straight line whose slope directly gives 1/Vm and the vertical-axis intercept is equal to Km/Vm (Fig. 5.11 below).
s and (s0−s) are measured in a series of defined time-points and on a graph we plot (2.3 log s0/s)/t as a function of (s0−s)/t; a straight line should be obtained if the kinetics follow the classical law well. This line has a slope of −1/Km and the horizontal-axis intercept is Vm (Fig. 5.12). This plot is therefore applicable in principle even for cases where the previous plots are not suitable as a result of the imprecision in the estimation of the initial rates. Furthermore, only a single experiment is required in principle, although it is necessary to check that the same straight line is obtained with several substrate concentrations.
Fig. 5.12
Graphical plot from the integrated Michaelis equation
This method involves plotting, for each experiment, v on the vertical axis versus –s on the horizontal, then drawing the corresponding lines. According to the Michaelis equation, these straight lines intersect at the same point having coordinates of Vm and Km. Figure 5.13a illustrates the graphical procedure. However, the effect of experimental error means that the intersection point is not unique. The number of intersection points is 1/2n(n − 1). For the five experimental curves presented in Fig. 5.13b there are 10intersection points. Each of these points gives an estimation of Km and Vm, and then the average value of these is determined.
Fig. 5.13
Direct plot of Eisenthal and Cornish-Bowden
(a) determination of Vmand Km. Each straight slope represents one experiment for a substrate concentrations, giving an initial ratev. The intersection point gives Vmand Km− (b) the intersection point can regress if there is an error in the straight line. Each intersection gives an estimation of Kmand Vm. For each group, the average value is considered to be the best value for the parameters
The problem of which method is the best to analyse the experimental data and evaluate the kinetic parameters from the Michaelis equation is a very old problem. The direct plot of v against s0 is an equilateral hyperbola passing through the origin and having the asymptotes v = Vm and s0 = −Km. But it is practically impossible to obtain Vm and Km with precision from this plot because:
only finite and positive values of s are measurable,
it is rarely possible to use sufficiently high substrate concentrations to determine precisely the plateau corresponding to the maximal rate.
In the Michaelis-Menten plot (or semi-logarithmic plot) there is an inflexion point at s = Km, and the maximum slope at this point is 0.576. This method is statistically correct, however, the maximum rate must be measured experimentally with great precision. Consequently, most enzymologists prefer to use one of the linear plots that have just been described. The validity of the kinetic constants estimated from these diverse plots has been widely discussed, in particular by Wilkinson (1961), Johansen and Lumry (1961), Dowd and Riggs (1965), Colquhoun (1971), and Cornish-Bowden and Eisenthal (1974).
As analysed by Dowd and Riggs, the frequency distribution of the Km and Vm values derived from the three linear plots show clearly the great inferiority of the Lineweaver-Burk plot. This is evident by examining the diagrams in Fig. 5.14a opposite, which give the frequency distribution of Vm and Km for these three plots, based on 500identical experiments; the error in v is assumed to be constant and relatively high. 40 values estimated for Vm and 35 values for Km were superior to 100 or inferior to 0 in the Lineweaver-Burk plot. An analogous conclusion can be drawn from the same diagram, but making the assumption that the error in v increases practically proportionally to v (Fig. 5.14b). Even allowing a small error in v, the Lineweaver-Burk plot leads to the least correct evaluation, since in this case the authors obtained a non-negligible number of negative values in estimating the parameters.
Fig. 5.14
Frequency distribution for Kmand Vmvalues for the three linear plots
The two other linear plots: v against v/s0 or s0 against s0/v, are therefore superior to the Lineweaver-Burk plot. Nevertheless, they do not permit a statistically significant analysis since the values plotted on both axes are not independent variables. For this reason, as shall be analysed in the last paragraph of this chapter, we prefer to employ statistical methods of analysis based on the non-linear Michaelis-Menten plot and then to evaluate the parameters by a non-linear regression method, especially since today’s computer programs enable direct data processing.
Whatever the case, the marked inferiority of the Lineweaver-Burk plot compels us to advise against its use for the estimation of kinetic parameters.
3 Kinetics of Enzymatic Reactions in the Presence of Effectors (Inhibitors or Activators)
3.1 Kinetics of Enzymatic Reactions in the Presence of Inhibitors
The kinetics of enzymatic reactions can be considerably modified by the presence of inhibitors in the reaction medium. The phenomenon of inhibition is very frequent in enzymology and the most diverse chemical components are capable of inhibiting enzymatic reactions; this, of course, depends on the enzyme and reactant.
As we shall see in Part VI, the regulation of cellular metabolism relies for the most part on physiological mechanisms of inhibiting enzyme activity.
Furthermore, it is interesting to provoke inhibition of an enzymatic reaction using substances of known structure in order to obtain information about the mechanism of enzyme action. The inhibition of enzymes in vivo increasingly underpins the numerous chemotherapeutic procedures. The study of inhibitory phenomena is therefore of primary importance in enzymology.
Some types of inhibition result from the reversible association of an inhibitor to an enzyme. Others are the consequence of irreversible binding; the action of irreversible inhibitors will be considered in Parts III and V.
Different types of reversible inhibition exist. The most simple and the most classic are competitive inhibition, non-competitive inhibition and uncompetitive inhibition or inhibition by blocking the intermediate complex. In these types of inhibition, the presence of the inhibitor on the enzyme totally abolishes its activity, although partial inhibition also exists. We shall now examine successively total and partial inhibition.
3.1.1 Total Inhibition
3.1.1.1 Competitive Inhibition
There is competitive inhibition when the binding of an inhibitor molecule, I, to the enzyme prevents substrate binding, and reciprocally, the inhibitor cannot practically bind to the ES complex; there is exclusive binding of either the inhibitor or the substrate. Competitive inhibition occurs in particular when the inhibitor and substrate are structurally analogous and bind to the same site on the enzyme, but this is not the only example. The scheme for an enzymatic reaction subjected to competitive inhibition is written thus:
There is competition between substrate and inhibitor molecules with respect to the enzyme; an excess of substrate displaces the inhibitor. The rate equation at steady state is obtained by taking the equation for enzyme conservation:
Furthermore, when (I) » e0, the free-inhibitor concentration is practically equal to the total-inhibitorconcentration; this condition generally arises in experiments in vitro in which the inhibitor, like the substrate, is in excess relative to the enzyme concentration.
The maximal reaction rate, Vm, remains unchanged whatever the inhibitor concentration. An excess of substrate displaces the inhibitor.
If we use the Eadie plot to determine the kinetic parameters, we obtain a series of straight lines whose slopes increase in absolute value as a function of the inhibitor concentration by a factor of \(\left[ {{{1 + \left( {\rm{I}} \right)} \mathord{\left/ {\vphantom {{1 + \left( {\rm{I}} \right)} {{\rm{K}}_{\rm{i}} }}} \right. \kern-\nulldelimiterspace} {{\rm{K}}_{\rm{i}} }}} \right]\), but which converge to the same point on the vertical axis, i.e. Vm (Fig. 5.15a below). The Lineweaver-Burk plot also leads to a beam of straight lines coinciding at a point on the vertical axis that has the value1/Vm (Fig. 5.15b). From these K′m values for different inhibitor concentrations, it is straightforward to obtain Km and Ki with a secondary plot(Fig. 5.15c).
Fig. 5.15
Competitive inhibition
(a)Eadie plot – (b) Lineweaver-Burk plot – (c) secondary plot of K′mas a function of (I) permitting the determination of Kmand Ki– (d)Dixon plot of 1/vias function of (I) for two substrate concentrations. In (a) and (b) unbroken lines: reactions in the presence of inhibitor; dashed lines: reactions in the absence of inhibitor
Another graphical plot, suggested by Dixon, to determine directly the constant Ki involves plotting 1/v as a function of (I) according to the equation:
For different concentrations of s0, we obtain the graph depicted in Fig. 5.15d.
When Ki is very small relative to (I) and Km, i.e. when the inhibitor has a high affinity for the enzyme, it can be difficult to determine the nature of the inhibition. Indeed, in the expression for v, we have (I)/Ki » 1 and Km(I)/Ki » s0.
The rate expression is thus simplified to the following:
In this case, it is indistinguishable from non-competitive inhibition (see the next paragraph).
3.1.1.2 Non-competitive inhibition
In this type of inhibition, substrate and inhibitor binding are not exclusive; they are independent. The inhibitor binds without altering the affinity of the enzyme for its substrate. A ternary ESI complex can thus be formed, but it is inactive. The presence of the inhibitor alone may also lead to the formation of an inactive EI complex. This can be represented by the following scheme:
The Eadie plot gives rise to a series of parallel lines with decreasing values for the vertical-axis intercept as the inhibitor concentration increases (Fig. 5.16a).
Fig. 5.16
Non-competitive inhibition
(a) Eadie plot – (b) Lineweaver-Burk plot – (c) secondary plot of 1/V ′ m as a function of (I) – (d) Dixon plot. The symbols are the same as in Fig.5.15
The Lineweaver-Burk plot gives a series of straight lines which converge at a point on the horizontal axis whose values is −1/Km (Fig. 5.16b). The changes in apparent maximum rate as a function of the inhibitor concentration permits determination of the constant Kiwith a secondary plot (Fig. 5.16c).
the Dixon plot, 1/v against (I), produces the graphs indicated in Fig. 5.16d. The straight lines thus obtained for different values of s converge at the point −Ki on the horizontal axis.
