To understand the mechanisms of enzyme catalysis, that is the mechanisms by which the enzyme lowers the energy barrier of the reaction that it catalyses, it is not sufficient to study only the catalytic act, but it is important to analyse the diverse events implied over the course of an enzymatic reaction which condition this catalytic act. The formation of intermediate complexes including non-covalent associations takes part in the catalytic function as does their decomposition. The formation of one or more intermediate complexes between the enzyme and the substrate is a necessary but not sufficient condition for the reaction to take place. It is therefore important to present the mechanisms of enzyme-substrate association; it is only for reasons of clarity that this first step of enzymatic reactions constitutes a chapter in itself.
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
To understand the mechanisms of enzyme catalysis, that is the mechanisms by which the enzyme lowers the energy barrier of the reaction that it catalyses, it is not sufficient to study only the catalytic act, but it is important to analyse the diverse events implied over the course of an enzymatic reaction which condition this catalytic act. The formation of intermediate complexes including non-covalent associations takes part in the catalytic function as does their decomposition. The formation of one or more intermediate complexes between the enzyme and the substrate is a necessary but not sufficient condition for the reaction to take place. It is therefore important to present the mechanisms of enzyme-substrate association; it is only for reasons of clarity that this first step of enzymatic reactions constitutes a chapter in itself.
The first authors emphasised the importance of the structural complementarity between the enzyme and its substrate to ensure their association, the enzyme constituting a sort of matrix (template) in which the substrate would come to insert itself. This rigid notion of the “lock and key” mechanism reflected well the static conception which was assumed during nearly a half century following studies of E. Fischer on the stereospecificity of enzymatic reactions.
Flexibility and mobility of certain regions in proteins, in particular those that are localised at the active centre, currently bring about a much more dynamic vision of enzyme-substrate interactions. This is currently the notion of structural complementarity between the enzyme and the transition state of the substrate that is particularly underlined, because it is responsible for both specificity and catalytic efficiency. The affinity of enzymes is indeed much larger for the transition state than for the fundamental state of the substrate; this represents an important thermodynamic factor in enzyme catalysis.
This chapter includes first a presentation of the nature of forces implicated in enzyme-substrate complex formation, then an energy aspect, and finally a discussion on the mechanisms of formation of these complexes.
1 Nature of Forces Involved in Enzyme-Substrate Associations
The forces responsible for the association between an enzyme and its substrate are the same as those present in associations between simple molecules. However, the complexity of the enzyme protein structure is such that multiple groups can be implicated and different types of forces can intervene simultaneously; in addition, the presence of a solvent such as water brings about by its nature an important contribution. The different forces susceptible to be involved in enzyme-substrate associations consist of:
Electrostatic interaction forces that are produced between atoms or groups of atoms that possess a permanent electric charge; this concerns interactions between two ions, between an ion and a dipole or even between two permanent dipoles.
Induction forces that result in the induction of a dipole in a non-polar group submitted to an electrostatic field with a permanent charge. One can distinguish the interactions between an ion and an induced dipole, or even between a permanent dipole and an induced dipole. Such interactions are sometimes classified under Van der Waals interactions. Some authors include them in electrostatic interactions; at the limit the diverse forces considered are of an electrostatic nature.
Electrokinetic interactions or Londondispersion forces. These are inter-actions between non-polar groups due to the mutual induction of fluctuating dipoles.
Short-range repulsions which correspond to electrostatic repulsions between electrons following the recovery of electron clouds of two atoms.
Hydrogen bonds that form when two electronegative atoms are joined by the intermediate of a hydrogen atom; they are also of an electrostatic nature.
The covalent bond resulting from a shared pair of electrons between two atoms. The covalent bond is sometimes implicated in enzyme-substrate complexes; it appears then in a step consecutive to the formation of the Michaelis complex which by definition is non-covalent.
2 Electrostatic Interaction Forces
2.1 Interactions Between Two ions or Coulomb Interactions
A point charge in a homogeneous media possesses an electric field whose inten-sity diminishes as a function of the distance.
The force of the field is given by the equation:
In this expression q is the charge, r is the distance from the charge to the point where the field is measured, Z is the valence of the ion and e the electron charge. The dielectric constant D depends on the medium in which the charge is found and in particular on the polar nature of molecules that constitute the media. In vacuum, the dielectric constant is equal to one; it is 74.1 in water at 37.5ºC.
Ions in finite dimensions can be considered like point charges with the total charge being concentrated in their centre. If the charge is expressed in electrostatic units (esu) and the distance in cm, the force of the electric field is in dynes/esu. At a distance of 5 Å from an electron in vacuum, the field is equal to −4.8×10−10 esu/ (5×108 cm)2, or −1.95×10−5 dynes/esu, the field being the same sign as the charge. Knowing that 1 dyne/esu equals 300 V.cm−1, the field is −5.76×107 V.cm−1. This example shows the intensity of the electric field neighboring an ion.
