
Abstract
Targeted therapies against cancer have become more and more important. In particular, the inhibition of tumor angiogenesis and vascular targeting have been the focus of new treatment strategies. Numerous new substances were developed as angiogenesis inhibitors and evaluated in clinical trials for safety, tolerance, and efficacy. With positive study results, some of these molecules have already been approved for clinical use. For example, this is true for the vascular endothelial growth factor neutralizing antibody bevacizumab (BEV) in metastatic colorectal cancer, nonsmall cell lung cancer, renal cancer, and breast cancer. The tyrosine kinase (TK) inhibitors sorafenib and sunitinib have been approved for metastatic renal cancer as well as for hepatocellular carcinoma, and sunitinib has also been approved for gastrointestinal stroma tumors. In this chapter we try to give an overview of the substances currently investigated in Phase III studies and beyond with regard to antiangiogenesis in cancer therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Vascular Endothelial Growth Factor
- Overall Survival
- Epidermal Growth Factor Receptor
- Median Overall Survival
- Advanced NSCLC
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Besides surgery and radiation, chemotherapy has been the cornerstone of cancer treatment for decades. Over the past ten years, a new generation of substances has come into focus targeting molecular pathways in the malignant cell itself or in cells supporting tumor growth, more specifically. For example, strategies aiming at tumor angiogenesis have been extensively studied, following observations that the growth and metastasis of tumors depend on the development of vascular supply. This research led to the isolation of an array of mediators that are capable of inhibiting tumor angiogenesis. Possibly the most pivotal positive regulator of angiogenesis is vascular endothelial growth factor (VEGF). Strategies to either block binding of VEGF to its receptors or to block intracellular signaling events in the downstream cascade represent the basis of many new developments in antiangiogenic cancer therapy (Ferrara et al. 2003).
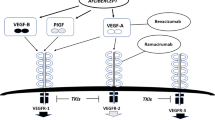
During the 1990s, the first angiogenesis inhibitors entered clinical trials for cancer therapy. The first drug in this class that was granted approval by the Food and Drug Administration (FDA) in the United States was the anti-VEGF antibody BEV in 2004 (Ferrara et al. 2004). Soon, broad-spectrum receptor tyrosine kinase (TK) inhibitors (RTKI) targeting the VEGF/VEGFR pathway followed in clinical development. The idea behind the development of these compounds was partly based on the rather modest activity of the BEV when used as monotherapy, giving a rationale for higher efficacy when aiming at more than one target. In fact, most agents currently investigated in clinical studies work by mechanisms illustrated in Fig. 9.1. By now at least the RTKIs sorafenib and sunitinib are also approved for treatment of certain cancers, and more than 40 other drugs that were preclinically screened and selected for their antiangiogenic activity are listed in clinical trials of the National Cancer Institute’s (NCI) database.

Therapeutic strategies to target the VEGF/VEGF receptor (VEGFR) system. (a) Tumors secrete VEGF in response to hypoxia, VEGF binds to VEGFR-2 on endothelial cells, thereby switching on the intracellular tyrosine kinase (TK) activity. Subsequent signal transduction steps promote proliferation, migration, invasion and tumor angiogenesis. Downregulation of VEGF secretion on the tumor side can be achieved by antagonists of the Epidermal Growth Factor Receptor (EGFR) and other “accidental” antiangiogenic drugs. (b) VEGF-TRAP (Aflibercept), fusion protein of the second IgG domain of VEGFR-1, the third IgG domain of VEGFR-2 and the Fc region of human IgG functions as decoy receptor. (c) Mab against VEGF (bevacizumab; Avastin™) prevents binding to VEGFR. (d) Small molecule Receptor tyrosine kinase inhibitors (RTKI) suppress kinase activity of VEGFR after VEGF binding; e.g., sorafenib, sunitinib, axitinib, vatalanib, vandetanib, cediranib
This chapter summarizes some of the substances currently approved or investigated as antiangiogenic cancer drugs in Phase III studies and beyond.
Anti-VEGF Antibody (Bevacizumab, Avastin™)
The humanized monoclonal anti-VEGF antibody BEV is the first VEGF targeting drug which has been officially approved for cancer therapy (Ferrara et al. 2004). In particular, BEV is approved, in combination with intravenous 5-fluorouracil-based (5-FU) chemotherapy, for first- or second-line treatment of patients with metastatic carcinoma of the colon or rectum and in combination with carboplatin and paclitaxel for the first-line treatment of patients with unresectable, locally advanced, recurrent, or metastatic nonsquamous nonsmall cell lung cancer (NSCLC). Furthermore, it has been approved in combination with paclitaxel for first-line treatment of patients with metastatic breast cancer and in combination with interferon α-2a for first-line treatment of patients with advanced and/or metastatic renal cell cancer (RCC). The original FDA approval for BEV in 2004 was based on data from a large, placebo-controlled, randomized study demonstrating prolongation in the median survival of patients with metastatic colorectal cancer (CRC) treated with BEV in addition to a combination chemotherapy regimen containing 5-FU, Leucovorin, and Irinotecan (IFL) by approximately five months, compared to patients treated with the IFL chemotherapy regimen alone (20.3 vs. 15.6 months). At that time, this study represented one of the largest improvements in survival ever reported in a randomized, Phase III study of patients with metastatic CRC (Hurwitz et al. 2004). The following approval for second-line therapy was based on results of a randomized, controlled, multicenter Phase III trial (E3200) of 829 patients with advanced or metastatic CRC who had received previous treatment with irinotecan and 5-FU as initial therapy for metastatic disease or as adjuvant therapy (Giantonio et al. 2007). In detail, it could be shown that patients who received BEV plus the 5-FU-based chemotherapy regimen known as FOLFOX4 (oxaliplatin/5-FU/leucovorin) had a 33% improvement in overall survival (OS), compared to patients who received FOLFOX4 alone (hazard ratio (HR) 0.75). Median OS for patients receiving BEV plus FOLFOX4 was 13.0 months, compared to 10.8 months for those receiving FOLFOX4 alone. The third approval and first in another cancer type was based on results from E4599, a randomized, controlled, multicenter trial that enrolled 878 patients with unresectable, locally advanced, recurrent, or metastatic nonsquamous NSCLC (Sandler et al. 2006). Results showed that patients receiving BEV plus paclitaxel and carboplatin chemotherapies had a 25% improvement in OS, the trial’s primary end point, compared to patients who received chemotherapy alone (hazard ratio (HR) 0.80). One-year survival was 51% in the BEV arm vs. 44% in the chemotherapy-alone arm. Median OS of patients treated with BEV plus chemotherapy was 12.5 months, compared to 10.2 months for patients treated with chemotherapy alone. Notably, a pilot study including NSCLC patients with squamous histology prior to E4599 showed an increased death rate in the BEV arm due to fatal pulmonal hemorrhages, leading to exclusion of this subtype for further studies (Johnson et al. 2004). Subsequently, BEV has been approved in first-line therapy of metastatic breast cancer, which was based on an improvement in progression-free survival (PFS) only. In this study 722 patients who had not received chemotherapy for locally recurrent or metastatic breast cancer were randomized to receive either paclitaxel alone or in combination with BEV (Miller et al. 2007). The addition of BEV to paclitaxel resulted in an improvement in PFS (11.3 vs. 5.8 months; p < 0.0001) with no significant improvement in OS (26.5 vs. 24.8 months; p = 0.14). Partial response (PR) rates in patients with measurable disease were higher with BEV plus paclitaxel, but no complete responses (CR) were observed. This has led to a limited approval of BEV not including patients with breast cancer that has progressed following anthracycline and taxane chemotherapy administered for metastatic disease. The most recent approval for BEV has been granted for the combination with interferon-α (IFN-α) as first-line treatment of patients with advanced and/or metastatic clear RCC. This was based on a multicentre, randomized, Phase III study, randomizing 649 patients with previously untreated metastatic RCC to receive IFN-α plus BEV or placebo (Escudier et al. 2007a). Median duration of PFS was significantly longer in the BEV plus IFN-α group than it was in the control group (10.2 vs. 5.4 months; p < 0.0001). With regard to OS, only a trend in favor of the BEV group could be observed.
Currently, BEV is listed in 47 clinical phase III trials, mostly evaluating new combinations for approved indications such as breast cancer, CRC, and NSCLC, but also for other entities such as lymphoma or osteosarcoma (see Table 9.1).
Aflibercept (VEGF – Trap)
Another approach to target the VEGF/VEGF receptor system is to deliver a soluble decoy for VEGF. To this end, a recombinant fusion protein was constructed from the second Ig domain of VEGFR-1 and the third Ig domain of VEGFR-2, fused to the Fc region of human IgG (Aflibercept; VEGF – trap, Regeneron in cooperation with Sanofi-Aventis). The resulting decoy receptor possesses an affinity for all VEGF isoforms that is significantly higher than that of the monoclonal antibody. In addition, aflibercept binds Placental Growth Factor (PLGF), which has also been implicated in tumor angiogenesis. Numerous preclinical models demonstrated significant inhibition of angiogenesis and tumor growth (Holash et al. 2002; Kim et al. 2002a). The first clinical Phase II study published is an open-label, multicenter, two-stage trial in patients with metastatic CRC with at least one prior systemic therapy and good performance status (Tang et al. 2008). Prior treatment with a VEGF or VEGFR inhibitor other than BEV was not allowed. Aflibercept (4 mg/kg) was administered every 2 weeks intravenously. In total, 51 patients were included (BEV naive = 24 patients; prior BEV = 27; median age = 59). During 287 therapy cycles, most common adverse events (AE) of any grade were fatigue (n = 40), hypertension (n = 28), proteinuria (n = 25), headache (n = 22), voice alteration (n = 16), anorexia (n = 12), and joint pain (n = 9). Serious AE (Grade 3/4) consisted of hypertension (n = 4), proteinuria (n = 4), fatigue (n = 3), headache (n = 3). One patient died during treatment due to progressive disease (PD). In the BEV naïve group (n = 24), disease control rate defined as either PR or stable disease (SD) for at least 16 weeks was 29% (95% Confidence Interval (CI) 13–51%), and median PFS was 2.0 months (95% CI 1.7 – not reached). In the group with prior BEV treatment (n = 27), disease control rate was 30% (95% CI 14–50%) and median PFS was 3.4 months (95% CI 1.9 – not reached). The authors concluded that aflibercept is well tolerated in pretreated patients with CRC and shows single agent activity (Tang et al. 2008). Based on the study results, aflibercept is now tested in a randomized Phase III study in combination with irinotecan based second-line chemotherapy for patients with CRC. Other currently listed Phase III studies for aflibercept evaluate its efficacy in prostate cancer, NSCLC, advanced ovarian and pancreatic cancer (see Table 9.1).
Sorafenib (Nexavar™)
Sorafenib (Nexavar™; Bayer Pharmaceuticals) represents the class of small-molecule compounds with activity against a broad spectrum of receptor tyrosine kinases. Originally developed as RAF-1 inhibitor in a high-throughput screening program, sorafenib later was found to be active against VEGFR-1/-2/-3; platelet derived growth factor (PDGF) -β receptor; Fms-like tyrosine kinase-3 (FLT-3); c-Kit protein and RET receptor tyrosine kinases (Adnane et al. 2006). Thereby, sorafenib inhibits tumor growth by targeting the endothelial cell as well as the tumor cell and was shown to inhibit proliferation, promote apoptosis, and disrupt angiogenesis. In preclinical mechanism of action studies, sorafenib demonstrated a potent antiangiogenic effect in nearly all models tested, resulting in significant reduction of micro-vessel density (Strumberg 2005; Wilhelm et al. 2006). It also showed promising activity in tumor xenograft models in nude mice in combination with chemotherapy. The first clinical entity in which sorafenib was tested again was clear-cell RCC (Kane et al. 2006). This cancer is special due to its loss of the von Hippel-Lindau tumor suppressor gene, which results in overexpression of hypoxia inducible factors (HIF) – 1 and – 2, subsequently upregulating pro-angiogenic factors (e.g., VEGF). The tumors are usually hypervascularized and increased RAF-1 activity also is found. All these pathways are within the target range for sorafenib providing a strong rationale for testing it in RCC.
