
Abstract
In this chapter, we summarise the current thinking about the nature of endocannabinoids. In describing the life cycle of these agents, we highlight the synthetic and catabolic enzymes suggested to be involved. For each of these, we provide a systematic analysis of information on sequence, subcellular and cellular distribution, as well as physiological and pharmacological substrates, enhancers and inhibitors, together with brief descriptions of the impact of manipulating enzyme levels through genetic mechanisms (dealt with in more detail in the chapter “Genetic Models of the Endocannabinoid System” by Monory and Lutz, this volume). In addition, we describe experiments investigating the stimulation of endocannabinoid synthesis and release in intact cell systems.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- 2-arachidonoylglycerol
- Anandamide
- Diacylglcyerol lipase
- Endocannabinoid turnover
- Fatty acid amide hydrolase
- N-acylphosphatidylethanolamine phospholipase D
1 Cannabinoid Signalling in the CNS
The widely accepted phenomenon of synaptic plasticity highlights the fact that the efficiency of synaptic transmission can alter, dependent on the local environment. Two relevant aspects of synaptic plasticity involving cannabinoid receptors are depolarization-evoked suppression of excitation and inhibition (Gerdeman and Lovinger 2003; Diana and Marty 2004). Although dealt with in more detail in the chapter “Endocannabinoid Signaling in Neural Plasticity” by Brad Alger in this volume, in brief, these phenomena are proposed to result from transmitter-mediated post-synaptic depolarization of neurones leading to an elevation of intracellular calcium ions, resulting in the generation of a retrograde messenger which acts on the presynaptic neurone to alter neurotransmitter release. The involvement of the CB1 cannabinoid receptor has been identified through the use of the relatively selective antagonists, rimonabant (Wilson and Nicoll 2001) and AM251 (Kreitzer and Regehr 2001), as well as animal models with disruption of the gene encoding CB1 cannabinoid receptors (Varma et al. 2001; Wilson et al. 2001; Kim et al. 2002; Yoshida et al. 2002). The majority of evidence favours the involvement of ester endocannabinoids (ECBs) in mediating these retrograde effects. In contrast, the amide ECBs have been proposed to act in an anterograde fashion, subserving a more conventional neurotransmitter-like role (Egertova et al. 2008).
From these studies and previous investigations (Di Marzo et al. 1994), the hypothesis has emerged that ECBs are made “on demand” as a result of heightened neuronal activity. In this chapter, we will look at synthetic pathways which appear to be consistent with this hypothesis and more recent developments, which suggest alternative strategies for endocannabinoid biosynthesis, potentially of more relevance to the pathophysiological state. In addition, we will examine metabolic pathways which inactivate, or potentially transform, ECBs.
2 What Are Endocannabinoids?
Anandamide (N-arachidonoylethanolamine, AEA) is the archetypal ECB described first by Raphael Mechoulam, Roger Pertwee and colleagues in 1992 (Devane et al. 1992). It was identified in the classical fashion by screening solvent extracts of brain in a cannabinoid receptor radioligand binding assay, with subsequent determination of structure by GC-MS and re-synthesis. N-Palmitoylethanolamine (PEA) was identified in the same experiments, but was not considered to be an ECB since only one cannabinoid receptor (CB1) had been identified at the time and PEA had negligible affinity for this. The situation is much more complex now with a variety of putative ECB receptors of the G-protein-coupled, ion channel and nuclear receptor families proposed (see the chapter “Endocannabinoid Receptor Pharmacology” by Mackie and Yao, this volume) along with chemically related agents having affinities for one or more of these. As a note of caution, it is not always clear whether endogenous levels of some of these agents in different tissues are sufficient to activate cognate receptors allowing them to be labelled as true ECBs (Oka et al. 2003). 2-Arachidonoylglycerol (2AG) has been suggested to be the most biologically important ECB, as it occurs in greater concentrations in tissues, and shows greater efficacy at these targets, than AEA (Sugiura et al. 1997, 1999; Sugiura and Waku 2000, 2002). Although AEA and 2AG are considered the principal ECBs, the range of endogenous agents active at cannabinoid receptors is certainly not limited to these two (Hanus et al. 1993). As a pair, they are closely structurally related in that they are both based on the polyunsaturated fatty acid arachidonate. They are both hydrophobic entities, with partition coefficients (XlogP values, indices of hydrophobicity) of 5.5 and 5.4, respectively. In comparison, conventional neurotransmitters like dopamine, glutamate and GABA have XlogP values of 0.9, −3.3 and −0.7, and are considerably more hydrophilic, partitioning readily into aqueous solutions. This hydrophobicity is also considerably more marked than that of prostaglandin E2 (2.8) and more similar to leukotriene A4 (5.0). In comparison to arachidonic acid (6.5), however, the endocannabinoids AEA and 2AG are less hydrophobic. Similarly, the precursor molecules 1-stearoyl-2-arachidonoylglycerol (14.3) and N-arachidonoyl-1-stearoyl-2-arachidonoylglycerolphosphoethanolamine (XlogP likely in excess of 20) are likely only to be found dissolved in membranes. 1-Stearoyl-2-arachidonoylglycerol and 1-palmitoyl-2-oleoylglycerol, in particular, partition into enriched domains of membranes (Basanez et al. 1996; Jimenez-Monreal et al. 1998). This hydrophobicity has a marked influence on the life cycle of ECBs. For example, it has been hypothesised that AEA is able to merge into the phospholipid bilayer as an extended conformation with the ethanolamine headgroup protruding, and access the receptor binding site by lateral diffusion without leaving the plane of the membrane (Tian et al. 2005), suggesting a role as an autocrine messenger without the need for a specific release mechanism.
Despite sharing similar structural features, the turnovers of AEA and 2AG follow parallel pathways with little overlap in selectivity. A convenient division of the ECBs is into ester or amide derivatives (see the chapter “Pharmacological Tools in Endocannabinoid Neurobiology” by Mor and Lodola, this volume).
3 Ester Endocannabinoids
Given the levels of 2AG in rodent brain (in our assays of rat brain, between 10 and 30 nmol g−1), and the relative ability of isolated astrocytes and neurones to generate 2AG, it has been suggested that astrocytes are the major source of 2AG in the brain (Walter et al. 2004).
3.1 Synthesis of Ester Endocannabinoids
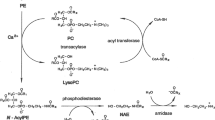
The “classical” pathway for 2AG synthesis is through the sequential activation of phospholipase C and diacylglycerol lipase (DGL) enzymes (Figs. 1 and 2). The intermediate in 2AG synthesis through this pathway, diacylglycerol (DAG), is more widely associated as a second messenger in phosphoinositide turnover, activating protein kinase C. Enzymes competing for DAG include DAG kinase (seven isoforms), which is able to generate phosphatidic acid, and DAG acyltransferases (two isoforms prominent in adipose tissue), which generate triacylglycerols.

Reaction scheme for phospholipase C action. Phosphatidylinositol-4,5-bisphosphate, accumulated primarily in plasma membranes, is cleaved to form the water-soluble second messenger inositol 1,4,5-trisphosphate, which causes calcium release from intracellular stores. The co-product, diacylglycerol, shown here as 1-stearoyl-2-arachidonoylglycerol, remains inserted in the plasma membrane and can activate protein kinase C, be recycled to form phospholipids or be hydrolysed to generate monoacylglycerols

Reaction scheme for diacylglycerol lipase action. Membrane-associated diacylglycerol, shown here as 1-stearoyl-2-arachidonoylglycerol, is hydrolysed to generate free fatty acid and 2-acylglycerol, both of which are much more water soluble than the parent and may have multiple cellular actions
DGL is a membrane-associated enzyme generated as two separate gene products, DGLα and DGLβ (Table 1). The β isoform (694 aa) is a truncated paralogue of the α isoform (1,004 aa), although both show similar topology, with a short intracellular N-terminus and four transmembrane domains in the first 10% of the molecule. The remainder of the protein encompasses the active site and putative sites for regulation. Analysis of the protein sequence suggests two consensus sequences in the cytoplasmic C-terminus of the DGLα isoform for serine/threonine phosphorylation, one of which (S-727) is a potential target for both protein kinases A and C.
3.1.1 Regulation of Phospholipase C Activity
Although PLC appears capable of hydrolysing a variety of phosphoinositides in vitro, phosphatidylinositol-4,5-bisphosphate (PIP2) appears to be the physiological substrate (Fig. 1). This substrate and one of the products (DAG) are sufficiently hydrophobic to be retained in the plasma membrane, while the second product of PLC action, inositol-1,4,5-trisphosphate, is much more hydrophilic (XlogP of –7) and so can migrate away from the membrane. Currently, 13 isoforms of PLC have been identified, which are widely distributed in the body (Suh et al. 2008). Within the cell, PLC-β isoforms (β1-β4) are membrane-associated and activated by G-proteins of the Gq family, while PLC-γ isoforms (γ1, γ2) are recruited to membranes by activation by tyrosine kinase-linked receptors of the growth factor family. PLC-δ isoforms (δ1, δ3, δ4) associate with PIP2 in the plasma membrane and are activated by elevated concentrations of intracellular calcium ions leading to the view that PLC-δ is a calcium amplifier. PLC-ε1 is activated by the low molecular weight G-proteins Ras and Rho, as well as the exchange protein activated by cyclic AMP (Epac). Much less is known about the regulation of the ζ1, η1 and η2 isoforms (Suh et al. 2008). Gene expression of all of these isoforms appears abundant in CNS tissues, with the exception of the ζ1 isoform, which appears to have a crucial role in oocyte fertilisation. Clearly, therefore, the apparent potential for regulation of this route of ECB synthesis is huge.
U73122 is an aminosteroid which has been used to inhibit PLC activity, although its activity has not been assessed against all 13 isoforms. It has been shown to inhibit 2AG synthesis in a macrophage cell line (Berdyshev et al. 2001), 3T3 mouse fibroblasts (Parrish and Nichols 2006) and rat brain synaptosomes (Oka et al. 2007a), as well as inhibiting DSI in the hippocampus (Edwards et al. 2006). However, U73122 has also been shown to interfere with 2AG-evoked regulation of excitability in rat microglial cells (Carrier et al. 2004) or rat hippocampal slices (Hashimotodani et al. 2008). Furthermore, using mice in which genes encoding three of the isoforms of phospholipase C (PLCδ1, PLCδ3 and PLCδ4) were disrupted failed to alter cannabinoid-induced DSI responses (Hashimotodani et al. 2008).
The role of phospholipase C in 2AG generation in the CNS is, therefore, inconclusive.
3.1.2 Regulation of DGL Activity
DGL (Table 1) hydrolyses DAG to generate monoacylglycerol and free fatty acid (Fig. 2) with some selectivity for the sn-1 position (Bisogno et al. 2003). The substrate specificity is not well understood, but a dually monounsaturated DAG appeared better hydrolysed than a mixed monounsaturated/saturated or monounsaturated/polyunsaturated DAG (Bisogno et al. 2003). DGL action, therefore, takes a predominantly membrane-associated substrate and generates two products, both of which are much more able to migrate away from the membrane. The recombinant enzymes are activated by calcium at supra-physiological concentrations of 100 μM or above, albeit to levels less than those evoked by glutathione (Bisogno et al. 2003). Whether these modulations are replicated with the enzyme in situ awaits further investigation.