The force exerted on an ion by another ion situated at a distance r is equal to the force of the field exerted by one multiplied by the charge of the other:
The most important quantity for problems of association is the interaction energy between two separated charges. This energy can be evaluated by integration of the force between the charges from an infinite distance to the distance r:
The interaction energy between the charges varies with the inverse of their distance. If the charges have the same sign, the energy is positive, and there is a repulsion. If they are of opposite signs, there is an attraction. As an example, to show the role of the dielectric constant, the interaction energy between two monovalent ions, an anion and a cation situated 5 Å apart in vacuum, is −66.3 kcal.mol−1, whereas in water it is −0.85 kcal.mol−1, that is very slightly higher than thermal motion.
2.1.1 Interactions between an ion and a Dipole
An electric dipole results in the presence of two equal charges of opposite sign separated by a distance l. The size of a dipole is given by its moment:
The electrostatic interaction between a dipole and an ion is the sum of the interactions of the ion with each dipole charge. The potential energy is given by the equation:
When the length l of the dipole is less than the distance r between the ion and the dipole centre, which is generally the case, the general equation can be simplified and becomes:
The potential energy depends on the orientation. The scheme below shows the interaction energy between an ion and a dipole with moment µ in the different positions, aligned and perpendicular:
If the dipole is free to rotate rapidly and uniformly, the average of the potential energy over time is zero. However, following the ion electric field, the dipole orients statistically towards the position of maximum attraction, meaning near the position of minimum potential energy. The degree of orientation depends on the relative sizes of the interaction energy and the thermal agitation, kT. A statistical dipole is produced with an average moment:
This relationship is particularly valid when the potential energy of electrostatic interaction is weaker than thermal motion. If it is much larger, and under the condition that the dipole is free to orient, the preceding expression reduces to:
This can be illustrated by some numerical examples. Let us consider a dipole in which the charges situated at a distance of 2 Å correspond to half of the electron charge. The dipole moment is:
The potential energy is expressed in ergs/molecule if q and µ are expressed in esu and r in cm. If a dipole of 1 debye is situated at a distance of 10 Å from a monovalent ion at 37.5ºC, the potential energy is:
or −0.269 kcal.mol−1 at 37.5ºC; at this temperature the thermal motion energy is 0.617 kcal.mol−1, larger than the interaction energy. If the dipole is not free to rotate, but is found fixed in the position of optimal attraction, the interaction energy is then 0.691 kcal.mol−1. The interaction energy between an ion and a dipole when the dipole is free to rotate only represents therefore 40% of the interaction energy when the dipole is fixed in its position of optimal attraction.
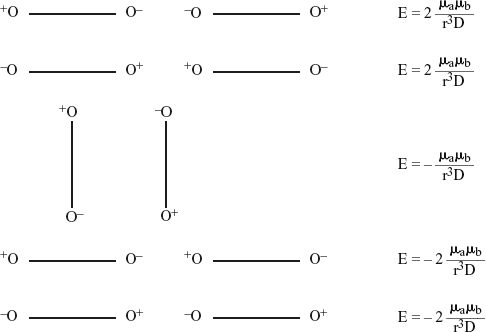
2.1.2 Interactions Between Permanent Dipoles
The potential energy of two dipoles depends not only on their distance, but also on their relative orientation. It is convenient therefore to consider first the case of two fixed dipoles, and then the case of two dipoles with rotational freedom.
Let us consider first the case of two fixed dipoles placed a distance r apart from each other, this distance being greater than the length l of the dipole. The potential energy is given by the Moelwyn-Hughes equation:
The scheme below indicates the different possible orientations:
General case
Aligned or parallel dipoles
In summary the potential energy of electrostatic interaction between two fixed dipoles not having any freedom of rotation is given by the general expression:
The minimum potential energies are obtained when the dipoles are aligned with their opposite sign charges opposing.
When two dipoles are free to rotate, two situations can arise. In the first, dipole A is fixed and dipole B is free to rotate; this can correspond with the case of a dipole on the surface of an enzyme and a small molecule in solution. The potential energy is:
θ being the angle that aligns the centres with the axis of the dipole. In the second situation, the two dipoles are free to rotate and their interaction is due to the polarisation of mutual orientation that they exert. The potential energy is of the form:
This expression is only valid when the potential interaction energy is less than the thermal motion; otherwise the interaction would be sufficiently strong to orient the dipoles in the position of minimal energy, meaning the maximal attraction. Therefore, when the potential energy is weak or on the order of thermal motion, it is expressed by the general expression:
These interactions are Keesom interactions; their energy varies as 1/r6. They become therefore very weak as the distance increases.
2.2 Induction Interactions
2.2.1 Interaction Between an Ion and An Induced Dipole
All the molecules subject to an electric field are polarised and there results a dipole whose moment depends on the force of the field and on the properties of the mol-ecule. In the most general case, the total polarisation is given by the relation:
Pe is the electron polarisation due to the displacement of electrons in the field, Pa is the atom polarisation due to the displacement of atoms or groups of atoms in the molecule, and Pm is the orientation polarisation resulting from the preferential orientation in the electric field created by the ion of a molecule possessing a dipole. When the molecule does not possess a permanent dipole, the polarisation induced by the ion field reduces to Pe + Pa and is called the induced polarisation Pi. The induced dipole moment is proportional to the force of the field:
However, the energy is necessary for inducing the dipole (α0 F2/2), from where the expression of interaction energy between an ion and an induced dipole comes:
The values of polarisability α0 are determined by measures of optical refraction, since the latter depends on electron and atom distortions induced by the alternating electric field from the radiation. The refraction varies with the wavelength of light used.