In a number of Phase I/II trials, the optimal dose was determined to be 400 mg twice daily (b.i.d.) with dose-limiting toxicities (DLT) such as fatigue, skin rash, hand-foot syndrome, and diarrhea (Richly et al. 2006; Siu et al. 2006; Strumberg et al. 2006). Also, the postulated antiangiogenic effect was confirmed by diminished blood flow in dynamic contrast-enhanced magnetic resonance imaging (Flaherty et al. 2008). The most promising result was the significant increase in PFS in RCC patients treated with sorafenib (24 weeks) vs. placebo (6 weeks; p = 0.0087) (Ratain et al. 2006).
This prompted the Phase III treatment approach in RCC global evaluation trial (TARGET), which represents the biggest randomized treatment trial for this disease so far (Escudier et al. 2007b). From November 2003 to March 2005, the participating 117 centers in 19 countries randomized 903 patients with advanced or metastatic RCC who failed standard therapy to receive either continuous treatment with oral sorafenib (at a dose of 400 mg b.i.d.) or placebo; resulting in 451 patients who received sorafenib and 452 who received placebo. A single planned analysis of PFS in January 2005 already demonstrated a statistically significant benefit of sorafenib over placebo. Regarding this result, patients who were on placebo were allowed to crossover to the sorafenib arm later that year. In detail, the PFS was 5.5 months in the sorafenib group and 2.8 months in the placebo group (HR for disease progression in the sorafenib group = 0.44; 95% CI 0.35–0.55; p < 0.01). The first interim analysis of the primary end point OS in May 2005 indicated that sorafenib reduced the risk of death, as compared with placebo (HR = 0.72; 95% CI 0.54–0.94; p = 0.02). However, this survival benefit did not meet the previously specified criteria for statistical significance. Only in a preplanned placebo-censored analysis, excluding patients who crossed over to sorafenib, results showed a significant survival advantage (17.8 months median OS vs. 14.3 months; HR = 0.78; 95% CI 0.62–0.97; p = 0.0287) (Bukowski et al. 2007). Partial responses were reported as the best response in 10% of patients receiving sorafenib and in 2% of those receiving placebo (p < 0.001). Again, diarrhea, rash, fatigue, and hand-foot skin reactions were the most common AE associated with sorafenib. Hypertension and cardiac ischemia were more common in patients receiving sorafenib than in those receiving placebo. At the time of publication, the authors concluded that sorafenib prolongs PFS as compared with placebo in patients with advanced clear-cell RCC in whom previous therapy has failed. However, sorafenib therapy was associated with increased toxicities (Escudier et al. 2007b). In a concomitant quality of life (QOL) analysis it was shown that sorafenib had a positive effect on cancer-related symptoms and did not negatively impact QOL (Cella et al. 2006).
This result led to the approval of sorafenib for the treatment of patients with advanced RCC who have failed prior IFN-α or interleukin-2-based therapy, or are considered unsuitable for such therapy.
Apart from RCC, hepatocellular carcinoma (HCC) also represents a rather chemoresistant, but highly vascularized tumor with vast expression of VEGF. Furthermore, Raf-1 is constitutely overexpressed in HCC offering a rationale for treatment with sorafenib. A number of Phase I and II trials showed promising results for sorafenib either as monotherapy or in combination with doxorubicin (Abou-Alfa et al. 2006; Furuse et al. 2008; Gollob et al. 2007; Richly et al. 2008). This prompted a randomized Phase III trial for patients with advanced or metastatic HCC in which sorafenib was compared with placebo (SHARP-trial). Patients with advanced measurable HCC in good performance status and Child-Pugh status A were treated with either sorafenib 400 mg b.i.d. or placebo. Overall, 602 patients were randomized leaving 299 in the sorafenib arm and 303 in the placebo arm. Baseline characteristics were similar for both arms. Based on 321 deaths (Sorafenib n = 143; Placebo n = 178), the HR for OS was 0.69 (95% CI 0.55–0.87; p = 0.0006), representing a 44% improvement. This met early stopping criteria and median OS was 10.7 months for sorafenib vs. 7.9 for placebo. There was no accumulation of serious AE in the sorafenib arm. The most frequent grade 3/4 AE for sorafenib vs. placebo were diarrhea (11 vs. 2%), hand-foot skin reaction (8 vs. 1%), fatigue (10 vs. 15%), and bleeding (6 vs. 9%). At the time of presentation at the annual meeting of the American Society of Clinical Oncology (ASCO) in 2007, the authors concluded that sorafenib is the first drug to demonstrate a statistically significant improvement in OS for patients with advanced HCC (Llovet et al. 2007). Again, these findings led to the approval of sorafenib for this indication.
Currently, sorafenib is listed in ten active Phase III studies for treatment of RCC, HCC, NSCLC, unresectable melanoma and adenocarcinoma of the pancreas (see Table 9.1).
Sunitinib Malate (SU11248; Sutent™)
Sunitinib (SU11248; Sutent™; Pfizer Oncology) is a broad-spectrum orally available TK inhibitor of VEGFR, PDGFR, c-kit, and Flt-3 kinase activity. Just like sorafenib, it emerged from a drug-discovery program that was initiated to identify compounds with activity against selected receptor tyrosine kinases involved in tumor angiogenesis (Atkins et al. 2006; Roskoski 2007). Again, the highly vascularized clear cell RCC was one of the first diseases in which sunitinib was tested. After the promising Phase I and II results, a large multicentered, international randomized Phase III trial was started (Motzer et al. 2007). Single-agent sunitinib was compared with IFN-α in patients with treatment-naive advanced or metastatic RCC. Altogether, 750 patients were randomized (1:1) to receive either 50 mg sunitinib once daily in 6-week cycles (4 weeks on treatment, 2 weeks off) or to receive IFN-α administered subcutaneously at nine million units 3 times a week until disease progression or withdrawal from the trial.
Median duration of treatment was 11 months for sunitinib vs. 4 months for IFN-α. The 2008-updated response rate was 47% (95% CI 42–52%) for sunitinib vs. 12% (95% CI 9–16%) for IFN-α (p < 0.000001), including 11 CR for sunitinib and four for IFN-α (Figlin et al. 2008). Median PFS was significantly higher in the sunitinib arm (11 months vs. 5 months; p < 0.000001). Also the median OS was significantly longer for sunitinib (26.4 months; 95% CI 23.0–32.9) vs. IFN-α (21.8 months; 95% CI 17.9–26.9), which results in a HR of 0.821 (95% CI 0.673–1.001; p = 0.051). The most common grade 3/4 treatment-related AEs for the sunitinib group were hypertension (12%), fatigue (11%), diarrhea and hand-foot syndrome (both 8%), and for IFN-α fatigue (13%) and anorexia (2%).
These results led to the approval of sunitinib in patients with advanced or metastatic RCC as first-line therapy and based on two other Phase II studies also as second-line therapy after cytokine or interferon failure (Motzer et al. 2006a, b).
Another malignant disease for which sunitinib has been approved is the gastrointestinal stroma tumor (GIST). Based on its strong activity against the GIST driving c-kit receptor, there was an imminent rationale for the therapy with sunitinib. Also, in cases of advanced, unresectable, or metastatic disease, the other TK inhibitor imatinib was already established as standard first-line therapy for this indication. However, after failure of imatinib, there was no accepted standard therapy available in unresectable GIST tumors (Faivre et al. 2007; Heinrich et al. 2008; Liegl et al. 2008).
Sunitinib was first tested in a population of GIST patients in two multicenter randomized studies. The first one represented a two-sided, randomized, double-blind, placebo-controlled trial of sunitinib in patients with GIST who had disease progression during prior imatinib treatment or who were intolerant of imatinib. Altogether, 312 patients were randomized (2:1) to receive either 50 mg sunitinib (n = 207) or placebo orally (n = 105), once daily, on the same 4 weeks on and 2 weeks off schedule until disease progression or withdrawal from the study for another reason. Patients randomized to placebo were then offered to crossover to open-label sunitinib (Demetri et al. 2006).
Demographics were comparable between the sunitinib and placebo groups with regard to age (69 vs. 72% younger than 65 years for sunitinib vs. placebo, respectively), gender (male: 64 vs. 61%), performance status (ECOG 0: 44 vs. 46%, ECOG 1: 55 vs. 52% and ECOG 2: 1 vs. 2%). Prior treatment included surgery (94 vs. 93%) and radiotherapy (8 vs. 15%). Reasons for imatinib failure were also comparably balanced between both arms; being intolerance (4 vs. 4%), progression within 6 months of starting treatment (17 vs. 16%), or progression beyond 6 months (78 vs. 80%).
The trial was unblinded early after the preplanned interim analysis including the first 149 cases of disease progression or death revealed significantly longer time to tumor progression (TTP) in patients initially treated with sunitinib than in those who started with placebo. In detail, the primary study endpoint, median TTP, was more than 4 times as long with sunitinib (27.3 weeks; 95% CI 16.0–32.1) as with placebo treatment (6.4 weeks; 95% CI 4.4–10.0; HR 0.33, 95% CI 0.23–0.47; p < 0·0001). All other efficacy analyzes were uniformly statistically and clinically significant and confirmed the findings of the primary endpoint data. The median PFS was similar to TPP (24.1 weeks; 95% CI 11.1–28.3 for sunitinib; 6.0 weeks for placebo, respectively; HR 0.33; 95% CI 0.24–0.47; p < 0·0001). Moreover, 16% (33) of patients in the sunitinib group were progression-free for at least 26 weeks, compared with 1% (one) in the placebo group. As more than half the patients in the initial sunitinib group were still alive at the time of the interim analysis, OS data were not mature at the time of publication and a median OS was not calculated. However, there was a gain in OS in patients treated initially with sunitinib compared to those who started on placebo despite the availability of the crossover option (HR 0.49, 95% CI 0.29–0.83; p = 0.007). Later on, an update presented at the ASCO 2008 conventional analysis showed that OS converged in the two treatment groups (Sunitinib median 74.7 weeks; 95% CI 61.4–85.7; placebo 64.9 weeks, 45.7–98.4; HR 0.82, p = 0.128) as expected for the crossover design (Demetri et al. 2008). However, rank-preserving structural failure time analysis yielded an estimated median OS for placebo of 36.0 weeks (95% CI 25.9–51.0), revealing a significant sunitinib treatment effect (HR 0.46, p < 0.0001) comparable to that of the blinded phase. The most common treatment-related AEs throughout the entire study were fatigue, diarrhea, nausea, and skin discoloration, mainly grade 1/2; incidences increased slightly with extended duration of sunitinib treatment. In terms of best overall objective tumor response, 7% (14) of patients in the sunitinib group showed PR as the best response, 58% (120) had SD, and 19% (39) had PD, compared with rates of 0 48 (50), and 37% (39), respectively, for placebo. Six of fifty-nine patients who crossed over to sunitinib from the placebo group also had confirmed PR (10.2%, 95% CI 3.8–20.8). Four patients (7% overall) who crossed over to sunitinib from placebo had SD for at least 26 weeks after crossover. Based on these results, sunitinib is now approved for the therapy of GIST patients with advanced or unresectable disease after imatinib failure or intolerance (Goodman et al. 2007).