Tetrahydrolipstatin (THL, also known as orlistat), an agent used to target pancreatic lipase in the treatment of obesity, was also found to inhibit the recombinant enzymes with IC50 values of 60–100 nM (Bisogno et al. 2003), although the activity in bovine aorta was more sensitive by an order of magnitude (Lee et al. 1995). THL is ineffective at 25 μM against MGL or FAAH activities, but does show inhibition of NAPE-PLD (IC50 of 10 μM) and triacylglycerol lipase (IC50 of 10 μM) (Szabo et al. 2006). Intriguingly, it also shows some occupancy of cannabinoid receptors (CB1 IC50 of 4 μM vs. CB2 IC50 > 25 μM) (Szabo et al. 2006). RHC80267 shows low potency inhibition of DGL in platelets with an IC50 of 1–4 μM, with some selectivity vs. other enzymes expressed (no inhibition at 100 μM against phospholipase C or phospholipase A2 activities (Sutherland and Amin 1982) or MGL (Rindlisbacher et al. 1987)). A recent investigation of ‘activity-based protein profiling’ of mouse brain using fluorophosphonate probes indicated that these two agents interfered with multiple serine hydrolases (Hoover et al. 2008), including FAAH and ABHD12 (see below). Intriguingly, the two isoforms of DGL were not identified using this methodology, suggesting either low abundance in this tissue, or reduced activity against the fluorophosphonate substrate. Despite this apparent lack of selectivity, it was noted that very few enzyme activities were inhibited by both THL and RHC80267, leading the authors to suggest the use of both agents to identify the role of DGL in biological processes.
These inhibitors have been used to identify the essential role of DGL in 2AG accumulation in the action of the Ca2+ ionophores ionomycin in neuroblastoma cells (Bisogno et al. 1999; Szabo et al. 2006) and A23187 in RTMGL1 rat microglial cells (Carrier et al. 2004) as well as ATP in astrocytes (Walter et al. 2004).
Currently, there are no published reports of genetic interference with DGL expression.
3.1.3 Alternative Pathways of DAG and 2AG Synthesis
Although the best established route of 2AG biosynthesis described above involves a two-step process utilising sequential activities of PLC (Fig. 1) and DGL (Fig. 2) activities, at least three further routes are possible using phosphatidylinositol, phosphatidylcholine or phosphatidylserine as starting points (Fig. 3).

Alternative routes of 2AG formation. Aside from the canonical route of 2AG formation through PLC/DGL action, diacylglycerol can also be formed via phosphatidic acid generated by PLD action. Additionally, PLA1 activity can generate a lysophospholipid, which may be used to generate 2AG directly through a LPLC activity, or indirectly through a LPLD/LPLC sequence. Phospholipase A1 hydrolysis of phosphatidic acid can also allow generation of 2AG, through the intermediate 2AG-3-phosphate, which can then be hydrolysed by a phosphatase/lysophospholipase C activity
A Ca2+-independent phospholipase A1 (PLA1) activity in rat brain hydrolyses phosphatidylinositol to generate LPI, lysophosphatidylinositol (Kobayashi et al. 1996), which has recently been suggested to be the endogenous ligand for a cannabinoid-related receptor, GPR55 (Oka et al. 2007b, 2009). A PLA1 activity able to hydrolyse phosphatidylinositol in cytosolic and microsomal fractions of rat brain has been described, which was less active than a PLC-like phosphodiesterase activity (Hirasawa et al. 1981). Later reports described a PLA1 activity found in the soluble fraction of brains, which exhibited some selectivity for phosphatidylinositol over other phospholipid substrates (Ueda et al. 1993a, b).
In molecular terms, three isoforms of PLA1 have been identified (Aoki et al. 2007). PS-PLA1 (also known as PLA1A, ENSG00000144837) is a soluble enzyme released by activated platelets (Sato et al. 1997), which hydrolyses phosphatidylserine to produce lysophosphatidylserine and a fatty acid. Two further, membrane-associated PLA1 activities have been identified (mPA-PLA1α, LIPH or PLA1B, ENSG00000163898 and mPA-PLA1β, LIPI or PLA1C, ENSG00000188992), which appear to hydrolyse preferentially phosphatidic acid, giving rise to lysophosphatidic acid and a fatty acid (Hiramatsu et al. 2003). Other lipase activities, such as hepatic lipase (LIPC, ENSG00000166035) and endothelial lipase (LIPG, ENSG00000101670), have also been reported to exhibit phospholipase A1 activity when presented with phosphatidylcholine as a substrate (Gillett et al. 1993; Jaye et al. 1999).
Following PLA1 degradation of phospholipid, a lysophospholipase C (LPLC) activity of rat brain, with some selectivity for LPI, is able to generate 2AG (Tsutsumi et al. 1994). This enzyme, although not precisely identified at the molecular level, appears to be an integral membrane protein (Tsutsumi et al. 1995).
Two further alternative routes of 2AG synthesis, independent of the phosphatidylinositol/PLC pathway, involve phospholipase D (PLD) activity, which favours phosphatidylcholine as a substrate, generating phosphatidic acid. In mouse N18TG2 neuroblastoma cells stimulated by the calcium ionophore ionomycin, this appears to be the major synthetic route (Bisogno et al. 1999), with sequential formation of phosphatidic acid, DAG and then 2AG. The conversion of phosphatidic acid to DAG is catalysed by phosphatidic acid phosphatases or lipid phosphate phosphatases (Brindley 2004).
The phosphatidic acid phosphatase can be inhibited by high concentrations (100 μM) of the β-adrenoceptor antagonist propranolol, which has allowed identification of the involvement of this enzyme in 2AG biosynthesis in cultured neuroblastoma (Bisogno et al. 1999) and microglial (Carrier et al. 2004) cells. It has, however, not been widely applied to investigate mechanisms of ECB biosynthesis, presumably because of “non-specific” effects, for example, directly interfering with electrophysiological recordings (Hashimotodani et al. 2008) due to its local anaesthetic-like action.
A further alternative pathway for 2AG synthesis involves the generation of 2-arachidonoylglycerol-3-phosphate (2AG-3P), a lysophosphatidic acid (Nakane et al. 2002). This may theoretically be generated from phosphatidic acid by phospholipase A1 or from lysophospholipids by lysophospholipase D (LPLD). A phosphatidic acid-hydrolysing PLA1 activity was identified in porcine platelet membranes (Inoue and Okuyama 1984), and subsequently in rat liver (Kucera et al. 1988) and bovine brain (Higgs and Glomset 1994), leading to cloning of the enzyme from bovine testis (DDHD1, ENSG00000100523) (Higgs et al. 1998).
To date, a single isoform of LPLD has been identified at the molecular level. This is autotaxin (ENPP2, ENSG00000136960), a membrane-associated enzyme initially characterised as an ecto-nucleotide pyrophosphatase/phosphodiesterase. The primary physiological role of this enzyme, however, is thought to be the regulation of levels of lysophosphatidic acid, which it produces from lysophosphatidylcholine (Goding et al. 2003). It remains to be determined whether this entity is able to regulate ECB production, however; the fact that it contains extracellular enzymatic activity allows some speculation about a particular signalling role.
Although the enzymatic pathway involved in 2AG-3P synthesis has not been unequivocally defined, levels in rat brain of 2AG-3P (530 pmol g−1) were lower than those of 2AG (37,000 pmol g−1 (Artmann et al. 2008)), suggesting either lower rates of 2AG-3P synthesis or higher rates of 2AG-3P dephosphorylation. The rapid conversion of 2AG-3P to 2AG (70% in 2 min) by rat brain homogenate (Nakane et al. 2002) suggests that this may be a feasible route for 2AG synthesis in vivo.
3.2 Hydrolysis of Ester Endocannabinoids
FAAH appears to be the primary enzyme involved in amide ECB hydrolysis (see Sect. 4.3 below). Initial characterization of cell-free preparations from cells expressing recombinant FAAH (Goparaju et al. 1998) or endogenously expressing FAAH (Di Marzo et al. 1998) suggested that 2AG might also be hydrolysed through this route. However, tissues from mice with disruption of the gene encoding FAAH show an unchanged ability to hydrolyse 2AG, suggesting FAAH plays only a minor role in turnover of ester ECBs (Lichtman et al. 2002). The identification of monoacylglycerol lipase (MGL) as a serine hydrolase enzyme capable of hydrolysing ester ECBs in vitro drew attention to an alternative route of ECL turnover (Dinh et al. 2002). Although this enzyme was shown to have a central role in lipid turnover over 30 years ago (Tornqvist and Belfrage 1976), it appears to have an additional important role in the regulation of ester ECBs. It is generally thought to be a cytosolic enzyme, with the primary sequence consistent with a lack of predicted transmembrane domains. Experimentally, however, both soluble and membrane-associated activities are observed (Dinh et al. 2002; Saario et al. 2004; Vandevoorde et al. 2005), with some pharmacological evidence to indicate minor differences between the two (Vandevoorde et al. 2005; Duncan et al. 2008).
Primary sequence analysis indicated the possibility for phosphorylation of MGL and, presumably, regulation of activity by protein kinases, in particular calcium/calmodulin kinase II and cyclic AMP- and cyclic GMP-dependent protein kinases (Dinh et al. 2002). Although this has not been investigated directly, this suggests the possibility that 2AG hydrolysis can be regulated by intracellular levels of calcium and cyclic nucleotides. Immunostaining analysis suggested predominant expression of MGL in nerve fibres and cell bodies of brain regions rich in CB1 cannabinoid receptors (Dinh et al. 2002).
The substrate specificity of MGL showed hydrolytic activity towards 2AG, but not AEA (Dinh et al. 2002), but with little specificity between acylglycerols (Ghafouri et al. 2004; Vandevoorde et al. 2005).
The available MGL inhibitors described to date have little selectivity. Fluorophosphonate analogues, such as methylarachidonylfluorophosphonate (MAFP), are potent inhibitors of MGL activity in the nanomolar range (Saario et al. 2004; Duncan et al. 2008). However, they exhibit similar activity at FAAH (De Petrocellis et al. 1997), and are thus unhelpful in defining a role of MGL in intact tissues. URB754, on the other hand, appeared at first to have selectivity for MGL over FAAH (Makara et al. 2005). Subsequently, a retraction was published indicating that a contaminant of the preparation was found to be responsible (Makara et al. 2007). URB602 was initially described as a non-competitive selective inhibitor of MGL activity (Hohmann et al. 2005). However, this compound has been reported not to show selectivity over FAAH (Vandevoorde et al. 2007; Duncan et al. 2008). OMDM169, a recently reported analogue of the DAGL inhibitor THL, shows sub-micromolar potency at MGL activity and enhances levels of 2AG, but not AEA, in ionomycin-stimulated N18TG2 neuroblastoma cells, but still is only tenfold selective over FAAH (Bisogno et al. 2009). JZL184, a carbamate analogue, on the other hand, appears to be almost 1,000-fold selective for MGL over FAAH (Long et al. 2009). Intraperitoneal administration of this agent caused an elevation of 2AG, but not AEA, in KCl-perfused microdialysate of mouse nucleus accumbens.