By determinations at two or more wavelengths, it is possible to calculate the refraction at zero frequency where one obtains the effect of a constant field. The molar refraction at zero frequency, R0, relies on the polarisability by the equation:
N being Avogadro’s number. The values of R0 are known for most of the atoms and the molar refraction of a substance is approximately equal to the sum of the atomic refractions of its constitutive atoms as shown by Landolt in 1862.
2.2.2 Interactions Between a Dipole and an Induced Dipole or Debye Interactions
A permanent dipole can also polarise a neighboring molecule. The potential energy of a polarisable molecule in an electric field, as well as what was indicated above is driven by the equation:
A being a constant dependent on the orientation of the dipoles.
2.3 Electrokinetic Interactions or London Dispersion Forces
Non-polar molecules or groups are susceptible to attracting each other although they do not apparently possess any localised charge. The nature of these forces remained unknown until London in 1930 showed their relationship with optical dispersion and gave the expressions of these interactions derived from quantum mechanics.
A symmetrical molecule possesses at each instant a dipole moment following fluctuations in relative positions of the nucleus and electrons in the outer layer. This instantaneous dipole creates a field that polarises an adjacent molecule. The second molecule then possesses a fluctuating dipole that, in turn, polarises the first mol-ecule. Thus a coupling establishes between the oscillations of the electrons of the two molecules, so that the statistical distribution of electrons is continually in favor of the attraction. There is not a simple dipole in a molecule, but several oscillating dipoles; since these dipoles have different moments, the interaction energy is obtained by integration of the ensemble for each molecule.
The potential energy of two neutral molecules is given by the London equation:
α being the polarisability and h Planck’s constant (6.62×10−27 ergs.s−1); νi is the frequency of electric oscillation responsible for the polarisability. The term hνi represents the ionisation potential I of the molecule. Thus, the potential energy is given by the expression:
The existence of such forces implicates a tight contact between the two molecules because the energy varies as 1/r6.
2.4 Short-Range Repulsive Interactions
Besides attraction forces there are repulsive forces that maintain a separation between atoms and molecules. These non-specific repulsive forces become visible when atoms or molecules are found in very close contact. They result in repulsions between electron clouds of two molecules when they sufficiently approach each other. The repulsion energy is of the general form:
that is: \( {\rm{E}} = \,\frac{{{\rm{ - A}}}}{{{\rm{r}}^{\rm{a}} }} + \frac{{\rm{B}}}{{{\rm{r}}^{\rm{b}} }} \) a and b indicate the dependence as a function of the distance. Besides electrostatic interactions most of the previously described interactions vary as 1/r6, the coefficient a is therefore generally taken equal to 6. The value of the exponent b is large and difficult to evaluate with precision. There exist several approximations; the most utilised consists of attributing the value 12 to this exponent; this is the Lennard-Jonespotential or 6-12 potential, in which the potential energy is expressed by the equation:
the coefficients A and B were evaluated for all atom pairs. The energy curve as a function of distance is given in Fig. 10.1 opposite. A recapitulation of different interactions is presented in Table 10.1 opposite.
Fig. 10.1
Variation in potential energy as a function of the distance between reacting atoms
The hydrogen bond is established between two electronegative atoms bound by a hydrogen atom, which forms a covalent bond with one of the two electronegative atoms. It is written D—H…A, D being the donor and A the acceptor. In hydrogen bonds, the contact between the atoms is closer than in Van der Waals inter-actions between the same atoms; this is due to strong electrostatic interactions between donor and acceptor. Such bonds are formed more easily with hydrogen than with other atoms due to the weak size of the hydrogen atom that permits it to have a close contact with other electronegative atoms. The optimal energy configuration is obtained when the atoms are collinear; the variation in linearity brings about a decrease in this energy. The hydrogen bond force decreases when the distance H…A increases; the energy profile as a function of the distance is analogous to that of Van der Waals interactions represented in Fig. 10.1; it depends moreover on the angle formed by the atoms (Fig. 10.2).
Fig. 10.2
Variation in energy of the interamide hydrogen bond as a function of the N…O distance for different values of the hydrogen bond angle
Different theories were proposed to explain the nature of the hydrogen bond. The most common assumes an electrostatic interaction between the two bound dipoles. The electronegative atoms D and A possess a negative fractional charge when they are bound to other atoms. Therefore the D—H bond constitutes a dipole and can be written D−-H+, the charges being electron and proton charge fractions. Similarly the A—B bond can be assimilated to a dipole A−-B+. The interaction of these two bonds in their collinear position can be represented as follows:
The dotted lines represent a dipole interaction. If this representation corresponds to a real mechanism of the hydrogen bond, the bond energy can be evaluated approximately by a dipole interaction calculation. In the case of the hydrogen bond of water O—H…O, the length of the bond is 2.76 Å and the distance between the dipole centres is 2.40 Å. The potential energy found for the dipole interaction is −6.61 kcal.mol−1; it must be corrected for the repulsion that represents about 35% of the energy, resulting in a final energy of −4.5 kcal.mol−1. The value determined experimentally is −4.5 kcal.mol−1, therefore in agreement with the calculation. The hydrogen bond energy depends on the nature of the acceptor and the donor. It is generally between −2 and −10 kcal.mol−1 and is considerably weaker in water and polar solvents.