Currently, sunitinib is listed for 20 Phase III studies involving the approved indications RCC and GIST, as well as NSCLC, breast cancer, CRC, and pancreatic islet cell tumors (see Table 9.1).
Axitinib (AG-013736)
Axitinib (AG-013736; Pfizer Oncology) represents a potent small molecule TK inhibitor of all known VEGFRs at subnanomolar concentrations and PDGFR-ß and c-Kit in low nanomolar concentrations. Structurally, it is a substituted indazole derivative discovered by using a structure-based drug design. In vitro, axitinib selectively blocks VEGF stimulated receptor autophosphorylation leading to inhibition of endothelial cell proliferation and survival. In numerous preclinical models, axitinib inhibited tumor angiogenesis and the growth of human colorectal and murine lung tumors. In a transgenic mouse model of spontaneous islet cell tumors, axitinib eliminated suppressed vascular sprouting within 24 h. At 7 days, vascular density decreased more than 70%, and significant tumor shrinkage was seen at 21 days (Inai et al. 2004).
The first-in-human Phase I trial was conducted to test axitinib in patients with advanced solid malignancies in order to determine DLTs and the maximum-tolerated dose (MTD). Altogether, 36 patients received axtinib at doses ranging from 5 to 30 mg orally b.i.d. (Rugo et al. 2005). Similar to other TK inhibitors, observed DLTs included hypertension, hemoptysis, and stomatitis primarily seen at higher dose levels. All toxicities were manageable with medication or drug holidays. The MTD and recommended Phase II dose of AG-013736 was specified for 5 mg b.i.d. The trial demonstrated three confirmed partial responses and other evidence of clinical activity (Rugo et al. 2005). Subsequently, axitinib was tested in advanced or metastatic RCC in a multicenter, open-label, Phase II study (Dutcher et al. 2008). Altogether, 58 patients with sorafenib or sunitinib-refractory (progression or unacceptable toxicity) metastatic RCC, and measurable disease, regardless of additional prior therapies, were enrolled. All patients received a starting dose of axitinib 5 mg orally b.i.d., which was titrated to 7 mg b.i.d. and then to 10 mg b.i.d. according to tolerance. Stratification was performed by prior therapy into three groups: 14 patients were refractory to sunitinib and sorafenib (Group 1), 29 patients were refractory to cytokines and sorafenib (Group 2), and 15 patients were refractory to sorafenib alone (Group 3). With a median follow-up of 10.3 months, the overall response rate (ORR) was 7, 28, and 27% and the median PFS was 7.1, 9.0, and 7.7 months for groups 1, 2, and 3, respectively. Overall, grade 3/4 treatment-related AEs included fatigue (13%), hypertension (11%), hand-foot syndrome (11%), diarrhea (5%), and dyspnea (5%). The authors concluded, that axitinib appears to have antitumor activity in metastatic RCC refractory to sunitinib and sorafenib (Dutcher et al. 2008). To this end, a randomized Phase III trial is currently recruiting patients. The Axis-trial is a head-to-head comparison of axitinib (5 mg b.i.d.) and sorafenib (400 mg b.i.d.) for second-line therapy of metastatic RCC and is designed to enroll 540 patients until 2010 (NCT00678392).
The other malignant disease in which axitinib is currently evaluated in a Phase III trial is pancreatic cancer (see Table 9.1). So far, standard of care for patients with advanced pancreatic cancer is gemcitabine-based chemotherapy. Therefore, axitinib was tested in a Phase I/II trial in combination with gemcitabine in patients with pancreatic cancer (Spano et al. 2008). In detail, eight patients were treated on the Phase I part and 103 for the Phase II part of the trial. Prior gemcitabine or VEGF/VEGFR inhibitors were not allowed. The randomization took place between standard dose gemcitabine (1,000 mg/m2 over 30 min on days 1, 8, 15) plus axitinib (5 mg b.i.d.) or placebo. The median number of days on axitinib was 158 days (range: 57–330 days). The most commonly reported AEs were anemia (48%), alkaline phosphatase elevations (48%), leukopenia (45%), and thrombocytopenia (27%). The most common nonhematologic AEs were nausea (24%), vomiting (20%), fatigue (19%), diarrhea (18%), anorexia (18%), constipation (13%), dyspnea (12%), and fever (12%). In the axitinib group 66% of patients (n = 45) reached at least disease stabilization, including 7% PRs compared to 59% SD (n = 20) and no PR for gemcitabine plus placebo. This yielded a median OS of 210 days in the axitinib group in comparison to 169 days for gemcitabine plus placebo (HR 0.74; 95% CI 0.427–1.284) for the whole study group. For the subpopulation in very good performance status (ECOG PS 0/1), the calculated death risk reduction for axitinib was even bigger with 33% (HR 0.67; 95% CI 0.372–1.196). These results prompted the currently recruiting randomized, double-blind Phase III study of gemcitabine plus axitinib vs. gemcitabine plus placebo for the first-line treatment of patients in good performance status with locally advanced, unresectable, or metastatic pancreatic cancer. The trial is estimated to enroll more than 500 patients until planned completion date in September 2009 (NCT00471146).
Cediranib (AZD2171; Recentin™)
Another broad-spectrum kinase inhibitor is cediranib (AZD2171; Recentin™; AstraZeneca). Its predominant effect is directed against VEGFR-2 with additional potent inhibition of VEGFR-1 and -3, c-Kit, Flt-3 and to a lesser extent against epidermal growth factor receptor (EGFR). This broad activity range was determined in a wide range of cell lines (Wedge et al. 2005). Cediranib significantly inhibits VEGF driven vascular sprouting and demonstrated potent antitumor effects in a number of preclinical studies. It is orally bioavailable and was preclinically tested at a dose range of 1.5–6 mg/kg bodyweight per day. Based on these observations, a range of clinical Phase I studies were performed, the first being a dose-finding trial with 83 patients suffering of different solid tumors. In this study, cediranib was generally well tolerated at doses not higher than 45 mg/day and gave encouraging antitumor activity. Pharmacokinetic data revealed a half-life of approximately 20 h and the optimal dosing was determined to be 20–30 mg once daily (Drevs et al. 2007).
The next step was the initiation of Phase I/II study in conjunction with standard doses of carboplatin (AUC 6) and paclitaxel (200 mg/m²) in order to assess the tolerability, safety, and antitumor activity of this combination in patients with stage IIIB / IV NSCLC of any histology in first-line therapy (Laurie et al. 2006; Laurie et al. 2008). Cediranib was started on Day 2 of the first cycle at a dose of 30 mg p.o. daily. Of the 20 enrolled patients, nine received cediranib at 30 mg/day, 11 at 45 mg/day. Again, most common grade 3/4 toxicity was hypertension, other common toxicities were: fatigue, anorexia, mucositis, and diarrhea. Hematologic toxicity was not greater than that expected with chemotherapy alone. At time of presentation, 15 patients were evaluable for response, with 6 PR, 8 SD, and 1 PD. The authors concluded that, full single-agent dose of cediranib may be administered with standard chemotherapy. However, the subsequent started randomized Phase II/III trial CTG BR.24 comparing carboplatin/paclitaxel plus cediranib (30 mg/day) vs. this chemotherapy combination plus placebo did not reach Phase III. The National Cancer Institute of Canada Clinical Trials Group (NCIC-CTG) decided in 2008 that the BR.24 study should not continue into Phase III following the planned end of Phase II efficacy and tolerability analysis by the study’s data safety monitoring committee, mostly because of an imbalance in toxicity. Although cediranib gave evidence of clinical activity, the study did not meet the predefined criteria for automatic continuation into Phase III (Laurie et al. 2008). Instead, NCIC-CTG agreed to start a new randomized Phase III study evaluating this combination in advanced or metastatic NSCLC with a lower dose of cediranib (20 mg/day); this BR.29 trial is expected to enroll the first patients in 2009.
Another step in the clinical development of cediranib was the initiation of a two-stage, multicenter Phase II clinical trial in patients with recurrent ovarian, peritoneal, or fallopian tube cancer (Hirte et al. 2008). Of the 60 patients who were enrolled; 49 had ovarian, 8 peritoneal, and 3 fallopian tube cancer; follow up was available for 154 cycles of treatment given to 46 patients. As in other Phase I studies with cediranib, the most frequent AEs were fatigue (85%), diarrhea (80%), hypertension (72%), anorexia (57%). Hypertension (33%) and fatigue (20%) were the most frequent grade 3/4 AEs. The median TTP and median OS for all patients was 4.1 months (95% CI 3.4–7.6) and 11.9 months (95% CI 9.9-not reached). This prompted a randomized Phase III study evaluating the combination of carboplatin/paclitaxel with or without cediranib in treating women with relapsed ovarian epithelial cancer, fallopian tube cancer, or primary peritoneal cancer.
Altogether, cediranib is currently listed in five active Phase III studies, such as the HORIZON II Phase II/III study of chemotherapy with cediranib vs. placebo in first-line metastatic CRC and HORIZON III, which represents a head-to-head comparison with bevacizumab (Avastin™) for this indication. Both studies completed recruitment by the end of 2008 and results are eagerly awaited. Also, the Phase III REGAL trial, in recurrent glioblastoma comparing cediranib monotherapy vs. lomustine ± cediranib began enrolling patients in late 2008 (see Table 9.1).
Vandetanib (ZD6474; Zactima®)
Vandetanib (ZD6474; Zactima™; AstraZeneca), an orally bioavailable 4-anilinoquinazoline derivate, acts as selective and reversible inhibitor of ATP binding to TK receptors VEGFR-2, -3, RET, and EGFR. In comparison to other kinase inhibitors, vandetanib is somehow more selective, which is demonstrated by a lack of effect against structurally related receptors such as PDGFR or c-Kit. Its antiangiogenic and antitumor activity has been shown in a wide range of preclinical animal models (Herbst et al. 2007). The potent anti-EGFR activity gave a reasonable rationale for testing vandetanib in cancers in which EGFR antagonists have been proven effective.
The clinical development so far was focused on NSCLC. First, the antitumor activity of vandetanib monotherapy or vandetanib with paclitaxel and carboplatin was compared with paclitaxel and carboplatin in previously untreated patients with NSCLC in a partially blinded, placebo-controlled, randomized Phase II study (Heymach et al. 2008). Patients were randomly assigned 2:1:1 to receive vandetanib alone, vandetanib plus chemotherapy, or chemotherapy alone. Interestingly, the risk of progression was reduced for patients receiving vandetanib plus chemotherapy (n = 56) vs. chemotherapy alone (n = 52; HR = 0.76; p = 0.098); but median PFS differed only by 1 week (24 vs. 23 weeks). The vandetanib monotherapy arm (n = 73) was discontinued after a planned interim PFS analysis met the criterion for discontinuation. Also, the OS was not significantly different between groups. Rash, diarrhea, and hypertension were common adverse events. The authors concluded, that vandetanib could be safely administered to patients with NSCLC, including those with squamous cell histology and treated brain metastases. The slightly longer PFS for vandetanib met the prespecified study end point, but was not significant (Heymach et al. 2008).
