
Abstract
Over the past decade, the increasing availability of zirconium-89 has fueled a rise in the development and use of targeted positron emission tomography (PET) probes with relatively long serum half-lives, particularly radiolabeled antibodies. The stable coordination of 89Zr in radiopharmaceuticals is of critical importance to prevent its in vivo release and subsequent non-specific accumulation in nontarget organs, especially the bone. To date, the only chelator for 89Zr to be used in the clinic is the siderophore-derived desferrioxamine (DFO), though concerns persist regarding the in vivo stability of the [89Zr]Zr-DFO complex. In recent years, research into the development of alternative, more stable chelators for 89Zr has flourished. This chapter provides a general overview of zirconium-89, specifically its chemical properties, production, radiochemistry, and biomedical applications. Several examples of new chelators for 89Zr are discussed along with their strengths and weaknesses compared to DFO. Finally, a brief exploration of the future of 89Zr radiochemistry is included to provide the reader with awareness of the most recent progress in the field as well as what this relatively new positron-emitting radionuclide can offer going forward.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Fundamentals
Zirconium is a group IV transition metal with a diverse array of applications in metallurgy, gemology, nuclear power, food packaging, the steel industry, and—of course—nuclear medicine. Zirconium has no known role in biological systems and is generally regarded as non-toxic and environmentally benign. There are five naturally occurring isotopes of zirconium : 90Zr (stable; ~51%), 91Zr (stable; ~11%), 92Zr (stable; ~17%), 94Zr (stable; ~17%), and 96Zr (t1/2 ~ 2.0 × 1019 y; ~3%). In nature, the element is typically found as part of the mineral zircon (ZrSiO4). Over 25 different radioisotopes of zirconium have been synthesized, though the relevant radionuclide for nuclear medicine is zirconium-89, which is used for the positron emission tomography (PET) of diseases such as cancer.
PET imaging using 89Zr-labeled radiopharmaceuticals is typically focused on one of three aims: selecting patients who will most likely benefit from a targeted therapy, predicting a patient’s response to therapy, or monitoring a patient’s response to therapy. The overwhelming majority of preclinical and clinical investigations with 89Zr have involved the radiolabeling of monoclonal antibodies (mAbs). The radiometal’s multiday half-life (~3.3 days) extends the time-frame of imaging, improving image contrast by allowing the slow-circulating mAbs to accumulate at sites of disease and clear from the bloodstream. Typically, 89Zr is produced using a biomedical cyclotron via the 89Y(p,n)89Zr reaction and is isolated and purified using hydroxamate-functionalized resins. In biological systems, zirconium-89 exists as a +4 oxidation state and can form complexes with coordination numbers (CN) of up to 8. Desferrioxamine (DFO)—which coordinates Zr4+ in a hexadentate fashion, leaving two coordination sites available for exogenous water molecules—is the current “gold standard” chelator for 89Zr. For several years, the less-than-ideal in vivo stability of [89Zr]Zr-DFO has been tolerated in preclinical and clinical immunoPET despite the transchelation of the radiometal to the bone over time. The deposition of 89Zr in the bone poses a concern due to both the limited true-positive identification of osseous lesions and, just as importantly, the elevated radiation dose to the sensitive bone marrow. To resolve these issues, several new chelators have been explored to improve the stability of the 89Zr-chelator complex by optimizing both the number of ligands in the coordination sphere (i.e. CN = 8) as well as the geometry and morphology of the chelator itself.
Details
Biomedical Utility
The importance of zirconium-89 in PET lies in its ability to facilitate the in vivo tracking of targeting vectors that require extended amounts of time to achieve optimal target-to-background contrast ratios. Over the last decade, the standardization of its production and commercial availability has paved the way for the development of companion diagnostics for immunotherapy and antibody-based treatments. Indeed, the physical half-life of 89Zr (t1/2 = 78.4 h) complements the biological half-lives of variety of larger biomolecules, including monoclonal antibodies (mAbs), antibody fragments, proteins, and nanoparticles. These targeting vectors via typically labeled with zirconium-89 via a bifunctional chelator that is covalently attached to the biomolecule or nanoparticle. Currently, desferrioxamine B (DFO) is the most widely used chelator for Zr-89 in both preclinical and clinical studies. Zirconium-89 is biologically inert and is trapped within the cell once internalized, making it a residualizing radionuclide [1]. This enables better visualization of disease targets and higher target-to-background activity concentration ratios several days after the administration of the [89Zr]Zr-DFO-labeled tracer. However, a disadvantage of imaging with [89Zr]Zr-DFO-labeled biomolecules is the suboptimal stability of [89Zr]Zr-DFO complex, which leads to the decomplexation of [89Zr]Zr4+ in vivo and its subsequent accumulation in bone. This phenomenon has been observed in both rodents [2] and humans [3].
Coordination Chemistry
The zirconium cation exists predominantly in the +IV oxidation state in aqueous solutions, though lower atypical oxidation states (I, II, III) have been reported [4]. Zr(IV) lacks valence electrons with its [Kr]4d0 electronic configuration, and it has an ionic radius of 85 pm and a covalent radius of 1.48 pm [4, 5]. Because of its high charge and small ionic size, Zr is categorized as a hard Lewis acid with a strong affinity for hard Lewis bases such as oxygen donors. Zr forms metal complexes with varying coordination geometries and coordination numbers ranging from 4 to 12. That said, studies by Intorre and Martell suggest that eight-coordinate complexes of Zr(IV) are ideal [6, 7] . Complexes with lower coordination numbers typically recruit additional ligands to achieve an octacoordinate environment, thereby strengthening the metal complex against polymerization and hydrolysis [5]. Zr(IV) with eight-coordinating ligands conventionally forms a dodecahedral structure. Table 1 lists the geometries of Zr(IV) complexes with various coordination numbers.
Zirconium (IV) has the potential to undergo hydrolysis, forming polynuclear species (e.g. dimers, trimers, and tetramers) that are bridged by hydroxo- or oxo-donor groups [5, 8]. In very dilute solutions (10−4 M) at pH ~ 2, Zr(IV) exists in a hydrolyzed state in the form [Zr(OH)n](4−n)+ (note: n is pH-dependent) in very dilute solutions (~ 10-8 M) [5]. Hydrolyzed precipitates with very low solubility (~ 10−8 M) can also form at pH ~ 2 [8].