The hydrogen bond plays an important role in biological systems. The residues and side chains as well as the NH and C═O groups of the peptide backbone of an enzyme are often implicated in hydrogen bonds with the substrate. Hydrogen bonds sometimes stabilise the negative charges of the substrate developed in the transition state. For instance in serine proteases, the carbonyl from the oxygen of substrates interacts with two NH groups of the peptide backbone. Oxygen becomes negatively charged in the transition state and is found stabilised by dipole moments of two amide groups. All these aspects will be detailed in Chap. 12.
2.6 Hydrophobic Interactions
Non-polar interactions are established between non-polar groups in water; they represent the tendency of non-polar compounds to pass from an aqueous phase to an organic phase. They result in the reorganisation of the structure of water in the presence of a non-polar compound. The molecules of water around a non-polarised compound are organised in order to contract the maximum number of hydrogen bonds between them. The entropy increase due to the disorganisation of the water molecule network structure constitutes the major contribution to the energy of hydrophobic interactions (Néméthy & Schéraga, 1962).
The hydrophobic character of a molecule is measured by the sharing between organic and aqueous phases, the most often between n-octanol and water. By such determinations, it was shown that several substituents of a molecule contribute in an additive manner to its hydrophobic character. There exists an empirical correl-ation between the accessible surface of an amino acid side chain and its energy of transfer from water to an organic phase (Chothia, 1974). This corresponds to 20-25 kcal.mol−1 per Å2 of surface area. The accessible surface of a protein is defined by the area engendered by the centre of a water molecule rolling along its surface.
2.7 The Covalent Bond
A simple covalent bond or σ bond exists between two atoms when two electrons of opposite spin are shared between the two atoms. The electron density is greater between the two nuclei and the electron resonance in this region contributes the greatest part (resonance energy) to the bond energy that varies between 50 and 150 kcal.mol−1 according to the implied atoms. However, covalent bonds do not have obligatorily a character of electronic symmetry. Indeed, when they join two different atoms, the bond usually possesses an ionic character, the electrons being more or less attracted by one of these atoms, conferring upon it an electronegativity. It is not essential here to enter into detail the chemical bond. It is necessary simply to mention that the enzyme-substrate interactions can use simple bonds or σ bonds, and also π bonds like with Schiff bases that form with transaminase and aldolase substrates.
2.8 Determination of the Nature of Enzyme-Substrate Interactions
The different types of forces presented above are involved in the enzyme-substrate association. The first complex or Michaelis complex only implicates non-covalent interactions. It is however difficult to evaluate the energy contribution of hydrogen bonds and electrostatic and hydrophobic interactions. Indeed, in these associations, some interactions are disrupted, for example the interactions between water and the substrate, and water and the active site of the enzyme; others are established in such a way that the global energy represents a difference. These aspects are discussed in the following paragraph. In some enzymatic reactions, starting from the Michaelis complex, a covalent complex between a part of the substrate and a catalytic group of the enzyme is formed. This is the case with acyl-enzymes and phosphoryl-enzymes that were presented in Parts II and III.
The nature of interactions between an enzyme and its substrate can be analysed stepwise. The method consists of evaluating the particular contribution carried by a substrate analog, charged or not, or of a competitive inhibitor, then to compare the dissociation constants, and by consequence the differences in interaction energy in the formation of complexes between the enzyme and the substrates with and without substituents. One can use either direct binding studies when they are possible, or kinetic studies. The best conditions are achieved when it is possible to directly study the equilibrium of enzyme-substrate association by one of the procedures described in Part I and therefore evaluate the dissociation constant Ks. This is only achieved when there exist conditions under which the reaction does not interfere with the measurement, and this without the parameters of association being modified. Substrate analogs that behave like competitive inhibitors are frequently used. The test of values of Ki or Ks as a function of diverse factors, like size, shape, and charge of the molecule of the substrate or inhibitor, gives information on the dimensions and the topology of the binding site of substrates, as well as on the electrostatic and hydrophobic interactions that are involved in the formation of the Michaelis complex.
When the direct study of interactions is not achievable, kinetic methods can be used. In general, as we have underlined in Part II, the Michaelis constant obtained experimentally is a complex parameter. In particular cases however, it represents the dissociation constant of the enzyme-substrate complex. In this case, it is necessary to verify it; the knowledge of the parameter Km = Ks can give information on the enzyme-substrate association. With enzymatic reactions that proceed via the formation of an acyl-enzyme, the value of Ks is obtained by a kinetic study with diverse substrates bringing about the same acyl-enzyme intermediate (see Chap. 5). When it is not possible to attain Ks, one can often resort to substrate analogs playing the role of competitive inhibitors, the inhibition constant Ki being a true dissociation constant. The study of variations in this parameter as a function of pH permitted in some cases the determination of the nature of ionisable groups implicated in the formation of the Michaelis complex as well as the electrostatic contribution to the interaction energy. Examples are given in Chap. 7.