The next set of studies focused on vandetanib in second-line therapy of NSCLC. Eligible patients had locally advanced or metastatic (stage IIIB/IV) NSCLC after failure of first-line platinum-based chemotherapy (Heymach et al. 2007). First, a randomized Phase II study was initiated comparing vandetanib (100 or 300 mg/day) plus docetaxel (75 mg/m2 intravenous infusion every 21 days) vs. placebo plus docetaxel. After including 127 patients, median PFS was 18.7 weeks for vandetanib 100 mg plus docetaxel (n = 42; HR = 0.64; p = 0.037); 17.0 weeks for vandetanib 300 mg plus docetaxel (n = 44; HR = 0.83; p = 0.231); and 12 weeks for docetaxel (n = 41). There was no statistically significant difference in OS among the three treatment arms. Common AEs included diarrhea, rash, and asymptomatic prolongation of corrected QT (QTC) interval. At the time of publication, the authors concluded that the primary objective was achieved, with vandetanib 100 mg plus docetaxel demonstrating a significant prolongation of PFS compared with docetaxel in relation to the prespecified significance level. On the basis of these encouraging data, Phase III evaluation of vandetanib 100 mg plus docetaxel in second-line NSCLC (ZODIAC trial) was initiated in 2006. Until completion in September 2008, the study enrolled 1,391 patients previously treated with one prior anticancer therapy for advanced NSCLC. Median duration of follow-up was 12.8 months, with 87% patients progressed and 59% dead. Addition of vandetanib to docetaxel showed a statistically significant improvement in PFS versus docetaxel (HR 0.79, 97.58% CI 0.70-0.90; P<0.001). Significant advantages for vandetanib plus docetaxel were also seen for ORR (17% vs 10%, P<0.001). Overall survival showed a positive trend for vandetanib plus docetaxel that was not statistically significant (HR 0.91, 97.52% CI 0.78-1.07; P=0.196). The adverse event profile was consistent with that previously observed for vandetanib in NSCLC. Common AEs occurring more frequently in the vandetanib arm included diarrhea (42% vs 33%), rash (42% vs 24%) and neutropenia (32% vs 27%). (see Table 9.1).
Three other Phase III trials with vandetanib in second- and third-line therapy of NSCLC recently stopped recruiting patients and will complete data collection in 2009. The so-called ZEAL trial is a randomized, double-blind, placebo-controlled Phase III study evaluating the combination of vandetanib 100 mg with pemetrexed vs. pemetrexed alone. This study enrolled 534 patients previously treated with one prior anticancer therapy for advanced NSCLC. There were positive trends seen for vandetanib plus pemetrexed for both PFS (HR 0.86, 97.58% CI 0.69 -1.06; P=0.108) and OS (HR 0.86, 97.54% CI 0.65 -1.13; P=0.219). There was a statistically significant advantage for ORR (19.1% vs 7.9%, P<0.001) in the combination arm. The ZEST study also is a randomized, double-blind, Phase III study evaluating the efficacy of vandetanib 300 mg vs. erlotinib 150 mg, which enrolled 1,240 patients with locally advanced or metastatic NSCLC after failure of at least one prior anticancer therapy. There was no difference in PFS for patients treated with vandetanib versus erlotinib (HR 0.98, 95.22% CI 0.87 -1.10; P=0.721), and no difference in the secondary endpoints of OS (HR 1.01, 95.08% CI 0.89 -1.16; P=0.830) and ORR (both 12%). Finally, the ZEPHYR trial is a randomized Phase III study to assess the efficacy of vandetanib vs. best supportive care in patients with NSCLC (Stage IIIB-IV) after therapy with an EGFR inhibitor. This study is expected to enroll over 900 patients and data collection will be completed by April 2009.
At the time of writing this review, there was no active Phase III trial listed in the NCI’s database, but 26 Phase II and 17 Phase I studies, including trials on medullary thyroid carcinoma, breast cancer, and glioma, were ongoing.
Vatalanib (PTK787/ZK222584)
Vatalanib (PTK787/ZK222584; Bayer Schering Pharma AG, Berlin; Novartis, East Hanover, NJ) is an oral multitargeted kinase inhibitor that acts on VEGFR-1, -2, -3, c-KIT, and PDGFR (Wood et al. 2000). After oral administration, vatalanib reaches peak concentration in 1.0–2.5 h and has a half-life of 4.5 h, with no evidence of accumulation at steady state following once-daily dosing. Vatalanib demonstrated clinical activity in patients with several types of human cancer (Drevs et al. 2000; Roboz et al. 2006; Sharma et al. 2009; Thomas et al. 2005; Thomas et al. 2007). For further clinical development, vatalanib was investigated in two multinational randomized phase III studies in first- (CONFIRM-1) and second-line (CONFIRM-2) metastatic CRC. In CONFIRM-2, 855 patients were randomized to FOLFOX4 chemotherapy plus vatalanib (1,250 mg/day) or placebo (Kohne et al. 2007). Eligibility included histologically documented metastatic CRC, pretreatment for metastatic disease with irinotecan-/fluoropyrimidine- based therapy, measurable disease by Response Evaluation Criteria In Solid Tumors (RECIST), good performance status, and adequate organ function. In both trials, toxicities were similar. In detail, for the CONFIRM-2 trial, grade 3–4 AEs were hypertension (21% for vatalanib vs. 5% for placebo), diarrhea (16 vs. 8%), fatigue (15 vs. 7%), nausea (11 vs. 5%), vomiting (9 vs. 5%), and dizziness (9 vs. 1%). Thrombotic and embolic events of all grades occurred in 6% of the vatalanib treated patients vs. 1% of the placebo group. At the time of interim analysis in July 2005, OS was 12.1 months in the vatalanib and 11.8 months in the placebo group (HR 0.94; p = 0.511). PFS was significantly longer in the vatalanib arm (5.5 vs. 4.1 months; HR 0.83; p = 0.026). Interestingly, Lactat dehydrogenase (LDH), a rather unspecific marker related to poor prognosis in CRC, was predictive for the outcome in the vatalanib group. Especially patients with high LDH gained improvement in PFS when treated with vatalanib (5.6 vs. 3.8 months; HR 0.63; p < 0.001) and in OS (9.6 vs. 7.5 months; HR 0.78; p = 0.10). For CONFIRM-1, 1,168 patients were randomized to receive FOLFOX-4 plus vatalanib (1250 mg/day) or FOLFOX-4 plus placebo. The addition of vatalanib did not result in differences in the response rate (42% for FOLFOX-4 plus vatalanib vs. 46% for FOLFOX-4 plus placebo) or PFS time (7.7 months for FOLFOX-4 plus vatalanib vs. 7.6 months for FOLFOX-4 plus placebo). Thus, it was concluded that significant clinical benefits for vatalanib treatment in CRC seems to be limited to LDH-high patients, the reason for this remains unclear (Hecht et al. 2005; Kohne et al. 2007).
Currently, no active Phase III studies with vatalanib are listed, but six Phase II and four Phase I studies for therapy of glioma, multiple myeloma, pancreatic cancer, and melanoma are active.
Endostatin (rh-Endostatin, YH-16, Endostar™)
Endostatin, a 20-kiloDalton (kDa) fragment of collagen XVIII, is a group member of endogenous antiangiogenic proteins activated by proteolytic processing (Ferreras et al. 2000). Endostatin was shown to inhibit endothelial cell proliferation, migration, invasion, and vascular sprouting (O’Reilly et al. 1997). The reduction in endothelial cell survival induced by endostatin has been proposed to involve binding to the fibronectin receptor α5β1 (Sudhakar et al. 2003), interference with VEGF/VEGFR signaling (Hajitou et al. 2002; Kim et al. 2002b), inhibition of matrix metalloproteinases (MMP), e.g., MMP-2 (Kim et al. 2000), and downregulation of c-myc and cyclin-D1 (Hanai et al. 2002; Shichiri and Hirata 2001). Also, endostatin seems to downregulate a number of proteins essential to angiogenesis such as the Id1 and -3, HIF1-α and Ephrin B1 and B2 (Shichiri and Hirata 2001). Despite initial high hopes, the clinical development of endostatin came close to an unsuccessful end after treatment of about 160 cancer patients in Phase I and II studies when the sole manufacturer (EntreMed, Rockville, USA) announced the cease of production in 2003 due to lack of efficacy, difficult application scheme, and concerns about its production in yeast. Some years later, the Chinese protein chemist Luo may have solved the folding problem by adding nine amino acids to the endostatin molecule (Fu et al. 2008). This reformulation apparently made it possible to manufacture a soluble rh-endostatin (Endostar™, Simcere Pharmaceutical Co., Nanjing, China) using not yeast but bacteria and providing higher in vivo stability, now eligible for daily application once rather than twice. Phase I/II studies revealed that rh-endostatin was effective as single agent with good tolerance in clinical use. The first randomized study presented was designed to compare the response rate, median TTP, clinical benefit, and safety in patients with advanced NSCLC, treated with rh-endostatin (7.5 mg/m2 on days 1–14) plus standard dose vinorelbine (25 mg/m2 on day 1 and 5) and cisplatin (30 mg/m2 on Days 2–4), or placebo plus chemotherapy (Sun et al. 2005). Altogether, 493 NSCLC patients in good performance status were recruited for this double-blind study (326 in the rh-endostatin group, 167 as control). Of the 486 assessable patients, overall response rates were 35.4% for rh-endostatin and 19.5% in the control group (p = 0.0003). The median TTP were 6.3 and 3.6 months for rh-endostatin vs. control (p < 0.001), yielding a clinical benefit rate of 73.3 vs. 64.0% respectively (p = 0.035). Grade 3/4 neutropenia, anemia, nausea/vomiting were comparable in both arms. There was no data on OS reported. The authors concluded that the addition of rh-endostatin to standard chemotherapy resulted in significant improvement in response rate, median TTP, and clinical benefit rate compared with chemotherapy alone in advanced NSCLC patients (Sun et al. 2005). Subsequently, the national Food and Drug Administration of China approved Endostar™ for this setting. The currently listed Phase III studies involving rh-endostatin exclusively originate in China and enroll only NSCLC patients testing different combinations with chemotherapy or application in the adjuvant setting (see Table 9.1).
Thalidomide
One drug that exhibits an antiangiogenic effect by still not fully clarified mechanisms is thalidomide (D’Amato et al. 1994). Originally introduced as sedative and withdrawn due to deleterious side effects, today there is increasing evidence for the efficacy of thalidomide in cancer therapy. Thalidomide was developed in the 1950s and chiefly sold from 1957 to 1961 in almost 50 countries under at least 40 names to pregnant women, as an antiemetic to combat morning sickness and sleeping problems. Later, the teratogenic effects of thalidomide became clear when approximately 10,000 children mainly in Africa and Europe were born with severe malformations, including phocomelia in the late 1950s and early 1960s (Lenz 1967; Lenz and Knapp 1962). However, it was soon found that the teratogenicity caused by thalidomide was only associated with one particular optical isomer. Research continued, although the drug was not prescribed for decades, and finally the US FDA granted approval for treatment of erythema nodosum leprosum (ENL) in 1998. One year later, the first report was presented demonstrating activity of thalidomide in multiple myeloma (MM) tested in 180 patients with advanced disease (Singhal et al. 1999). Clinical development continued under strict regulations regarding the pregnancy status of patients and even their partners and finally the US FDA granted accelerated approval for thalidomide in combination with dexamethasone for the treatment of newly diagnosed MM in 2006. Since then, thalidomide was shown to be useful in a variety of tumors. Its mechanism of action in cancer is attributed to multiple, including direct cytotoxic, antiangiogenic, and antiinflammatory effects (Kumar 2006). The combination of temozolomide and thalidomide has shown promising activity in metastatic melanoma (Hwu et al. 2003), metastatic neuroendocrine tumors (Kulke et al. 2006), and unresectable or metastatic leiomyosarcoma (Boyar et al. 2008). Recently, the surprising effects of thalidomide have led to the development of a series of immunomodulatory drugs (IMiDs) and selective cytokine inhibitory drugs (SELCIDs) with even higher antiangiogenic potency (Dredge et al. 2005; List et al. 2005). The modulation of the immune system consists of stimulation of T – cells and NK – cells (Chang et al. 2006). In our own studies, thalidomide demonstrated biological and clinical activity in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) with ORR up to 56 and 25%, respectively (Steins et al. 2003; Steins et al. 2002). Responding patients experienced hematologic improvements including an increase in hemoglobin values and platelet counts. In four of 20 AML patients, a bone marrow blast clearance of at least 50% was achieved after treatment with thalidomide for at least 1 month (PR). Furthermore, we observed a long-term response in one AML patient of more than 20 months, subsequently meeting the criteria of complete remission. Interestingly, the decrease in leukemic blast infiltration in the bone marrow of responders was accompanied by a significant reduction of MVD. While it still remains unclear how exactly thalidomide inhibits angiogenesis, some data suggests a downregulation of VEGF as one possible mode of action (Komorowski et al. 2006; Li et al. 2003).