Nuclear Properties of Zr-89
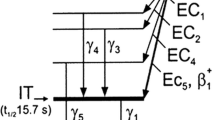
Zirconium-89 has a half-life of 78.4 h, decaying to stable 89Y via positron decay (23%) and electron capture (77%) (Fig. 1) [9]. This decay results in the emission of a characteristic gamma ray of 909 keV (99%), which allows for the fingerprint identification of this radionuclide using high-purity germanium gamma spectroscopy. Importantly, this high-energy gamma does not significantly interfere with imaging because it is not in coincidence with the ß+ decay [9]. 89Zr produces PET images with good spatial resolution due to its low positron energy (Eß[beta] + max = 0.9 MeV), a value comparable to that of the positron emitted by 18F (Eß[beta] + max = 0.64; t1/2 ~ 1.8 h) [9, 10]. 89Zr, however, has a much lower positron branching ratio than fluorine-18: 23% vs. 97%, respectively. A comparison of the nuclear properties of 89Zr and 18F is provided in Table 2 [11].

Simplified decay scheme of Zr-89
Production and Purification of 89Zr
Early investigations exploring the production of 89Zr were carried out by Link and colleagues [12, 13]. Both this pioneering work as well as more recently published endeavors have contributed to the simplification of the production and purification of 89Zr as well as standard operating procedures and semi-automated platforms to make this isotope accessible to researchers worldwide [14,15,16,17]. Zirconium-89 is currently produced using a medical cyclotron via the 89Y(p,n)89Zr reaction [16, 18]. The production of 89Zr using solid targets provides the highest yields and will be the main focus in this chapter; however, liquid targets using yttrium solutions such as YCl3 and Y(NO3)3 have been employed in cases wherein the cyclotron is incapable of solid targetry [19]. To produce the radiometal from a solid target, a Yttrium foil is secured onto an aluminum or niobium holder [16]. Yttrium-89 is 100% abundant and can be purchased commercially as a foil with varying thickness. In lieu of a foil, yttrium can also be deposited onto a niobium or copper support [18, 20]. To achieve optimal production of 89Zr with the highest possible yields, a number of variables need to be considered, including the energy of the incident protons, the thickness of the target, and the irradiation time. Table 3 lists examples of different production methods developed to optimize yields of 89Zr while minimizing the production of the long-lived 88Zr contaminant.
Proton-induced reactions on yttrium targets at energies relevant to the production of 89Zr can also result in the production of two Zr contaminants: 89mZr (t1/2 = 4.2 min) and 88Zr (t1/2 = 83.4 days). Zirconium-89m is short-lived and completely decays to 89Zr within an hour; thus, it is not considered a potential source of impurity [17, 18]. Zirconium-88 is long-lived and inseparable from 89Zr; however, the quantity of 88Zr produced is considered negligible (~ 0.0005%) at the energies (11–15 MeV) used to produce 89Zr (see Table 3) [20]. Other long-lived radioactive contaminants that could be produced from impurities in the yttrium target and/or the target holder are 65Zn, 48V, 56Co, and 156Tb. However, none of these metals bind to the hydroxamate resin used for the purification of 89Zr [20], and thus they can be separated easily from the desired product during the processing of the target.
The processing of the target is performed using a solid-phase extraction system. To this end, the target is dissolved in 2 M HCl, and the 89Zr is separated from the bulk target material via solid-phase extraction using a hydroxamate-functionalized resin. Hydroxamate functions as a bidentate ligand with a high affinity for 89Zr, while 89Y can be washed from the resin with 2 M HCl [16]. At 2–3 M concentrations of HCl, iron can be a challenging contaminant, as it is chemically similar to zirconium. However, it has low affinity for hydroxamate-based resin systems and can be chemically separated from 89Zr through multiple washings of the column.
The affinity of Zr4+ for the hydroxamate-functionalized resin was illustrated by the work of Guerard et al. [21]. As we have mentioned, [89Zr]Zr4+ is a hard acid and has a predilection for binding “hard” Lewis bases such as nitrogen and—especially—oxygen. The solid-state x-ray diffraction analysis of the complex formed by Zr and N-methyl acetohydroxamic acid (Me-AHA) shows a 1:4 stoichiometric ratio (CN = 8) between Zr and hydroxamate groups and a log K of 17.3. We adapted this crystal structure into a 2D structure for simplicity (Fig. 2). Based on this work, we can infer that [89Zr]Zr4+ binds the hydroxamate groups on the resin with similar affinity. Once it has bound the 89Zr, the resin must be washed with ample volume of 2 M HCl followed by water to remove yttrium, which has a low affinity for hydroxamates. Zirconium-89 is then eluted via transchelation with 1 M oxalic acid (H2C2O4), which binds in a 1:4 stoichiometric ratio (similar to the manner in which it coordinates acetohydroxamic acid) [22]. This separation process is depicted in Fig. 3 and is fully described in the seminal paper published by Holland et al. In this work, step-by-step procedures were established for preparing the hydroxamate-functionalized resin as well as for the separation of 89Zr from 89Y [16]. In addition, the purification process can be semi-automated to reduce radiation exposure to personnel, making it more feasible to safely isolate large quantities of 89Zr [17, 23].

Zr-Me-AHA forms octacoordinate species based on the crystal structure reported by Guerard et al.

Flow chart depicting the separation of Zr-89 from Y-89
Desferrioxamine: The Current Standard Chelator for 89Zr
Desferrioxamine B (DFO) (Fig. 4) was isolated and characterized by Bickel et al. in 1960 from Streptomyces pilosus [24]. DFO, marketed as Desferal® for the treatment of iron intoxication, acts as an iron-scavenging siderophore and contains three hydroxamate moieties linked together in a linear scaffold. This hexadentate ligand offers three neutral and three anionic oxygen donors. DFO complexes Fe(III) with a formation constant of 1 × 1030 M−1, forming an octahedral geometry. Other metals with a + 3 charge—e.g. Al(III), Ga(III), and Cr(III)—have been shown to bind to this ligand but with lower stability [25]. Importantly, because of its strong affinity and ability to encapsulate Zr(IV), DFO has been widely accepted as the “gold standard” chelator of this metal. However, decomplexation still remains an issue despite the moderate thermodynamic stability of the Zr-DFO complex. A detailed discussion can be found in the following sections.