Although a knockout mouse with the gene encoding MGL has not yet been described, siRNA silencing of the enzyme in HeLa human cervical carcinoma cells causes an elevation of cellular 2AG levels equivalent to those obtained in the presence of MAFP (Dinh et al. 2004).
A functional, activity-based protein profiling approach to studying the enzymes in rat brain responsible for 2AG hydrolysis indicated MGL accounted for the vast majority of activity. Two further, poorly characterised enzymes, abhd6 and abhd12, were identified as contributing up to 15% of 2AG hydrolysis, but evidence for their physiological significance is currently lacking.
4 Amide Endocannabinoids
The canonical pathway of AEA formation in neural tissues is thought to be via a two-step reaction – a transacylase-phosphodiesterase pathway. The intermediate involved is a low abundance phospholipid, which acts as a precursor for N-acylethanolamides (NAEs), including AEA. It is generally considered that the formation of this precursor, rather than its metabolism, is the rate-determining step in AEA synthesis.
4.1 Synthesis of NAPEs
N-Acylphosphatidylethanolamines (NAPEs) were described in plants about 40 years ago (Dawson et al. 1969), and observed to be mobilised during seed germination and to accumulate during stress. More recently, they were identified as precursors of the ethanolamide ECBs (Di Marzo et al. 1994). They are synthesised through the action of an acyltransferase (E.C. 2.3.1.-), which catalyses the lysophospholipase A1-style hydrolysis of a fatty acid from the sn-1 position of phosphatidylcholine and transfers it to the amine of phosphatidylethanolamine (PE) (Fig. 4). In mammalian systems, this activity was initially identified in dog heart and reported to be calcium-dependent (Natarajan et al. 1982; Reddy et al. 1983). In mouse cerebral cortical neurones, NAPE formation was also enhanced substantially in the presence of the calcium ionophore A23187 (Hansen et al. 1995). Intriguingly, although the adenylyl cyclase activator forskolin and the Gs-coupled receptor agonist vasoactive intestinal polypeptide both failed to enhance NAPE accumulation in cultured neurones, they potentiated the stimulatory effects of the calcium ionophore ionomycin (Cadas et al. 1996a). The protein kinase inhibitor, H89, was able to prevent this potentiation, indicating a role for phosphorylation of a key enzyme in this process. In a comparison of cell types, NAPE synthesis appeared restricted to cultured neurons rather than astrocytes (Cadas et al. 1996a). In cultured neurons, the use of exogenous PLD activities indicated that approximately half of cellular NAPEs were available for hydrolysis, indicating a likely accumulation in the plasma membrane (Cadas et al. 1996b). A molecular correlate for this calcium-dependent transferase has yet to be identified.

Reaction scheme for N-acyltransferase action. The two phospholipids, phosphatidylethanolamine and phosphatidylcholine are co-substrates for N-acyltransferase activity, where the sn-1 fatty acid (depicted here as arachidonic acid) from the phosphatidylcholine is transferred to the amine of the phosphatidylethanolamine
In contrast, a Ca2+-independent PE N-acyltransferase has recently been described (Jin et al. 2007). This predominantly cytosolic activity appears to be identical to a protein termed rat lecithin-retinol acyltransferase-like protein 1 (RLP-1, ENSG00000168004). Given that this enzyme is highly expressed in testis and pancreas, with much lower levels expressed in brain, it seems unlikely that it contributes significantly to ECB precursor formation in neural tissues.
Very recently, NAPEs have been described to have functions beyond acting simply as precursors for ECBs. Reportedly, NAPEs are synthesised in the gut, prompted by fat ingestion, and released into the circulation where they appear to have a hormonal function. Administration of exogenous NAPE led to reduced food intake which was independent of CB1 receptors (Gillum et al. 2008).
4.2 Synthesis of Amide Endocannabinoids
As the (perhaps inappropriately considered) archetypal ECB, AEA synthesis has received the most attention. In the chemistry lab, AEA can be synthesised as a simple condensation product of arachidonic acid and ethanolamine, but in vivo, generation of the ethanolamide ECBs is thought to occur mainly as a result of hydrolysis of a minor membrane phospholipid, N-arachidonoylphosphatidylethanolamine (Di Marzo et al. 1994). This is a substrate for a phospholipase D-type activity (NAPE-PLD, ENSG00000161048, Table 1) which can produce a wide range of endogenous fatty acid ethanolamides, including AEA (Okamoto et al. 2004).
4.2.1 Pharmacological and Biochemical Manipulation of NAPE-PLD Activity
An early report of crude preparations of rat heart homogenates identified a membrane-associated NAPE-PLD activity (Fig. 5) capable of hydrolysing NAPEs to generate phosphatidic acid and diacylglycerols (Schmid et al. 1983). With the inhibition of phosphatidic acid phosphatase activity, the production of diacylglycerol was inhibited indicating that the latter was produced in a two-step process. In addition to the phosphatidic acid, NAEs were produced apparently in equimolar quantities, and in the absence of synthesis of N-acylethanolamine phosphates (indicating the lack of involvement of PLC). Similar levels of lyso-N-acylphosphatidylethanolamine (lysoNAPE) to phosphatidic acid were observed, indicating activity of phospholipase A1 and/or A2 in this preparation. In this crude preparation, supplementation with calcium or magnesium ions at concentrations up to 5 mM was without effect (Schmid et al. 1983). The same crude preparations were also able to hydrolyse LNAPE and an ether analogue of NAPE with activities only slightly less than those with NAPE itself, although it is uncertain whether these activities reside in the NAPE-PLD activity or are present in parallel enzymes (Schmid et al. 1983).

Reaction scheme for NAPE-PLD action. The action of a selective phospholipase D activity allows cleavage of N-arachidonoylphosphatidylethanolamine to generate anandamide and phosphatidic acid. The latter is highly hydrophobic (dipalmitoylphosphatidic acid has an XlogP value of 12.9) and so stays associated with the membrane, while the anandamide can more readily move into the aqueous milieu
NAPE-PLD activity from rat brain microsomes was observed to generate AEA at a slightly lower rate compared to other shorter chain, more saturated NAEs (Sugiura et al. 1996). In the presence of calcium ions, the generation of these latter shorter chain, more saturated NAEs appeared enhanced, while AEA production was unchanged, although the mechanism for this selective action has not been elucidated. A further stimulus for NAPE-PLD activity is the presence of polyamines. Spermine, spermidine and putrescine were able to replace calcium ions or detergent (see below) as enhancers of NAPE-PLD activity at concentrations within the physiological range (Liu et al. 2002), although whether polyamine levels are a physiological influence on AEA levels has not been identified.
Intriguingly, addition of the non-ionic detergent Triton X-100 (up to 0.2%) led to a doubling of NAPE-PLD activity, while the same concentrations of an alternative non-ionic detergent, Tween 20, inhibited activity to 4% of the level in control preparations (Schmid et al. 1983). Ionic detergents, such as SDS or taurodeoxycholate, also inhibited NAPE-PLD activity in these preparations. It may be that these influences are more physical than biochemical, with the possibility that Triton X-100 allows a particular conformation of enzyme:substrate interaction to occur, which the other detergents are unable to facilitate. Recently, it was noted that solubilisation of NAPE-PLD from the membrane by detergents revealed a greater sensitivity to divalent cations, including calcium (Wang et al. 2008a), leading to the suggestion that a membrane component was able to substitute for calcium. A heat-stable membrane fraction was able to enhance enzyme activity, which was later suggested to be the phospholipid PE. It was surmised that membrane components, including PE, were able to maintain activity of NAPE-PLD in a tonically active state, implying that formation of the NAPE precursor was the rate-determining state in amide ECB synthesis (Wang et al. 2008a).
Although this is dealt with by Monory and Lutz in the chapter “Genetic Models of the Endocannabinoid System” in this volume, it is pertinent to consider briefly the impact of genetic manipulation of NAPE-PLD activity. Cloning of the gene encoding this enzyme allowed identification of a primary sequence distinct from classical phospholipase D activities, with characteristics of a metallo-β-lactamase family (Okamoto et al. 2004), including the obligate incorporation of a zinc atom (Wang et al. 2006). Subsequently, it was observed that disruption of the gene encoding NAPE-PLD leads to increased levels of many forms of the precursor NAPE and decreased levels of the cognate NAEs (Leung et al. 2006). In particular, OEA, PEA and SEA levels were reduced, while their cognate precursors were enhanced. In comparison, AEA and DHEA, as well as their precursors, were unaltered. This has been taken as evidence for alternative pathways for synthesis of NAEs, particularly AEA (see below).
Using viral over-expression of NAPE-PLD activity in HeLa cells, it was observed that cellular levels of OEA and PEA were increased, without altering AEA levels (Fu et al. 2008). This suggests either that AEA synthesis can be selectively driven by synthesis of the precursor NAPE, or that AEA synthesis does not involve NAPE-PLD activity.
4.2.2 Alternative Pathways of Amide ECB Generation
In NAPE-PLD knockout mice, lower brain levels of saturated N-acylethanolamines were detected but concentrations of polyunsaturated NAEs, including AEA, were essentially unchanged (Leung et al. 2006), indicating the existence of more than one synthetic pathway (Fig. 6). Indeed, a further three routes for AEA synthesis have been proposed, although their roles in the physiological generation of AEA in neural preparations is unclear.

Alternative routes of AEA formation. Aside from the canonical pathway of NAPE hydrolysis to form AEA, through NAPE-PLD action, phospholipase C action can generate anandamide-phosphate, which can subsequently be hydrolysed by at least two phosphatases, PTPN22 and SHIP1, to generate AEA. Phospholipase A2 hydrolyses NAPE to generate a lysoNAPE, which can then be hydrolysed by a lysophospholipase D activity to generate AEA. A third route, utilising a phospholipase B-like action of abhd4 generates glycerophospho-AEA. A membrane-associated glycerophosphodiesterase, GDE1, is able to then generate AEA
Studies by Natarajan et al. (1984) provided in vitro evidence for multi-step enzymatic activities capable of producing NAEs from NAPEs. This involved the hydrolysis of one or both acyl chains from NAPEs followed by cleavage of the phosphodiester bond of the resulting lysoNAPE or glycerophospho (GP)-NAE, respectively. A secreted PLA2 has been shown to catalyse the deacylation of NAPE to yield lysoNAPE in vitro (Sun et al. 2004), although this enzyme was primarily expressed in the gut, with little expression in the brain. This suggests the existence of additional enzymes responsible for the calcium-independent NAPE hydrolase activity detected in NAPE-PLD knockout mouse brain.