This kind of approach has been used for a long time in enzymology. Among the first studies, one must cite those that were carried out with enzyme-substrate interactions in the case of carboxypeptidase. The research groups of Neurath, Schwert, Smith, Lumry, and Poleglase studied the effect of the structure of various substrates and inhibitors on kinetic parameters of the enzymatic reaction. These studies showed first of all that the enzyme specificity brings about an attack on peptide bonds in the C-terminal position under the condition that the last amino acid R″ is a glycine, an alanine a phenylalanine, a tryptophan, a tyrosine, a leucine or an isoleucine and that it possesses a free carboxylate:
The residue R′ must belong to a glycine, an alanine, a glutamate, a methionine or a tryptophan. If R is replaced by H, the substrate is no longer hydrolysed.
Table 10.2 below gathers the values of Ki obtained by these authors for different competitive inhibitors of this enzyme. The inhibitory activity of the first three compounds decreases as the distance between the indole ring and the carboxyl group increases. With another series, the benzoic, phenylacetic, β-phenylpropionic, and γ-phenyl butyric acids, this distance is still very large with a maximum effect for β-phenyl propionic acid. On the contrary, neither the cis form nor the trans form of cinnamic acid (C6H5—CH=CH—COOH) produce inhibition. Taking into account the preceding results, the absence of inhibition cannot be explained by the distance between the phenyl group and the carboxylate, but rather by the fact that the cinnamic acids have a rigid planar structure such that the enzyme cannot associate at the same time with the phenyl group and the carboxylate; a non-planar configuration of the substrate must be necessary for its association with the enzyme. In the aliphatic series, the inhibitory ability increases with the length of the chain. The action of these different inhibitors is not sensitive to the ionic strength of the media and can only be interpreted by Van der Waals interactions between the substrate and the active centre of the enzyme.
Table 10.2
Values of Kifor carboxypeptidase inhibitors
An analogous study with aspartate transcarbamylase and diverse inhibitors has shown that the two carboxyls of aspartate or its analogs must be in the cis config-uration for the interaction with the active centre to be optimal; thus, contrary to maleate, fumarate is a good competitive inhibitor.
One particularly interesting example is that of acetylcholinesterase. This enzyme plays an important role in the transmission of the nerve influx since it catalyses the hydrolysis of the neurotransmitter acetylcholine. Well before the enzyme structure was known, the kinetic analysis of the enzymatic reaction had brought about an elaboration on a scheme of enzyme-substrate interactions implicating an anionic site where the quaternary ammonium of the substrate would interact, as well as the esterase site containing the serine that is acetylated in the first stage. The enzyme pres-ents a great catalytic efficiency approaching that of a diffusion-controlled reaction. The three-dimensional structure of acetylcholinesterase of Torpedo californica was determined in 1991 with a resolution of 2.8 Å (Sussman et al., 1991). The enzyme is a homodimer bound to the plasma membrane by a covalent bond that implicates the C-terminal extremity of each protomer. After solubilisation by a phospholipase, the crystallised form stays homodimeric. The active site is localised at the bottom of a narrow deep groove 20 Å long. Like with serine proteases, it involves a catalytic triad with serine 200 and histidine 440, but the aspartate is replaced by glutamate 327. In addition, the orientation of the catalytic triad is the mirror image of that of chymotrypsin. The most striking phenomenon is that the quaternary ammonium is not bound to an “anionic site” possessing a negative charge, but interacts with some of fourteen aromatic residues that line the groove. The charge of the quaternary ammonium is interacting with the π electrons of the aromatic residues.
By a kinetic approach using the nucleophilic competition analysis on the hydrolysis of different substrates by trypsin with a series of primary aliphatic alcohols, CH3(CH2)n–1CH2OH, Seydoux et al. (1969) determined the hydrophobic interactions between the aliphatic chain of these alcohols and an apolar site of the enzyme; the interaction energy was evaluated as a function of the chain length (Fig. 10.3).
Fig. 10.3
Schematic representation of hydrophobic interactions between an apolar site of trypsin and the aliphatic alcohol chain
The crystallographic data, when they are known with sufficient resolution, or the NMR data give precise information on the nature of enzyme-substrate interactions. As an example Fig. 10.4 below shows the binding of NAD+ to glyceraldehyde-3-phosphate dehydrogenase (from Biesecker et al., 1977). The hydrogen bonds between the enzyme and the coenzyme as well as the residues of the enzyme in Van der Waals contact with NAD+ are indicated in this figure. The residues Leu 187 and Pro 188 form such a contact with the ribose part of adenosine. The binding sites of Pi and Ps anions correspond very probably to the binding sites of inorganic phosphate implicated in the stage of phosphorylation, and to the binding site of 3-phosphate of the substrate, respectively. Pi interacts by hydrogen bonds with the amino acids Ser148, Thr150 and 208, and Ps, with residues Thr179, Asn181 and Arg231 that form hydrogen bonds with the substrate.