Desferrioxamine B and its three hydroxamate groups
The emergence of zirconium-89 as an ideal radionuclide for antibodies and large molecules prompted the need to create bifunctional variants of DFO to facilitate bioconjugation reactions to the amines and cysteines of these biomolecules. Several bifunctional derivatives of DFO are now available through commercial sources, and conjugation techniques are discussed below in the section on “Tricks of the Trade.”
Tricks of the Trade
Neutralizing the [89Zr]Zr-Oxalate
89Zr is typically supplied as [89Zr]Zr-oxalate in a 1 M solution of oxalic acid. The pH of the [89Zr]Zr-oxalate must be neutralized to pH 6.8–7.4 in order to achieve optimal labeling efficiencies with DFO-bearing conjugates [26]. Several different techniques are employed for neutralization. Choosing a method is often informed by the starting volume of the solution of [89Zr]Zr-oxalate, as the neutralization process can significantly dilute this solution, leading to poor radiolabeling efficiencies. Vosjan et al. recommend that the total volume of [89Zr]Zr-oxalate (37–185 MBq) to be neutralized should be 200 μL. If the desired activity for [89Zr]Zr-oxalate is less than 200 μL, then 1 M oxalic acid should be added to obtain this volume. The [89Zr]Zr-oxalate solution can then be neutralized by adding 90 μL of 2 M Na2CO3 and 300 μL of 0.5 M HEPES (pH 6.8–7.4) [27]. However, using the carbonate buffer to neutralize larger volumes (>200 μL) of the [89Zr]Zr-oxalate can lead to the formation of insoluble precipitates. Not surprisingly, this phenomenon lowers the amount of 89Zr available in solution for radiolabeling. Alternatively, diluting the starting volume of [89Zr]Zr-oxalate with an equal volume of 1 M HEPES buffer (pH 7.1) prior to adjusting the pH with 2 M NaOH keeps the 89Zr in solution [28].
Conjugation of DFO to Antibodies
Meijs and colleagues were the first to develop an approach to the conjugation of DFO to mAbs. This strategy employed a two-step procedure in order to tag the lysine residues (ε-amines) within mAbs [29]. Lysines were first modified to carry maleimide groups (mAb-SMCC). Then a thioester-bearing variant of DFO (Df-SATA) was converted to a free thiol, facilitating the Michael addition between the bifunctional chelator and the modified antibody to form with the DFO-modified immunoconjugate: mAb-SMCC-SATA-Df (Fig. 5). Radiolabeling with 89Zr was achieved by sublimating [89Zr]Zr-oxalate and subsequently adding the Df-mAb immunoconjugate, producing a radiochemical yield of 90% and a specific activity of 185 kBq/μg (5 μCi/μg).

DFO was derivatized with N-succinimidyl-S-acetylthioacetate (SATA), which reacts with maleimide-modified lysines on the mAb
Years later, Verel and colleagues improved upon this SMCC-SATA conjugation chemistry by modifying DFO with an amine-reactive moiety to facilitate the formation of an amide linkage between the chelator and the ε-amine of a lysine residue. This bioconjugation was performed using a five-step procedure (Fig. 6) [20]: (i) the modification of DFO with succinic anhydride, (ii) the chelation of Fe(III) to protect the hydroxamate groups, (iii) the addition of 2,3,5,6-tetrafluorophenol (TFP) to form an amine-reactive TFP ester, (iv) the attachment of the amine-reactive bifunctional chelator to the lysines of the biomolecules, and (v) the removal of the Fe(III). Furthermore, the authors established optimal conditions for radiolabeling with 89Zr without the need for the sublimation of the [89Zr]Zr-oxalate.

The attachment of desferrioxamine to antibodies proceeds through a five-step procedure involving (1) the conjugation of succinic anhydride to DFO mesylate, (2) the coordination of Fe(III) to protect the hydroxamate groups, (3) the addition of 2,3,5,6-tetrafluorophenol (TFP), (4) the attachment of the bifunctional chelator to the free amines of the mAb, and (v) the removal of Fe(III) to prepare the chelator for the coordination of [89Zr]Zr4+
In an effort to simplify this chemistry further, Perk and colleagues developed isothiocyanato-p-benzyl-desferrioxamine (p-NCS-Bn-DFO) , a bifunctional variant of DFO that is currently commercially available [26]. p-NCS-Bn-DFO reacts with the lysines of biomolecules to form thiourea linkages that are similar in stability to the amide bonds formed by other bifunctional variants of DFO (Fig. 7) [26]. The reaction conditions for the conjugation of p-NCS-Bn-DFO to a biomolecule—most often an antibody—are mild. The two species are typically incubated in a basic buffer such as sodium carbonate (pH ~ 9) for 30 min at 37 °C, though different buffers (e.g. phosphate buffered saline) and longer incubation times have also been used [26]. p-NCS-Bn-DFO is not soluble in the aqueous buffer used for conjugation, so it should be dissolved in dimethyl sulfoxide (DMSO) prior to its addition to the reaction mixture containing the biomolecule. We recommend preparing a relatively concentrated stock solution of p-NCS-Bn-DFO in DMSO (5–10 mg/mL) so as to limit the amount of DMSO in the conjugation reaction and thus lower the likelihood of the precipitation of the biomolecule. Generally speaking, the final concentration of DMSO in the reaction mixture should not exceed 10% v/v.

The conjugation of DFO-Bz-SCN to an amine forms a thiourea linkage
Other conjugation methods have been employed to facilitate the site-specific modification of biomolecules with DFO [30, 31]. For example, several studies have used a maleimide-bearing variant of DFO to facilitate Michael additions to engineered cysteine residues in antibodies (Fig. 8) [30, 32]. The buffers used for these ligations are similar to those used with p-NCS-Bn-DFO, though they generally have a lower pH (~6.5–7) due to the lower pKa of the sulfhydryl groups of cysteines (pKa = 8.3) compared the ε-amines of lysines (pKa = 10.5). Furthermore, a reducing agent such as tris(2-carboxyethyl)phosphine (TCEP) is necessary to reduce disulfide bridges and produce thiols available for conjugation. It is important to note that sulfhydryl-containing reductants such as dithiothreitol (DTT) and beta-mercaptoethanol (BME) should be avoided in these cases because of their ability to react with the DFO-bearing maleimide.