The lysoNAPE evolved following a PLA2-mediated hydrolysis of NAPE can itself be hydrolysed by a LPLD activity which produces NAEs and lysophosphatidic acid (LPA). This activity was, however, found to be enriched in brain and testis (Sun et al. 2004). Given the profound biological actions of LPA, it is interesting to speculate on the dual functions of products of this enzyme.
More recently, Simon and Cravatt (2006) identified a novel enzyme αβ-hydrolase 4 (abhd4, ENSG00000100439) as a lysophospholipase/phospholipase B that selectively hydrolyzes NAPEs and lysoNAPEs to yield GP-NAE. This enzyme is indeed present in the brain and probably represents the NAPE-PLD-independent route for NAE biosynthesis observed in both NAPE-PLD-knockout and wild-type mice. Currently, very little is known about the distribution of abhd4 between neuronal and glial populations, as well as its subcellular location. The enzyme shows little selectivity between acyl groups, generating PEA at an equal rate to AEA, and is inhibited by fluorophosphonates, with 5 μM MAFP proving an effective inhibitor (Simon and Cravatt 2006). As yet, no selective inhibitors have been described. However, since NAPE-PLD is insensitive to MAFP up to 100 μM (Petersen and Hansen 1999), it is possible that this agent allows some discrimination of the two routes of AEA synthesis.
GDE1 (ENSG00000006007) is an integral membrane protein which has been identified as a glycerophosphodiesterase (Zheng et al. 2000). Initial characterization indicated an interaction with RGS16, a regulator of G-protein signalling, implying the possibility that enzyme activity might be modulated by cell-surface receptors. Indeed, in a recombinant system, the enzyme was able to hydrolyse glycerophosphoinositol preferentially (compared to glycerophosphocholine) and this activity was enhanced by isoprenaline and reduced by phenylephrine (Zheng et al. 2003). Recently, the substrate profile of this enzyme was extended to include GP-NAE, including a glycerophospho derivative of AEA (Simon and Cravatt 2008). Since the enzyme activity is stimulated by magnesium ions and inhibited by calcium ions, chelation of these allowed accumulation of several GP-NAEs in a rat brain membrane fraction, including saturated, mono-unsaturated and polyunsaturated fatty acid derivatives. Analysis of multiple recombinant glycerophosphodiesterase activities suggested identity with GDE1. Taken together, these data suggest a role for abhd4 and GDE1 in calcium-independent generation of AEA (and other NAEs) in neural tissue.
Another route that has been identified is the PLC hydrolysis of NAPE and the consequent production of acylethanolamine-O-phosphates, which may subsequently be hydrolysed by the phosphatases, PTPN22 or SHIP1 (Liu et al. 2008). Intriguingly, the PLC route was suggested to react more rapidly (<10 min) than the PLB route (≥1 h). Whether the PLC activity which is able to hydrolysis NAPE is a member of the conventional phosphoinositide-specific PLC activities described earlier is as yet unknown.
The physiogical role of PTPN22 and SHIP1 in ECB turnover in neural tissues is, as yet, almost completely unexplored.
4.3 Hydrolysis of Amide Endocannabinoids
The best characterised and investigated pathway of ECB turnover is the hydrolysis of amide ECBs. This is partly because of the relative ease of assay and synthesis of substrates, but also because inhibitors of ECB hydrolysis show some promise as therapeutic agents.
4.3.1 FAAH1 Activity
FAAH was cloned from rat tissues on the basis of identifying the enzyme responsible for hydrolysis of oleamide, an ECB-related fatty acid amide (Cravatt et al. 1996). Expression of human and rat enzymes in recombinant systems indicated intracellular location of both enzymes, although the two appeared to have distinct patterns. The rat enzyme appeared to associate with Golgi and ER membranes, predominantly in perinuclear regions, while the human enzyme appeared more associated with cytoskeletal elements (Cravatt et al. 1996). In neural tissues, FAAH-like immunoreactivity is associated primarily with neurons, in a pattern extensively (although not completely) complementary to the expression of CB1 cannabinoid receptors (Egertová et al. 1998; Tsou et al. 1998).
Hydrolysis rates of AEA were greater than those of oleamide, OEA and PEA in mouse brain and liver, but were diminished by ~99% in both tissues in mice in which the faah gene was disrupted (Lichtman et al. 2002) indicating the predominant role for FAAH in the hydrolysis of AEA, at least in “normal” neural tissues. It is, therefore, easy to understand the focus on development of FAAH inhibitors as therapeutic alternatives to receptor agonists. Intriguingly, there is a possibility that endogenous inhibitors of FAAH are able to regulate ECB turnover. Thus, N-arachidonoyl amino acids, such as N-arachidonoylglycine and N-arachidonoylalanine show species-dependent inhibition of FAAH activity (Grazia Cascio et al. 2004), although whether these are physiological regulators of ECB hydrolysis is unknown. Of potentially more direct influence is the observation that FAAH has a wide substrate profile, such that many endogenous fatty acid amides, including OEA and PEA, but not limited to NAEs, are also substrates for the enzyme and are present in quantities up to 100 times those of AEA. This observation led to the hypothesis that these compounds act as “entourage” compounds. That is, although they have no direct activity at CB1 or CB2 cannabinoid receptors, they are able to slow the hydrolysis of AEA through competition for FAAH activity sufficiently so that they can indirectly influence cannabinoid activity. The issue is complicated further by studies of the putative ECB-like receptor GPR119, which suggested that it was activated by OEA, PEA and SEA (Overton et al. 2006). Whether there is a convergence between GPR119 and conventional cannabinoid receptors awaits further investigation.
Synthetic inhibitors of FAAH abound and can be divided into two broad groups. One group is based around mimicking endogenous ligands, while the second is structurally unrelated compounds. Although α-keto ethyl esters and trifluoromethylketone analogues of AEA were effective FAAH inhibitors, the overlap in pharmacophore meant that activity at CB1 receptors and other eicosanoid-metabolising enzymes reduced their applicability (Koutek et al. 1994). Assessment of a number of carbamate analogues identified an irreversible inhibitor with nanomolar potency, URB597 (Kathuria et al. 2003). Although this compound has some “off-target” activity (Zhang et al. 2007), including activation of TRPA1 channels (Niforatos et al. 2007), the profile of its action in vitro and in vivo is consistent with a predominant action to elevate NAEs. Although systemic administration of URB597 has been demonstrated to increase levels of AEA, OEA and PEA in rat CNS tissues (Gobbi et al. 2005; Russo et al. 2007). Moise et al. (2008) reported that it elevated brain levels of OEA and PEA but not AEA in the hamster brain, indicating the possibility of species-selective effects of the inhibitor on multiple enzyme activities.
The observation that some, but not all, non-steroidal anti-inflammatory drugs, previously thought to exert their therapeutic effects through inhibition of cyclooxygenase activity, were also able to inhibit FAAH activity at relevant concentrations (Fowler et al. 1997) raised the possibility that some of the therapeutic effects of these agents might be mediated through cannabinoid receptors.
4.3.2 FAAH2 Activity
A second isoform of FAAH, FAAH2 (ENSG00000165591), has a limited species distribution in mammals, being found in man, other primates, elephants and rabbits, but not mice, rats, pigs, dogs, sheep or cows (Wei et al. 2006). Although the subcellular distribution of this isoform has not been precisely identified, it was predicted to be membrane-associated with the active site oriented towards the luminal side of the membrane. Whether FAAH2 regulates ECB levels in the extracellular medium or in subcellular organelles is, as yet, unknown. Although FAAH2 appears to hydrolyse the conventional fatty acid ethanolamine ECB-like compounds, the activity against AEA, OEA and PEA is greatly reduced, while ODA hydrolysis is similar to that evoked by FAAH1 (Wei et al. 2006). Unlike FAAH1, FAAH2 appears not to be expressed in brain or small intestine, but in contrast to FAAH1, shows low expression in heart, muscle and ovary (Wei et al. 2006). Both isoforms show high expression in kidney, liver, lung and prostate. In comparison with FAAH1, there appear to be no inhibitors of FAAH2 with greater than 100-fold selectivity (Wei et al. 2006), although both URB597 and OL135 show more than tenfold selectivity. In contrast, JNJ1661010 appears to be 100-fold selective for FAAH1 (Karbarz et al. 2009). It appears unlikely, therefore, that FAAH2 is a major regulator of AEA levels in human neural tissues.
4.3.3 NAAA Activity
N-Acylethanolamine acid amidase (NAAA, ENSG00000138744) activity is a lysosomal enzyme, with an acid pH optimum, and structural similarity to acid ceramidase. The mature enzyme is glycosylated and requires proteolysis for activation (Wang et al. 2008b). It appears to hydrolyse preferentially PEA compared to AEA in cell-free systems (Ueda et al. 1999). In intact cells, however, NAAA appeared capable of hydrolysing a variety of fatty acid ethanolamides, including AEA (Sun et al. 2005), but not 2AG (Tsuboi et al. 2005). The enzyme is expressed to relatively high levels in lung, spleen and large intestine, but in contrast to FAAH activity, is less well expressed in liver, testis and brain (Tsuboi et al. 2005). Under normal circumstances, therefore, it appears unlikely to contribute significantly to AEA turnover in neural tissues.
In counterpoint to FAAH activities, NAAA is not inhibited by MAFP concentrations up to 10−5 M (Ueda et al. 1999). The enzyme is also insensitive to URB597, but can be inhibited by a retroamide, N-cyclohexylcarbonylpentadecylamine, in the micromolar range (Tsuboi et al. 2004). As yet, genetic disruption of this enzyme has not been reported.
5 Other Routes of ECB Transformation
Other than FAAH, NAAA and MGL, the most prominent route of ECB inactivation appears to be through oxidative metabolism. Intriguingly, there is the possibility that this is not simply an inactivation, but rather a transformation to metabolites, which may themselves be active, albeit not only at canonical cannabinoid receptors.
Aside from oxidative metabolism, a further form of transformation of NAEs was identified using tissue from FAAH–/– mice (Mulder and Cravatt 2006). O-Phosphorylcholine derivatives of NAEs (PC-NAEs) were identified in the brain and/or spinal cord of FAAH–/– mice, but not wild-type mice. Intriguingly, although AEA levels were elevated in FAAH–/– mice, there were no detectable levels of PC-AEA, although PC-PEA and PC-OEA were detectable. Whether these metabolites are generated in other species or under pathological conditions or indeed whether they have biological activity in their own right is unknown. The mechanism of PC-NAE formation is also unknown; however, an enzyme activity has been identified which is capable of hydrolysing PC-NAEs to generate O-phosphorylcholine and NAE (Mulder and Cravatt 2006). This is ENPP6 (ENSG00000164303), a membrane-associated member of the nucleotide pyrophosphate/phosphatase family (Sakagami et al. 2005), which is expressed highly in human (although not mouse) brain. ENPP6 exhibits LPLC activity with some selectivity for lysophosphatidylcholine over any other lysophospholipid, including LPI (Sakagami et al. 2005). Although PC-NAEs were poor substrates for FAAH activity, they were efficiently hydrolysed by recombinant ENPP6 (Mulder and Cravatt 2006).