Currently, thalidomide and the subsequently developed IMiDs Revlimid™ (lenalidomide, CC-5013) and Actimid™ (CC-4047) are listed in 27 active Phase III trials in the NCI database. Apart from hematological malignancies, these substances are tested for treatment of poor liver function HCC and RCC (see Table 9.1).
Vascular Disrupting Agents
While classic inhibitors of tumor angiogenesis mostly compromise the formation of new blood vessels, occlusion of the existing tumor vasculature by inducing thrombosis or extensive endothelial damage leading to severe hemorrhagic necrosis is the main goal of the substances referred to as vascular disrupting agents (VDA). The largest group of VDAs already in clinical stage of development is the family of combretastatins, which act as microtubulin destabilizing drugs, and the structurally distinct flavonoid 5,6-dimethylxanthenone-4-acetic acid (DMXAA).
The first agent extensively studied was Combretastatin-4 (CA-4), which demonstrated rapid and extensive vascular disruption concomitant with hemorrhagic necrosis within the first hour of treatment in preclinical models (Dark et al. 1997). The fast onset of action is attributed to cytoskeletal shift changes including contraction of actinomyosin and the malformed assembly of stress fibers (see Fig. 9.2). Subsequently, this leads to disruption of the endothelial monolayer with increased permeability for macromolecules and shear-stress activation of platelets with intravascular thrombosis (Galbraith et al. 2001; Kanthou and Tozer 2002; Tozer et al. 1999). Finally, this endothelial disruption and platelet aggregation results in rapid almost complete vascular obstruction and tumor necrosis.

Proposed mechanism of vascular disrupting agents. The lead compounds of this class, Combretastatin A-4 (Zybrestat™) or 5,6-dimethylxantheonone-4-acetic acid (DMXAA; ASA404) induce rapid vascular obstruction within the tumor by acting on the endothelial cytoskeleton. In detail, shape changes and intracellular damage is seen leading to subsequent disruption of the endothelial monolayer. Increased vascular permeability and high interstitial fluid pressure in the tumor adds up to vascular collapse and obstruction. Due to direct exposure of the basement membrane to blood cells, platelets and plasmatic coagulation are activated leading to rapid tumor vessel thrombosis within minutes of exposure
For the clinical setting, Combretastatin-A4P is developed by OXiGENE (Waltham, MA, USA) as Zybrestat™. In July 2007, the company initiated a 180-patient pivotal registration study with Zybrestat™ for the treatment of anaplastic thyroid cancer, under a Special Protocol Assessment (SPA) agreement with the US FDA. For this study, the FDA granted Zybrestat™ “Fast-Track” status as potential cancer therapy. This is a randomized open-label Phase II/III study in which the experimental drug is tested in combination with conventional chemotherapeutics carboplatin and paclitaxel (NCT00507429).
In a previous Phase II study with 18 patients suffering of advanced anaplastic thyroid cancer Zybrestat™ as monotherapy achieved a median PFS of 7.4 weeks (range 2–84 weeks); with five patients remaining progression-free for more than 3 months (Cooney et al. 2006). The median OS in this study was approximately 20 weeks. Zybrestat™ also demonstrated activity in other Phase I studies for treatment of advanced solid neoplasms, such as NSCLC and ovarian cancer (Anderson et al. 2003; Bilenker et al. 2005; Dowlati et al. 2002; Rustin et al. 2003a; Stevenson et al. 2003). It is also the first VDA that has been clinically tested in combination with classic antiangiogenic drugs such as bevacizumab (Avastin™) (Nathan et al. 2008). In this study, 9 of 14 patients with advanced solid tumors experienced disease stabilization for more than 12 weeks. Three patients experienced SD for more than 24 weeks. Besides, DCE-MRI demonstrated statistically significant reductions in tumor perfusion. This effect rapidly reversed after Zybrestat™ alone, but was sustained following the combination of Zybrestat™ and BEV (Nathan et al. 2008). This observation and other preclinical evidence have prompted another randomized Phase II study in which the combination of carboplatin, paclitaxel, and BEV is evaluated with and without Zybrestat™ for patients with advanced NSCLC (Stadium IIIb and IV) as first-line therapy. Like in other studies involving antiangiogenic drugs, patients with predominant squamous cell histology are excluded. This study started in March 2008 and is aimed for enrollment of 60 patients until 2010 (NCT00653939).
The current lead compound of the structurally and mechanistically distinct flavonoids is DMXAA (AS1404, Antisoma Research Limited, London, UK) (Rewcastle et al. 1991). In contrast to combretastatins, cytoskeletal effects of DMXAA seem to be confined to actin assembly leaving interphase microtubules unharmed. In experimental models, DMXAA has been shown to enhance antitumor effects of melphalan and other cytotoxic agents as well as hyperthermia and radiation (Murata et al. 2001; Pruijn et al. 1997; Siim et al. 2003).
The first clinical Phase I study was presented in 2003 (Rustin et al. 2003b). DMXAA was applied to 46 patients for a total of 247 infusions of over 15 dose levels ranging from 6 to 4,900 mg/m². The MTD was reached at 3,700 mg/m2 with DLTs observed in form of urinary incontinence, visual disturbance, and anxiety at the highest dose level (4,900 mg/m2). Dose-dependent increases in the serotonin metabolite 5-hydroxyindoleacetic acid were found at dose levels of 650 mg/m2 and above. There was one unconfirmed PR at 1,300 mg/m2.
Phase II studies have been published for ovarian cancer, NSCLC, and hormone refractory prostate cancer (HRPC). The first randomized study evaluated DMXAA in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m2) in NSCLC with histologically confirmed stage IIIb or IV NSCLC previously untreated with chemotherapy (McKeage 2006). Until 2006, 77 patients were randomized to receive up to six cycles of carboplatin/paclitaxel with or without DMXAA (1,200 or 1,800 mg/m²). Thirty-five patients received chemotherapy alone, 36 chemotherapy plus 1,200 mg/m² DMXAA and 6 plus 1,800 mg/m² DMXAA. The safety profile in the control arm and DMXAA arm was comparable. Twenty-three of thirty patients in the control arm achieved disease control and seven progressed, for the 1,200 mg/m² DMXAA arm 28 of 33 patients reached disease control and five progressed. Finally, all six patients receiving 1,800 mg/m² DMXAA achieved disease control, with three confirmed PRs. The encouraging updated survival data was presented in 2008 (McKeage and Jameson 2008); median OS for chemotherapy alone being 8.8 months (n = 36), 14.0 months for chemotherapy plus 1,200 mg/m² DMXAA (n = 34) and 14.9 months for the 1,800 mg/m2 DMXAA group (n = 30). It is noteworthy that in this trial, patients with squamous NSCLC were also benefited.
These data prompted the currently recruiting international multicenter, randomized Phase III trial ATTRACT-1 (Antivascular Targeted Therapy Researching ASA404 in Cancer Treatment; NCT00662597). Previously untreated patients with advanced NSCLC (St. IIIb or IV) are randomized to receive standard chemotherapy carboplatin/paclitaxel in combination with 1,800 mg/m² DMXAA (AS1404, now licensed to Novartis, Basel, Switzerland) or placebo. It is planned to recruit 1,200 patients with comparison of OS as primary objective. Safety data from the previous studies described earlier indicates a different toxicity profile in contrast to other antiangiogenic drugs. Typical vascular effects such as proteinuria, arterial hypertension and thrombosis, pulmonary hemorrhage, wound healing or other bleeding complications were almost not observed with DMXAA both in squamous and nonsquamous histology. This allows also patients with predominant squamous histology to be recruited, a group that is usually ruled out in other trials involving most other antiangiogenic agents. Also, a second randomized Phase III study (ATTRACT-2) evaluating the efficacy of DMXAA in second-line therapy of NSCLC was initiated in late 2008 (see Table 9.1).
Accidental Antiangiogenesis Agents
Apart from the aforementioned agents, some already FDA-approved anticancer drugs were later to be shown to have antiangiogenic activity as well. For example, the FDA-approved EGFR antibodies cetuximab (Erbitux™; Merck) and panitumumab (Vectibix™; Amgen) as well as the EGFR antagonists erlotinib (Tarceva™; Genentech, OSI Pharmaceuticals in collaboration with Genentech and Roche) and gefitinib (ZD1839; Iressa™; AstraZeneca) were shown to inhibit tumor angiogenesis by partly blocking the VEGF receptor and downregulation of various pro-angiogenic factors such as VEGF (Ciardiello et al. 2001; Hoffmann et al. 2007; Huang et al. 2002; Perrotte et al. 1999; Pore et al. 2006), basic fibroblast growth factor (bFGF) (Albanell et al. 2001), HIF1-α (Li et al. 2008) and transforming growth factor- (TGF) (Pino et al. 2006). Also, the proteasome inhibitor bortezomib (Velcade™; Millennium Pharmaceuticals), approved for multiple myeloma, demonstrated potent antiangiogenic activity in clinical and preclinical models (Galimberti et al. 2008; Nawrocki et al. 2002; Williams et al. 2003). Even drugs like celecoxib (Celebrex™; Pfizer Inc.) originally approved not for therapy of malignant disease but for treatment of rheumatoid arthritis have been shown to increase production of endogenous angiogenesis inhibitors like endostatin and demonstrated clinical anticancer activity as well. Celecoxib is currently listed in four randomized Phase III trials for therapy of metastatic CRC, NSCLC, pancreatic, and prostate cancer (NCT00268476, NCT00295035, NCT00300729, NCT00486460). Also, the concept of metronomic chemotherapy was introduced meaning the inhibition of tumor angiogenesis by simply changing the dose and frequency of a cytotoxic chemotherapeutic agent like cyclophosphamide (Browder et al. 2000; Hanahan et al. 2000). Vice versa to these “accidental” antiangiogenic drugs, it became clear that “classic” antiangiogenic drugs affect not only endothelial but also tumor cells directly (Beaudry et al. 2008). Especially pancreatic and breast cancer cells were shown to express VEGFR-2 offering the possibility to directly target them with VEGF/VEGFR antagonists (Higgins et al. 2006a; Higgins et al. 2006b).