3.1.1.3 “Uncompetitive” Inhibition or Inhibition by Blocking the Intermediate Complex
A type of inhibition called “uncompetitive” inhibition exists which involves a mechanism of inhibition by blocking the intermediate complex. In this case actually, the inhibitor binds to the enzyme-substrate complex and not to the free enzyme, giving an inactive ternary complex. The reaction scheme can be represented as follows:
The apparent Michaelis constant, K′m, and the apparent maximum rate, V′m, vary as a function of the inhibitor concentration; they become proportionately smaller:
It seems as though the presence of the inhibitor facilitates the formation of an intermediate complex, since K′m decreases as (I) increases, yet at the same time the reaction is prevented from taking place.
In this type of inhibition the Eadie plot produces a series of straight lines whose slopes decrease while the inhibitor concentration increases (Fig. 5.17a below). These lines converge to a point on the horizontal axis whose value is equal to the ratioVm/Km and which does not alter. The Lineweaver-Burk plot gives a series of parallel lines since the slope is equal to Km/Vm (Fig. 5.17b).
Fig. 5.17
“Uncompetitive” inhibition
(a) Eadie plot – (b) Lineweaver-Burk plot – (c) secondary plot of 1/V ′ m or 1/K ′ m as a function of (I) – (d) Dixon plot. The symbols are the same as in Fig.5.15
The intercept with the horizontal axis is −Ki(1+Km/s0) and with the vertical, (Km+s0)/Vms0; its slope is 1/VmKi (Fig. 5.17d).
3.1.1.4 Inhibition by the Binding of the Inhibitor to the Substrate
In some enzymatic reactions, in particular in proteolysis reactions where the substrate can be a molecule with large dimensions, the inhibitor may bind to the substrate and not to the enzyme. This does not happen frequently, but ought to be borne in mind. The inhibition may either be competitive or non-competitive. For each of these types of inhibition, we end up with the same rate equation whether the inhibitor binds to the enzyme or the substrate, on the condition that free (S) stays large compared to e0 (Yon, 1961).
Consequently, it is not always enough to know the type of inhibition in order to determine the inhibitor’s mode of action. Sometimes it is necessary to do a study with different substrates and possibly with other enzymes that recognise the same substrate.
3.1.1.5 Inhibition by High Substrate Concentrations
Some enzymatic reactions obey the law of Henri-Michaelis at low substrate concentrations, but at high concentrations the rate, after having reached a maximum, diminishes. For such reactions, the plot of the initial reaction rate as a function of the substrate concentration gives rise to the curves shown in Fig. 5.18. This occurs when the enzyme is liable to bind several substrate molecules in the active site. Only the complex to which the substrate binds in a favourable orientation is active. We might imagine that, when the substrate concentration increases, two or more molecules may bind at each of the enzyme’s sub-sites, and since none at this point is in a favourable orientation to ensure a reaction, the ternary (or higher order) complex remains inactive.
Fig. 5.18
Inhibition by excess substrate
(a) change in the rate as a function of the substrate concentration – (b) Lineweaver-Burk plot – (c) Eadie plot – (d) change in 1/v as a function of s
This situation, which is a true case of non-competitive inhibition by the substrate itself, has been theoretically studied by Haldane, and corresponds to the following scheme:
K′s represents the dissociation constant of the ternary complex.
In this case, however, it is possible to determine the Michaelis constant of the system by using sufficiently low substrate concentrations that the inhibition is negligible. Furthermore, we can determine the inhibition constant K′s if we use conditions in which the substrate concentration is sufficiently high relative to Km such that the term Km/s0 becomes negligible. In these conditions, the rate equation simplifies to:
The plot of 1/v as a function of s0 leads to a straight line in the region corresponding to high substrate concentrations where this simplification is valid. The horizontal-axis intercept gives a value of –K′s (Fig. 5.18d). When the substrate concentration diminishes, the equation ceases to be valid and we observe a curvature lead-ing to a minimum and 1/v increases. This sort of inhibition arises with urease or acetylcholinesterase, for example.
3.1.1.6 Inhibition by The Reaction Products
Another frequently observed case in enzymology is inhibition by the reaction products, even when the reverse reaction cannot take place. Indeed, the reaction products often have very similar structures to that of the substrate or a part of the substrate, and thus are capable of forming a specific complex with the enzyme. Again, this is competitive inhibition, but none of the plots described above are able to show this. In fact, these diagrams rely on the determination of initial rates, and under the initial reaction conditions the inhibition is not yet apparent, as the product concentration is negligible. However, if the reaction products were to be added at timezero (t = 0), then inhibition would be observed. Inhibition by the reaction products can be schematised as follows:
with (P) = s0 − s and e0 = (E) + (ES) + (EP), and Ki being the dissociation constant of the enzyme-product complex, EP. The solution of this system leads to the following rate equation:
and yet the first-order constant varies with the initial substrate concentration. If Km = Ki, the plot obtained from the integrated rate equation gives a series of lines whose slopes vary as a function of the initial substrate concentration.
If we plot 2.3/t × log (s0/s) as a function of (s0 − s)/t, we obtain a series of straight lines. The intercept of these on the horizontal axis decreases as s0 increases and the absolute values of their gradients diminish as a function of s0 (Fig. 5.19).
Fig. 5.19
Inhibition by the reaction product
Plot based on the integrated Michaelis equation for different substrate concentrations
The slope of these lines can, incidentally, be positive or negative depending on the respective values of Km and Ki. If Km>Ki, then the value of [1−(Km/Ki)] is negative and the slope is positive. If Km<Ki, the slope is negative, which represents the most general case – the enzyme more often than not has a greater affinity for its substrate than for the reaction products. The different curves obtained converge to the same point on the horizontal axis whose value corresponds to:
Harmon and Niemann demonstrated inhibition of this type for the tryptic hydrolysis of benzoyl-L-arginine amide. One of the hydrolysis products, benzoyl-L-arginine, inhibits the reaction. If this product is added to the reaction at time0, it behaves like a classic competitive inhibitor, as would be the case in general, and benzoyl-L-arginine has a Ki value of 6.5×10−3M (Bechet et al., 1956).
When we come to study two-substrate reactions, we shall analyse in detail inhibition by the reaction products, as it provides important information about the reaction scheme and often enables distinguishing between two possible schemes.
3.1.2 Partial Inhibition
To address partial inhibition, we shall present first of all a generalised formulation for diverse types of reversible inhibition, in the case of an approximation to a quasi-equilibrium, i.e. when all complexes are in rapid equilibrium with their components. We always assume conditions where e0 « 0 and (I). The general scheme is written:
α represents the change in the dissociation constant of the enzyme-substrate complex under the effect of the inhibitor, or in the dissociation constant of the enzyme-inhibitor complex under the effect of substrate binding. The coefficient β is the factor of change in the rate constant when the inhibitor is present. The constants of dissociation equilibrium are as follows:
For the diverse types of inhibition, generally α>1 and β<1, although the first condition is not absolutely necessary; the rate may decrease even if α<1 on the condition that β is sufficiently small
.
3.1.2.1 Partial Competitive Inhibition
In total competitive inhibition \({\rm{\alpha }} \to \infty \) and we have the expression given previously (see the paragraph on competitive inhibition). In partial competitive inhibition ∞>α>1 and β=1. The inhibitor only partially prevents substrate binding and does not affect the degradation rate of the complex. The rate equation is:
The maximum rate remains identical in the presence of inhibitor. Only the apparent Michaelis constant varies. The graphs obtained using the Eadie or Lineweaver-Burk plots do not differ from those when the inhibition is totally competitive. However, the Dixon plot of 1/v against (I) is no longer linear, but gives a series of curves (Fig. 5.20 above).
Fig. 5.20
Partial competitive inhibition for different values of the coefficient α
In total non-competitive inhibition, α=1 and β=0. The equations are as previously indicated. In partial non-competitive inhibition α=1 and 0<β<1. The rate equation is:
K′m alone varies as the inhibitor concentration increases. As before, 1/v as a function of (I) is no longer linear. Figure 5.21 illustrates how 1/v changes as a function of (I) for different values of β.
Fig. 5.21
Partial non-competitive inhibition for different values of β
In general, there is mixed inhibition when α>1 and β<1. The apparent affinity of the enzyme for its substrate is reduced in the presence of inhibitor and the rate of degradation of the ESI complex is less than that of the ES complex. At the same time, V′m and K′m are modified by the presence of inhibitor. The behaviour is intermediate between partial competitive and partial non-competitive inhibition.
If 0<β<1, the inhibition is partially mixed. If β=0, the inhibition is totally mixed. The ESI complex can no longer be converted. The reaction rate is given by the relationship:
We have outlined above the different types of simple inhibition, either total or partial. In very general terms, we term competitive inhibition all those types that only modify the Michaelis constant, and non-competitive inhibition all those that modify the maximum reaction rate. The importance of this generalisation will be revealed in the analysis of inhibition by the reaction products, for reactions with several substrates.
