
Abstract
Astragalus and Oxytropis represent one of the largest angiosperm genus complexes. It is widely discussed why - and if at all - it contains so many species. Here, we discuss how the genetic divergence in a postglacial relict species, Oxytropis pilosa, can provide insights into the relative genetic variance of taxa. Methods are introduced to obtain sequence clusters of similar genetic diversity and to detect statistically significant differences. These approaches provide objective means to identify taxa that deserve the same protection efforts, independent of a fixed species concept. O. pilosa ITS sequences display an inter- and intra individual variance, comparable to clusters comprising numerous accepted species. Regarding genetic diversity, some O. pilosa populations represent relicts that deserve the same or a higher protection status than some species of Astragalus. We discuss the need of objectivity in identifying the evolutionary hot spots as well as the measures to define and protect them.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Conservation Effort
- Internal Transcribe Space Sequence
- Corrected Entropy
- Astragalus Species
- Relict Species
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Astragalus (∼2.500 species; Podlech 1986) and Oxytropis (∼300 species) are closely related genera, representing one of the largest angiosperm genus complexes. Taxonomic instability within the genus complex comprising both Astragalus and Oxytropis results in a high number of similarities, which may differ in their assignment to the genus. In fact, the separation of both genera is based on a single morphological criterion only: Astragalus exhibits a mucronate keel along the adaxial side of the legume, whereas Oxytropis is characterized by an abaxial carinate legume (Candolle 1802; Barneby 1964; Chater 1968). Molecular studies encountered difficulties in providing sufficient resolution at the species level, independent of the sequence marker used (nuclear and/or chloroplast DNA spacer: Sanderson 1991; Sanderson and Doyle 1993; Wojciechowski et al. 1993, 1999; Kazempour Osaloo et al. 2003). Efforts in chemotaxonomy were promising (Williams and Barneby 1977), but have been only sporadically applied (Tin et al. 2007).
It has been widely discussed why so many species are recognized for Astragalus (Sanderson and Wojciechowski 1996), but it has seldom been addressed whether this large number of species, which is continuing to increase in local floras, optimally represent the natural relationships. Astragalus underwent a considerable radiation in all its main centers of biodiversity: Eurasia (mainly Southwest Asia), Africa, North America, and South America. The high number of morphologically defined species - and varieties - is caused by numerous endemics, often referred to as “relict” species because of their isolated mountainous stands and small populations. It is rare to find widespread Astragalus species. Astragalus covers various climates within its biogeographical areas, such as continental to arctic steppes, stony alpine slopes, and Mediterranean mountainous rocky places or even tropical African highland (Barneby 1964; Gillett 1964; Chater 1968). The preference for mountainous habitats, in conjunction with (post)glacial rapid environmental changes, may have facilitated differentiation processes leading to a split up into numerous species and varieties.
Oxytropis shows a quite similar distribution area in Eurasia and North America compared to Astragalus, but is absent from South America. This distribution is comparable to that of another large genus - Pedicularis (Sauer et al. 1990). Oxytropis exceeds more to the (arctic) North of the Northern Hemisphere than Astragalus, and some remarkable species such as O. campestris with its distinct sub species can be found on both continents. Oxytropis pilosa (Fig. 1) is a rare relict species exhibiting a highly fragmented distribution area. It is mainly found in the Pannonian Basin (Hungary to Austria) and in the Alps with prompt occurrences in Scandinavia (including the seashore of Gotland), the Baltic as well as in the Abruzzo (Hegi 1924; Meusel et al. 1965; M. Schlee, pers. observation). In a number of other accounts including O. pilosa (M. Schlee, G.W. Grimm, M. Göker, V. Hemleben, in prep.), the internal transcribed spacers (ITS1 and ITS2) of the nuclear ribosomal DNA (nrDNA) allowed reconstruction of detailed evolutionary pathways (Schlee et al. 2003; Denk and Grimm 2005; Grimm et al. 2007) as they are biparentally inherited, part of a multicopy gene region and undergo (to some degree incomplete) concerted evolution as well as frequent intragenomic recombination (Arnheim et al. 1980; Hemleben et al. 1988; Baldwin et al. 1995; Volkov et al. 2007). We detected significant intraspecific and intraindividual genetic variability in O. pilosa by cloning and sequencing a large number of nrDNA internal transcribed spacer sequences (ITS1 and ITS2; M. Schlee, G.W. Grimm, M. Göker, V. Hemleben, in prep.). The variability found allows discussion of migratory and evolutionary patterns for this species, which will form the subject of an upcoming study.