Conclusions and Future Perspectives
Taken together, the classic concept in cancer therapy that a drug is either directed exclusively against the tumor cell or against the vascular cell in tumor angiogenesis has been replaced by a far more complex model of tumor-stroma interactions. Thus, (multi-)targeted therapies against cancer have become more and more important. Up to date, numerous new substances were developed as angiogenesis inhibitors and evaluated in clinical trials for safety, tolerance, and efficacy. Yielding positive study results, some of these molecules have already been approved for clinical use as described earlier. Although the clinical benefit for patient groups studied is only in the range of few months, the benefit for single patients can be considerably more long-lasting. Treatment results begin to change even in diseases where no therapeutic advances could be made for decades. Today, the wide array of available agents offers the clinician multiple treatment choices.
However, the question of the optimal antiangiogenic approach is still an open debate and subject to a number of clinical studies described in this chapter; for example, which combinations for what tumors, treatment in early stage vs. advanced stage or maintenance?
In particular, the concept of tumor dormancy induced or maintained by angiogenesis inhibitors is widely discussed in the expert field. One intriguing observation for this model is the vast difference between the prevalence of clinically presenting cancer and unapparent malignant tumors found in autopsy studies. Besides, tumor dormancy may be a clinically relevant phenomenon in patients who have been treated for primary cancer and relapse after a long disease-free period (Demicheli et al. 2005; Uhr et al. 1997). Hypothetically, a small number of remaining malignant cells are able to re-activate their tumorigenic potential even years later. Most often, this phenomenon has been explained as consequence of a complex and poorly understood shift in the balance between host and tumor, the angiogenic switch. Historically, the failure to induce the angiogenic switch has been proposed as one of the mechanisms that may be responsible for tumor dormancy (Brem and Folkman 1975; Folkman and Kalluri 2004; Hanahan and Folkman 1996).
However, it is not clear whether a sustained production of angiogenic factors is required to finally break the balance or a short-term angiogenic burst may suffice to break dormancy. To this end, it is absolute speculative that the lower incidence of clinically apparent breast cancer in women with Down syndrome might be due to constant elevated serum values of endogenous angiogenesis inhibitor endostatin regulated on chromosome 21 (Retsky et al. 2009; Zorick et al. 2001). So far, it is still just an outlook into the future when hopefully our diagnostic tools are sensitive enough to detect recurrent disease before it leaves the dormant state and becomes symptomatic again or even better it might be possible to actively halt the tumor dormancy by antiangiogenic maintenance. However, the challenge for both basic researchers and clinicians will remain to integrate these numerous novel treatment approaches into existing protocols to eventually improve individual patient outcome.
References
Abou-Alfa GK, Schwartz L, Ricci S, Amadori D, Santoro A, Figer A, De Greve J, Douillard JY, Lathia C, Schwartz B, Taylor I, Moscovici M, Saltz LB (2006) Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol 24:4293–4300
Adnane L, Trail PA, Taylor I, Wilhelm SM (2006) Sorafenib (BAY 43–9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol 407:597–612
Albanell J, Codony-Servat J, Rojo F, Del Campo JM, Sauleda S, Anido J, Raspall G, Giralt J, Rosello J, Nicholson RI, Mendelsohn J, Baselga J (2001) Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor trea. Cancer Res 61:6500–6510
Anderson HL, Yap JT, Miller MP, Robbins A, Jones T, Price PM (2003) Assessment of pharmacodynamic vascular response in a phase I trial of combretastatin A4 phosphate. J Clin Oncol 21: 2823–2830
Atkins M, Jones CA, Kirkpatrick P (2006) Sunitinib maleate. Nat Rev Drug Discov 5:279–280
Beaudry P, Nilsson M, Rioth M, Prox D, Poon D, Xu L, Zweidler-Mckay P, Ryan A, Folkman J, Ryeom S, Heymach J (2008) Potent antitumor effects of ZD6474 on neuroblastoma via dual targeting of tumor cells and tumor endothelium. Mol Cancer Ther 7:418–424
Bilenker JH, Flaherty KT, Rosen M, Davis L, Gallagher M, Stevenson JP, Sun W, Vaughn D, Giantonio B, Zimmer R, Schnall M, O’Dwyer PJ (2005) Phase I trial of combretastatin a-4 phosphate with carboplatin. Clin Cancer Res 11: 1527–1533
Boyar MS, Hesdorffer M, Keohan ML, Jin Z, Taub RN (2008) Phase II study of temozolomide and thalidomide in patients with unresectable or metastatic leiomyosarcoma. Sarcoma 2008:412503
Brem H, Folkman J (1975) Inhibition of tumor angiogenesis mediated by cartilage. J Exp Med 141:427–439
Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O’Reilly MS, Folkman J (2000) Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res 60:1878–1886
Bukowski RM, Eisen T, Szczylik C, Stadler WM, Simantov R, Shan M, Elting J, Pena C, Escudier B (2007) Final results of the randomized phase III trial of sorafenib in advanced renal cell carcinoma: survival and biomarker analysis. J Clin Oncol 25(Suppl):5023 (abstract)
Cella D, Yount S, Du H, Dhanda R, Gondek K, Langefeld K, George J, Bro WP, Kelly C, Bukowski R (2006) Development and validation of the functional assessment of cancer therapy-kidney symptom index (FKSI). J Support Oncol 4:191–199
Chang DH, Liu N, Klimek V, Hassoun H, Mazumder A, Nimer SD, Jagannath S, Dhodapkar MV (2006) Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: therapeutic implications. Blood 108:618–621
Ciardiello F, Caputo R, Bianco R, Damiano V, Fontanini G, Cuccato S, De Placido S, Bianco AR, Tortora G (2001) Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res 7:1459–1465
Cooney MM, Savvides P, Agarwala S, Wang D, Flick S, Bergant S, Bhakta S, Lavertu P, Ortiz J, Remick SC (2006) Phase II study of combretastatin A4 phosphate (CA4P) in patients with advanced anaplastic thyroid carcinoma (ATC). J Clin Oncol 24(Suppl):5580 (abstract)
D’Amato RJ, Loughnan MS, Flynn E, Folkman J (1994) Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA 91:4082–4085
Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ (1997) Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res 57:1829–1834
Demetri GD, Huang X, Garrett CR, Schoeffski P, Blackstein ME, Shah MH, Verweij J, Tassell V, Baum CM, Casali PG (2008) Novel statistical analysis of long-term survival to account for crossover in a phase III trial of sunitinib (SU) vs. placebo (PL) in advanced GIST after imatinib (IM) failure. J Clin Oncol 26(Suppl):10524 (abstract)
De Boer R, Arrieta O, Gottfried M, Blackhall FH, Raats J, Yang CH, Langmuir P, Milenkova T, Read J, Vansteenkiste J (2009) Vandetanib plus pemetrexed versus pemetrexed as second-line therapy in patients with advanced non-small cell lung cancer (NSCLC): A randomized, double-blind phase III trial (ZEAL). J Clin Oncol 27: 8010
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG (2006) Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368: 1329–1338
Demicheli R, Miceli R, Moliterni A, Zambetti M, Hrushesky WJ, Retsky MW, Valagussa P, Bonadonna G (2005) Breast cancer recurrence dynamics following adjuvant CMF is consistent with tumor dormancy and mastectomy-driven acceleration of the metastatic process. Ann Oncol 16:1449–1457
Dowlati A, Robertson K, Cooney M, Petros WP, Stratford M, Jesberger J, Rafie N, Overmoyer B, Makkar V, Stambler B, Taylor A, Waas J, Lewin JS, McCrae KR, Remick SC (2002) A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res 62: 3408–3416
Dredge K, Horsfall R, Robinson SP, Zhang LH, Lu L, Tang Y, Shirley MA, Muller G, Schafer P, Stirling D, Dalgleish AG, Bartlett JB (2005) Orally administered lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitro. Microvasc Res 69:56–63
Drevs J, Hofmann I, Hugenschmidt H, Wittig C, Madjar H, Muller M, Wood J, Martiny-Baron G, Unger C, Marme D (2000) Effects of PTK787/ZK 222584, a specific inhibitor of vascular endothelial growth factor receptor tyrosine kinases, on primary tumor, metastasis, vessel density, and blood flow in a murine renal cell carcinoma model. Cancer Res 60:4819–4824
Drevs J, Siegert P, Medinger M, Mross K, Strecker R, Zirrgiebel U, Harder J, Blum H, Robertson J, Jurgensmeier JM, Puchalski TA, Young H, Saunders O, Unger C (2007) Phase I clinical study of AZD2171, an oral vascular endothelial growth factor signaling inhibitor, in patients with advanced solid tumors. J Clin Oncol 25:3045–3054
Dutcher JP, Wilding G, Hudes GR, Stadler WM, Kim S, Tarazi JC, Rosbrook B, Rini BI (2008) Sequential axitinib (AG-013736) therapy of patients (pts) with metastatic clear cell renal cell cancer (RCC) refractory to sunitinib and sorafenib, cytokines and sorafenib, or sorafenib alone. J Clin Oncol 26(Suppl):5127 (abstract)
Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, Chevreau C, Filipek M, Melichar B, Bajetta E, Gorbunova V, Bay JO, Bodrogi I, Jagiello-Gruszfeld A, Moore N (2007a) Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet 370: 2103–2111
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM (2007b) Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 356:125–134
Faivre S, Demetri G, Sargent W, Raymond E (2007) Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov 6:734–745
Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9:669–676
Ferrara N, Hillan KJ, Gerber HP, Novotny W (2004) Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 3:391–400
Ferreras M, Felbor U, Lenhard T, Olsen BR, Delaisse J (2000) Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett 486:247–251
Figlin RA, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Negrier S, Huang J, Kim ST, Chen I, Motzer RJ (2008) Overall survival with sunitinib versus interferon (IFN)-alfa as first-line treatment of metastatic renal cell carcinoma (mRCC). J Clin Oncol 26(Suppl):5024 (abstract)
Flaherty KT, Rosen MA, Heitjan DF, Gallagher ML, Schwartz B, Schnall MD, O’Dwyer PJ (2008) Pilot study of DCE-MRI to predict progression-free survival with sorafenib therapy in renal cell carcinoma. Cancer Biol Ther 7:496–501
Folkman J, Kalluri R (2004) Cancer without disease. Nature 427:787
Fu Y, Wu X, Han Q, Liang Y, He Y, Luo Y (2008) Sulfate stabilizes the folding intermediate more than the native structure of endostatin. Arch Biochem Biophys 471:232–239
Furuse J, Ishii H, Nakachi K, Suzuki E, Shimizu S, Nakajima K (2008) Phase I study of sorafenib in Japanese patients with hepatocellular carcinoma. Cancer Sci 99:159–165