3.2 Kinetics of Enzymatic Reactions in the Presence of an Activator
While certain effectors have an inhibitory effect on enzymatic reactions, conversely, others are able to activate them. Diverse types of activator exist: metal ions, anions, various natural molecules and the substrate itself can behave as an activator. These phenomena also arise in the regulation of cellular metabolism; in particular, they help to coordinate the regulation of several metabolic pathways. Coenzymes have sometimes been considered to be activators. Bearing in mind the kinetic mechanisms involved, we shall treat dissociable coenzymes as second substrates.
It is also important to consider the activators’ modes of action. The mechanisms by which a substance is capable of activating an enzyme reaction can be as varied as the inhibitory mechanisms: either there is total activation (the enzyme has no activity in the absence of the activator), or there is partial activation (the enzyme has a low, but non-zero, activity in the absence of the activator); this only serves to increase the reaction rate.
3.2.1 Total Activation
We shall distinguish several cases depending on whether the activator and the substrate bind to the enzyme randomly or in a sequential manner.
Independent binding of the substrate and activator
This situation is described by the following scheme:
The ES complex is inactive; only the ESA complex is capable of giving rise to the reaction products. In this case, there are no interactions between the substrate and activator binding sites. Ka represents the dissociation constant of the enzyme-activator complex or the enzyme-substrate-activatorcomplex:
Similarly, the dissociation constant of the enzyme-substrate complex, Ks, is the same as the dissociation constant of the enzyme-activator-substrate complex:
where a is the activator concentration, having the condition that a » e0. Only V′m increases as a function of the activator concentration.
3.2.1.1 Dependent Binding of the Activator and Substrate
In this case, the binding of one of the ligands, activator or substrate, affects the affinity of the enzyme for the other. Assuming a quasi-equilibrium, we have the following relationship:
This system could be treated as a two-substrate reaction (see further on) approximating a quasi-equilibrium and with a single reaction product. It would therefore exhibit a random Bi-Uni mechanism. The rate equation is exactly the same:
Both of the parameters K′m and V′m are affected by the presence of the activator.
3.2.2 Partial Activation
There are a great number of enzyme reactions in which the ES complex reacts only weakly but whose turnover rates can be considerably enhanced by the presence of an activator. As before, for inhibition in which both the catalytic constant and the affinities are affected, the situation can be described in a general way using the following scheme:
As for partial inhibition, assuming that the system is in a state of quasi-equilibrium, the reaction rate is given by:
with β>1; α can be >1 if β is large enough for the rate to be increased in the presence of the activator. The apparent kinetic parameters are as follows:
Furthermore, if β is very large, then βk2 » k2, which becomes again a case of independent substrate and activator binding.
3.2.3 Examples of Enzymatic Activation
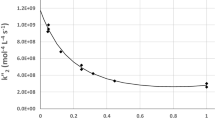
We shall give the example of the activation of β-galactosidase from E.coli by Mg++ ions although its properties are a little more complex than in the simple systems described in this section. The enzyme is a tetramer with a molecular weight of 540000daltons, formed from four identical protomers, and catalyses the hydrolysis of β-D-galactosides. β-galactosidase is sensitive to the action of diverse cations. Na+ ions are indispensable for its activity. Certain divalent cations, in particular Mg++ and Mn++, are activators; on the contrary, others like Be++ and Ca++, are inhibitors. The enzyme possesses a weak activity, yet significant in the absence of Mg++. The properties of the enzyme deprived of Mg++ were determined by Tenu et al. (1972).
This quantitative study of activation by Mg++ was carried out at the optimal reaction pH, in well-controlled conditions, in particular by keeping the Na+ concentration (0.145 M) and the ionic strength (0.17±0.02) constant. Phenylgalactoside was chosen as the substrate; with this substrate, the first chemical step is rate-limiting, which facilitated the interpretation. Additionally, the conditions were chosen such that the substrate concentration was much above Km. Figure 5.23 shows the activation by different concentrations of Mg++. The latent phase observed when Mg++ is added to the reaction at time0 shows that activation by magnesium is a slow process. After a variable time-period dependent on the Mg++ concentration, the reaction reaches a steady state, which corresponds to the linear kinetic phase; the rate becomes constant. If the enzyme is first incubated in the presence of magnesium, this latent phase is no longer observed. The slow activation by magnesium is independent of the substrate’s nature. Therefore, the activation must result from the binding of Mg++ to the enzyme.
In this section, we shall only present the results obtained when the steady phase of the activation is reached, as represented by the linear parts of the curves in Fig. 5.23.
By studying the change in the catalytic constant as a function of the Mg++ concentration (M), we can determine an apparent dissociation constant for the enzyme-Mg++ complex, using this type of representation:
\(\Delta {\rm{k}}_{{\rm{cat}}} \) represents the difference in values of the catalytic constant kcat for a given Mg++ concentration and for zero Mg++ concentration (kcat being different from 0 under these conditions); \(\left( {\Delta {\rm{k}}_{{\rm{cat}}} } \right)_{\max } \) is the corresponding difference at saturating Mg++. K is the dissociation constant of the enzyme-Mg++ complex. Figure 5.24 reveals that the saturation curve is Michaelian. The constant K has a value of (0.65±0.05)×10−6M, which corresponds to a strong apparent affinity of the enzyme for the metal ion. Direct measurements of Mg++ binding to the enzyme showed that a single Mg++ ion binds per enzyme protomer.
There are numerous examples of substrate inhibition described in the literature, much rarer are cases describing activation. However, this phenomenon has been observed for some enzymes. Alberty et al. (1954) noted the activation of fumarase by its substrate. Béchet and Yon (1964) described a similar phenomenon for the hydrolysis of several substrates by trypsin (Fig. 5.25 below).
The activation by the substrate can be described by a phenomenological rate equation of the form:
where a change in s to the second degree appears in the numerator and denominator; a, b, c, and d are constants that depend on the experimental kinetic parameters of the reaction.
If this phenomenological equation remains valid, diverse mechanisms may be involved in the apparent activation of an enzyme by high substrate concentrations. Clearly, the significance of these parameters depends on the mechanisms involved. In this paragraph, we shall only explore the simplest mechanism, involving the existence of two substrate-binding sites. One is the normal site where the substrate is converted and the other is an activator site where the substrate undergoes no modification. The scheme is shown below:
If α=1, the sites are equivalent and independent. This scheme thus reminds us of the general scheme given for inhibition; but with activation, β>1. The reaction rate is:
Figure 5.25 illustrates the activation by an excess of substrate during the hydrolysis of benzoyl-L-arginine methyl ester by trypsin. The Eadie plot is no longer linear. It is possible to define two values for Vm and Km in the zones of extreme substrate concentrations.
Fig. 5.25
Activation by the substrate during hydrolysis by trypsin
It is important to note that other types of mechanism lead to kinetic profiles of this sort as we shall see later on, and this is particularly true with, for example, mnemonic, hysteretic and allosteric enzymes.
4 Enzymatic Reactions with one Substrate and Several Intermediate Complexes
4.1 Kinetics at the Steady State
The numerous data currently available, studied in detail for diverse enzymatic reactions, in many cases generally show that the enzymatic reaction pathway includes several intermediate complexes. However, the number of intermediate complexes remains limited.
By way of example, we shall discuss the following scheme, for the general case of a reversible reaction. We shall then turn to the reaction in a single direction when the reverse reaction is negligible:
Several methods exist for determining the rate equation at steady state, i.e. when d(X)/dt=d(Y)/dt=0. The equations for the system under these conditions are:
The last two equations are, respectively, the equations for the conservation of enzyme and substrate.
Under conditions in which only the two intermediate complexes are present, these equations are readily solvable. However, when the number of intermediates increases, the equations become more complex but there are adequate methods to solve them. In particular some simple methods for kinetic analyses exist, such as:
by determinants,
by graphical methods, including that of King and Altman.
These methods allow the determination of the reaction rate which, in the direction of product appearance, is given by the relationship:
The rules of Cramer enable the determination of (X), (Y) and (E) by writing the determinants of X(Dx), Y(Dy) and E(De) and the determinant of the coefficients (D).
4.1.2 Analysis by the Graphical Method of King and Altman
To solve this system, we write that (E)/e0, (X)/e0 and (Y)/e0 are the sums of all allowed combinations leading to E, X and Y respectively, divided by the total number of all combinations. A closed loop is a forbidden loop. Thus, for the free enzyme the allowed combinations are the following configurations:
Σ represents the sum of the numerators of the three expressions above. Solving this system, of course, leads to the same rate equation as before.
4.1.3 Analysis by other Graphical Methods
Other graphical methods have been suggested for deriving rate equations for complex kinetic schemes. The theory of graphs has been applied to solve enzyme kinetics at steady state and to problems involving simple inhibition by Volkenstein and Goldstein (1966). Chou and Forsen (1980), and Chou (1980, 1981) applied the method to solve enzyme reactions composed of branched schemes. With all these methods, including that of King and Altman, there is a fundamental formula that expresses the concentration of the mth form of the enzyme in the reaction:
n being the total number of enzyme forms in the reaction scheme. The different methods involve obtaining Ni (i=1, 2, 3…m…n) in the simplest way.