5.1 Oxidative Metabolism of ECBs
5.1.1 Cyclooxygenase Activity
Cyclooxygenase (COX) activities are membrane-bound enzymes responsible for the production of prostanoids (prostaglandins and thromboxanes) from arachidonic acid. Of the two isoforms, COX-1 is generally held to be constitutively expressed and responsible for the “house-keeping” roles of prostanoids, while COX-2 is generally inducible, although it is constitutively expressed in some tissues (e.g. spinal cord) and thought to be responsible for the inflammatory, pyrexic and hyperalgesic prostanoids. Given that the two major ECBs, AEA and 2AG, are arachidonate derivatives, it is, in retrospect, not too surprising that COX metabolises ECBs to produce prostanoid-like molecules. Intriguingly, AEA and 2AG appear to be poor substrates for COX-1, but are readily metabolised by COX-2 (Yu et al. 1997; Kozak et al. 2000). Perhaps more intriguing is the observation that ECBs, through the CB1 receptor, are able to induce COX-2 expression in the cerebral microvasculature (Chen et al. 2005), indicating the possibility of diversion of ECBs through alternative metabolic routes following repeated administration.
The products of COX-2 oxidative metabolism of ECBs are biologically active (Sang et al. 2006, 2007; Hu et al. 2008) and so COX-2 metabolism represents transformation of ECBs rather than inactivation. The prostanoid ethanolamides and glyceryl esters appear not to be active at conventional cannabinoid or prostanoid receptors, however, but rather through separate targets, as yet undefined at the molecular level (Fowler 2007; Woodward et al. 2008). The major route of prostaglandin inactivation, via 15-hydroxyprostaglandin dehydrogenase, appears to be less effective for oxidation of COX-2 metabolites of ECBs (Kozak et al. 2001). In parallel, FAAH or MGL hydrolysis of the COX-2 metabolites of AEA or 2AG, respectively, was reduced in comparison to the untransformed parent ECB (Vila et al. 2007). It appears, therefore, that whilst the ECBs themselves are transient species, COX-2 metabolism is able to generate derivatives which are far more long-lasting.
Numerous cellular and tissue preparations have been shown to be able to metabolise administered ECBs through the COX-2 pathway (Kim and Alger 2004; Patsos et al. 2005; Ahn et al. 2007; Ho and Randall 2007; Rockwell et al. 2008; Jhaveri et al. 2008; Bajo et al. 2009); however, definitive evidence for COX-2 metabolism of endogenous ECBs is currently lacking. Intriguingly, however, the observation that typical antibody-based assays for prostanoids fails to distinguish prostaglandins from prostamides suggests that there is more to be elucidated from the COX-2 metabolism of ECBs (Glass et al. 2005). The picture is further obscured by the observation that many, but not all, non-steroidal anti-inflammatory drugs, previously thought to target COX activities selectively, are also able to inhibit FAAH activity at pharmacologically relevant concentrations (Fowler et al. 1997, 1999, 2003). The possibility exists, therefore, that clinical efficacy of some of these agents may be due to a combination of preventing the accumulation of inflammatory prostanoids, as well as promoting the accumulation of anti-inflammatory ECBs (Jhaveri et al. 2008).
5.1.2 Lipoxygenase Activity
Mammalian lipoxygenases (LOXs) are bound to membranes inside the cell, including the nuclear membrane, and generate hydroperoxides of unsaturated fatty acids (typically of the 1Z,5Z pentadiene structure) by introducing molecular oxygen at the points of unsaturation. For arachidonate, 5-, 12- and 15-hydroperoxidation generates hydroperoxyeicosatetraenoic acids (HPETEs). 5-LOX metabolism of arachidonate, primarily in white blood cells, generates leukotrienes, while sequential oxidative metabolism of arachidonate by 5- and 15-LOXs generates lipoxins. Although a sequence of metabolic steps allows AEA to be metabolised to 12-oxygenated species in splenocytes, this appears to be due to FAAH-mediated hydrolysis of AEA generating arachidonate, thereafter metabolised by 12-LOX (Bobrov et al. 2000). Both 12- and 15-LOX, but not 5-LOX, appear capable of metabolising AEA and 2AG (Hampson et al. 1995; Ueda et al. 1995; Edgemond et al. 1998; Moody et al. 2001; Kozak et al. 2002). Indeed, incubation of AEA with plant-derived 5-LOX generates a 12-hydroperoxide derivative (Van Zadelhoff et al. 1998).
In the brain, the majority of LOX activity appears to reside in the pineal gland (Nishiyama et al. 1993; Hada et al. 1994), through which enzyme activity a product consistent with 12-hydroxyAEA was identified (Hampson et al. 1995). Rat brain homogenates are able to generate both 12-hydroxyAEA and 15-hydroxyAEA (Veldhuis et al. 2003). Human platelets are able to convert AEA to 12(S)-hydroxyAEA, while both 12(S)-hydroxyAEA and 15(S)-hydroxyAEA appeared following incubation of AEA with human polymorphonuclear lymphocytes (Edgemond et al. 1998).
As with COX activity, LOX metabolism represents transformation, rather than inactivation, of ECBs, since LOX products are active at CB1 and CB2 receptors (Edgemond et al. 1998), as well as TRPV1 (Craib et al. 2001) and PPAR (Kozak et al. 2002) receptors.
5.1.3 Cytochrome P450s and Epoxygenase Activity
Arachidonic acid is subject to an additional form of oxidative metabolism, in which 5,6-epoxyeicosatrienoic acid (EET), 8,9-EET, 11,12-EET or 14,15-EET may be formed. These epoxides are thought to be important signalling molecules in the vascular system and are metabolised by epoxide hydrolases to form diols. Human liver microsomes, containing multiple cytochrome P450 activities, were found to catalyse epoxide formation at all four unsaturations of AEA (Snider et al. 2007). One isoform of cytochrome P450, 4X1, generates 14,15-EET ethanolamide from AEA (Stark et al. 2008), while a second, 2D6, is not only able to generate all four epoxides from AEA, but also oxidises them further to generate diol analogues of AEA (Snider et al. 2008).
One isoform of cytochrome P450, 4F2, oxidises the ω-carbon of the fatty acid chain to produce N-20-hydroxyarachidonoylethanolamine.
The potential for oxidation at the terminal alcohol of AEA has recently been demonstrated, with alcohol dehydrogenase metabolism generating N-arachidonoylglycine (Aneetha et al. 2009), which may prove to be the endogenous ligand for the putative ECB-like receptor GPR18 (Kohno et al. 2006).
6 Stimulation of ECB Synthesis and Release
It is widely thought that AEA and 2AG (which are very lipophilic compounds) are produced from their precursor membrane phosphoglycerides via Ca2+-sensitive biosynthetic pathways, activated on demand, rather than being pre-synthesised and stored in secretory vesicles awaiting exocytosis, as is the case for many neurotransmitters. Hence, it is likely that ECB agents act largely as local (autocrine/paracrine) mediators rather than conventional hormones. ECBs can, however, be detected in the plasma, although their tissues of origin are not clear, and a longer range hormonal action should not be completely disregarded. AEA and 2AG are regarded as retrograde mediators in the brain where post-synaptic depolarisation leads to the elevation of intracellular Ca2+ from intracellular stores, entry through receptor/voltage-operated Ca2+ channels (or both). This is assumed to stimulate Ca2+-sensitive enzymes such as NAPE-PLD catalysing the biosynthesis of AEA and, particularly, 2AG. The released ECB mediators retrogradely traverse the synapse to activate presynaptic CB1 receptors resulting in inhibition of voltage-activated calcium channels, activation of K+ channels and inhibition of neurotransmitter release.
However, in spite of the massive research effort expended on the ECBs in recent years, there have been remarkably few direct studies of stimulated ECB synthesis and release, particularly in native cells and tissues. The chemical nature of the ECBs probably explains the difficulty in measuring extracellular concentrations, their high lipophilicity suggesting that they are unlikely to exist alone in the aqueous extracellular medium for any length of time. However, elegant electrophysiological studies by Brad Alger (see the chapter “Endocannabinoid Signaling in Neural Plasticity” by Alger, this volume) have unambiguously shown that depolarisation and agonist-mediated Ca2+ mobilisation stimulates release of ECBs, indicated by depolarisation-induced suppression of inhibition and excitation in the CA1 region of the hippocampus (Kim et al. 2002). More direct studies (Bisogno et al. 1997) showed that stimulation of mouse neuroblastoma cells with the Ca2+ ionophore, ionomycin, caused the synthesis, release and subsequent degradation of 2AG. Stella and Piomelli (2001) also demonstrated Ca2+ mobilising receptor-mediated enhancement of 2AG, OEA and PEA, but not AEA, in rat cortical neurones. These authors (Stella et al. 1997) had previously reported that high frequency stimulation of hippocampal slices had increased the synthesis of 2AG but not AEA.
Despite the weight of evidence favouring the Ca2+-mediated formation of 2AG but less so of AEA, Di Marzo’s group have proposed that AEA acts as a kind of intracellular “volume switch” for Ca2+; van der Stelt et al. (2005) reported that in dorsal root ganglion (DRG) cells, purinoceptor or muscarinic cholinergic receptor activation leads to AEA synthesis which acts on TRPV1 channels, gating extracellular Ca2+ allowing more AEA synthesis, thus providing a feed-forward mechanism. This potentially vicious cycle is suggested to be interrupted by released AEA acting on extracellular facing CB1 receptors and inhibiting TRPV1 function, as demonstrated by Millns et al. (2001). In more recent studies, Vellani et al. (2008) again suggested a central role for the TRPV1 channel in the control of DRG and, by extension, sensory nerve activity. They showed that, in addition to activation by AEA, TRPV1 channels were activated and/or sensitised by stimulating protein kinases A or C leading to enhanced AEA but not 2AG or PEA levels. This indicates that, in addition to Ca2+ mobilisation, the generation of other second messengers following receptor activation has the potential to modulate ECB synthesis and release.
In our own studies of ECB synthesis and release in rat cerebral cortical slices in vitro, we have found little evidence for Ca2+-mobilising stimuli elevating the levels of ECBs but it is notable that the FAAH inhibitor URB597 (see above) causes a robust increase in tissue levels and accompanying release, suggesting that, in this preparation, there is a high on-going turnover of ECBs independent of calcium ions (Fig. 7).