Here, we make use of the large amount of sequence data obtained for O. pilosa to conduct a comparison of the genetic diversity of clades by objective means. This is done to find an alternative to classical methods, such as morphotaxonomy-based approaches for the identification of units that represent hot spots of biodiversity (here, genetic resources). We apply non-hierarchical single-linkage clustering to the combined Astragalus and Oxytropis ITS dataset after determining the distance threshold that optimally delimits O. pilosa. Thus, the resulting clusters represent estimates for species boundaries obtained by the use of an operational and fully-automated method treating O. pilosa as the standard. The clusters are expected to be uniform, given the distance threshold and the clustering algorithm (i.e., non-hierarchical single-linkage, NHSL). The obtained clusters are discussed with respect to the taxonomic affiliation (assigned species) of the comprised sequences. A character-based approach is used to determine whether the genetic diversity of selected clusters is significantly distinct. If one agrees that in conservation management major emphasis needs to be laid on equal protection efforts for taxa that are equally diverse (Watt 1947; Stock et al. 1999; Potthast 2000; Schlee 2004), it is crucial to compare diversity by appropriate means. Taking into account that taxonomic affiliations and taxonomic ranks often only poorly reflect relative genetic variance, we wonder whether they are sufficient to determine valuable genetic resources. Here, we thus address the question if biodiversity assessment based on substantial molecular data is superior to estimating diversity from data linked to the recognition of morphospecies and if the proposed techniques are valuable to estimate the relative need for conservation efforts as a means to discriminate protection units. Finally, we briefly discuss the conservation measures that are appropriate in the case of O. pilosa, once the most valuable genetic resources have been identified.
Material and Methods
In addition to our own dataset of Oxytropis ITS clones comprising the ITS1, 5.8S rDNA, and ITS2 regions, and flanking 18S and 25S rDNA (also including some European Astragalus; EMBL accession numbers AM401376 to AM401574; AM943374 to AM943384; FM205750 to FM205773), we added all ITS sequences stored in DDBJ, EMBL, and NCBI gene banks (sequences usually obtained by direct sequencing, not by cloning; downloaded on January 2008) for the genera Astragalus and Oxytropis. Solitary ITS1 and ITS2 sequences were combined if they belonged to the same source according to the original literature/description. The sequences were aligned using the POA program (Lee et al. 2002). The 5.8S rDNA was excluded from the analyses. Phylogenetic inference was performed under the maximum likelihood (ML) optimality criterion using RA x ML 7.0 (Stamatakis 2006; Stamatakis et al. 2008). The program implements a new fast ML bootstrapping and subsequent search for the best topology. Duplicate sequences were eliminated prior to ML analyses. Tree inference and 100 bootstrap replicates were conducted under the CAT approximation (Stamatakis 2006), but final parameter optimization was done under a GTR+Γ model. A plain text (NEXUS) file containing the complete alignment, GenBank accession numbers, and information on the sets of identical sequences reduced by RAxML to a single one, respectively, is available at http://www.goeker.org/mg/clustering/.
To obtain a non-nested sequence classification based on the given distance (or similarity) threshold, we use NHSL clustering, as in the case of the popular tool blastclust (ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.html; regarding hierarchical single-linkage clustering, see, e.g., Legendre and Legendre 1998, pp. 308-312). NHSL is based on the notion of a “link”. A link is defined as any distance between two objects (here, sequences) that is smaller than or equal to the predefined threshold. NHSL starts by assigning the first object to the first cluster. For each of following objects in turn, it assigns that object to the same cluster than a previously clustered object if the distance between them is a link. If no such previously classified object is found, the current object is assigned to a new cluster. If several such previously classified objects are found that belong to distinct clusters, these clusters are joined.