Let us consider the scheme comprising the two intermediates X and Y. In the first step, we draw a graph of the enzyme’s states in which each point represents one species of the enzyme; the arcs joining them up represent the paths for going from one species to another (Fig. 5.26a below). In the second step, each point on the graph is associated with a loop whose weight is equal to the sum of the arcs that leave that point. Next, the sign is changed for each arc on the graph. GraphD is thus transformed into graphD+ (Fig. 5.26b). In the third step, we take a reference point, for example the species Y, and we trace every graph that comprises a path going from Y to E and all cycles which neither have an intersection with them nor with the paths (Fig. 5.26c). For each sub-graph, we take the weight of all the points multiplied by the sign (−1)n+c+1, n being the number of points, and c, the number of cycles in the corresponding sub-graph.
Fig. 5.26
Solving a system comprising two reaction intermediates by the graphical method
In the last step, to avoid omitting a sub-graph and to facilitate verification, the authors recommend the us of methods that involve construction of a matrix A = aij from the transformed graph D+:
\(\left| {{\rm{a}}_{{\rm{ij}}} } \right| = 1\) if a path exists between Ei and Ej,
\(\left| {{\rm{a}}_{{\rm{ij}}} } \right| = 0\) if a path does not exist between Ei and Ej.
Thus, for a point Em, if we take Eq as a reference point, the number of sub-graphs is:
and we find once again the rate expression obtained by the previous methods.
The graphical method is useful because it enables a certain number of simplifications by direct operations on the graphs. Thus:
parallel branches can be added. The value of the resulting branch is equal to the sum of the values of the branches;
the graph can be simplified if some branches fuse together by using the graph’s symmetry;
the number of nodes can be reduced.
In order to illustrate these operations, let us look again at the example of the action of a modifierM (a partial inhibitor or activator) on the enzymatic reaction that we discussed in the previous section. Taking the scheme:
Let E1 be the free enzyme, E2 the ES complex, E3 the EM complex and E4 the ternary complex EMS. We can sketch the schemaD (Fig. 5.27a). The first property–addition of parallel branches– can be used to write a simplified form of the graph (Fig. 5.27b). The number of nodes can be reduced.
Fig. 5.27
Graph of an enzyme system containing a modifier, M
To calculate E3 by taking E2 as a reference point, two paths exist to go from one to the other: one is E2→E4→E3, the other E2→E1→E3, which, by applying the principles outlined above from the transformed graph (Fig. 5.28a), would give the two sub-graphs indicated in Fig. 5.28b. However, each of these paths can be condensed into a single point as indicated in Fig. 5.28c. As a result:
It is interesting to note that the reaction parameters are related to the complex constants. So, the equilibrium constant Keq=N1/N2. The kinetic parameters of the forward reaction are related to the constants by the relationships:
The kinetic parameters of the forward reaction are obtained from the general rate equation when p is zero; those of the reverse reaction are obtained when s is zero.
The expression for Km clearly shows that this parameter has a complex value; thus the Michaelis constant with respect to the substrate S is:
It is clear that this parameter, which experimentally always represents the substrate concentration that leads to the half-maximal reaction rate, is a complex parameter; it does not reflect the inverse of the enzyme’s affinity for its substrate.
4.2 Example: Enzymatic Reactions Catalysed by Serine Proteases
It has been demonstrated that hydrolytic enzymes possessing a serine in the active site form an acyl-enzyme intermediate with the substrate, an acyl-serine (see Chap. 12), according to the scheme:
This is a covalent complex between part of the substrate and a serine, the other part of the substrate or “leaving group” is liberated after this step in the reaction. In the following step, the acyl-enzyme is hydrolysed with the incorporation of a water molecule and the enzyme is regenerated.
The reaction scheme is written as follows:
ES1 corresponds to the classic Michaelis complex, ES2 is the acyl-enzyme, P1 is the leaving group and P2 the second reaction product. The leaving group can be a non-specific part of the substrate as in the case of trypsin, chymotrypsin, elastase, or a specific part as seen in acetylcholinesterase.
Letting p and q be the respective concentrations of ES1 and ES2, at steady state we can write the following equations:
The kinetic parameters of the Michaelis equation, Km and Vm, become the apparent parameters that we may determine experimentally and which correspond to these complex expressions:
If k−2 is very small relative to k3, i.e. if the acylation process is practically irreversible under the experimental reaction conditions, these expressions may be simplified to:
4.4 Determination of The Elementary Kinetic Constants
Generally, the studies carried out under steady state conditions do not permit the determination of experimental parameters with a complex significance. The parameters of individual steps can only be obtained in extreme cases. It is however possible for particular enzymatic systems, e.g. reactions catalysed by serine proteases, to determine all system parameters including the individual rate constants by using a range of carefully chosen substrates.
Let us take the reactions catalysed by trypsin as examples. In the biological context, this enzyme hydrolyses peptide bonds where the amino acid at the positionα to the carboxyl is L-lysine or L-arginine. But it can also hydrolyse ester and amide bonds requiring the same specificity. The tryptic hydrolysis of ester and amide derivatives of L-benzoyl arginine and tosyl-L-argininehas been studied:
The corresponding ester and amide, e.g. benzoyl-L-arginine ethyl ester (BAEE) and benzoyl-L-arginine amide (BAA), give the same acyl-enzyme intermediate, benzoyl-L-arginine trypsin. These two substrates will, therefore, have the same rate constants for deacylation, k3.
A kinetic study of the ester and amide hydrolyses gave the following kinetic parameters for the two substrates:
It is of note, upon inspection of these values, that:
the deacylation step cannot be rate-limiting for the hydrolysis of these two substrates because the values of kcat are not identical;
the acylation step does not seem to be rate-limiting for the two substrates. If this were the case, then Km,app would be equal to Km. Now, there is a difference of 103 between the Km,app values of the two substrates whose structures differ extremely little. Nevertheless, no conclusions can be drawn and further experiments are necessary.
Complementary information was obtained by studying the inhibition by the reaction product, benzoyl-L-arginine:
Benzoyl-L-arginine behaves as a competitive inhibitor with respect to the enzyme, with an inhibition constant Ki=2.5×10−3M. Comparing the Km of the amide to the Ki of the inhibitor analogue, which has the same value, it is reasonable to think that for amide hydrolysis Km=Ks, and therefore that acylation is rate-limiting; consequently, kcat=k2.
On the other hand, the difference between Ks and Km,app for ester hydrolysis is a factor of 103, suggesting that deacylation is rate-limiting, and so:
Thus, it is possible to obtain the kinetic parameters for all the elementary steps; k3 is identical for the two substrates and k2=1000 k3 for the ester. Table 5.1 summarises the results of this study.
Table 5.1
Values for the kinetic parameters corresponding to the tryptic hydrolysis of a benzoyl-L-arginine ester and amide
A large number of hydrolytic enzymes do not possess a very narrow specificity for their second substrate, namely, water. In certain cases, it is possible to substitute other more potent nucleophilic agents for water. Analysis of the reaction products therefore provides a means to understand how these nucleophilic agents participate in an enzymatic reaction. This method suggested by Koshland and Herr (1957) has been applied with success to a great number of systems such as ribonuclease (Findlay et al., 1960), diverse cholinesterases (Wilson et al., 1950), papain and subtilisin (Glazer, 1966), chymotrypsin (Bender et al., 1966), and to trypsin and β-galactosidase (Yon et al., 1967; 1973).
We have just seen that during reactions catalysed by trypsin and chymotrypsin a covalent acyl-enzyme intermediate is formed. This entity reacts with water to liberate the second reaction product, the first product having been liberated during the formation of the acylenzyme. Such a mechanism, involving a covalent intermediate, has been established for several enzymes, including serine proteases and esterases, cysteine proteases and glycosidases. Yon and co-workers demonstrated this mechanism in reactions catalysed by β-galactosidase (1973).
For serine proteases, competition between water and other nucleophilic agents takes place at the acyl-enzyme step; for β-galactosidase, the competition arises with the galactosyl-enzyme. The nucleophile acts as an acceptor, the part of the substrate covalently linked to the enzyme can thus be transferred to this acceptor. This property is used industrially in peptide synthesis, in which the nucleophile is an amino acid or a peptide that can be linked to a part of a protein or to another peptide.
In kinetic studies of nucleophilic competition we must distinguish two situations according to whether or not the enzyme presents a significant affinity for water and the acceptor.
4.5.1 Study of Nucleophilic Competition in the case Where no Binding Site exists for Water and its Analogues
Bender et al. (1966) studied the kinetic consequences of nucleophilic competition in hydrolysis reactions of esters using chymotrypsin. The kinetic scheme that was proposed is shown below:
where S is a substrate of the form: R−CO−X−R′ with X = O in the case of esters, or X = NH in the case of amides and peptides. If the substrate is a peptide bond and the nucleophile another amino acid, a transpeptidation reaction occurs. Such reactions, as well as transglycosylations, take place in a biological environment. P1, the first reaction product, is R′−XH; P2 is the hydrolysis product of the acyl-enzyme, R−COOH; P3, the deacylation product from the nucleophilic agent, N=R′′YH, has the form: R−CO−Y−R′′. Ks is the dissociation constant of the ES complex, k2 is the acylation constant and k4 is the deacylation constant from the nucleophile N. If P1 is itself utilised as a nucleophile, k4 is identical to k−2, the reverse acylation constant. The constant k′3 is assumed to be equal to k3(W), (W) being the water concentration and may be considered to be constant if, in the reaction medium, it changes negligibly.