Accumulation of ECBs in rat brain cerebral cortex in vitro. Brain slices were incubated for 30 min in the absence and presence of ligands and/or calcium-free Krebs’ ringer solution before extraction and quantification of ECBs (Sarmad et al. 2008)
There have been a few attempts to monitor in vivo ECB release using microdialysis coupled with LC/MS analysis. Béquet et al. (2007) reported that, in the rat hypothalamus, local depolarisation following high K+ or glutamate perfusion enhanced AEA and 2AG release independently of Ca2+. Their experiments supported a release-modulating role for CB1 receptors in that the antagonist rimonabant enhanced, while the CB agonist WIN55212-2 reduced, AEA release, although, intriguingly, the same treatments induced opposite changes in 2AG. The mechanisms underlying the control of release clearly require further investigation. At the present time, even basic questions such as whether the release process is an active, energy-dependent mechanism or simply a passive flow down concentration gradients remain unanswered.
7 Conclusion
Despite a massive research effort over the last two decades, there is still a plethora of questions to be addressed concerning the ECB system. There has been a probably unwarranted concentration on AEA, given its archetypal status, and the challenge now is to clarify the roles of the many related fatty acids and their interactions, not only with CB1 and CB2 receptors but with the ever-growing family of G-protein-coupled, nuclear and ion channel receptors responsive to ECBs. The immense complexity of the synthetic and metabolic pathways followed by the ECBs provides a great challenge but also an opportunity for the development of selective therapeutic agents to tackle some of the diseases involving the ECB system which are described in later chapters in this volume.
Abbreviations
- 2AG:
-
2-Arachidonoylglycerol
- 2AG-3P:
-
2-Arachidonoylglycerol-3-phosphate
- AEA:
-
Anandamide, N-arachidonoylethanolamine
- COX:
-
Cyclooxygenase
- DAG:
-
Diacylglycerol
- DGL:
-
Diacylglycerol lipase
- DSI:
-
Depolarization-evoked suppression of inhibition
- ECB:
-
Endocannabinoid
- EET:
-
Epoxyeicosatrienoic acid
- Epac:
-
Exchange protein activated by cyclic AMP
- FAAH:
-
Fatty acid amide hydrolase
- LOX:
-
Lipoxygenase
- LPI:
-
Lysophosphatidylinositol
- LPLC:
-
Lysophospholipase C
- LPLD:
-
Lysophospholipase D
- lysoNAPE:
-
Lyso-N-acylphosphatidylethanolamine
- MAFP:
-
Methylarachidonylfluorophosphonate
- MGL:
-
Monoacylglycerol lipase
- NAAA:
-
N-Acylethanolamine acid amidase
- NAE:
-
N-Acylethanolamine
- NAPE:
-
N-Acylphosphatidylethanolamine
- ODA:
-
Oleamide
- OEA:
-
N-Oleoylethanolamine
- PE:
-
Phosphatidylethanolamine
- PEA:
-
N-Palmitoylethanolamine
- PIP2 :
-
Phosphatidylinositol-4,5-bisphosphate
- PLA1 :
-
Phospholipase A1
- PLA2 :
-
Phospholipase A2
- PLB:
-
Phospholipase B
- PLC:
-
Phospholipase C
- PLD:
-
Phospholipase D
- SEA:
-
N-Stearoylethanolamine
- THL:
-
Tetrahydrolipstatin
References
Ahn DK, Choi HS, Yeo SP et al. (2007) Blockade of central cyclooxygenase (COX) pathways enhances the cannabinoid-induced antinociceptive effects on inflammatory temporomandibular joint (TMJ) nociception. Pain 132:23–32
Aneetha H, O'Dell DK, Tan B et al. (2009) Alcohol dehydrogenase-catalyzed in vitro oxidation of anandamide to N-arachidonoyl glycine, a lipid mediator: Synthesis of N-acyl glycinals. Bioorg Med Chem Lett 19:237–241
Aoki J, Inoue A, Makide K et al. (2007) Structure and function of extracellular phospholipase A1 belonging to the pancreatic lipase gene family. Biochimie 89:197–204
Artmann A, Petersen G, Hellgren LI et al. (2008) Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim Biophys Acta-Mol Cell Biol L 1781:200–212
Bajo M, Roberto M, Schweitzer P (2009) Differential alteration of hippocampal excitatory synaptic transmission by cannabinoid ligands. J Neurosci Res 87:766–775
Basanez G, Nieva JL, Goni FM et al. (1996) Origin of the lag period in the phospholipase C cleavage of phospholipids in membranes. Concomitant vesicle aggregation and enzyme activation. Biochemistry 35:15183–15187
Bequet F, Uzabiaga F, Desbazeille M et al. (2007) CB1 receptor-mediated control of the release of endocannabinoids (as assessed by microdialysis coupled with LC/MS) in the rat hypothalamus. Eur J Neurosci 26:3458–3464
Berdyshev EV, Schmid PC, Krebsbach RJ et al. (2001) Activation of PAF receptors results in enhanced synthesis of 2-arachidonoylglycerol (2-AG) in immune cells. FASEB J 15:2171–2178
Bisogno T, Maurelli S, Melck D et al. (1997) Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem 272:3315–3323
Bisogno T, Melck D, De Petrocellis L et al. (1999) Phosphatidic acid as the biosynthetic precursor of the endocannabinoid 2-arachidonoylglycerol in intact mouse neuroblastoma cells stimulated with ionomycin. J Neurochem 72:2113–2119
Bisogno T, Howell F, Williams G et al. (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163:463–468
Bisogno T, Ortar G, Petrosino S et al. (2009) Development of a potent inhibitor of 2-arachidonoylglycerol hydrolysis with antinociceptive activity in vivo. Biochim Biophys Acta 1791:53–60
Bobrov MY, Shevchenko VP, Yudushkin IA et al. (2000) Hydrolysis of anandamide and eicosapentaenoic acid ethanolamide in mouse splenocytes. Biochemistry (Mosc) 65:615–619
Brindley DN (2004) Lipid phosphate phosphatases and related proteins: Signaling functions in development, cell division, and cancer. J Cell Biochem 92:900–912
Cadas H, Gaillet S, Beltramo M et al. (1996a) Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci 16:3934–3942
Cadas H, Schinelli S, Piomelli D (1996b) Membrane localization of N-acylphosphatidylethanolamine in central neurons: studies with exogenous phospholipases. J Lipid Mediat Cell Signal 14:63–70
Carrier EJ, Kearn CS, Barkmeier AJ et al. (2004) Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol Pharmacol 65:999–1007
Chen P, Hu S, Yao J et al. (2005) Induction of cyclooxygenase-2 by anandamide in cerebral microvascular endothelium. Microvasc Res 69:28–35
Craib SJ, Ellington HC, Pertwee RG et al. (2001) A possible role of lipoxygenase in the activation of vanilloid receptors by anandamide in the guinea-pig bronchus. Br J Pharmacol 134:30–37
Cravatt BF, Giang DK, Mayfield SP et al. (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87
Dawson RMC, Clarke N, Quarles RH (1969) N-acylphosphatidylethanolamine, a phospholipid that is rapidly metabolized during the early germination of pea seeds. Biochem J 114:265–267
De Petrocellis L, Melck D, Ueda N et al. (1997) Novel inhibitors of brain, neuronal, and basophilic anandamide amidohydrolase. Biochem Biophys Res Commun 231:82–88
Devane WA, Hanus L, Breuer A et al. (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949
Di Marzo V, Fontana A, Cadas H et al. (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372:686–691
Di Marzo V, Bisogno T, Sugiura T et al. (1998) The novel endogenous cannabinoid 2-arachidonoylglycerol is inactivated by neuronal- and basophil-like cells: connections with anandamide. Biochem J 331:15–19
Diana MA, Marty A (2004) Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). Br J Pharmacol 142:9–19
Dinh TP, Carpenter D, Leslie FM et al. (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA 99:10819–10824
Dinh TP, Kathuria S, Piomelli D (2004) RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol Pharmacol 66:1260–1264
Duncan M, Thomas AD, Cluny NL et al. (2008) Distribution and function of monoacylglycerol lipase in the gastrointestinal tract. Am J Physiol – Gastrointest Liver Physiol 295:G1255–G1265
Edgemond WS, Hillard CJ, Falck JR et al. (1998) Human platelets and polymorphonuclear leukocytes synthesize oxygenated derivatives of arachidonylethanolamide (anandamide): their affinities for cannabinoid receptors and pathways of inactivation. Mol Pharmacol 54:180–188
Edwards DA, Kim J, Alger BE (2006) Multiple mechanisms of endocannabinoid response initiation in hippocampus. J Neurophysiol 95:67–75
Egertova M, Simon GM, Cravatt BF et al. (2008) Localization of N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) expression in mouse brain: A new perspective on N-acylethanolamines as neural signaling molecules. J Comp Neurol 506:604–615
Egertová M, Giang DK, Cravatt BF et al. (1998) A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc R Soc Lond B Biol Sci 265:2081–2085
Fowler CJ (2007) The contribution of cyclooxygenase-2 to endocannabinoid metabolism and action. Br J Pharmacol 152:594–601
Fowler CJ, Tiger G, Stenstrom A (1997) Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure–activity relationship. J Pharmacol Exp Ther 283:729–734
Fowler CJ, Janson U, Johnson RM et al. (1999) Inhibition of anandamide hydrolysis by the enantiomers of ibuprofen, ketorolac, and flurbiprofen. Arch Biochem Biophys 362:191–196
Fowler CJ, Holt S, Tiger G (2003) Acidic nonsteroidal anti-inflammatory drugs inhibit rat brain fatty acid amide hydrolase in a pH-dependent manner. J Enzyme Inhib Med Chem 18:55–58
Fu J, Kim J, Oveisi F et al. (2008) Targeted enhancement of oleoylethanolamide production in proximal small intestine induces across-meal satiety in rats. Am J Physiol Regul Integr Comp Physiol 295:R45–R50
Gerdeman GL, Lovinger DM (2003) Emerging roles for endocannabinoids in long-term synaptic plasticity. Br J Pharmacol 140(5):781–789
Ghafouri N, Tiger G, Razdan RK et al. (2004) Inhibition of monoacylglycerol lipase and fatty acid amide hydrolase by analogues of 2-arachidonoylglycerol. Br J Pharmacol 143:774–784
Gillett MP, Vieira EM, Dimenstein R (1993) The phospholipase activities present in preheparin mouse plasma are inhibited by antiserum to hepatic lipase. Int J Biochem 25:449–453
Gillum MP, Zhang D, Zhang XM et al. (2008) N-acylphosphatidylethanolamine, a gut-derived circulating factor induced by fat ingestion, inhibits food intake. Cell 135:813–824
Glass M, Hong JW, Sato TA et al. (2005) Misidentification of prostamides as prostaglandins. J Lipid Res 46:1364–1368
Gobbi G, Bambico FR, Mangieri R et al. (2005) Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA 102:18620–18625
Goding JW, Grobben B, Slegers H (2003) Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta – Mol Basis Dis 1638:1–19
Goparaju SK, Ueda N, Yamaguchi H et al. (1998) Anandamide amidohydrolase reacting with 2-arachidonoylglycerol, another cannabinoid receptor ligand. FEBS Lett 422:69–73
Grazia Cascio M, Minassi A, Ligresti A et al. (2004) A structure–activity relationship study on N-arachidonoyl-amino acids as possible endogenous inhibitors of fatty acid amide hydrolase. Biochem Biophys Res Commun 314:192–196
Hada T, Hagiya H, Suzuki H et al. (1994) Arachidonate 12-lipoxygenase of rat pineal glands: catalytic properties and primary structure deduced from its cDNA. Biochim Biophys Acta 1211:221–228
Hampson AJ, Hill WAG, Zanphillips M et al. (1995) Anandamide hydroxylation by brain lipoxygenase: metabolite structures and potencies at the cannabinoid receptor. Biochim Biophys Acta – Lip Lip Met 1259:173–179
Hansen HS, Lauritzen L, Strand AM et al. (1995) Glutamate stimulates the formation of N-acylphosphatidylethanolamine and N-acylethanolamine in cortical neurons in culture. Biochim Biophys Acta – Lip Lip Met 1258:303–308
Hanus L, Gopher A, Almog S et al. (1993) Two new unsaturated fatty acid ethanolamides in brain that bind to the cannabinoid receptor. J Med Chem 36:3032–3034