Obviously, larger threshold values will result in larger but less numerous clusters, whereas small thresholds will lead to numerous small clusters. A biologically sensible threshold is usually not known a priori. However, a particularly well studied (and monophyletic) taxon (here, O. pilosa), can be used as a standard by determining the lowest possible threshold that results in all sequences obtained from that taxon being assigned to a single cluster. It is easy to implement an algorithm that calculates this value for a predefined group. Wirth et al. (1966, p. 61) who used similarities instead of distances define “…a similarity value, c, which is the largest fixed linking similarity value for which the cluster is still an interlinked aggregate of specimens”. For each of the n objects within the group, the distance to the least distant object that belongs to the same group is determined; the largest of these n values represents the result, analogous to c. This is due to the fact that in NHSL, a single link is sufficient for an object to be assigned to a cluster. This algorithm and NHSL have been implemented in the program optsil (Göker et al. 2009) which is available upon request. The determination of the standard threshold and NHSL relied on uncorrected distances (also called “Hamming” or “p” distances; e.g., Swofford et al. 1996, p. 455), which represent the relative number of deviations between two sequences. For downloaded sequences, we relied on the NCBI taxonomy provided in the same files to assign them to genera and species. The optimal threshold calculated for O. pilosa was then applied in NHSL.
A disadvantage of the NHSL approach is that clustering methods such as single-linkage clustering cannot be considered as valid methods of phylogenetic inference, mainly because lower pair wise distances (or higher similarities) do not necessarily indicate a closer phylogenetic relationship (e.g., Felsenstein 2004, pp. 165-167). This issue has led to the widespread avoidance of UPGMA clustering (Sokal and Michener 1958) in phylogenetic studies. However, the difficulties may be tempered or even disappear if non-nested clustering is applied and if species are to be distinguished, because within-species sequence dissimilarities are expected to be rather low. The same rationale applies to the usual arguments against uncorrected distances because unobserved, superimposed nucleotide substitutions only play a role if distances are large (e.g., Felsenstein 2004, p. 158).
Göker and Grimm (2008) used the well-known Shannon entropy formula (Shannon 1948) to calculate the character data of hosts (plant individuals) from the character data of their associates (cloned sequences obtained from the respective plant individuals). For all sequences belonging to the same individual, the entropy of each alignment column was calculated to represent the amount of genetic divergence within each individual using the program g2cef designed and implemented by M. Göker (downloadable from http://www.goeker.org/mg/distance/). An alignment of length n will thus result in n corrected entropy values per group. Because the variance in nucleotide characters may depend on the number of sequences, each entropy value was corrected by division through the maximum possible entropy for the given number of associates, which is 0 in the case of a single associate. Accordingly, the corrected entropy values for individuals present with a single sequence only are undefined.
For each pair of groups, n differences between the n corresponding corrected entropy values can be determined. Subsequently, a non-parametric Wilcoxon signed-rank test or a parametric t-test (or any other appropriate statistical test) can be applied to assess whether the distribution of these differences significantly deviates from 0 and, thus, the two original entropy distributions are significantly different from each other. This procedure is similar to paired-site tests used to assess whether the scores of two phylogenetic trees are significantly different, given a sequence alignment of the same taxa (Felsenstein 2004, p. 364 ff.). Pair wise tests were conducted with R (R Development Core Team 2005) and restricted to those clusters obtained by NHSL that comprised more than five sequences.