4.5.1.1 General Equations
Solving the system at steady state, for the initial rates of product appearance Pi (i = 1, 2, 3), leads to the following equations:
From the previous equations, it is possible to distinguish three important cases depending on the relative values of the rate constants, k2 and k′3, and assuming that k4(N) is the same order of magnitude as k′3, which is always achievable by choosing a suitable concentration of the added nucleophile N.
Case I– If k2 » k′3, i.e. if the deacylation step is rate-limiting, expressions [5], [6], [7] and [8] can be simplified. The apparent Michaelis constant is written:
with kcat,1 and Km,app being linear functions of (N) and possibly (W). The rate of appearance of product P1 increases with (N) when s » Km,app; when s « Km,app, no further effect is observed.
The rate of appearance of product P2 (case I-P2):
$${\rm{k}}_{{\rm{cat,}}2} = {\rm{k^\prime}}_3$$
is independent of (N). This is strictly identical to competitive inhibition with:
Case II– If k′3 » k2, i.e. if the acylation step is rate-limiting, the expressions [5], [6], [7] and [8] may also be simplified. The apparent Michaelis constant Km,app becomes equal to Ks, the dissociation constant of the first enzyme-substrate complex.
The rate of appearance of product P1 (case II-P1) is:
$${\rm{k}}_{{\rm{cat,}}1} = {\rm{k}}_2$$
In this case, (N) has no effect on kcat,1 nor on Km,app.
The rate of appearance of product P2 (case II-P2) is:
The inverse of the appearance rate of product P2 is a linear function of (N), irrespective of the concentration of s. This case is identical to non-competitive inhibition with:
The appearance rate of product P3 increases with (N), and tends towards the limit valuek2 when k4(N) » k′3.
Case III– If k′3 ∼ k2, i.e. if both acylation and deacylation are partially rate-limiting, the previous expressions can no longer be simplified, and (N) affects both Km,app and kcat. Km,app is a complex function of (N) which tends towards a limit value equal to Ks when k4(N) becomes very large relative to k2.
The rate of appearance of product P1 (case III-P1), kcat,1 increases with (N) and tends towards a limit value equal to k2 when k4(N) » k2. For s « Km,app, dP1/dt is independent of (N).
Regarding the appearance of product P2 (case III-P2), 1/kcat,2 is a linear function of (N):
The inhibition decreases when the concentration of s increases, but does not cancel out, even when the enzyme is saturated by the substrate, which distinguishes it from what is observed in caseI-P2. This form of inhibition is formally identified as partial competitive and partial non-competitive inhibition.
The appearance rate of product P3 (caseIII-P3), kcat,3, increases with (N) and tends towards a limit value equal to k2 when k4(N) becomes very large relative to k3 and k′3. CaseIII-P3 is interesting as it is possible to obtain all of the system’s kinetic constants by studying the effect of the concentration of (N) on the kinetic parameters of the reaction. For example, in caseIII-P2, we can determine all of the system’s kinetic constants from the experimental parameters.
4.5.1.2 Interpretation of Experimental Data
Thus, by studying the initial rate of product appearance P2 in the presence of an added nucleophile as well as water, and depending on the kinetic characteristics of the hydrolysis reaction of the substrate studied, it is possible to observe various types of inhibition. These range from competitive pseudo-inhibition to non-competitive pseudo-inhibition. By following the release of product P1 under the same conditions, we observe a more or less complex activation of the reaction in favourable cases; this activation disappears when s « Km,app.
Figure 5.29 below summarises the effect of (N) on the kinetic parameters kcat,1, kcat,2 and Km,app for the different cases I, II and III. The corresponding expressions for these parameters are given in Table 5.2 opposite.
Fig. 5.29
Change in kinetic parameters as a function of the nucleophile concentration and depending on the rate-limiting step of the reaction
4.5.2 Determination of the Kinetic Parameters in the case Where a Binding Site exists for Water and its Analogues
In Scheme 5.1 (pp. 153), it is assumed that the deacylation reactions are bimolecular, i.e. of the form k3(ES′)(W) or k4(ES′)(N). It may, however, be necessary to take into account non-covalent interactions between the enzyme and the water molecule or its analogues preceding the actual reaction event. This would imply the existence of a binding site for water and other nucleophiles in the active site of the enzyme.
It is reasonable to think that these sites are not independent. In this instance Scheme 5.1 becomes:
In this scheme ES′W and ES′N are the complexes formed, respectively, between the acyl-enzyme and water, W, and the acyl-enzyme and the nucleophile N. k*3 and k*4 are the monomolecular deacylation constants relating to the degradation of the complexes ES′W and ES′N, respectively.
KW and KN are the dissociation constants for the complexes ES′W and ES′N, respectively. The kinetic analysis of this scheme at steady state leads to the following equations for kcat,1, kcat,2, kcat,3 and Km,app.
The expressions dP1/dt, dP2/dt and dP3/dt can be explained as for the previous ones. In general, the forms of the functions kcat,i = fi[(W),(N)] and Km,app = g[(W),(N)] depend on the order of magnitude of the quantities KW/(W) and KW(N)/KN(W) compared to unity. One particularly interesting case arises where these quantities are negligible relative to 1, when KW « (N) and (N) ∼ KN, the molar H2O concentration of an aqueous solution being about 55.5 M and the concentration of N rarely exceeding 5 M. If the structure of the nucleophile N resembles that of water, we have additionally, KN ∼ KW. In this case, the equations are written:
If the water concentration (W) can be considered as a constant, these expressions are identical in form to the expressions obtained in the Scheme5.1. It is possible to pass from one scheme to the other by using the relations:
If (W) is likely to vary, for example, when the nucleophile concentration becomes very high, the variable (N) must be replaced by the ratio (N)/(W). Thus, even if a binding site exists for water and its analogues in the enzyme’s active site, it may not be possible to observe an effect of analogue saturation in a wide concentration range. This can be explained by the fact that the water concentration in the reaction medium is very high and the water and analogue sites are not independent.
The significance of the kinetic constants experimentally attained differs however between the reaction Schemes 5.1 and 5.2. With an equal intrinsic reactivity (identical k*4 values), two nucleophiles would be able to display different apparent reactivities if their affinities for the enzyme’s receptor site are not the same (different KN values); in this case, Schema 5.1 is insufficient. Thus, for example, the apparent reactivities of a series of normal primary aliphatic alcohols towards acyl-trypsins and chymotrypsins can vary considerably as a function of the hydrocarbon chain of the alcohol, whereas their “intrinsic” reactivities, evaluated by means of non-enzymatic alcoholysis reactions, remain appreciably constant.
If we use a nucleophile that reveals no saturation effect in a wide concentration range, we can apply the relations from Table 5.2, which give the graphs seen in Fig. 5.29. The analysis of these parameters as a function of nucleophile concentration enables the determination of the rate-limiting step of the reaction and allows us to obtain the values of the elementary rate constants for each reaction when no step is rate-limiting.
This method has been used successfully during the study of tryptic hydrolysis of some substrates (Seydoux & Yon, 1967). Along with the previously described method this has made possible the determination of the elementary steps corresponding to the tryptic hydrolysis of various substrates as indicated in Table 5.3.
Table 5.3
Kinetic parameters corresponding to the tryptic hydrolysis of different substrates
A comparable analysis undertaken by Viratelle and Yon (1973) for reactions catalysed by the E.coli β-galactosidase demonstrated for the first time that these reactions also proceed via the formation of an intermediate chemical component which may degrade by reacting with water or another nucleophilic component. Figure 5.30 below indicates the change in the parameterskcat,i and Km,app for the hydrolysis of two substrates, o-phenyl galactoside and m-nitrophenyl galactoside as a function of the nucleophile methanol. This profile is characteristic of a reaction in which k2 and k3 are of the same order of magnitude. By an analogous study, it was possible to determine the kinetic parameters, including the rate constants for the elementary steps, for diverse substrates that were more or less specific to the enzyme. Table 5.4 lists these values.
Fig. 5.30
Study of nucleophilic competition by methanol in the hydrolysis of m-nitrophenyl galactoside (●) and of o-nitrophenyl galactoside (■) by β-galactosidase from E.coli
Table 5.4
Kinetic parameters corresponding to the hydrolysis of different substrates of E.coli β-galactosidase at pH 7.0, 25°C, 10−3M MgSO4, 0.146 M NaCl (from Yon, 1976)
4.6 Kinetic Study of the Pre-Steady State: Titration of Enzyme Active Sites
The analysis of the preceding kinetic scheme (Sect. 5.4.1) assumed a steady state, i.e. d(ES1)/dt = d(ES2)/dt = 0. In pre-steady state conditions, P1 appears before P2. When the steady state is reached, the rates of appearance of P1 and P2 are the same, as indicated in Fig. 5.31.
Fig. 5.31
Appearance of P1and P2over time in the pre-steady state and steady state phases
The kinetic treatment at the pre-steady state for the appearance of product P1 in conditions where s » e0, i.e. s = s0 with k2<k1, leads to the following expression:
Π represents the initial burst of the product extrapolated to time 0 of the reaction. When s0 » Km,app and k3 « k2, then: Π = e0.