Hashimotodani Y, Ohno-Shosaku T, Maejima T et al. (2008) Pharmacological evidence for the involvement of diacylglycerol lipase in depolarization-induced endocanabinoid release. Neuropharmacol 54:58–67
Higgs HN, Glomset JA (1994) Identification of a phosphatidic acid-preferring phospholipase A1 from bovine brain and testis. Proc Natl Acad Sci USA 91:9574–9578
Higgs HN, Han MH, Johnson GE et al. (1998) Cloning of a phosphatidic acid-preferring phospholipase A1 from bovine testis. J Biol Chem 273:5468–5477
Hiramatsu T, Sonoda H, Takanezawa Y et al. (2003) Biochemical and molecular characterization of two phosphatidic acid-selective phospholipase A1s, mPA-PLA1α and mPA-PLA1β. J Biol Chem 278:49438–49447
Hirasawa K, Irvine RF, Dawson RMC (1981) The catabolism of phosphatidylinositol by an EDTA-insensitive phospholipase A1 and calcium-dependent phosphatidylinositol phosphodiesterase in rat brain. Eur J Biochem 120:53–58
Ho W-SV, Randall MD (2007) Endothelium-dependent metabolism by endocannabinoid hydrolases and cyclooxygenases limits vasorelaxation to anandamide and 2-arachidonoylglycerol. Br J Pharmacol 150:641–651
Hohmann AG, Suplita RL, Bolton NM et al. (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435:1108–1112
Hoover HS, Blankman JL, Niessen S et al. (2008) Selectivity of inhibitors of endocannabinoid biosynthesis evaluated by activity-based protein profiling. Bioorg Med Chem Lett 18:5838–5841
Hu SSJ, Bradshaw HB, Chen JSC et al. (2008) Prostaglandin E-2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFκB activity. Br J Pharmacol 153:1538–1549
Inoue M, Okuyama H (1984) Phospholipase A1 acting on phosphatidic acid in porcine platelet membranes. J Biol Chem 259:5083–5086
Jaye M, Lynch KJ, Krawiec J et al. (1999) A novel endothelial-derived lipase that modulates HDL metabolism. Nat Genet 21:424–428
Jhaveri MD, Richardson D, Robinson I et al. (2008) Inhibition of fatty acid amide hydrolase and cycloxygenase-2 increases levels of endocannabinoids and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacol 55:85–93
Jimenez-Monreal AM, Villalain J, Aranda FJ et al. (1998) The phase behavior of aqueous dispersions of unsaturated mixtures of diacylglycerols and phospholipids. Biochim Biophys Acta 1373:209–219
Jin XH, Okamoto Y, Morishita J et al. (2007) Discovery and characterization of a Ca2+-independent phosphatidylethanolamine N-acyltransferase generating the anandamide precursor and its congeners. J Biol Chem 282:3614–3623
Karbarz MJ, Luo L, Chang L et al. (2009) Biochemical and biological properties of 4-(3-phenyl-[1, 2, 4] thiadiazol-5-yl)-piperazine-1-carboxylic acid phenylamide, a mechanism-based inhibitor of fatty acid amide hydrolase. Anesth Analg 108:316–329
Kathuria S, Gaetani S, Fegley D et al. (2003) Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 9:76–81
Kim J, Alger BE (2004) Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci 7:697–698
Kim J, Isokawa M, Ledent C et al. (2002) Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci 22:10182–10191
Kobayashi T, Kishimoto M, Okuyama H (1996) Phospholipases involved in lysophosphatidylinositol metabolism in rat brain. J Lipid Mediat Cell Signal 14:33–37
Kohno M, Hasegawa H, Inoue A et al. (2006) Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem Biophys Res Commun 347:827–832
Koutek B, Prestwich GD, Howlett AC et al. (1994) Inhibitors of arachidonoyl ethanolamide hydrolysis. J Biol Chem 269:22937–22940
Kozak KR, Rowlinson SW, Marnett LJ (2000) Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J Biol Chem 275:33744–33749
Kozak KR, Crews BC, Ray JL et al. (2001) Metabolism of prostaglandin glycerol esters and prostaglandin ethanolamides in vitro and in vivo. J Biol Chem 276:36993–36998
Kozak KR, Gupta RA, Moody JS et al. (2002) 15-lipoxygenase metabolism of 2-arachidonylglycerol: Generation of a PPARα agonist. J Biol Chem 277:23278–23286
Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727
Kucera GL, Sisson PJ, Thomas MJ et al. (1988) On the substrate specificity of rat liver phospholipase A1. J Biol Chem 263:1920–1928
Lee MW, Kraemer FB, Severson DL (1995) Characterization of a partially purified diacylglycerol lipase from bovine aorta. Biochim Biophys Acta 1254:311–318
Leung D, Saghatelian A, Simon GM et al. (2006) Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry 45:4720–4726
Lichtman AH, Hawkins EG, Griffin G et al. (2002) Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J Pharmacol Exp Ther 302:73–79
Liu Q, Tonai T, Ueda N (2002) Activation of N-acylethanolamine-releasing phospholipase D by polyamines. Chem Phys Lipids 115:77–84
Liu J, Wang L, Harvey-White J et al. (2008) Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacol 54:1–7
Long JZ, Li W, Booker L et al. (2009) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5:37–44
Makara JK, Mor M, Fegley D et al. (2005) Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci 8:1139–1141
Makara JK, Mor M, Fegley D et al. (2007) Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci 10(1):134
Millns PJ, Chapman V, Kendall DA (2001) Cannabinoid inhibition of the capsaicin-induced calcium response in rat dorsal root ganglion neurones. Br J Pharmacol 132:969–971
Moise AM, Eisenstein SA, Astarita G et al. (2008) An endocannabinoid signaling system modulates anxiety-like behavior in male Syrian hamsters. Psychopharmacol 200:333–346
Moody JS, Kozak KR, Ji C et al. (2001) Selective oxygenation of the endocannabinoid 2-arachidonylglycerol by leukocyte-type 12-lipoxygenase. Biochemistry 40:861–866
Mulder AM, Cravatt BF (2006) Endocannabinoid metabolism in the absence of fatty acid amide hydrolase (FAAH): discovery of phosphorylcholine derivatives of N-acyl ethanolamines. Biochemistry 45:11267–11277
Nakane S, Oka S, Arai S et al. (2002) 2-Arachidonoyl-sn-glycero-3-phosphate, an arachidonic acid-containing lysophosphatidic acid: occurrence and rapid enzymatic conversion to 2-arachidonoyl-sn-glycerol, a cannabinoid receptor ligand, in rat brain. Arch Biochem Biophys 402:51–58
Natarajan V, Reddy PV, Schmid PC et al. (1982) N-Acylation of ethanolamine phospholipids in canine myocardium. Biochim Biophys Acta 712:342–355
Natarajan V, Schmid PC, Reddy PV et al. (1984) Catabolism of N-acylethanolamine phospholipids by dog brain preparations. J Neurochem 42:1613–1619
Niforatos W, Zhang X-F, Lake MR et al. (2007) Activation of TRPA1 channels by the fatty acid amide hydrolase inhibitor 3'-carbamoylbiphenyl-3-yl cyclohexylcarbamate (URB597). Mol Pharmacol 71:1209–1216
Nishiyama M, Watanabe T, Ueda N et al. (1993) Arachidonate 12-lipoxygenase is localized in neurons, glial cells, and endothelial cells of the canine brain. J Histochem Cytochem 41:111–117
Oka S, Tsuchie A, Tokumura A et al. (2003) Ether-linked analogue of 2-arachidonoylglycerol (noladin ether) was not detected in the brains of various mammalian species. J Neurochem 85:1374–1381
Oka S, Arai S, Waku K et al. (2007a) Depolarization-induced rapid generation of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand, in rat brain synaptosomes. J Biochem 141:687–697
Oka S, Nakajima K, Yamashita A et al. (2007b) Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun 362:928–934
Oka S, Toshida T, Maruyama K et al. (2009) 2-Arachidonoyl-sn-glycero-3-phosphoinositol: A Possible Natural Ligand for GPR55. J Biochem 145:13–20
Okamoto Y, Morishita J, Tsuboi K et al. (2004) Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279:5298–5305
Overton HA, Babbs AJ, Doel SM et al. (2006) Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab 3:167–175
Parrish JC, Nichols DE (2006) Serotonin 5-HT2A receptor activation induces 2-arachidonoylglycerol release through a phospholipase C-dependent mechanism. J Neurochem 99:1164–1175
Patsos HA, Hicks DJ, Dobson RR et al. (2005) The endogenous cannabinoid, anandamide, induces cell death in colorectal carcinoma cells: a possible role for cyclooxygenase-2. Gut 54:1741–1750
Petersen G, Hansen HS (1999) N-acylphosphatidylethanolamine-hydrolysing phospholipase D lacks the ability to transphosphatidylate. FEBS Lett 455:41–44
Reddy PV, Natarajan V, Schmid PC et al. (1983) N-Acylation of dog heart ethanolamine phospholipids by transacylase activity. Biochim Biophys Acta – Lip Lip Met 750:472–480
Rindlisbacher B, Reist M, Zahler P (1987) Diacylglycerol breakdown in plasma membranes of bovine chromaffin cells is a two-step mechanism mediated by a diacylglycerol lipase and a monoacylglycerol lipase. Biochim Biophys Acta 905:349–357
Rockwell CE, Raman P, Kaplan BLF et al. (2008) A COX-2 metabolite of the endogenous cannabinoid, 2-arachidonyl glycerol, mediates suppression of IL-2 secretion in activated Jurkat T cells. Biochem Pharmacol 76:353–361
Russo R, LoVerme J, La Rana G et al. (2007) The fatty-acid amide hydrolase inhibitor URB597 (cyclohexyl carbamic acid 3'-carbamoyl-biphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther 322:236–242
Saario SM, Savinainen JR, Laitinen JT et al. (2004) Monoglyceride lipase-like enzymatic activity is responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Biochem Pharmacol 67:1381–1387
Sakagami H, Aoki J, Natori Y et al. (2005) Biochemical and molecular characterization of a novel choline-specific glycerophosphodiester phosphodiesterase belonging to the nucleotide pyrophosphatase/phosphodiesterase family. J Biol Chem 280:23084–23093
Sang N, Zhang J, Chen C (2006) PGE2 glycerol ester, a COX-2 oxidative metabolite of 2-arachidonoyl glycerol, modulates inhibitory synaptic transmission in mouse hippocampal neurons. J Physiol 572:735–745
Sang N, Zhang J, Chen C (2007) COX-2 oxidative metabolite of endocannabinoid 2-AG enhances excitatory glutamatergic synaptic transmission and induces neurotoxicity. J Neurochem 102:1966–1977
Sarmad S, Patel A, Barrett DA et al. (2008) Calcium-independent formation of endocannabinoids in rat brain slices. Proceedings of the British Pharmacological Society, p 111P
Sato T, Aoki J, Nagai Y et al. (1997) Serine phospholipid-specific phospholipase A that is secreted from activated platelets. A new member of the lipase family. J Biol Chem 272:2192–2198
Schmid PC, Reddy PV, Natarajan V et al. (1983) Metabolism of N-acylethanolamine phospholipids by a mammalian phosphodiesterase of the phospholipase D type. J Biol Chem 258:9302–9306
Simon GM, Cravatt BF (2006) Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for α/β hydrolase 4 in this pathway. J Biol Chem 281:26465–26472
Simon GM, Cravatt BF (2008) Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-n-acyl ethanolamine precursors in mouse brain. J Biol Chem 283:9341–9349
Snider NT, Kornilov AM, Kent UM et al. (2007) Anandamide metabolism by human liver and kidney microsomal cytochrome P450 enzymes to form hydroxyeicosatetraenoic and epoxyeicosatrienoic acid ethanolamides. J Pharmacol Exp Ther 321:590–597
Snider NT, Sikora MJ, Sridar C et al. (2008) The endocannabinoid anandamide is a substrate for the human polymorphic cytochrome P450 2D6. J Pharmacol Exp Ther 327:538–545
Stark K, Dostalek M, Guengerich FP (2008) Expression and purification of orphan cytochrome P450 4X1 and oxidation of anandamide. FEBS J 275:3706–3717
Stella N, Piomelli D (2001) Receptor-dependent formation of endogenous cannabinoids in cortical neurons. Eur J Pharmacol 425:189–196
Stella N, Schweitzer P, Piomelli D (1997) A second endogenous cannabinoid that modulates long-term potentiation. Nature 388:773–778
Sugiura T, Waku K (2000) 2-Arachidonoylglycerol and the cannabinoid receptors. pp. 89-106.