Oxytropis pilosa, xerothermic habitat on gypsum, Ascoli Piceno, Marche, Italy
Results
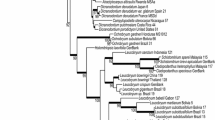
Our final data matrix comprised 437 ITS sequences of Astragalus and Oxytropis species comprising 464 characters from ITS1 and ITS2. Of these 437 sequences, 184 were exact sequential duplicates of others; 291 of the 464 characters were variable and 195 were parsimony-informative. After elimination of duplicates, the alignment analyzed under ML comprised 358 distinct alignment patterns, including a proportion of gaps and completely undetermined characters of 7.48%. The best ML tree obtained is shown in Fig. 2. The tree was rooted along an unresolved backbone polytomy giving respect to a bipartition that separated all Astragalus s.str. from the remaining Astragalus lineages and Oxytropis (in 98% of ML bootstrap replicates). The remaining backbone could be considered to be rather well-resolved, indicating several lineages with bootstrap support (BS) between 70 and 100. Towards the tip of the tree, a general decrease in BS was observed (details not shown). Particularly, the well-known Neo-Astragalus clade received little BS. Multiple accessions of the same Astragalus species (also listed in Table 1) were found within the same clade (with varying support), as were all ITS sequences of O. pilosa (BS = 74, new cloned data plus one directly sequenced NCBI GenBank accession). In contrast, the two other extensively sampled species of Oxytropis, O. arctica and O. campestris, were intermixed with each other and with several other Oxytropis species.

ML phylogram based on ITS data of Astragalus and Oxytropis, with major clusters (see text) and clades annotated. Numbers at nodes indicate non-parametric bootstrap support based on 100 ML bootstrap replicates. Clusters (see text) are highlighted by distinct background shading, clade names are given at right. A, A. vogelii; B, A. epiglottis and A. pelecinus; C, clade of Eurasian Astragalus; D, A. corrugatus and allies; E, clade of Eurasian and North American Astragalus; F, A. alpinus; G, A. adsurgens (cf. Table 1)
Optimal distance thresholds calculated for the present species are shown in Table 1. We obtained an optimal value for O. pilosa of about 0.0069. Results of NHSL obtained with the optimal threshold are shown together with the ML tree in Fig. 2.
The largest clusters and their properties are shown in Table 2. Oxytropis campestris appeared inseparable from O. arctica as well as from a number of less intensively sampled Oxytropis species. The clusters 39, 42, 44 and 45 each contained a number of distinct Astragalus species, indicating that genetic divergence within many accepted Astragalus species is much lower than within either O. pilosa or the O. arctica-O campestris complex.
The results of the Wilcoxon signed-rank and t-tests, which are shown in Table 3, confirmed this conclusion. According to the Wilcoxon test, genetic divergence of O. pilosa was not significantly different from the O. arctica-O. campestris complex, but significantly different (lower) from Astragalus clusters 44 and 45 (species numbers given in Table 2), and significantly different (higher) from Astragalus cluster 42 and Astragalus cluster 39, respectively. According to the t-test, genetic divergence of O. pilosa was not significantly different from any of the other clusters, whereas the O. arctica-O. campestris complex was significantly more divergent than Astragalus clusters 42 and 39, respectively.
Discussion and Conclusion
Genetic variability of populations within the Astragalus-Oxytropis complex has most often been addressed by techniques, such as allozyme polymorphisms (Karron et al. 1988; two endemic and two widespread species), RFLP (Kulshreshtha et al. 2004; one Astragalus species comprising six varieties, two Oxytropis species, pea as outgroup), AFLP, or RAPD (Travis et al. 1996; one variety). Only a few studies used cloned ITS sequences to compare populations (Dong et al. 2003, eight widespread species, one comprising two varieties, another comprising two sub species; Yip and Kwan 2006, one widespread species from 23 localities divided into at least two undefined varieties). Both studies found the marker highly conserved, and proposed the use of 5S rDNA spacers for the future. In contrast, our results indicate that if a sufficiently large number of cloned sequences are retrieved and if populations are sampled from a broad biogeographic range, valuable insights into the genetic divergence of populations can be obtained. Such substantially sampled taxa (here, O. pilosa) can be used as standards to optimize distance thresholds for non-hierarchical clustering algorithms. The adapted clustering techniques will result in a division of sequences into subsets of comparable genetic divergence as a basis to define conservation units. Statistical tests based on multiple sequence alignments allow us to compare the molecular diversity of different groups by objective means and to identify the significant differences regarding genetic variance. In contrast, traditional approaches such as AFLP, RAPD, RFLP, and microsatellite studies are often methodologically restricted to the species level and are statistically difficult to interpret (Felsenstein 2004, pp. 240f, 246f).