In these conditions, the initial burst of the product P1represents the quantity of enzyme that has effectively reacted with the substrate. This provides a means for stoichiometric titration of the active enzyme.
Such active-site titrants of trypsin exist: p-nitrophenyl acetate and p-nitrophenyl guanidinobenzoate. To titrate the active sites, the nitrophenol burst phase is followed by spectrophotometry.
When it is impossible to reach conditions of saturating substrate concentration, for example, when the substrate’s solubility is low relative to Km,app, it is still possible to evaluate the number of active sites by determining the initial burst of P1 at several substrate concentrations. Indeed, the relation:
The change in \({1 \mathord{\left/ {\vphantom {1 {\sqrt {\Pi _{} } }}} \right. \kern-\nulldelimiterspace} {\sqrt {\Pi _{} } }}\) as a function of 1/s0 gives a linear plot, which, upon extrapolation to the vertical axis, yields the value\({1 \mathord{\left/ {\vphantom {1 {\sqrt {e_0 } }}} \right. \kern-\nulldelimiterspace} {\sqrt {e_0 } }}\). It is however desirable to obtain a sufficient number of points in order for the extrapolation to be precise. When k3 is not really rate-limiting, analysis of the coefficientk from the exponential as a function of the substrate concentration allows the rate constants to be obtained for each individual step, by using the plot shown in Fig. 5.32.
Fig. 5.32
Change in the exponential constant as a function of the substrate concentration
By way of example, the kinetics for the appearance of paranitrophenol during the titration of a β-galactosidase mutant are presented in Fig. 5.33a opposite. The initial burst is analysed as a function of the enzyme and substrate concentrations as shown, respectively, in Fig.5.33b and c.
Fig. 5.33
Hydrolysis of p-nitrophenyl galactoside by a point mutant (13PO) ofβ-galactosidase from E.coli
These few examples illustrate the diverse experimental approaches that enable the rate constants to be obtained for the elementary steps in a single-substrate reaction involving two intermediates. When the reaction products appear in a sequential order, the appearance of P1 under pre-steady state conditions can incidentally lead to the stoichiometric titration of the active site.This method is of the utmost importance as much in fundamental studies as in applied aspects of enzymology.
4.7 Generalisation for N Intermediates
The rate equation can be generalised for the case of n intermediate complexes. Let us take the following reaction scheme:
Disallowed combinations are those that contain subscripts whose algebraic sum is equal to 0 or 1; i.e. constants having equal subscripts, opposite signs or consecutive subscripts (e.g. k1k−1 or k−1k2). The number of intermediates, n, is never very large, but it can exceed 2. We see, therefore, the usefulness of the graphical method to solve the rate equation when the reaction scheme involves more than two intermediates.
5 Enzymatic Reactions with Two Substrates
Enzymes nearly always catalyse reactions having several substrates, frequently two:
Certain enzymes require the presence of a dissociable coenzyme. For kinetic analysis, the coenzyme can be formally considered as a second substrate. For example, alcohol dehydrogenase catalyses the reaction:
Commonly, the concentration of one of the substrates is in large excess and will not be significantly modified over the course of the reaction. In this case, when analysing the kinetics, only a single substrate need be taken into account. Enzymatic hydrolysis reactions use water as a second substrate. When they take place in aqueous solution, the second substrate does not contribute to the kinetics, as has been previously mentioned. In another solvent, the water concentration may be limiting and the kinetic analysis must take this into account.
Enzyme reactions that involve several substrates can follow very different reaction pathways. Their kinetics thus differ, and their analyses may reveal information about the reaction scheme.
5.1 Nomenclature
Before commencing study of these reactions, we shall indicate some elements of the nomenclature employed. A scheme is first of all defined by the number of substrates and the number of products:
a Uni Bi mechanism corresponds to one substrate and two products;
a Bi Bi mechanism has two substrates and two products;
a Ter Bi mechanism has three substrates and two products;
• a Ter Quad mechanism has three substrates and four products.
Sequential mechanisms are those in which the reaction substrates associate to the enzyme in a defined order; for random mechanisms, there is no defined order. A ping-pong mechanism refers to a reaction scheme in which the enzyme oscillates between two forms. Furthermore, Iso designates a mechanism that involves enzyme isomerisation. Here, the discussion is limited to mechanisms involving two substrates.
Before detailing these systems, it is appropriate to define the various kinetic parameters. There is one Michaelis constant with respect to substrateA and another with respect to substrateB; these are complex parameters. A dissociation constant can also be defined for the first substrate, equal to (E)(A)/(EA). Different rules of nomenclature have been suggested to define these parameters and they are given in Table 5.5. Throughout the remainder of this book we shall adopt the nomenclature of Cleland (1963).
In reaction schemes involving two substratesA and B and two products P and Q, it is useful to distinguish linear and branched schemes. For the sake of simplicity and because they are the most frequently encountered, we shall only look at a few examples of these types. The discussion is not exhaustive, but the general principles raised may be applied to solve other reaction schemes. According to the Cleland plot, different states of the enzyme are represented by a horizontal line and the substrates and products by vertical arrows.
5.2 Linear Schemes
5.2.1 Ordered Bi Bi Mechanism
Substrates associate to the enzyme in a defined order; B can only bind to the EAcomplex and the reaction products are released in a defined sequence:
5.2.2 Iso-ordered Bi Bi Mechanism
This is the same mechanism as before, but during the reaction the enzyme undergoes isomerisation. In the final step it once again adopts its initial, active conformation:
In these two types of mechanism the ternary complex is the reactive complex.
5.2.3 Ping-pong Bi Bi Mechanism
In this mechanism the enzyme oscillates between two states, E and F:
5.2.4 Theorell-Chance Mechanism
This mechanism was proposed by Chance and Theorell to account for the kinetics of reactions catalysed by alcohol dehydrogenase. Substrate B, upon binding, ejects the first reaction product and converts it:
In the last two schemes the reaction complex is a binary complex between the enzyme and a single substrate.
5.3 Branched Schemes: Random Bi Bi Mechanism
In this scheme, the two substrates can bind independently to the enzyme. The reactive complex is the ternary complex EAB:
5.4 Kinetic Study of Some Two-Substrate Reactions
5.4.1 Ordered Bi Bi Mechanism
In order to solve this system under steady state conditions, we shall use the rules of King and Altman. The scheme can be written:
Considering the general case where the reverse reaction is not negligible, under steady state conditions we obtain the following rate equation:
$$\frac{v}{{e_0 }}\; = \;\frac{{N_{ab} ab - N_{pq} pq}}{{D_0 + D_a a + D_b b + D_{ab} ab + D_p p + D_q q + D_{qb} qb + D_{pq} pq + D_{abp} abp + D_{bpq} bpq}}$$
The values of the coefficients are given in Table 5.6. The values of the rate parameters are:
The reaction rate in the forward direction can be deduced from the rate equation by putting p = q = 0 in the initial conditions of the forward reaction. In its linear form:
Primary plots – The primary plots of Lineweaver-Burk (1/v versus 1/a or 1/b) yield concurrent straight lines; both substrates have symmetrical behaviour (Fig. 5.34). The intersection of the lines may be either above, below or even on the horizontal axis. The intersection of the linear curve 1/v as a function of 1/a has the horizontal-axis value of −1/Kia; when the graph 1/v is plotted against 1/b, this value becomes −Ka/KiaKb.
Fig. 5.34
Primary plots for an ordered Bi Bi mechanism at steady state
Secondary plots – From the primary plots, the slopes and ordinate (vertical-axis) intercepts as a function of the concentration of the other substrate provide secondary plots (Fig. 5.35 opposite), which enable the kinetic parameters of the reaction to be obtained. Thus, the slope of the curves in the plot 1/v versus 1/a is:
The constants Kia and Kib are the true dissociation constants for the enzyme-substrate complexes. The primary plots differ slightly from those in the previous case (Fig. 5.36) and allow V1 and Kia to be obtained. The secondary plots give the value of Kib.
Fig. 5.36
Primary plots in an ordered Bi Bi mechanism when approximating a quasi-equilibrium
Solving a system under steady state conditions is achieved using the rule of King and Altman. To this end, we write the scheme in the form shown below:
Under conditions that require taking the reverse reaction into account, the general rate equation is:
$$\frac{v}{{e_0 }}\;\; = \;\;\frac{{N_{ab} ab - N_{pq} pq}}{{D_a a + D_b b + D_{ab} ab + D_p p + D_q q + D_{pq} pq + D_{ap} ap + D_{bq} bq}}$$
The values of the coefficients are provided in Table 5.7.
Table 5.7
Values for the parameters in a ping-pong Bi Bi mechanism
It contains no term for 1/ab, which is due to the fact that the binary complexes are the active ones. In this expression, the Michaelis constants with respect to two substratesA and B have complex values, respectively equal to:
5.4.3 A Branched Scheme: The Random Bi Bi Mechanism
The scheme is represented as follows:
A scheme of this type represents the superposition of four ordered Bi Bi pathways. In other words, an ordered Bi Bi mechanism is a special case of this scheme in which the other pathways have a negligible probability of being followed.