Fig. 10.4
Interactions between NAD+and glyceraldehyde-3-phosphate dehydrogenase, according to the crystallographic data
The radiocrystallographic study of aspartate transcarbamylase in the laboratory of W. Lipscomb permitted also the determination of the amino acid side chains implicated in the interaction with an analog of two substrates, N-phosphonacetyl-L-aspartate (PALA). This complex interaction which involves two neighboring subunits is presented in Fig. 10.5 opposite. The oxygen atoms of the substrate phosphate group contract hydrogen bonds with OH groups of Ser52 and Thr55, the amide groups of the main chain of residues 53, 54 and 55 of the catalytic C1 subunit, and also with Ser80 and Lys84 of the C2 subunit. Likewise, the oxygens of the carboxylate group are interacting with groups of each of the subunits, Lys84 of the C2 subunit, Arg229 and Gln231 of the C1 subunit. The oxygen of the carbonyl interacts with His134 of the C1 subunit. Thus PALA contracts numerous interactions involving two catalytic subunits. Some other examples will be given in the following chapters pertinent to catalytic mechanisms.
Fig. 10.5
Side chains implicated in the association of N-phosphonacetyl-L-aspartate (PALA) with aspartate transcarbamylase
When the dissociation constant of the enzyme-substrate complex Ks is known, it is possible to evaluate the thermodynamic constants that correspond to the formation of this complex. The free energy of complex formation is reliant on the constant Ks by the equation:
The values of ΔG obtained for the formation of the Michaelis complex are generally weak and always negative; the formation of the first enzyme-substrate complex is accompanied by a decrease in free energy of the system, it is an exergonic process and therefore spontaneous.
The study in variations in Ks as a function of temperature permits the evaluation of the variation in enthalpy, ΔH, corresponding to the formation of the complex, according to the equation:
Knowing ΔG at a given temperature and ΔH, it is possible to calculate ΔS, the variation in entropy corresponding to the formation of the intermediate complex.
The same parameters are obtained from the inhibition constants Ki, permitting thus the determination of the contribution of a substituent or a charge to the energy of enzyme-substrate interaction. Table 10.3 gives the energy parameters of enzyme-substrate or enzyme-inhibitor association for some enzymatic systems.
Table 10.3
Energy parameters corresponding to enzyme-substrate or enzyme-inhibitor associations
In many enzymatic reactions, it is difficult to determine the dissociation constant of the complex; however it is possible to obtain information from the parameter kcat/Km. Its significance will be analysed in detail in the following chapter. The comparison of the kcat/Km values for two substrates that differ only by a substituent or charge permits deducing the energy contribution due to this substituent or charge.
This parameter includes both the binding energy of the substrate and the activation energy of the reaction:
ΔGo≠ is the activation energy characteristic of bond breaking.
For similar substrates, the differences in ΔGo≠ can be considered to be negligible; in other cases, they can be corrected by comparison with reactivities observed in non-enzymatic reactions. ΔGb is the intrinsic binding energy of substrates (Fig. 10.6).
Therefore, for two substrates s1 and s2 differing by a substituent or a charge, the difference in binding energy in the transition state is given by the following equation:
Diverse evaluations of enzyme-substrate interaction energy were carried out on these bases. In the case of aminoacyl tRNA synthetases, the differences between the binding energy of specific and non-specific substrates and competitive inhibitors were obtained by comparing the values of kcat/Km for the phosphate exchange reaction. Thus, in comparing kcat/Km for the association of isoleucyl tRNA synthetase with isoleucine and valine, the association of valyl tRNA synthetase with valine and amino butyric acid, it was shown that a supplementary methylene group contributes 3 to 3.8 kcal.mol−1 to the association energy, which is much larger than the simple transfer of a methylene group of water to n-octanol (0.68 kcal.mol−1), as Table 10.4 shows. The comparison of parameters for valine and alanine indicates a difference of 5.41 kcal.mol−1 between isopropyl and methyl groups whereas the corresponding value for the transfer from water to n-octanol is 4.6 kcal.mol−1. It seems therefore that the hydrophobic interaction energies with the enzyme are greater than those observed in studies in solution. The same observation was made with chymotrypsin. Fersht (1984) interprets the situation with chymotrypsin by the presence of at least sixteen water molecules at the binding site of the substrate and around the substrate. All happens as if the hydrophobic interaction for this enzyme corresponds with two normal interactions since there are two interfaces, one for the enzyme and the other one for the substrate, associated with water in an energetically unfavorable manner. In the case of synthetases, the explanation consists moreover of the formation of a cavity in the enzyme-substrate complex when valine occupies the binding site of isoleucine in isoleucine tRNA synthetase.
Table 10.4
Transfer energy values of selected groups
The role of hydrogen bonds in the association of tyrosyl tRNA synthetase with its specific substrate, tyrosine, was evaluated by comparison with the binding of phenylalanine. The affinity of the enzyme is at least 28 000 times stronger for tyrosine than for phenylalanine (δΔ G > 7 kcal.mol−1). The hydroxyl group forms two hydrogen bonds with the enzyme as well as Van der Waals interactions. In the absence of the substrate, there very likely exists a water molecule bound to this site; it is displaced by tyrosine. For the association with phenylalanine, it is not known if the water molecule is displaced or if, from an energy point of view, it is more favorable for the enzyme to undergo some distortions to bind phenylalanine.