Results of our molecular comparison approach are in agreement with previous studies. For instance, the fact that O. campestris and O. arctica are hardly separable has been addressed by Jorgensen et al. (2003). Likewise, the considerably high genetic variance observed in our O. pilosa data confirms the interpretation of that species as a relict species. Particularly, the comparison of O. pilosa and the Neo-Astragalus species underlines how much divergence is preserved within the former. In fact, single populations of O. pilosa are as divergent as or even considerably more divergent than some Astragalus species. As a consequence, they deserve the same conservation efforts as the latter. This result is striking regarding the tendency in literature to suggest the protection of even single alleles within a variety of A. cremnophylax (Travis et al. 1996; for further examples, see the discussion in Soltis and Gitzendanner 1999), one of the numerous species included in our cluster 44 (Fig. 2 and Tables 1-3). Already the limited data available on Astragalus indicates that some species within the Neo-Astragalus clade (Table 1) are genetically more diverse than others, which should be taken into account in course of conservation.
In our view, not each unique individual and/or morphotype should per se be considered a relict or endemic. Biodiversity hotspots and genetic resources can be detected by objective means, similar to the approach presented here. Suitable nature conservation efforts should, therefore, also rely on molecular markers and algorithms that identify those relict stocks with the highest potential for ongoing speciation processes, and those stocks that are endangered to become extinct. A highly adaptive species complex will frequently evolve (and lose) new, unique, and locally restricted morphotypes, but will not necessarily significantly increase its genetic diversity (Table 3). As a consequence, such a species complex will show a high biodiversity as reflected by the number of accepted (endemic) “species”, with only little genetic divergence between them. On the other hand, true relict species such as O. pilosa are morphologically and ecologically restricted or conservative (Hegi 1924), but can harbor a high genetic diversity (cf. Tables 1-3).
Focusing on the genetic diversity within the ITS regions may only be part of the story. Most likely, amplifying additional genes will result in additional insight into ecology and morphology. However, because of a lack of both time and funding, it is hardly realistic to do such multi gene sequencing for a broad sample size, and to detect and protect each individual that is a potential founder organism. Rather, one should focus on the identification and protection of at least some of the remaining true relicts in our rapidly changing world. Even though the cost of sequencing is continuously decreasing (because of recent advances in high-throughput techniques), there will always remain a trade-off between the number of loci amplified per individual and the number of individuals examined. As demonstrated above, it is easy to quantitatively determine units with similar genetic diversity, as long as a sufficient number of sequences is amplified for an appropriate reference taxon (here, O. pilosa). Accordingly, focusing on a single molecular locus, preferably one for which a large number of sequences are already deposited in public databases, is likely to remain the method of choice in many future studies. Once the most valuable genetic resources have been identified, using the methods described above (or any other appropriate means), protection measures for these resources can be laid out. Phytosociological studies can provide valuable insights into a species’ habitat needs. Such studies are necessary, particularly if the genetic divergence within single individuals is so high that they deserve considerable conservation efforts, as confirmed by the present study for O. pilosa. In such a case, conservation should aim at preserving as much of the estimated genetic richness as is possible. The monitoring of relicts should be intensified, in general, using the combination of molecular and phytosociological methods to detect the evolutionary hotspots and endangered genetic resources as well as the means to protect them. Phytosociological surveys were conducted for O. pilosa using the Braun-Blanquet approach to quickly obtain detail-rich relevées (Schlee et al. 2003; M. Schlee, G.W. Grimm, M. Göker, V. Hemleben, in prep.). As a conclusion from these surveys, conservation efforts need to include more suitable land-use of the habitats. A management system should prefer a grazing over a cutting regime. The toxicity of Astragalus and Oxytropis species, in general, is here of lesser importance because food aversions can be conditioned if they do not already exist in grazing animals (Ralphs and Provenza 1999). Hairiness of the plants (e.g., O. pilosa) or spines (many Astragalus species) then remain the species’ natural means of protection. If disturbances are too weak, networking concepts for habitats (e.g., NATURA 2000 for Europe) fail. However, if the natural linkage between habitats cannot be restored, networking is not necessarily desirable when speciation is in progress because it might be thwarted by artificial hybridization or homogenization among populations. The protection of ecological processes is, therefore, highly recommended instead of pursuing a networking concept at all cost. As a consequence, conservation efforts must hastily be improved to protect the disjunct populations of O. pilosa and to retain their genetic diversity as a valuable natural resource.