DFO-maleimide undergoes a Michael addition with engineered cysteine residues to form thioether bonds
Radiolabeling with 89Zr
Over 20 years ago, Meijs and colleagues first demonstrated the superior stability of [88Zr]Zr-DFO compared to [88Zr]Zr-DTPA by attaching each chelator to a solid support [33]. At the time, 88Zr (t1/2 = 83.4 days) was used primarily as a surrogate for 89Zr in proof-of-concept studies in vitro. The [88Zr]Zr-DFO was formed in ~90% radiochemical yields after 2 h of incubation using citrate, acetate, or PBS buffers and pH values of 4, 5, 6, or 7. Stability studies in human serum clearly illustrated that DFO provided a more stable coordination environment than DTPA; only 80% of [88Zr]Zr-DTPA remained intact after 24 h, while [88Zr]Zr-DFO proved nearly 100% stable over the same time period [33].
The radiolabeling of DFO-bearing mAbs and biomolecules with neutralized [89Zr]Zr-oxalate can be achieved within 30–60 min in a variety of buffers. Regardless of the buffer, it is critical to ensure that the pH of the reaction lies between pH 6.8–7.2 in order to achieve optimal transchelation of [89Zr]Zr-oxalate to DFO and thus produce high radiochemical yields [20, 26]. The buffers typically used for radiolabeling with 89Zr can be 0.9% saline, 0.25 M sodium acetate, 0.25–1 M HEPES, or 5 mM sodium citrate with 0.5 M HEPES. In our hands, the use of phosphate-buffered saline lowered the yields of 89Zr-labeled mAbs, presumably due to the presence of phosphate anions that can compete with DFO for the coordination of the metal. After the radiolabeling reaction, DTPA (pH 7; final concentration = 1 mM) may be added to the crude solution of [89Zr]Zr-DFO-mAb and incubated for 5 min at room temperature in order to sequester any free [89Zr]Zr4+ prior to purification. In addition, if subsequent in vitro assays suggest that the reactivity of the [89Zr]Zr-labeled biomolecule has been compromised during radiosynthesis, gentisic acid (5 mg/mL) may be added to the radiolabeling solution to protect the biomolecule from radiolysis [26, 34].
Purification of a DFO-Modified mAb and a [89Zr]Zr-DFO-Labeled Radioimmunoconjugate
Size exclusion chromatography can be used to remove any unreacted bifunctional chelator or free [89Zr]Zr4+ from DFO-modified immunoconjugates and their 89Zr-labeled analogs, respectively. To this end, desalting gravity flow columns (e.g. PD-10) or spin columns (e.g. Zeba, Amicon, GE Vivaspin) with molecular weight cutoffs of 10–50 kDa may be used. It is important to note that adding a relatively high concentration of the conjugate (≥0.5 mg/mL) to the column aids in the recovery of high yields of the conjugate after purification. In some cases, the radiolabeled antibody can bind too strongly to the PD10 column, making it “sticky” and difficult to elute. If this occurs, washing the column with 1% bovine serum albumin (BSA) or human serum albumin is suggested to prevent the binding of residual tracer to the resin and thus maximize the recovery of the purified 89Zr-labeled biomolecule.
Non-immunoglobulin Vectors
Relatively Small Molecules
Novel vectors based on small molecules may be radiolabeled with 89Zr to assess their biodistribution over several days. For example, Kuda-Wedagedara et al. conjugated DFO to cobalamin (vitamin B12) and subsequently labeled this vector with 89Zr using radiolabeling procedures similar to those employed for the labeling of antibodies [27, 35] The resulting [89Zr]Zr-DFO-cobalamin was used to image the nutrient demand of breast cancer xenografts via its interaction with the transcobalamin-CD320 receptor. An important finding in this study was that neither increasing the overall molecular weight of the cobalamin vector (MW = 1190 g/mol) by the addition of the DFO chelator (+752 g/mol) nor changing the overall charge of the molecule to +1 due to the contribution by Zr4+ affected the binding of the 89Zr-labeled cobalamin to the transcobalamin receptor.
Other relatively small molecules labeled with 89Zr via DFO-Bz-NCS include peptides such as exendin-4 for the imaging of insulinomas [36]. An aminohexanoic acid spacer was used to link exendin-4 to DFO, producing a final construct with a total molecular weight of ~5 kDa. This strategy was employed to prevent the chelator from interfering with exendin-4’s binding to its receptor, GLP-1. Interestingly, however, the 89Zr-labeled exendin-4 had a twofold lower affinity (Kd = 28 nM) than the 68Ga-labeled analog of this peptide (Kd = 11 nM). This difference could be due to the overall charge of the radiolabeled peptide: −1 for the 89Zr-labeled version and neutral for the 68Ga-labeled variant. Thus, considerations for radiolabeling smaller molecules with [89Zr]Zr4+ must include how the potential changes to the overall charge of the molecule and the steric hindrance created by the chelator may affect the affinity of the radiotracer to its target.
Cell Labeling
A variety of different cell types—including bone marrow cells and immune cells—have been labeled with zirconium-89 to take advantage of the residualizing properties of the radiometal. The delivery of zirconium-89 into the cell can be accomplished by complexing zirconium-89 with oxine ligands in a 1:4 stoichiometric ratio [37]. Ferris et al. developed an approach to the 89Zr-labeling of oxines using a biphasic system in which zirconium-89 is in a neutralized aqueous solution and the ligands are in chloroform [37]. The radiolabeling occurs in the interphase of the aqueous and organic layer. The chloroform was then dried to isolate [89Zr]Zr-oxine, which can be resuspended in dimethyl sulfoxide and diluted in PBS for biological studies. The cell permeability of [89Zr]Zr-oxine allows for its rapid, ex vivo cellular internalization and subsequent retention [38]. 89Zr-labeled cells can then be administered in vivo for PET imaging. An interesting study outlining this concept was conducted by Asiedu et al. to image the trafficking of bone marrow cells in an animal model of hematopoietic stem cell transplantation (HSCT) [39]. The radiolabeling efficiency was determined to be 26–30%, producing cells with a specific activity of 16.6 kBq/106 cells. Critically, this study revealed that labeling the bone marrow cells did not alter their cellular phenotype, cellular function, or survival. Interestingly, the authors report that the 89Zr-labeled cells quickly accumulated in the lungs, bone marrow, spleen, and liver immediately after bone marrow transfer.