Galbraith SM, Chaplin DJ, Lee F, Stratford MR, Locke RJ, Vojnovic B, Tozer GM (2001) Effects of combretastatin A4 phosphate on endothelial cell morphology in vitro and relationship to tumour vascular targeting activity in vivo. Anticancer Res 21:93–102
Galimberti S, Canestaro M, Ciancia E, Fazzi R, Marasca R, Petrini M (2008) Bortezomib is able to reduce angiogenesis in half of patients affected by idiopathic myelofibrosis: an ex vivo study. Leuk Res 32:1324–1325
Giantonio BJ, Catalano PJ, Meropol NJ, O’Dwyer PJ, Mitchell EP, Alberts SR, Schwartz MA, Benson AB 3rd (2007) Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol 25:1539–1544
Gollob JA, Rathmell WK, Richmond TM, Marino CB, Miller EK, Grigson G, Watkins C, Gu L, Peterson BL, Wright JJ (2007) Phase II trial of sorafenib plus interferon alfa-2b as first- or second-line therapy in patients with metastatic renal cell cancer. J Clin Oncol 25:3288–3295
Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, Booth BP, Verbois SL, Morse DE, Liang CY, Chidambaram N, Jiang JX, Tang S, Mahjoob K, Justice R, Pazdur R (2007) Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res 13: 1367–1373
Hajitou A, Grignet C, Devy L, Berndt S, Blacher S, Deroanne CF, Bajou K, Fong T, Chiang Y, Foidart JM, Noel A (2002) The antitumoral effect of endostatin and angiostatin is associated with a down-regulation of vascular endothelial growth factor expression in tumor cells. FASEB J 16:1802–1804
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86:353–364
Hanahan D, Bergers G, Bergsland E (2000) Less is more, regularly: metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest 105:1045–1047
Hanai J, Dhanabal M, Karumanchi SA, Albanese C, Waterman M, Chan B, Ramchandran R, Pestell R, and Sukhatme VP (2002) Endostatin causes G1 arrest of endothelial cells through inhibition of cyclin D1. J Biol Chem 277: 16464–16469
Hecht JR, Trarbach T, Jaeger E, Hainsworth J, Wolff R, Lloyd K, Bodoky G, Borner M, Laurent D, Jacques C (2005) A randomized, double-blind, placebo-controlled, phase III study in patients (Pts) with metastatic adenocarcinoma of the colon or rectum receiving first-line chemotherapy with oxaliplatin/5-fluorouracil/leucovorin and PTK787/ZK 222584 or placebo (CONFIRM-1). J Clin Oncol 23(Suppl):3 (abstract)
Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB, Fletcher JA, Fletcher CD, Huang X, Cohen DP, Baum CM, Demetri GD (2008) Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 26:5352–5359
Herbst RS, Heymach JV, O’Reilly MS, Onn A, Ryan AJ (2007) Vandetanib (ZD6474): an orally available receptor tyrosine kinase inhibitor that selectively targets pathways critical for tumor growth and angiogenesis. Expert Opin Investig Drugs 16:239–249
Herbst RS, Sun Y, Korfee S, Germonpre P, Saijo N, Zhou C, Wang J, Langmuir P, Kennedy SJ, Johnson BE (2009) Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small cell lung cancer (NSCLC): A randomized, double-blind phase III trial (ZODIAC). J Clin Oncol 27: CRA8003
Heymach JV, Johnson BE, Prager D, Csada E, Roubec J, Pesek M, Spasova I, Belani CP, Bodrogi I, Gadgeel S, Kennedy SJ, Hou J, Herbst RS (2007) Randomized, placebo-controlled phase II study of vandetanib plus docetaxel in previously treated non small-cell lung cancer. J Clin Oncol 25:4270–4277
Heymach JV, Paz-Ares L, De Braud F, Sebastian M, Stewart DJ, Eberhardt WE, Ranade AA, Cohen G, Trigo JM, Sandler AB, Bonomi PD, Herbst RS, Krebs AD, Vasselli J, Johnson BE (2008) Randomized phase II study of vandetanib alone or with paclitaxel and carboplatin as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol 26:5407–5415
Higgins KJ, Abdelrahim M, Liu S, Yoon K, Safe S (2006a) Regulation of vascular endothelial growth factor receptor-2 expression in pancreatic cancer cells by Sp proteins. Biochem Biophys Res Commun 345:292–301
Higgins KJ, Liu S, Abdelrahim M, Yoon K, Vanderlaag K, Porter W, Metz RP, Safe S (2006b) Vascular endothelial growth factor receptor-2 expression is induced by 17beta-estradiol in ZR-75 breast cancer cells by estrogen receptor alpha/Sp proteins. Endocrinology 147:3285–3295
Hirte HW, Vidal L, Fleming GF, Sugimoto AK, Morgan RJ, Biagi JJ, Wang L, McGill S, Ivy SP, Oza AM (2008) A phase II study of cediranib (AZD2171) in recurrent or persistent ovarian, peritoneal or fallopian tube cancer: final results of a PMH, Chicago and California consortia trial. J Clin Oncol 26:5521 (abstract)
Hoffmann S, Burchert A, Wunderlich A, Wang Y, Lingelbach S, Hofbauer LC, Rothmund M, Zielke A (2007) Differential effects of cetuximab and AEE 788 on epidermal growth factor receptor (EGF-R) and vascular endothelial growth factor receptor (VEGF-R) in thyroid cancer cell lines. Endocrine 31:105–113
Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, Ioffe E, Huang T, Radziejewski C, Bailey K, Fandl JP, Daly T, Wiegand SJ, Yancopoulos GD, Rudge JS (2002) VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA 99:11393–11398
Huang SM, Li J, Armstrong EA, Harari PM (2002) Modulation of radiation response and tumor-induced angiogenesis after epidermal growth factor receptor inhibition by ZD1839 (Iressa). Cancer Res 62:4300–4306
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350:2335–2342
Hwu WJ, Krown SE, Menell JH, Panageas KS, Merrell J, Lamb LA, Williams LJ, Quinn CJ, Foster T, Chapman PB, Livingston PO, Wolchok JD, Houghton AN (2003) Phase II study of temozolomide plus thalidomide for the treatment of metastatic melanoma. J Clin Oncol 21: 3351–3356
Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, Hu-Lowe DD, Shalinsky DR, Thurston G, Yancopoulos GD, McDonald DM (2004) Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol 165:35–52
Johnson DH, Fehrenbacher L, Novotny WF, Herbst RS, Nemunaitis JJ, Jablons DM, Langer CJ, DeVore RF 3rd, Gaudreault J, Damico LA, Holmgren E, Kabbinavar F (2004) Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol 22:2184–2191
Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, Liang C, Booth B, Chidambaram N, Morse D, Sridhara R, Garvey P, Justice R, Pazdur R (2006) Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res 12:7271–7278
Kanthou C, Tozer GM (2002) The tumor vascular targeting agent combretastatin A-4-phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood 99:2060–2069
Kim ES, Serur A, Huang J, Manley CA, McCrudden KW, Frischer JS, Soffer SZ, Ring L, New T, Zabski S, Rudge JS, Holash J, Yancopoulos GD, Kandel JJ, Yamashiro DJ (2002a) Potent VEGF blockade causes regression of coopted vessels in a model of neuroblastoma. Proc Natl Acad Sci USA 99:11399–11404
Kim YM, Hwang S, Pyun BJ, Kim TY, Lee ST, Gho YS, Kwon YG (2002b) Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem 277:27872–27879
Kim YM, Jang JW, Lee OH, Yeon J, Choi EY, Kim KW, Lee ST, Kwon YG (2000) Endostatin inhibits endothelial and tumor cellular invasion by blocking the activation and catalytic activity of matrix metalloproteinase. Cancer Res 60: 5410–5413
Kohne C, Bajetta E, Lin E, Valle JW, Van Cutsem E, Hecht JR, Moore MJ, Germond CJ, Meinhardt G, Jacques C (2007) Final results of CONFIRM 2: a multinational, randomized, double-blind, phase III study in 2nd line patients (pts) with metastatic colorectal cancer (mCRC) receiving FOLFOX4 and PTK787/ZK 222584 (PTK/ZK) or placebo. J Clin Oncol 25(Suppl):4033 (abstract)
Komorowski J, Jerczynska H, Siejka A, Baranska P, Lawnicka H, Pawlowska Z, Stepien H (2006) Effect of thalidomide affecting VEGF secretion, cell migration, adhesion and capillary tube formation of human endothelial EA.hy 926 cells. Life Sci 78:2558–2563
Kulke MH, Stuart K, Enzinger PC, Ryan DP, Clark JW, Muzikansky A, Vincitore M, Michelini A, Fuchs CS (2006) Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol 24:401–406
Kumar S (2006) Progress in the treatment of multiple myeloma. Lancet 367:791–792
Laurie SA, Arnold A, Gauthier I, Chen E, Goss G, Ellis PM, Shepherd FA, Matthews S, Robertson J, Seymour L (2006) Final results of a phase I study of daily oral AZD2171, an inhibitor of vascular endothelial growth factor receptors (VEGFR), in combination with carboplatin (C) + paclitaxel (T) in patients with advanced non-small cell lung cancer (NSCLC): a study of the National Cancer Institute of Canada Clinical Trials Group (NCIC CTG). J Clin Oncol 24:3054 (abstract)
Laurie SA, Gauthier I, Arnold A, Shepherd FA, Ellis PM, Chen E, Goss G, Powers J, Walsh W, Tu D, Robertson J, Puchalski TA, Seymour L (2008) Phase I and pharmacokinetic study of daily oral AZD2171, an inhibitor of vascular endothelial growth factor tyrosine kinases, in combination with carboplatin and paclitaxel in patients with advanced non-small-cell lung cancer: the National Cancer Institute of Canada clinical trials group. J Clin Oncol 26:1871–1878
Lenz W (1967) Perthes-like changes in thalidomide children. Lancet 2:562
Lenz W, Knapp K (1962) Thalidomide embryopathy. Arch Environ Health 5:100–105
Li X, Lu Y, Liang K, Pan T, Mendelsohn J, Fan Z (2008) Requirement of hypoxia-inducible factor-1alpha down-regulation in mediating the antitumor activity of the anti-epidermal growth factor receptor monoclonal antibody cetuximab. Mol Cancer Ther 7:1207–1217
Li X, Liu X, Wang J, Wang Z, Jiang W, Reed E, Zhang Y, Liu Y, Li QQ (2003) Thalidomide down-regulates the expression of VEGF and bFGF in cisplatin-resistant human lung carcinoma cells. Anticancer Res 23:2481–2487
Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, Fletcher CD, Corless CL, Fletcher JA (2008) Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol 216:64–74
List A, Kurtin S, Roe DJ, Buresh A, Mahadevan D, Fuchs D, Rimsza L, Heaton R, Knight R, Zeldis JB (2005) Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med 352:549–557
Llovet J, Ricci S, Mazzaferro V, Hilgard P, Raoul J, Zeuzem S, Poulin-Costello M, Moscovici M, Voliotis D, Bruix J (2007) Sorafenib improves survival in advanced hepatocellular carcinoma (HCC): results of a phase III randomized placebo-controlled trial (SHARP trial). J Clin Oncol 25(18 S):LBA1 (abstract)
McKeage M (2006) Phase Ib/II study of DMXAA combined with carboplatin and paclitaxel in non-small cell lung cancer (NSCLC). J Clin Oncol 24(suppl):7102 (abstract)
McKeage MJ, and Jameson MB (2008) Comparison of safety and efficacy between squamous and non-squamous non-small cell lung cancer (NSCLC) patients in phase II studies of DMXAA (ASA404). J Clin Oncol 26(suppl):8072 (abstract)
Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE (2007) Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 357:2666–2676
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA (2007) Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 356:115–124