References
Arnheim N, Krystal M, Schmickel R, Wilson G, Ryder O, Zimmer E (1980) Molecular evidence for genetic exchanges among ribosomal genes on nonhomologous chromosomes in man and apes. Proc Natl Acad Sci USA 77(12):7323–7327
Baldwin BG, Sanderson MJ, Porter JM, Wojciechowski MF, Campbell CS, Donoghue MJ (1995) The ITS region of nuclear ribosomal DNA: a valuable source of evidence of angiosperm phylogeny. Ann Mo Bot Gard 82(2):247–277
Barneby RC (1964) Atlas of North American Astragalus. Mem NY Bot Gard 13(1–2):1–1188
Candolle AP de (1802) [Astragalogia] Augustini-Pyrami Decandolle, Astragalogia nempe Astragali, Biserrulae et Oxytropidis, nec non Phacae, Colutae et Lessertiae, Historia Iconibus Illustrata. Joann. Bapt. Garnery, Parisiis
Chater AO (1968) Astragalus. In: Tutin TG, Heywood VH, Burges NA, Moore DM, Valentine DH, Walters SM, Webb DA (eds) Flora europaea, vol 2, 1st edn, Rosaceae to Umbelliferae. Cambridge University Press, Cambridge, pp 108–124
Denk T, Grimm GW (2005) Phylogeny and biogeography of Zelkova (Ulmaceae s.str.) as inferred from leaf morphology, ITS sequence data and the fossil record. Bot J Linn Soc 147(2):129–157
Dong TTX, Ma XQ, Clarke C, Song ZH, Ji ZN, Lo CK, Tsim KWK (2003) Phylogeny of Astragalus in China: molecular evidence from the DNA sequences of 5S rRNA spacer, ITS, and 18S rRNA. J Agric Food Chem 51(23):6709–6714
Felsenstein J (2004) Inferring phylogenies. Sinauer Associates, Sunderland, MA
Gillett JB (1964) Astragalus L. (Leguminosae) in the highlands of Tropical Africa. Kew Bull 17(3):413–423
Göker M, Grimm GW (2008) General functions to transform associate data to host data, and their use in phylogenetic inference from sequences with intra-individual variability. BMC Evol Biol 8:86
Grimm GW, Denk T, Hemleben V (2007) Evolutionary history and systematics of Acer section Acer - a case study of low-level phylogenetics. Plant Syst Evol 267(1–4):215–253
Hegi G (1924) Flora von Mitteleuropa. IV. Band, 3. Teil: Dicotyledones 2. Teil Leguminosae - Tropaeolaceae, 1st edn. Carl Hanser, München
Hemleben V, Ganal M, Gerstner J, Schiebel K, Torres RA (1988) Organization and length heterogeneity of plant ribosomal RNA genes. In: Kahl G (ed) Architecture of eukaryotic genes. VCH, Weinheim, pp 371–383
Jorgensen JL, Stehlik I, Brochmann C, Conti E (2003) Implications of ITS sequences and RAPD markers for the taxonomy and biogeography of the Oxytropis campestris and O. arctica (Fabaceae) complexes in Alaska. Am J Bot 90(10):1470–1480
Karron JD, Linhart YB, Chaulk CA, Robertson CA (1988) Genetic structure of populations of geographically restricted and widespread species of Astragalus (Fabaceae). Am J Bot 75(8):1114–1119
Kazempour Osaloo S, Maassoumi AA, Murakami N (2003) Molecular systematics of the genus Astragalus L. (Fabaceae): Phylogenetic analyses of nuclear ribosomal DNA internal transcribed spacers and chloroplast gene ndhF sequences. Plant Syst Evol 242(1–4):1–32
Kulshreshtha S, Creamer R, Sterling TM (2004) Phylogenetic relationships among New Mexico Astragalus mollissimus varieties and Oxytropis species by restriction fragment analysis. Weed Sci 52(6):984–988