The radiolabeling of the surface of cells is an alternative to intracellular labeling with [89Zr]Zr-oxine. Bansal et al. used DFO-Bz-NCS to covalently tag random lysine groups on the surface of stem cells to facilitate labeling with 89Zr [40]. In this study, the cell labeling efficiency was 30–50%, producing cells with specific activities of up to 0.5 MBq/106 cells. Efflux studies showed that the zirconium-89 remains associated with the cells after 7 days of incubation in 10% fetal bovine serum in cell medium. Furthermore, the authors conducted a subcellular fractionation study to confirm that the 89Zr remained associated with the membrane. The 89Zr-labeled stem cells in this study accumulated in the lungs (50 ± 27 %ID/g), liver (27 ± 19 %ID/g), and bone (16 ± 5 %ID/g). The authors also comment that the exploration of more stable chelators warrants further investigation.
Label-Free Vectors
The term “label-free” refers to the radiolabeling of vectors without the need for conjugating chelators such as DFO. For 89Zr, iron-complexing molecules or nanoparticles containing hard oxygen donors may be used to directly coordinate this radionuclide within the vector. For example, melanin is a familiar molecule, known for contributing to the pigment of human skin, eyes, and hair. Interestingly, it also contributes to various biological functions, including chelating metal ions such as iron. For this reason, melanin was developed as a water-soluble nanoparticle (7 nm) as an alternative to DFO for the treatment of iron overload [41]. Zhang et al. reasoned that the large molecular weight of melanin nanoparticles (MP) could combine with their intrinsic ability to coordinate iron to produce a slowly eliminated platform for the in vivo scavenging of excess iron. These nanoparticles would stand in stark contrast to DFO, which must be administered frequently during treatment due to its rapid clearance (t1/2 = 5 min in mice). Thus, the goal of these researchers was to determine the pharmacokinetic properties of melanin nanoparticles via radiolabeling them with zirconium-89 for PET imaging [41]. In this study, the MP were labeled with zirconium-89 at pH 5 at 37 °C for 30 min, yielding [89Zr]Zr-MP with a radiochemical yield of 90%, a specific activity of 190 MBq/μmol, and a radiochemical purity of >98%. In vitro stability studies in human plasma and PBS determined that the [89Zr]Zr-MP were stable up to 48 h. Subsequent in vivo pharmacokinetic studies using a two-compartment open model determined that the elimination half-life of [89Zr]Zr-MP in mice was ~16 h. Notably, [89Zr]Zr-MP had significant accretion in the liver and spleen at 48 h post-injection, promising result given that in patients, excess iron is predominantly deposited in the liver, spleen, and bone marrow.
Another example of a “label-free” vector is Feraheme ® (FH) , an ultrasmall superparamagnetic iron oxide (USPIO) nanoparticle that is FDA-approved for the treatment of anemia. FH (MW ~ 796 kDa; 17–31 nm) can be labeled directly with various radiometals—including 89Zr—under high temperature with thermodynamic and kinetic stability [42]. The radiolabeling of FH with zirconium-89 was performed in aqueous solution at pH 8.0 via heating at 120 °C for less than 1 h, ultimately producing the radiolabeled particles in 93 ± 3% radiolabeling yield with radiochemical purity of >98%. Boros et al. observed that loadings of Zr greater than a Zr:FH molar ratio of 125:1 resulted in the aggregation of the particles. However, keeping this ratio below this threshold allowed the particles to retain their physical, chemical, and magnetic properties. This radiotracer was investigated in animal models of inflammation in the muscle, producing results which suggest that [89Zr]Zr-FH is a promising radiotracer for the detection of inflammation and other diseases in which activated macrophages play a crucial role. However, more investigation is needed to elucidate the mechanism of the binding of zirconium-89 to FH. Other nanoparticles that have been used as platforms for label-free 89Zr-radiopharmaceuticals include paramagnetic octreotide-liposomes [43], nanodots [44], and mesoporous silica nanoparticles [45].
New Chelators for 89Zr
The Zr-DFO complex is stable against decomplexation when monitored for 7 days in PBS and serum at 37 °C [46]. However, the in vivo decomplexation of the radiometal from DFO-bearing biomolecules has been observed in several cases. When free in the body, [89Zr]Zr4+ is a bone-seeking radiometal, a trait which undermines its ability to resolve true-positive osseous lesions [2, 3]. The deposition of Zr-89 in bone tissue further restricts the dose of the imaging probe that can be administered due to the potential radiation dose to the radiosensitive bone marrow [3]. For this reason, several laboratories have developed new chelators for 89Zr with the goal of improving the in vivo kinetic and thermodynamic stability of the 89Zr-chelator complex. In theory, the ideal chelator for 89Zr should be octadentate, provide hard Lewis acid donor atoms, and be able to “wrap” or encapsulate around the cation’s 85 pm radius [1, 47].
Several novel chelators for [89Zr] are reviewed in the following section. They are divided into three categories according to their donor groups and structure: (i) linear hydroxamates, (ii) macrocyclic hydroxamates, and (iii) non-hydroxamate ligands. The intrinsic properties of the chelators—such as their hydrophilicity, their affinity for [89Zr]Zr4+, and their susceptibility to the transchelation of [89Zr]Zr4+ in the presence of EDTA, DTPA, and serum—are discussed (Table 4). In addition, their performance when conjugated to a protein or peptide (when available from the literature) is addressed as well (Table 5).