Motzer RJ, Rini BI, Bukowski RM, Curti BD, George DJ, Hudes GR, Redman BG, Margolin KA, Merchan JR, Wilding G, Ginsberg MS, Bacik J, Kim ST, Baum CM, Michaelson MD (2006a) Sunitinib in patients with metastatic renal cell carcinoma. JAMA 295:2516–2524
Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI (2006b) Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol 24: 16–24
Murata R, Siemann DW, Overgaard J, Horsman MR (2001) Improved tumor response by combining radiation and the vascular-damaging drug 5, 6-dimethylxanthenone-4-acetic acid. Radiat Res 156:503–509
Natale RB, Thongprasert S, Greco FA, Thomas M, Tsai CM, Sunpaweravong P, Ferry D, Langmuir P, Rowbottom JA, Goss GD (2009) Vandetanib versus erlotinib in patients with advanced non-small cell lung cancer (NSCLC) after failure of at least one prior cytotoxic chemotherapy: A randomized, double-blind phase III trial (ZEST). J Clin Oncol 27: 8009
Nathan PD, Judson I, Padhani AR, Harris A, Carden CP, Smythe J, Collins D, Leach M, Walicke P, Rustin GJ (2008) A phase I study of combretastatin A4 phosphate (CA4P) and bevacizumab in subjects with advanced solid tumors. J Clin Oncol 26(Suppl):3550 (abstract)
Nawrocki ST, Bruns CJ, Harbison MT, Bold RJ, Gotsch BS, Abbruzzese JL, Elliott P, Adams J, McConkey DJ (2002) Effects of the proteasome inhibitor PS-341 on apoptosis and angiogenesis in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther 1:1243–1253
O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88:277–285
Perrotte P, Matsumoto T, Inoue K, Kuniyasu H, Eve BY, Hicklin DJ, Radinsky R, Dinney CP (1999) Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin Cancer Res 5:257–265
Pino MS, Shrader M, Baker CH, Cognetti F, Xiong HQ, Abbruzzese JL, McConkey DJ (2006) Transforming growth factor alpha expression drives constitutive epidermal growth factor receptor pathway activation and sensitivity to gefitinib (Iressa) in human pancreatic cancer cell lines. Cancer Res 66:3802–3812
Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A (2006) EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res 66:3197–3204
Pruijn FB, van Daalen M, Holford NH, Wilson WR (1997) Mechanisms of enhancement of the antitumour activity of melphalan by the tumour-blood-flow inhibitor 5, 6-dimethylxanthenone-4-acetic acid. Cancer Chemother Pharmacol 39:541–546
Ratain MJ, Eisen T, Stadler WM, Flaherty KT, Kaye SB, Rosner GL, Gore M, Desai AA, Patnaik A, Xiong HQ, Rowinsky E, Abbruzzese JL, Xia C, Simantov R, Schwartz B, O’Dwyer PJ (2006) Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 24: 2505–2512
Retsky MW, Hrushesky WJ, Gukas ID (2009) Hypothesis: primary antiangiogenic method proposed to treat early stage breast cancer. BMC Cancer 9:7
Rewcastle GW, Atwell GJ, Li ZA, Baguley BC, Denny WA (1991) Potential antitumor agents. 61. Structure-activity relationships for in vivo colon 38 activity among disubstituted 9-oxo-9H-xanthene-4-acetic acids. J Med Chem 34: 217–222
Richly H, Schultheis B, Adamietz IA, Kupsch P, Grubert M, Hilger RA, Ludwig M, Brendel E, Christensen O, Strumberg D (2009) Combination of sorafenib and doxorubicin in patients with advanced hepatocellular carcinoma: results from a phase I extension trial. Eur J Cancer 45(4): 579–587
Richly H, Henning BF, Kupsch P, Passarge K, Grubert M, Hilger RA, Christensen O, Brendel E, Schwartz B, Ludwig M, Flashar C, Voigtmann R, Scheulen ME, Seeber S, Strumberg D (2006) Results of a phase I trial of sorafenib (BAY 43–9006) in combination with doxorubicin in patients with refractory solid tumors. Ann Oncol 17:866–873
Roboz GJ, Giles FJ, List AF, Cortes JE, Carlin R, Kowalski M, Bilic S, Masson E, Rosamilia M, Schuster MW, Laurent D, Feldman EJ (2006) Phase 1 study of PTK787/ZK 222584, a small molecule tyrosine kinase receptor inhibitor, for the treatment of acute myeloid leukemia and myelodysplastic syndrome. Leukemia 20:952–957
Roskoski R Jr (2007) Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun 356:323–328
Rugo HS, Herbst RS, Liu G, Park JW, Kies MS, Steinfeldt HM, Pithavala YK, Reich SD, Freddo JL, Wilding G (2005) Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. J Clin Oncol 23: 5474–5483
Rustin GJ, Galbraith SM, Anderson H, Stratford M, Folkes LK, Sena L, Gumbrell L, Price PM (2003a) Phase I clinical trial of weekly combretastatin A4 phosphate: clinical and pharmacokinetic results. J Clin Oncol 21:2815–2822
Rustin GJ, Bradley C, Galbraith S, Stratford M, Loadman P, Waller S, Bellenger K, Gumbrell L, Folkes L, Halbert G, PIITCoCR UK (2003b) 5, 6-dimethylxanthenone-4-acetic acid (DMXAA), a novel antivascular agent: phase I clinical and pharmacokinetic study. Br J Cancer 88:1160–1167
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH (2006) Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 355:2542–2550
Sharma S, Freeman B, Turner J, Symanowski J, Manno P, Berg W, Vogelzang N (2009) A phase I trial of PTK787/ZK222584 in combination with pemetrexed and cisplatin in patients with advanced solid tumors. Invest New Drugs 27:63–65
Shichiri M, Hirata Y (2001) Antiangiogenesis signals by endostatin. FASEB J 15:1044–1053
Siim BG, Lee AE, Shalal-Zwain S, Pruijn FB, McKeage MJ, Wilson WR (2003) Marked potentiation of the antitumour activity of chemotherapeutic drugs by the antivascular agent 5, 6-dimethylxanthenone-4-acetic acid (DMXAA). Cancer Chemother Pharmacol 51:43–52
Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, Munshi N, Anaissie E, Wilson C, Dhodapkar M, Zeddis J, Barlogie B (1999) Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med 341:1565–1571
Siu LL, Awada A, Takimoto CH, Piccart M, Schwartz B, Giannaris T, Lathia C, Petrenciuc O, Moore MJ (2006) Phase I trial of sorafenib and gemcitabine in advanced solid tumors with an expanded cohort in advanced pancreatic cancer. Clin Cancer Res 12:144–151
Spano J, Chodkiewicz C, Maurel J, Wong RP, Wasan HS, Pithavala YK, Bycott PW, Liau KF, Kim S, Rixe O (2008) A randomized phase II study of axitinib (AG-013736) and gemcitabine versus gemcitabine in advanced pancreatic cancer, preceded by a phase I component. J Clin Oncol 25:4551 (abstract)
Steins MB, Bieker R, Padro T, Kessler T, Kienast J, Berdel WE, Mesters RM (2003) Thalidomide for the treatment of acute myeloid leukemia. Leuk Lymphoma 44:1489–1493
Steins MB, Padro T, Bieker R, Ruiz S, Kropff M, Kienast J, Kessler T, Buechner T, Berdel WE, Mesters RM (2002) Efficacy and safety of thalidomide in patients with acute myeloid leukemia. Blood 99:834–839
Stevenson JP, Rosen M, Sun W, Gallagher M, Haller DG, Vaughn D, Giantonio B, Zimmer R, Petros WP, Stratford M, Chaplin D, Young SL, Schnall M, O’Dwyer PJ (2003) Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: magnetic resonance imaging evidence for altered tumor blood flow. J Clin Oncol 21:4428–4438
Strumberg D (2005) Preclinical and clinical development of the oral multikinase inhibitor sorafenib in cancer treatment. Drugs Today (Barc) 41: 773–784
Strumberg D, Awada A, Hirte H, Clark JW, Seeber S, Piccart P, Hofstra E, Voliotis D, Christensen O, Brueckner A, Schwartz B (2006) Pooled safety analysis of BAY 43–9006 (sorafenib) monotherapy in patients with advanced solid tumours: is rash associated with treatment outcome? Eur J Cancer 42:548–556
Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R (2003) Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci USA 100:4766–4771
Sun Y, Wang J, Liu Y, Song X, Zhang Y, Li K, Zhu Y, Zhou Q, You L, Yao C (2005) Results of phase III trial of endostarTM (rh-endostatin, YH-16) in advanced non-small cell lung cancer (NSCLC) patients. J Clin Oncol 23:7138 (abstract)
Tang P, Cohen SJ, Bjarnason GA, Kollmannsberger C, Virik K, MacKenzie MJ, Brown J, Wang L, Chen AP, Moore MJ (2008) Phase II trial of aflibercept (VEGF Trap) in previously treated patients with metastatic colorectal cancer (MCRC): a PMH phase II consortium trial. J Clin Oncol 26: 4027 (abstract)
Thomas AL, Morgan B, Horsfield MA, Higginson A, Kay A, Lee L, Masson E, Puccio-Pick M, Laurent D, Steward WP (2005) Phase I study of the safety, tolerability, pharmacokinetics, and pharmacodynamics of PTK787/ZK 222584 administered twice daily in patients with advanced cancer. J Clin Oncol 23:4162–4171
Thomas AL, Trarbach T, Bartel C, Laurent D, Henry A, Poethig M, Wang J, Masson E, Steward W, Vanhoefer U, Wiedenmann B (2007) A phase IB, open-label dose-escalating study of the oral angiogenesis inhibitor PTK787/ZK 222584 (PTK/ZK), in combination with FOLFOX4 chemotherapy in patients with advanced colorectal cancer. Ann Oncol 18:782–788
Tozer GM, Prise VE, Wilson J, Locke RJ, Vojnovic B, Stratford MR, Dennis MF, Chaplin DJ (1999) Combretastatin A-4 phosphate as a tumor vascular-targeting agent: early effects in tumors and normal tissues. Cancer Res 59:1626–1634
Uhr JW, Scheuermann RH, Street NE, Vitetta ES (1997) Cancer dormancy: opportunities for new therapeutic approaches. Nat Med 3:505–509
Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, Smith NR, James NH, Dukes M, Curwen JO, Chester R, Jackson JA, Boffey SJ, Kilburn LL, Barnett S, Richmond GH, Wadsworth PF, Walker M, Bigley AL, Taylor ST, Cooper L, Beck S, Jurgensmeier JM, Ogilvie DJ (2005) AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res 65:4389–4400
Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S (2006) Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 5:835–844
Williams S, Pettaway C, Song R, Papandreou C, Logothetis C, McConkey DJ (2003) Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Mol Cancer Ther 2: 835–843
Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J, Hofmann F, Mestan J, Mett H, O’Reilly T, Persohn E, Rosel J, Schnell C, Stover D, Theuer A, Towbin H, Wenger F, Woods-Cook K, Menrad A, Siemeister G, Schirner M, Thierauch KH, Schneider MR, Drevs J, Martiny-Baron G, Totzke F (2000) PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res 60: 2178–2189
Zorick TS, Mustacchi Z, Bando SY, Zatz M, Moreira-Filho CA, Olsen B, Passos-Bueno MR (2001) High serum endostatin levels in Down syndrome: implications for improved treatment and prevention of solid tumours. Eur J Hum Genet 9:811–814
Acknowledgment
Supported by a grant of the Deutsche Forschungsgemeinschaft (SPP1190) and the Institute for Innovative Medical Research (IMF KE 110715; IMF LI 110633)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Kessler, T., Bayer, M., Schwöppe, C., Liersch, R., Mesters, R.M., Berdel, W.E. (2010). Compounds in Clinical Phase III and Beyond. In: Liersch, R., Berdel, W., Kessler, T. (eds) Angiogenesis Inhibition. Recent Results in Cancer Research, vol 180. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-78281-0_9
Download citation
DOI: https://doi.org/10.1007/978-3-540-78281-0_9
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-78280-3
Online ISBN: 978-3-540-78281-0
eBook Packages: MedicineMedicine (R0)