Lee C, Grasso C, Sharlow M (2002) Multiple sequence alignment using partial order graphs. Bioinformatics 18(3):452–464
Legendre P, Legendre L (1998) Numerical ecology, 2nd English edn. Elsevier, Amsterdam
Meusel H, Jäger E, Weinert E (1965) Vergleichende Chorologie der zentraleuropäischen Flora. Band 1, Teil 1: Text; Band 1, Teil 2: Karten. Gustav Fischer, Jena
Podlech D (1986) Taxonomic and phytogeographical problems in Astragalus of the Old World and South-West Asia. Proc R Soc Edinb B 89:37–43
Potthast T (2000) Funktionssicherung und/oder Aufbruch ins Ungewisse? Anmerkungen zum Prozeßschutz. In: Jax K (ed) Funktionsbegriff und Unsicherheit in der Ökologie. Beiträge zu einer Tagung des Arbeitskreises “Theorie” in der Gesellschaft für Ökologie vom 10. bis 12. März 1999 im Heinrich-Fabri-Institut der Universität Tübingen in Blaubeuren (= Theorie in der Ökologie; 2). Peter Lang, Frankfurta a.M, pp 65–81
Ralphs MH, Provenza FD (1999) Conditioned food aversions: principles and practices, with special reference to social facilitation. Proc Nutr Soc 58(4):813–820
R Development Core Team (2005) R: A language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria
Sanderson MJ (1991) Phylogenetic relationships within North American Astragalus L. (Fabaceae). Syst Bot 16(3):414–430
Sanderson MJ, Doyle JJ (1993) Phylogenetic relationships in North American Astragalus (Fabaceae) based on chloroplast DNA restriction site variation. Syst Bot 18(3):395–408
Sanderson MJ, Wojciechowski MF (1996) Diversification rates in a temperate legume clade: are there “so many species” of Astragalus (Fabaceae)? Am J Bot 83(11):1488–1502
Sauer W, Gebert S, Shu-Kun C, Li H (1990) Beitrag zur Kenntnis chinesischer Pedicularis-Arten (Scrophulariaceae). Razprave IV. razreda SAZU [Razprave / Slovenska Akademija Znanosti in Umetnosti, Razred za Naravoslovne Vede = Dissertationes / Academia Scientiarum et Artium Slovenica, Classis 4]. Ljubljana 31(19):291–316
Schlee M (2004) Probleme der Erhaltung biologischer Vielfalt in der Kulturlandschaft - Ökologische Schäden durch verfehlte Pflegekonzepte. In: Potthast T (ed) Ökologische Schäden - begriffliche, methodologische und ethische Aspekte (= Theorie in der Ökologie; 10). Peter Lang, Frankfurt a.M, pp 95–120
Schlee M, Sauer W, Hemleben V (2003) Molekulare und pflanzensoziologische Analyse von pontisch-pannonischen Reliktarten aus wärmebegünstigten Saum-Gesellschaften Süddeutschlands und benachbarter Gebiete. Nova Acta Leopold NF 87(328):379–387
Shannon C (1948) A mathematical theory of information. Bell System Tech J 27:379–423
Sokal RR, Michener CD (1958) A statistical method for evaluating systematic relationships. Univ Kansas Sci Bull 38(Pt 2, No 22):1409–1438
Soltis PS, Gitzendanner MA (1999) Molecular systematics and the conservation of rare species [Review]. Conserv Biol 13(3):471–483
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22(21):2688
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML web-servers. Syst Biol 57(5):758–771
Stock M, Eskildsen K, Gätje C, Kellermann A (1999) Evaluation procedure for nature conservation in a national park - a proposal for the protection of ecological processes. Z Ökologie Naturschutz 8(1):81–95