Linear Hydroxamates
Desferrioxamine★ (DFO★)
DFO★ was developed in response to DFO’s inability to provide an octadentate coordination environment for Zr4+ [48]. In DFO★, an additional hydroxamic acid is appended onto the amine terminus of DFO. This creates a ligand with four hydroxamic acid moieties that can satisfy Zr(IV)’s preference for a coordination number of 8 (Fig. 9). Issues of solubility were reported for DFO★, but they were largely resolved by using organic cosolvents. The complexation of [89Zr]Zr4+ by DFO★ is driven by incubation at elevated temperatures, resulting in a compound with a 1:1 ratio of Zr:DFO★. Isomerization was observed, though this is typical of linear chelators which are not as rigid as macrocycles. After conjugating DFO★ to the GRPR-targeting peptide bombesin and radiolabeling the resulting construct with 89Zr, the authors challenged the stability of [89Zr]Zr-DFO★-bombesin against 300- and 3000-fold excesses of DFO mesylate. Remarkably, the [89Zr]Zr-DFO★ complex remained stable against transchelation after 24 h.

Structure of Zr-DFO★ complex
In this proof-of-concept work, the bifunctional chelator DFO★-p-phenyl-isothiocyanate (DFO★-p-Phe-NCS) was prepared --> and attached to three different antibodies: trastuzumab (anti-HER2), cetuximab (anti-EGFR), and rituximab (anti-CD20) [47]. The insolubility of DFO★-p-Phe-NCS in aqueous solution necessitated its dissolution in dimethyl sulfoxide (DMSO), which in turn led to modifications to the conjugation protocol aimed at reducing protein aggregation due to the presence of the organic solvent. In addition, rather than adding the DFO★ to the solution of antibody, the antibody was added to the DMSO solution of DFO★. Perhaps as a result, lowered chelator:mAb ratios were achieved for DFO★ compared to analogous conjugation reactions with DFO: 0.6 ± 0.1 vs. 0.9 ± 0.1 for trastuzumab and 0.8 ± 0.1 vs. 1.3 ± 0.1 for rituximab).
Importantly, however, the radiochemical yields obtained with each were comparable (>80%). The stability of [89Zr]Zr-DFO★-trastuzumab was interrogated by monitoring the radiochemical purity and immunoreactivity of the radioimmunoconjugate after storage in 20 mM histidine/240 mM sucrose (93% and 90%, respectively, after 168 h at 4 °C), 0.9% NaCl (89% and 82%, respectively, after 168 h at 4 °C), and serum (96% and 88%, respectively, after 168 h at 37 °C). In all conditions, [89Zr]Zr-DFO★-trastuzumab outperformed [89Zr]Zr-DFO-trastuzumab. The blood-pool residence times and tumoral activity concentrations in mice-bearing N87 xenografts were found to be similar between the two radioimmunoconjugates. Critically, however, decreased bone uptake was observed with the DFO★-bearing radioimmunoconjugate. One interesting observation discussed by Vugts et al. is the attenuated ability of 89Zr-labeled p-NCS-Bn-DFO to covalently bind to antibodies. The authors postulate that this may result from the metal center’s recruitment of the isothiocyanate to fill its coordination sphere, leaving the isothiocyanate unavailable to react with free amines on the antibody.
DFO-HOPO
The addition of a 1,2-hydroxypyridinone (HOPO) moiety to DFO was reported in 1988 by White et al. as a ligand for the octadentate complexation of Pu(IV) [49]. The rationale behind the choice of HOPO stemmed from its relatively high acidity, monoprotic nature, and aqueous solubility compared to catechols and hydroxamates, properties that should facilitate facile radiometallation [49]. Because it offers eight possible donor atoms, DFO-HOPO is an appealing ligand for Zr-89. Allott et al. reported the investigation of alternative synthetic routes for this chelator and explored the radiochemistry and stability of its complex with zirconium-89 (Fig. 10) [50]. The authors employed radiolabeling conditions similar to those used with DFO, forming [89Zr]Zr-DFO-HOPO in ~99% radiolabeling efficiency and a specific activity of ~ 20 MBq/nmol after incubation at room temperature and neutral pH (~7.4). However, two species—with Rf values of 0.1 and 0.6—were observed upon analysis of the radiolabeling reaction mixture via instant thin-layer chromatography (iTLC). The species at Rf ≈ 0.1 was identified as the positively charged kinetic product of the complexation reaction, while the species at Rf ≈ 0.6 was determined to be the neutral, more thermodynamically stable product of the reaction. The conversion of this kinetic product to its more thermodynamically stable cousin seemed to be related to both time and specific activity. More specifically, while heating the reaction to 80 °C does not prevent the formation of the kinetic product, lowering the specific activity of the complex and increasing the incubation time (~24 h at ambient temperature) yielded the neutral species exclusively.

DFO-HOPO coordinates Zr4+ with eight donors
The stability of the [89Zr]Zr-DFO-HOPO complex was assessed via incubation in solutions of EDTA, DFO, and mouse serum at 37 °C. Over 7 days, no demetallation of [89Zr]Zr-DFO-HOPO was observed under all three conditions, while [89Zr]Zr-DFO was observed to decompose in the presence of both EDTA and mouse serum. DFO-HOPO was also able to effectively transchelate [89Zr]Zr4+ from [89Zr]Zr-DFO, removing over 60% of the radiometal in only an hour of incubation. In vivo biodistribution studies revealed that [89Zr]Zr-DFO-HOPO undergoes both rapid renal clearance and slow biliary and intestinal elimination. The authors believe that the inclusion of a hepatobiliary clearance pathway can be attributed to the increased hydrophobicity of [89Zr]Zr-DFO-HOPO (log DpH 7.4 ~ −0.9 ± 0.3) compared to [89Zr]Zr-DFO (log DpH 7.4 ~ −3.0 ± 0.01).