The energy contribution associated with the formation of salt bridges between the enzyme and the substrate was obtained by comparing the association energies of amino acids and the corresponding deaminated acids for aminoacyl tRNA synthetases. Table 10.5 shows that the presence of the NH3+ group contributes about 4 kcal.mol−1 to the enzyme-substrate association energy. This is interpreted by the formation of a salt bridge with a carboxylate group of the enzyme.
Table 10.5
Role of the NH3+group in the interaction energy of aminoacyl tRNA synthetase(from Fersht, 1985)
Knowing the atomic coordinates of an enzyme-substrate complex or enzyme-substrate analog, it is conceivable to determine the interaction energies by a theor-etical approach. To attempt this, it is necessary to know the protein structure at a very high resolution and to possess accurate data on the protein-ligand complex. The problem of the solvent however renders the approach very difficult. Diverse theoretical methods were proposed to analyse protein-ligand interactions. Molecular modelling (Bash et al., 1983) with the example of drug binding, methotrexate to dihydrofolate reductase, permitted specification of interactions between the two molecules, but without determining the interaction energy. The methods of conformational energy minimisation (Blaney et al., 1982) were applied to determine differences in interaction energy between prealbumin and thyroxine and different analogs. Mc Cammon and collaborators (1984, 1986) proposed a theoretical approach, perturbation of the thermodynamic cycle, to calculate the difference in association energy of a receptor with two different ligands. The method takes into account problems of solvatation.
The examination of energy parameter values of Table 10.3 calls several remarks. The formation of enzyme-substrate and enzyme-inhibitor complexes is always exergonic; however the value of ΔG is generally weak. The value of enthalpy of associ-ation, ΔH, is generally not very high; it is sometimes zero. On the contrary, the enzyme-substrate association is always accompanied by great variations in entropy, sometimes positive and sometimes negative. The global variation in entropy is the result of at least three contributions. The first corresponds to the formation of a single molecule, the complex, between two molecules, the enzyme and the substrate (or the inhibitor); consequently it must be accompanied by a decrease in entropy (δΔS1 < 0). The second contribution results from the departure of the solvent around substrate molecules and from the active centre of the enzyme upon associ-ation, meaning a modification of the structure of water that goes from a structured state to a less structured state in liquid water (δΔS2 > 0). Yet these two factors are insufficient to account for the observed entropy variations. Indeed, each time it was possible to evaluate the contribution due to the departure of water molecules upon association of the protein with a ligand, it did not suffice to account for the observed association entropy variation. This effect had been evaluated by Pauling in the study of antibody-hapten association that is a good model of enzyme-substrate interactions. It is necessary therefore to consider another contribution which results in conformational variations in the protein induced by its association with the substrate. The variation in corresponding entropy (δΔS3) can thus be negative or positive.
The consideration of these energy parameters has brought about progressively, beginning from the 1950s, a revision in the representation of enzyme-substrate interactions.
4 Mechanisms of Enzyme-Substrate Association
A more dynamic image of enzyme-substrate association than the “lock and key” mechanism invoked by the first authors was progressively elaborated. However, many hypotheses were proposed, some resting on the model of a flexible enzyme and others conserving still a relatively rigid model.
4.1 Induced Fit Theory
The induced fit theory formulated by Koshland is a dynamic model that implicates a certain flexibility of the enzyme. It can be stated by the three following postulates:
the association of the substrate with the enzyme induces in the geometry of the latter a reversible change in amplitude variable according to the proteins;
for the enzyme activity to be effective, a very precise and very delicate orientation of the catalytic groups in relation to the corresponding substrate groups is necessary;
the substrate induces its own orientation according to the change that it provokes in the enzyme geometry.
Koshland supports his theory by diverse arguments. The values of thermodynamic parameters discussed above and in particular the values of enzyme-substrate association entropy assume that conformational protein variations accompany the formation of the complex.
The phenomenon of non-competitive inhibition that brings about the loss in activity can only be understood by a conformational enzyme modification. Indeed, a non-competitive inhibitor does not bind to the active centre; consequently its simple association with the enzyme should not bring about a loss in activity if the conformation of the latter is not modified.
The sequential mechanisms in the reactions of two substrates (see Part II) assumes a rearrangement in the local conformation of the enzyme upon binding of the first substrate, exposing the binding site of the second substrate not yet formed in the free enzyme.
The mechanisms of competitive inhibition of β-amylase by cycloamyloses and the formation of non-productive complexes were interpreted by Thoma and Koshland (1960) by the induced fit theory. β-amylase hydrolyses the glycosidic bonds after the two last glucoses, meaning after maltose unity of linear chains of starch. Cyclohexa- and cyclopenta-amyloses behave like competitive inhibitors, but are not hydrolysed by the enzyme. Thoma and Koshland interpreted this difference in behaviour as proof of the induced fit theory. The complex between the enzyme and the substrate is productive when the association occurs in such a way that the non-substituted hydroxyl in C4 forms a hydrogen bond with group X of the protein; this situation induces a change in conformation permitting catalytic groups A and B to be placed in the optimal position with respect to the glycosidic bond to be broken. This is not achieved in the non-productive complex and cannot occur with cycloamyloses (Fig. 10.7).