Swofford DL, Olsen GJ, Waddell PJ, Hillis DL (1996) Phylogenetic inference. In: Hillis DM, Moritz C, Mable BK (eds) Molecular systematics. Sinauer Associates, Sunderland, MA, pp 407–514
Tin MMY, Cho C-H, Chan K, James AE, Ko JKS (2007) Astragalus saponins induce growth inhibition and apoptosis in human colon cancer cells and tumor xenograft. Carcinogenesis 28(6):1347–1355
Travis SE, Maschinski J, Keim P (1996) An analysis of genetic variation in Astragalus cremnophylax var. cremnophylax, a critically endangered plant, using AFLP markers. Mol Ecol 5(6):735–745
Volkov R, Komarova N, Hemleben V (2007) Ribosomal DNA in plant hybrids: inheritance, rearrangement, expression. Syst Biodiver 5(3):261–276
Watt AS (1947) Pattern and process in the plant community. J Ecol 35(1/2):1–22
Williams MC, Barneby RC (1977) The occurrence of nitro-toxins in North American Astragalus (Fabaceae). Brittonia 29(3):310–326
Wirth M, Estabrook GF, Rogers DJ (1966) A graph theory model for systematic biology, with an example for the Oncidiinae (Orchidaceae). Syst Zool 15(1):59–69
Wojciechowski MF, Sanderson MJ, Baldwin BG, Donoghue MJ (1993) Monophyly of aneuploid Astragalus (Fabaceae): Evidence from nuclear ribosomal DNA internal transcribed spacer sequences. Am J Bot 80(6):711–722
Wojciechowski MF, Sanderson MJ, Hu JM (1999) Evidence on the monophyly of Astragalus (Fabaceae) and its major subgroups based on nuclear ribosomal DNA ITS and chloroplast DNA trnL intron data. Syst Bot 24(3):409–437
Yip PY, Kwan HS (2006) Molecular identification of Astragalus membranaceus at the species and locality levels. J Ethnopharmacol 106(2):222–229
Acknowledgments
Special thanks go to Angela Dressel and Karin Stögerer for technical assistance, and to Wilhelm Sauer for discussion. Reiner Schmahl, Eberhard Koch, Natalie Schmalz and Matthias Kropf, Ludmilla Barjaewa, Ludmilla Mantschenkowa, and Erich Kalmbach, Friedrich Schlee, Evgenia Zeidler-Schewzowa, and the University of Barnaul (Altai, Siberia) are kindly acknowledged for contributing plant material, GZU, LY, M, and MA for sending specimens for revision. Hospitality of the visited herbaria BC, BOLO, C, DR, E, FB, FI, FR, G, HBG, K, LE, LI, LJU, M, MPU (incl. SIGMA), P, RO, S, STU, TUB, UPS, W, WU, and Z was enjoyed with great pleasure. The project was supported by a grant of the “Landesgraduiertenförderung Baden-Württemberg”; grants for travelling were obtained from “Reinhold-und-Maria-Teufel-Stiftung” and the German Academic Exchange Service (DAAD).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer-Verlag Berlin Heidelberg
About this paper
Cite this paper
Schlee, M., Göker, M., Grimm, G.W., Hemleben, V. (2010). Relicts Within the Genus Complex Astragalus/Oxytropis (Fabaceae), and the Comparison of Diversity by Objective Means. In: Habel, J.C., Assmann, T. (eds) Relict Species. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-92160-8_5
Download citation
DOI: https://doi.org/10.1007/978-3-540-92160-8_5
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-92159-2
Online ISBN: 978-3-540-92160-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)