3,4,3-(L1-1,2-HOPO) (L1)
Deri et al. investigated the zirconium-chelating properties of L1, a ligand composed of four HOPO groups attached to a spermine backbone (Fig. 11) [51]. The complexation of [89Zr]Zr4+ with L1 is achieved under relatively standard conditions: pH ~ 7 and room temperature within 10 min. As with DFO-HOPO, the authors observed distinct kinetic and thermodynamic products whose formation seemed dependent on the concentration of the ligand. The authors proposed that this phenomenon could be due to either the formation of polynuclear dimers or isomers. Interestingly, the kinetic product was not observed when using macroscale amounts during the synthesis of cold natZr-L1. X-ray diffraction of the solid-state structure of natZr-L1 established that the metal was surrounded by an octacoordinate coordination environment composed of the eight oxygen donor atoms of the HOPO [52]. The stability of [89Zr]Zr-L1 was tested against a 100-fold excess EDTA, and the radiolabeled ligand remained >99% over 7 days; [89Zr]Zr-DFO, in contrast, remained only 90 ± 5% intact at 7 days. This EDTA challenge experiment was also performed at pH values of 5–8, and in each case, the stability of [89Zr]Zr-L1 proved superior to that of [89Zr]Zr-DFO. In vivo biodistribution assays revealed that [89Zr]Zr-DFO and [89Zr]Zr-L1 boast almost identical pharmacokinetic profiles, though [89Zr]Zr-L1 produced higher activity concentrations in the hepatobiliary tissues (e.g. liver, gallbladder and intestines), a newly observed route of clearance. The uptake of the radiometal in the bone at 24 h post-injection was also slightly elevated for [89Zr]Zr-L1 (0.2 ± 0.03 %ID/g) compared to [89Zr]Zr-DFO (0.06 ± 0.01 %ID/g), an observation the authors believe to be due to longer blood residency of [89Zr]Zr-L1.

3,4,3-(L1–1,2-HOPO) (L1)—composed of four HOPO moieties linked to a spermine backbone—is an octadentate ligand for Zr
The functionalization of L1 with p-benzyl-isothiocyanate created the bifunctional chelator—p-SCN-Bz-HOPO—that could be easily attached to antibodies and other proteins [52]. As a proof-of-concept, the authors conjugated p-SCN-Bz-HOPO to trastuzumab and subsequently radiolabeled the immunoconjugate with 89Zr. The radiochemistry was performed according to standard protocols, and comparable specific activities (74 MBq/mg) and immunoreactivities were achieved for both [89Zr]Zr-L1-trastuzumab (92 ± 6.8%) and [89Zr]Zr-DFO-trastuzumab (89 ± 2.1%). However, in vitro serum stability studies revealed that the stability of [89Zr]Zr-L1-trastuzumab was slightly lower than that of [89Zr]Zr-DFO-trastuzumab (89% vs. 95% respectively). Next, in vivo and pharmacokinetic studies in HER2-positive BT-474 xenografts were conducted to compare the in vivo behavior of both constructs. Tissue distribution data showed that the HOPO-containing radiotracer produced lower tumor uptake values than the DFO-bearing construct (61.9 ± 26.4 %ID/g vs. 138 ± 35.3 %ID/g at 336 h p.i.). The authors offered no discussion for this variability, though it is worth pointing out that L1 appears to be more hydrophobic than DFO due to the presence of the aromatic HOPO. This hydrophobicity may affect the pharmacokinetics, tumor penetration, and vascular extravasation of the radioimmunoconjugate depending on the number of L1 chelators conjugated to the antibody. However, the activity concentrations in the bone were significantly lower for [89Zr]Zr-L1-trastuzumab (2 ± 0.3 %ID/g at 336 h p.i.; tumor:bone ratio ~ 26) compared to the [89Zr]Zr-DFO-trastuzumab (17 ± 4.1 %ID/g at 336 h p.i.; tumor:bone ratio ~ 8) h p.i. Taken together, these data—most notably the lowered activity concentrations in the bone—suggest that L1 may be worth exploring as a potential alternative to DFO.
BPDET-LysH22,2-3-HOPO (L2)
L2 is an octadentate, bi-macrocyclic bifunctional ligand that incorporates two 3,2-HOPO moieties that form a “clam shell”-like structure (Fig. 12) [53]. Its design was predicated on combining the rigidity of a macrocyclic chelator with the 89Zr-binding properties of HOPO moieties. The incubation of the radiometal and the ligand for 15 min at ambient temperature successfully produced [89Zr]Zr-L2 with a specific activity of ~ 0.9 GBq/μmol and a Log P value of ~ 1.5 ± 0.03, making [89Zr]Zr-L2 more hydrophilic than [89Zr]Zr-DFO (Log P ~ −2.8 ± 0.04). Stability challenge experiments were conducted using both diethylenetriaminepentaacetic acid (DTPA, 50 mM) and human serum for 7 days at 37 °C. [89Zr]Zr-L2 was stable after 1 day of incubation in the presence of DTPA, but only 78% of the complex remained intact after 7 days. The demetallation of [89Zr]Zr-L2 was also studied in serum: 94% of the complex remained intact after 1 day, a value that decreased to 86% after 7 days. Biodistribution studies in healthy mice revealed that the blood clearance of [89Zr]Zr-L2 was relatively slower than the DFO complex. Furthermore, the retention of [89Zr]Zr-L2 in the kidneys was substantially elevated compared to [89Zr]Zr-DFO: 30 ± 7 %ID/g and 15 ± 2 %ID/g for the former and 1.0 ± 0.1 %ID/g and 0.7 ± 0.1 %ID/g for the latter at 1 and 7 days post-injection, respectively. The activity concentrations in the bone were also higher for [89Zr]Zr-L2 compared to [89Zr]Zr-DFO, with the former producing 0.3 ± 0.1 %ID/g in the bone after 1 day p.i., a value which was sustained after 7 days (0.3 ± 0.1 %ID/g) (see Table 3).

BPDET-LysH22,2–3-HOPO (L2) is a bi-macrocyclic ligand that incorporates 3,2-HOPO groups and displays a “clam shell” appearance. When bound to Zr, an eight-coordinate complex is formed
The authors proceeded to characterize the in vitro and in vivo stability of L2 as part of a trastuzumab-based radioimmunoconjugate . [89Zr]Zr-L2-trastuzumab displayed a 50% decrease in the amount of intact radioimmunoconjugate after 24 h in mouse serum. Biodistribution studies revealed that [89Zr]Zr-L2-trastuzumab had a shorter blood residence time than [89Zr]Zr-DFO-trastuzumab, as well as elevated levels of liver uptake (13 ± 4.4 %ID/g and 6.5 ± 2.2 %ID/g, respectively, at 6 days p.i.). Furthermore, the PET images produced by [89Zr]Zr-L2-trastuzumab revealed that the radioimmunoconjugate produces higher activity concentrations in the bone (15 ± 2.7 %ID/g at 6 d pi.) compared to [89Zr]Zr-DFO-trastuzumab (11 ± 1.0% ID/g, p = 0.0003). Clearly, the substandard in vivo performance of L2 makes it a less-than-desirable chelator of 89Zr.