5.4.3.1 Solving the System under Steady State Conditions
By employing King and Altman’s rules, we can solve the system under steady state conditions, i.e. when:
Even if we only consider the reaction in the direction of the formation of products P and Q, and assuming that k6 is negligible, we end up – in the initial reaction conditions– with a non-linearisable rate equation having the general form:
$$\frac{v}{{e_0 }}\;\; = \;\;\frac{{N_1 ab + N_2 a^2 b + N_3 ab^2 }}{{D_4 ab + D_5 b + D_6 a + D_7 a^2 + D_8 b^2 + D_9 a^2 b + D_{10} ab^2 + D_{11} }}$$
The values of the coefficients are listed in Table 5.8 opposite. This complex equation cannot be linearised. This system therefore leads to an equation that deviates from Michaelis’ law.
Table 5.8
Parameter values for a random Bi Bi mechanism, approximating the steady state(forward reaction where k6is negligible)
5.4.3.2 Solving a System under Conditions of Quasi-Equilibrium
We can readily solve this system by assuming the existence of a quasi-equilibrium between the free enzyme and the different complexes. For this approximation we consider that the constantsk5 and k6 are very small relative to all other rate constants in the system. This hypothesis is validated by the experimental results when the graphical plots (Lineweaver-Burk) are linear. Indeed, in this case, the rate equation takes on a Michaelian form. Two cases must be considered:
either the binding of A and B to the enzyme is dependent, i.e. the binding of A modifies the affinity of the enzyme for B and vice versa;
or it is independent; the binding of one substrate occurs in the same way in the presence or absence of the second.
5.4.3.2.1 Dependent Binding
Dependent binding is the most general case. If the binding is dependent, we must define for the system in equilibrium the following four equilibrium constants:
Ka for the dissociation of the complex EA: Ka = (E)(A)/(EA),
Kb for the dissociation of the complex EB: Kb = (E)(B)/(EB),
K’a for the dissociation of A from the complex EAB: K′a = (EB)(A)/(EAB),
K’b for the dissociation of B from the complex EAB: K′b = (EA)(B)/(EAB).
Since the system is at equilibrium, the four constants are related by the thermodynamic relationship:
which is strictly analogous to that describing the ordered Bi Bi mechanism assuming a steady state. These two schemes, which can be represented by the same phenomenological equation, are said to be“homeomorphic”.
Primary plots − The primary plots, as for sequential mechanisms, are represented by rays of lines converging to a point whose intercepts with the horizontal axis are −1/Kb and −1/Ka; and with the vertical axis, (Kb−K′b)/V1Kb and (Ka−K′a)/V1Ka, for the graphs 1/v = f(1/b) and 1/v = f(1/a), respectively. Now:
This shows clearly that when the dependence between the substrate-binding sites is positive (i.e. when the binding of the first substrate increases the enzyme’s affinity for the second), the linear plots intersect at a point above the horizontal axis (Fig. 5.39a). On the contrary, if the interactions between the sites are negative, the lines intersect at a point below the horizontal axis (Fig. 5.39b).
Fig. 5.39
Primary plots in a random Bi Bi mechanism approximating a quasi-equilibrium
(a) positive dependence between the substrate-binding sites (b) negative dependence between the substrate-binding sites (c) independent substratebinding sites.
If the binding sites of the two substrates are sufficiently distinct and if there is no interaction between them, the binding of one substrate will not affect the binding of the other. In this instance, the reaction scheme contains no more than two equilibrium constants, Ka = K′a and Kb = K′b. The rate equation is simplified to:
The linear plots intersect at points on the horizontal axis whose values are equal to −1/Ka and −1/Kb, respectively, in the graphs 1/v=f(1/a) and 1/v=f(1/b), as indicated in Fig.5.39c.
5.5 Homeomorphic Schemes: How is the Ambiguity of the Kinetic Response Removed?
In the few schemes just described, it clearly appears that a kinetic study, though necessary and indicative, is not in itself sufficient to determine unequivocally the type of kinetic scheme. We noted, for example, that the random Bi Bi mechanism treated under quasi-equilibrium conditions and the ordered Bi Bi mechanism treated at steady state lead to the same phenomenological expression for the reaction rate and constitute homeomorphic schemes.
It is useful therefore to choose alternative criteria in order to remove the ambiguity of the kinetic response. Among the methods used, one relies on analysing the inhibition kinetics of the reaction products or “dead-end” inhibitors. Another involves studying the binding equilibria of each substrate; lastly, fast kinetic methods may be used to study the reaction. The latter will be examined in the following chapter, which deals with experimental methods for studying kinetics.
5.5.1 Studying Inhibition by the Reaction Products: Cleland’s Rules
The products that are formed during a reaction are inhibitors of their own formation by virtue of the microscopic reversibility principle. Cleland determined several practical rules that follow directly from the analysis of the general expression for the reaction rate in the presence of two reaction products:
if a reaction product and a substrate bind to the same form of the enzyme, the inhibition is competitive (change in the slope of the linear plot v = f(v/s) as a function of the inhibitor concentration);
if a reaction product and a substrate bind to two different forms of the enzyme separated by reversible steps, the inhibition is non-competitive (change in the value of the vertical-axis intercept).
Let us consider the preceding different schemes. We shall examine successively the inhibition by the products P and Q with respect to each substrate, A and B. In an ordered Bi Bi mechanism, inhibition by the product P with respect to A is non-competitive (P binds to EQ; A to E); it is also non-competitive with respect to B. Inhibition by the productQ is competitive with respect to A, yet non-competitive with respect to B.
In a ping-pong Bi Bi mechanism, inhibition by the product P is non-competitive with respect to A and competitive with respect to B.
In a random Bi Bi mechanism, in a state of quasi-equilibrium, each reaction product behaves as a competitive inhibitor with respect to each substrate. This is valid if the diverse states of the enzyme are separated by reversible steps. If one of the steps separating two forms of the enzyme – one of which binds the substrate, the other the product – is irreversible, the situation is more complex.
In addition to the reaction products, other terminal or dead-end inhibitors exist that produce an abortive or dead-end complex with one of the enzyme forms. One part of the enzyme is thus trapped in an inactive form. If such an inhibitor binds to a form of the enzyme that has bound one of the substrates, the inhibition is uncompetitive with respect to this substrate. If it binds to the same enzyme form as the substrate, the inhibition is competitive. For example, in a sequential Bi Bi mech-anism, if there is a dead-end inhibitor I such that:
the inhibition by I with respect to A is competitive.
In the following case:
the inhibition by I with respect to A is uncompetitive.
Thus, based on this theoretical analysis, it is possible experimentally to obtain information about the kinetic mechanism of an enzymatic reaction by studying the influence of the reaction products and the dead-end inhibitors. In practice, the latter are generally substrate analogues. Table 5.9 indicates the types of inhibition by the reaction products for the reaction schemes that have been analysed above.
Table 5.9
Inhibition by the reaction products for the principal types of kinetic mechanism
A complementary study to that discussed above involves analysing the equilibrium of each substrate binding separately to the enzyme. This requires the use of either equilibrium dialysis or dynamic dialysis developed by Colowick and Kaplan (see Chap.2).
5.5.3 Study of Transitory Steps by Rapid Kinetics
It is clear that the precise determination of a reaction path requires the use of fast kinetic methods: flow and (or) relaxation. These methods are presented in the following chapter.
This enzyme, still sometimes called glutamate oxaloacetate transaminase, catalyses the following reaction:
The enzyme catalyses the transfer of the amino group from L-aspartate to α-ketoglutarate. As with all transaminases, this enzyme uses pyridoxal phosphate as a coenzyme, which is transformed into pyridoxamine phosphate during the reaction. The majority of these studies initially used the enzyme from pig heart; it was in fact the first transaminase to be purified. Its sequence and three-dimensional structure are known, as well as those of transaminases from other organisms. The kinetic studies were carried out around the 1960s by the teams of Banks and Vernon in the UK, Velick and Vavra in the USA, Braustein in the former USSR, Fasella in Italy (Rome), and Yon in France (Paris). The total reaction equilibrium was studied and the value of the equilibrium constant:
is of the order of 3–5. (α–KG) represents the concentration of α-ketoglutarate and (OAA) that of oxaloacetate.
Different reaction mechanisms could be invoked a priori to describe the reaction; they imply the formation of either a ternary complex, or a binary complex, with the associations being sequential or non-sequential. The kinetic studies have permitted the derivation of a reaction scheme for both the forward and reverse reactions.
For the forward reaction, the experiments were performed in the initial reaction conditions by following the appearance of oxaloacetate by spectrophotometry. This compound is in equilibrium with its enolic form which absorbs strongly:
In the experimental conditions used, the last reaction was not rate-limiting. Fig. 5.40 shows the change in 1/v as a function of the inverse of the α-ketoglutarate concentration for different aspartate concentrations.
Fig. 5.40
Kinetic study of the forward reaction catalysed by aspartate amino transferase
(a) 0.1 M imidazole/HCl buffer, at pH 8.0 and 25°C – (b) at pH 7.1 and 25°C, for different aspartate concentrations. (Reprinted from Chemical and Biological Aspects of Pyridoxal Catalysis, Banks B.E.C. et al., Kinetic studies of glutamic-aspartic transaminase, 205, 1963, with authors’ permission)
Figure 5.41 opposite depicts the change in 1/v as a function of the inverse of the glutamate concentration for different concentrations of oxaloacetate. The parallel nature of the straight lines obtained for the two series of experiments indicates that the reaction takes place according to a ping-pongmechanism; the reaction is described by the corresponding equation and can be split into two half-reactions:
Fig. 5.41
Kinetic study of the reverse reaction catalysed by aspartate amino transferase at pH 7.8 and 25°C, for different oxaloacetate concentrations
(Reprinted from Chemical and Biological Aspects of Pyridoxal Catalysis, Banks B.E.C. et al., Kinetic studies of glutamic-aspartic transaminase, 206, 1963, with authors’ permission)
PLP and PMP are the pyridoxal and pyridoxamine forms of the coenzyme, respectively.