Fig. 10.7
Competitive inhibition of β-amylase by cyclodextrane
The induced fit theory can be described by the scheme below:
E is the inactive form of the enzyme, EȲ the active form predominant in the presence of the substrate, but not significantly present in its absence; K1 << 1 and K4 >> 1, therefore K2 > K3, meaning −Δ G2 > −Δ G3. Since K1K2 = K3K4, one has K4 > K1; in the presence of the substrate the form E′S is favored. This scheme will be discussed in detail in Chap. 12.
There are today many experimental facts indicating the existence of conformational variations of an enzyme consecutive to the binding of the substrate. The first arguments were often obtained by spectrophotometric measurements. For example, the association of an inhibitor like (NAG)3 to lysozyme is accompanied by a modification in the environment of tryptophan residues, the association of a substrate analog with carboxypeptidase brings about a difference spectrum in the region of tyrosines.
The structural data confirmed and specified many of these conclusions. For instance, upon binding of (NAG)3 to lysozyme, a small displacement of residues Trp62 and 63 is observed. In the case of carboxypeptidase, the binding of a substrate analog induces a rotation of 12 Å of Tyr248. For many enzymes, crystallog-raphic studies revealed an open structure in the absence of substrates and a closed structure when the substrates are associated with the enzyme. These enzymes generally have a polypeptide chain folded into two structural domains and the closed structure is generated by a relative movement of two domains (hinge bending motion) that represents a particular mode of induced fit. This situation was observed for several NAD dehydrogenases and kinases, as we will see in particular examples.
In reality the term induced fit is open for discussion. Such as the square scheme is represented, it would be more suitable to consider the stabilisation by the substrate of a conformation that was very minor in its absence. Due to the internal motions in proteins, there exist many slightly different conformations presenting weak energy differences or protein sub-states in fluctuating equilibrium. Some of these less populated states in the free enzyme would be stabilised by ligand binding. This would have a selective effect rather than an instructive effect.
4.2 “Rack” or “Strain” Theory (Distortions or Constraints in the Substrate)
This theory assuming a relative enzyme rigidity had been proposed by Haldane (1930) and then by Pauling (1946); it was reintroduced in 1954 by Lumry and Eyring. It consists of assuming that the active site has a structure complementary to the transition state rather than a fundamental state of the substrate or the product. The substrate, in binding to the enzyme, is found in a constrained state and the bond to break undergoes a distortion that labilises it. Thus Haldane, recovering the image of the key in the lock, assumes that the key is not correctly adapted to the lock that exerts on it a certain constraint; this constraint in the substrate geometry would have the effect of destabilising the bond to be cleaved. This concept is stated today under a slightly modified form of stabilisation by the enzyme of the transition state of the substrate, which in fact is not a new concept.
It is accepted today that the structure of an enzyme active site is complementary to the transition state of the substrate rather than of its fundamental state. There exists a great number of experimental and theoretical arguments to support this concept. The transition of the chair form of the saccharide ring in the fourth position in the substrates of lysozyme deduced from crystallography studies has often been pres-ented as an example of the constraint in the geometry of the substrate upon formation of the enzyme-substrate complex. More recent studies on the association of (NAG)6 with lysozyme by a theoretical approach use refined atomic coordinates and no longer use molecular models. Warshel and Levitt (1976) suggest that the substrate binds to the enzyme in a chair configuration and only takes the half-chair configuration in the transition state. For serine proteases, the structural data indicates that there is no conformational variation of the enzyme upon substrate binding, but a complementarity of the active site for the transition state. One currently possesses numerous data showing that enzymes have a greater affinity for the transition state analogs than for the substrate.
These examples as well as the energy importance of the complementarity of the active centre of an enzyme to the transition state rather than to the fundamental state of the substrate are detailed in the following chapter (Sect. 11.4.3) in the discussion on the particularities of enzyme catalysis.
4.3 “Dynamic Rack” Theory
The “dynamic rack” represents a compromise between the concept of the flexible enzyme and that of the rigid enzyme, meaning between the induced fit theory and the “constraints” theory in substrate geometry. It assumes that an enzyme sufficiently flexible would undergo conformational changes upon substrate binding that would, in turn, bring about constraints in the geometry of the substrates.
In summary, enzyme-substrate associations involve diverse interactions of weak energy, electrostatic interactions, hydrophobic interactions, formation of hydrogen bonds, or salt bridges. These are exergonic processes of which the principal contribution is entropic. They are frequently accompanied by the departure of water molecules and by reversible conformational variations of the enzyme.
The energy consequences of these different mechanisms for the efficiency of the enzyme catalysis are analysed in the following chapter.
Bibliography
Books
Fersht A.R. –1977– Enzyme, structure and function, 1st ed., W.H. Freeman and C°, New York.
Ramachandran G.N. –1973– Accuracy of Potential Functions in the Analysis of Biopolymer Conformation, in E.D. Bergmann & B. Pullman eds, Conformation of biological molecules and polymers (Jerusalem Symposia on Quantum Chemistry and Biochemistry, 5, 1), The Israel Academy of Sciences and Humanities, Jerusalem.