Sugiura T, Waku K (2002) Cannabinoid receptors and their endogenous ligands. J Biochem 132:7–12
Sugiura T, Kondo S, Sukagawa A et al. (1996) Transacylase-mediated and phosphodiesterase-mediated synthesis of N-arachidonoylethanolamine, an endogenous cannabinoid receptor ligand, in rat brain microsomes: comparison with synthesis from free arachidonic acid and ethanolamine. Eur J Biochem 240:53–62
Sugiura T, Kodaka T, Kondo S et al. (1997) Is the cannabinoid CB1 receptor a 2-arachidonoylglycerol receptor? Structural requirements for triggering a Ca2+ transient in NG108-15 cells. J Biochem (Tokyo) 122:890–895
Sugiura T, Kodaka T, Nakane S et al. (1999) Evidence that the cannabinoid CB1 receptor is a 2-arachidonoylglycerol receptor. Structure–activity relationship of 2-arachidonoylglycerol ether-linked analogues, and related compounds. J Biol Chem 274:2794–2801
Suh PG, Park JI, Manzoli L et al. (2008) Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep 41(6):415–434
Sun YX, Tsuboi K, Okamoto Y et al. (2004) Biosynthesis of anandamide and N-palmitoylethanolamine by sequential actions of phospholipase A2 and lysophospholipase D. Biochem J 380:749–756
Sun YX, Tsuboi K, Zhao LY et al. (2005) Involvement of N-acylethanolamine-hydrolyzing acid amidase in the degradation of anandamide and other N-acylethanolamines in macrophages. Biochim Biophys Acta 1736:211–220
Sutherland CA, Amin D (1982) Relative activities of rat and dog platelet phospholipase A2 and diglyceride lipase. Selective inhibition of diglyceride lipase by RHC 80267. J Biol Chem 257:14006–14010
Szabo B, Urbanski MJ, Bisogno T et al. (2006) Depolarization-induced retrograde synaptic inhibition in the mouse cerebellar cortex is mediated by 2-arachidonoylglycerol. J Physiol 577:263–280
Tian XY, Guo JX, Yao FM et al. (2005) The conformation, location, and dynamic properties of the endocannabinoid ligand anandamide in a membrane bilayer. J Biol Chem 280:29788–29795
Tornqvist H, Belfrage P (1976) Purification and some properties of a monoacylglycerol-hydrolyzing enzyme of rat adipose tissue. J Biol Chem 251:813–819
Tsou K, Nogueron MI, Muthian S et al. (1998) Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett 254:137–140
Tsuboi K, Hilligsmann C, Vandevoorde S et al. (2004) N-cyclohexanecarbonylpentadecylamine: a selective inhibitor of the acid amidase hydrolysing N-acylethanolamines, as a tool to distinguish acid amidase from fatty acid amide hydrolase. Biochem J 379:99–106
Tsuboi K, Sun YX, Okamoto Y et al. (2005) Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J Biol Chem 280:11082–11092
Tsutsumi T, Kobayashi T, Ueda H et al. (1994) Lysophosphoinositide-specific phospholipase C in rat brain synaptic plasma membranes. Neurochem Res 19:399–406
Tsutsumi T, Kobayashi T, Miyashita M et al. (1995) A lysophosphoinositide-specific phospholipase C distinct from other phospholipase C families in rat brain. Arch Biochem Biophys 317:331–336
Ueda H, Kobayashi T, Kishimoto M et al. (1993a) A possible pathway of phosphoinositide metabolism through EDTA-insensitive phospholipase A1 followed by lysophosphoinositide-specific phospholipase C in rat brain. J Neurochem 61:1874–1881
Ueda H, Kobayashi T, Kishimoto M et al. (1993b) The presence of Ca2+-independent phospholipase A1 highly specific for phosphatidylinositol in bovine brain. Biochem Biophys Res Commun 195:1272–1279
Ueda N, Yamamoto K, Yamamoto S et al. (1995) Lipoxygenase-catalyzed oxygenation of arachidonylethanolamide, a cannabinoid receptor agonist. Biochim Biophys Acta – Lip Lip Met 1254:127–134
Ueda N, Yamanaka K, Terasawa Y et al. (1999) An acid amidase hydrolyzing anandamide as an endogenous ligand for cannabinoid receptors. FEBS Lett 454:267–270
van der Stelt M, Trevisani M, Vellani V et al. (2005) Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J 24:3026–3037
Van Zadelhoff G, Veldink GA, Vliegenthart JFG (1998) With anandamide as substrate plant 5-lipoxygenases behave like 11-lipoxygenases. Biochem Biophys Res Commun 248:33–38
Vandevoorde S, Saha B, Mahadevan A et al. (2005) Influence of the degree of unsaturation of the acyl side chain upon the interaction of analogues of 1-arachidonoylglycerol with monoacylglycerol lipase and fatty acid amide hydrolase. Biochem Biophys Res Commun 337:104–109
Vandevoorde S, Jonsson KO, Labar G et al. (2007) Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br J Pharmacol 150:186–191
Varma N, Carlson GC, Ledent C et al. (2001) Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21:RC133
Veldhuis WB, van der Stelt M, Wadman MW et al. (2003) Neuroprotection by the endogenous cannabinoid anandamide and arvanil against in vivo excitotoxicity in the rat: Role of vanilloid receptors and lipoxygenases. J Neurosci 23:4127–4133
Vellani V, Petrosino S, De Petrocellis L et al. (2008) Functional lipidomics. Calcium-independent activation of endocannabinoid/endovanilloid lipid signalling in sensory neurons by protein kinases C and A and thrombin. Neuropharmacol 55:1274–1279
Vila A, Rosengarth A, Piomelli D et al. (2007) Hydrolysis of prostaglandin glycerol esters by the endocannabinoid-hydrolyzing enzymes, monoacylglycerol lipase and fatty acid amide hydrolase. Biochemistry 46:9578–9585
Walter L, Dinh T, Stella N (2004) ATP induces a rapid and pronounced increase in 2-arachidonoylglycerol production by astrocytes, a response limited by monoacylglycerol lipase. J Neurosci 24:8068–8074
Wang J, Okamoto Y, Morishita J et al. (2006) Functional analysis of the purified anandamide-generating phospholipase D as a member of the metallo-β-lactamase family. J Biol Chem 281:12325–12335
Wang J, Okamoto Y, Tsuboi K et al. (2008a) The stimulatory effect of phosphatidylethanolamine on N-acylphosphatidylethanolamine-hydrolyzing phospholipase D (NAPE-PLD). Neuropharmacol 54:8–15
Wang J, Zhao LY, Uyama T et al. (2008b) Amino acid residues crucial in pH regulation and proteolytic activation of N-acylethanolamine-hydrolyzing acid amidase. Biochim Biophys Acta 1781:710–717
Wei BQ, Mikkelsen TS, McKinney MK et al. (2006) A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem 281:36569–36578
Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592
Wilson RI, Kunos G, Nicoll RA (2001) Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31:453–462
Woodward DF, Carling RW, Cornell CL et al. (2008) The pharmacology and therapeutic relevance of endocannabinoid derived cyclo-oxygenase (COX)-2 products. Phamacol Ther 120(1):71–80
Yoshida T, Hashimoto K, Zimmer A et al. (2002) The cannabinoid CB1 receptor mediates retrograde signals for depolarization-induced suppression of inhibition in cerebellar Purkinje cells. J Neurosci 22:1690–1697
Yu M, Ives D, Ramesha CS (1997) Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem 272:21181–21186
Zhang D, Saraf A, Kolasa T et al. (2007) Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacol 52:1095–1105
Zheng B, Chen D, Farquhar MG (2000) MIR16, a putative membrane glycerophosphodiester phosphodiesterase, interacts with RGS16. Proc Natl Acad Sci USA 97:3999–4004
Zheng B, Berrie CP, Corda D et al. (2003) GDE1/MIR16 is a glycerophosphoinositol phosphodiesterase regulated by stimulation of G protein-coupled receptors. Proc Natl Acad Sci USA 100:1745–1750
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Alexander, S.P.H., Kendall, D.A. (2009). The Life Cycle of the Endocannabinoids: Formation and Inactivation. In: Kendall, D., Alexander, S. (eds) Behavioral Neurobiology of the Endocannabinoid System. Current Topics in Behavioral Neurosciences, vol 1. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-88955-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-540-88955-7_1
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-88954-0
Online ISBN: 978-3-540-88955-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)