This reaction scheme has been confirmed by inhibition studies with glutarate which binds to the pyridoxal form of the enzyme and not to the pyridoxamine form.
The results are presented in Fig. 5.42a and b. In reality this scheme is further complicated by the existence of other reaction intermediates. The kinetic constants for the system were determined:
(a) glutarate inhibition of the forward reaction catalysed by aspartate amino transferase, with respect to α-ketoglutarate (b) inhibition of the reaction by α-ketoglutarate with respect to aspartate
(Reprinted from Chemical and Biological Aspects of Pyridoxal Catalysis, Banks B.E.C. et al., Kinetic studies of glutamic-aspartic transaminase, 207, 1963, with authors’ permission)
It has been particularly studied in J. Ricard’s laboratory in Marseille (France). The authors showed that the reaction kinetics can be interpreted according to an ordered Bi Bi mechanism.
The primary plots for the forward reaction are given in Fig. 5.43. The secondary plots are given in Fig. 5.44a and b.
Fig. 5.43
Primary plots of the phosphorylation reaction of glucose by MgATP catalysed by yeast hexokinase
These results are compatible with several reaction schemes: either ordered Bi Bi or random Bi Bi when approximating a quasi-equilibrium. On the one hand, the study of the inhibition by MgADP indicates non-competitive inhibition with respect to two substrates (Fig. 5.45 opposite). On the other hand, inhibition by glucose-6-phosphate is competitive with respect to glucose and non-competitive with respect to MgATP (Fig. 5.46a and b). The results of these studies of product inhibition indicate an ordered Bi Bi mechanism.
Fig. 5.45
Non-competitive inhibition by the reaction product during glucose phosphorylation catalysed by hexokinase
The aim of experimental data processing, in particular with kinetic data in enzymology, is to estimate the degree of reliability of the constants determined from the experimental results by using statistical techniques. These are generally based on least-squares methods. This facilitates the evaluation of the quantitative values in the interpretation and verification of the hypotheses. This is the case for the analysis of the Michaelis equation by the method of non-linear regression which was employed from 1961 (Johansen & Lumry, 1961; Wilkinson, 1961; Cleland, 1963). Such an analysis is largely facilitated today by the use of computers and theexistence of appropriate software. We shall recall a few definitions, before succinctly outlining the principles of linear, multilinear and non-linear regression. For the reader who wishes to deepen his or her understanding of these aspects, we recommend the book by M.Desmadril: L’Analyse des données en C (Data analysis in C).
6.1 A Few Definitions
The arithmetic average of a variable is defined by the relation:
The covariance is nil if the variablesx and y are independent. It differs from 0 when they are linked.
6.2 Simple Linear Regression
Simple linear regression is applicable to all linear relationships of the form y = ax+b. For a given value of x, the observable is equal to the quantity Y = β0+β1x, plus a quantity ε, which represents the error, i.e. the deviation of the experimental value from the regression line:
The method of linear regression consists of estimating the values of β0 and β1 that best account for the experimental results, i.e. the values (xi,yi). The valueS, which represents the spread of the experimental points from the line β0+β1 x, is defined by the relationship:
where wi is the statistical weight associated with each value of yi, an experimental value of the variableY. It is distributed with a variance σ2, independent of the xi values.
p is the number of parameters to be determined, which here is equal to 2.
The estimates b0 and b1 of the parameters β0 and β1 which give the best Y values associated with (xi,yi) are those which lead to the smallest value of S. If we consider relationship [14] as a function of both β0 and β1, it is a minimum at the point S0(b0,b1) such that:
Linear regression with a constant term of the order n + 1 can always be adapted to regression of the nth order with the constant term by an appropriate transformation of the variables. We suppose that the observed or transformed variable yi follows the relationship:
such that \(\partial S/\partial \beta _k^{} = 0\) for k = 1, 2, 3… n. Once more we are solving a system of n linear equations with n unknowns. As before we put:
The principle of least squares is only applicable to a linear relationship having between two and several variables. More often in practice, however, we have to analyse non-linear relationships, in particular the Michaelis equation. In this situation two approaches are possible. Either, with the help of an appropriate transformation of several variables, we obtain a linear relationship, or when it is impossible to lin-earise, an iterative method must be used. One of the conditions for using the least-squares method is that, in the linear relationship, the variables must be independent.
the expression then becomes: \({\rm{y}}_{\rm{i}} = {\rm{\alpha }}_{1i} {\rm{x}}_{1{\rm{i}}} + {\rm{\alpha }}_{2{\rm{i}}} {\rm{x}}_{2{\rm{i}}} \) and we return to the former case. We determine α1 and α2 by minimising the function \(\sum {w_i (y_i - y)^2 }\), which is again multilinear regression. The values of α are given by the relationship:
From these α values, the new values of Km and Vm are thus used in subsequent multilinear regression. This iterative process is continued until there is negligible change in the parameter values between two successive iterations
.
6.5 Checking the Adequacy of the Fit
6.5.1 Examination of The Residual Values
Analysis of the results requires an initial model, for example, that described by the Michaelis equation, and thereafter those parameter values are determined that account most appropriately for the experimental results. Before determining the confidence intervals associated with each parameter, it is necessary to test whether or not the chosen model is correct. This is achieved by inspection of the residual values, and is a useful test whatever the regression method employed. The residual values comprise n differences:
yi being the experimental value and Y the calculated value of the function. The εi values correspond to the observed errors if the model is adequate. If so, the residuals must be independent and distributed randomly, their mean value must be nil and have a constant variance.
Let us look at the Michaelis-Menten equation. Figure 5.47a displays an example of fitting using a non-linear regression method; Fig. 5.47b gives the distribution of residues. First, there is good apparent agreement between the experimental and calculated values; second, it is clear that the residuals are distributed randomly.
Fig. 5.47
(a) fit to the Michaelis-Menten equation by a non-linear regression method. Convergence of the regression function as proved by the correspondence between the calculated (▲) and experimental (○) values of the reaction rate for different substrate concentrations – (b) distribution of the residual values
Certain anomalies may arise as depicted in Fig. 5.48. They correspond either to (a)insufficient parameters, (b) an inadequate model, or (c) to a variance that depends on Y. It is thus useful to weight the experimental values. Another important criterion is that, in linear regression, the covariance of x and y must be nil.
The variance is not always constant as can be seen by studying the residual values. To account for this, a statistical weight wi must be applied to each value of yi. It is inversely proportional to the variance of yi and even to the square of the absolute error: wi=1/(Dyi)2. If Dyi is constant, wi is constant and it cancels out in the various expressions. This is equivalent to putting it equal to 1. If the relative error Dyi/yi is constant, wi≠K/yi2. If the law that governs the change in error is not known in advance, it is helpful to carry out a polynomial regression with the pairs (εi,yi) in order to obtain the polynomial f(yi) which represents the error change in yi. We then let wi=1/f(yi)
.
6.5.3 General Strategy
As a general rule to analyse the kinetics of an enzyme reaction, we must first carry out a certain number of logical operations, and in a certain order. These are:
correctly define the problem, for example, the dependence of the rate on the reactant concentrations;
perform adequate experiments under optimal conditions for the intended purpose, which might be either the determination of parameters, or the choice of a better kinetic model;
collect the experimental data in the order that they are acquired as well as any information that might reveal systematic errors;
formulate the problem using a mathematical model, for example, the rate equation;
analyse the data while considering the agreement between the model and the results, either in the estimation of parameters, or the choice of a kinetic scheme;
interpretation of the results must lead to an understanding of the system, and to the possibility of generalising the conclusions. An important test is the experimental checking of predictions that result from the model.
Bibliography
General Works
Bertrandias F. & Bertrandiasj.P. –1997– Mathématiques pour les sciences de la vie, de la nature et de la santé, Grenoble Sciences Collection, EDP Sciences, Paris.
Banks B.E.C., LaurenceA.J., Vernon C.A. & Wooton J.F. –1963– Kinetic studies of glutamic-aspartic transaminase (from heart muscle), in Chemical and biological aspects of pyridoxal catalysis, E.E. Snell, P.M. Fasella, A.E. Braunstein & A. Rossi-Fanelli eds, 197–215, Pergamon Press, London.
Deschavannes P.J., Viratelle O.M. & Yon J.M. –1978– Functional properties of beta-galactosidase from mutant strain 13 PO of Escherichia coli, in Proc. Natl Acad. Sci. USA75, 1892–1896.
Hammes G.G. & Fasella P.M. –1963– in Chemical and biological aspects of pyridoxal catalysis, E.E. Snell, P.M. Fasella, A.E. Braunstein & A. Rossi-Fanelli eds, 185-195, Pergamon Press, London.