Tetrahydroxamate Chelators (L3 and L4)
A pair of tetrahydroxamate-based chelators with different linkers (L3 = −CH2; L4 = −CH2CH2−) were prepared by Rousseau et al. for the purpose of providing 89Zr with a symmetrical and compact coordination environment (Fig. 13) [54]. The two chelators were functionalized with a p-SCN-Bn group and conjugated to trastuzumab, and the in vitro and in vivo stability of the resulting 89Zr-labeled radioimmunoconjugates were investigated. Radiolabeling yields of >90% were obtained for [89Zr]Zr-L3-trastuzumab, [89Zr]Zr-L4-trastuzumab, and [89Zr]Zr-DFO-trastuzumab. Regrettably, stability assays in mouse plasma revealed that neither [89Zr]Zr-L3-trastuzumab nor [89Zr]Zr-L4-trastuzumab was particularly stable, with 75% and 54% of 89Zr released from L3 and L4, respectively, within 1 day of incubation. The demetallation of 89Zr from each chelator was also evident in vivo: [89Zr]Zr-L3-trastuzumab and [89Zr]Zr-L4-trastuzumab produced activity concentrations of 19.5 ± 3.6 %ID/g and 18.3 ± 2.9 %ID/g, respectively, in the bone after 3 days p.i., while the bone uptake from [89Zr]Zr-DFO-trastuzumab was only 7.6 ± 0.4 %ID/g in the same experiment. Ultimately, the authors attributed the release of the radiometal from both chelators to the steric constraints put upon the octacoordinate complex by the short spacer arms of L3 and L4.

Tetrahydroxamate chelators L3 (X = CH2) and L4 (X = CH2CH2)
Tripodal Tris(Hydroxypyridinone) (L5)
In another case, three 1,6-dimethyl-3-hydroxypyridin-4-one moieties were attached to form a tripodal HOPO ligand (Fig. 14) [55]. While this chelator was originally meant as a coordination scaffold for hard, trivalent metals such as Ga(III) and Fe(III), Ma et al. explored extending its utility to zirconium-89. The incubation of natZr4+ and L5 produced a mononuclear species—natZr-L5—with an overall +1 charge that could be observed via mass spectrometry [56]. A competition assay using a ten-fold excess Fe(III) was conducted against both [89Zr]Zr-L5 and [89Zr]Zr-DFO at room temperature for 20 min. Over this time period, >85% of the 89Zr dissociated from the [89Zr]Zr-L5 complex, compared to only ~7% of 89Zr from [89Zr]Zr-DFO. Not to be deterred, the authors functionalized L5 with maleimidopropionate and conjugated the bifunctional chelator to trastuzumab. The radiolabeling of [89Zr]Zr-L5-trastuzumab proceeded according to standard methods, and the incubation of [89Zr]Zr-L5-trastuzumab and [89Zr]Zr-DFO-trastuzumab in serum over 7 days revealed that both constructs remained >95% intact, though some aggregation was observed for the former. Surprisingly, in vivo biodistribution studies showed that [89Zr]Zr-L5-trastuzumab was cleared rapidly by the renal system, with the majority of the injected dose (55–75%) accreting in the bladder after 1 h p.i. [89Zr]Zr-L5-trastuzumab also produced very high activity concentrations in the bone: 29 ± 3.3 %ID/g at 3 days and 26 ± 0.6 %ID/g at 7 days p.i. The demetallation of the [89Zr]Zr-L5 complex is believed to result from the kinetic lability and fluxional behavior of the complex in solution, though the intraconversion of the complex between multiple isomers could also play a role in its rapid dissociation.

Tripodal tris(hydroxypyridinone) (L5) is composed of three 1,6-dimethyl-3-hydroxypyridin-4-one moieties that form a tripodal ligand that is a hexacoordinate chelator for zirconium-89
Macrocyclic Hydroxamates
Fusarinine (L6)
L6 (Fig. 15a) and its triacetylated analog TAFC (L6-COCH3) are produced by the fungus Aspergillus fumigatus for the remediation of iron [57]. The 36-membered cyclic backbone of FSC contains three hydroxamic acid moieties for the coordination of metal centers. Both L6 and TAFC formed hexadentate complexes with [89Zr]Zr4+ after incubation at room temperature at pH ~6.8–7.2 between 30 and 90 min. A specific activity of ~25 GBq/μmol was achieved for [89Zr]Zr-TAFC. L6 was subsequently conjugated to a succinic anhydride-functionalized cyclic RGD peptide (L6-(RGD), Fig. 15b) by forming an amide bond with a free amine on L6’s backbone. The radiolabeling of L6-RGD with zirconium-89 proceeded in a similar fashion to that of TAFC, yielding the product in >90% radiochemical yield after 60 min of incubation at room temperature. No specific activities were reported for either [89Zr]Zr-L6 or the [89Zr]Zr-RGD construct. The stability of [89Zr]Zr-TAFC and [89Zr]Zr-L6-RGD were explored via challenge experiments against EDTA and DFO and compared to that of [89Zr]Zr-DFO as the standard [58]. [89Zr]Zr-TAFC showed high stability in the presence of a 1000-fold excess of EDTA, with ~97% of the metal complex remaining intact after 7 days of incubation. In the presence of DFO at pH~6, however, the transchelation of [89Zr]Zr4+ from [89Zr]Zr-TAFC occurred as early as 4 h post-incubation. After this period, ~92% of the complex remained intact, a value which decreased to ~74% after a full day and ~40% after a week. [89Zr]Zr-L6-RGD similarly demonstrated stability in EDTA (pH~7) even after 7 days of incubation (~94% of the metal complex intact), though no studies were performed using [89Zr]Zr-DFO-RGD for the sake of comparison. No data were provided on the performance of the radiolabeled L6 in the EDTA and DFO challenge experiments. Furthermore, no DFO challenge was reported for radiolabeled L6-RGD construct. [89Zr]Zr-TAFC demonstrated fast in vivo pharmacokinetics with minimal blood residency (0.05 ± 0.01 %ID/g) at 6 h p.i. The complex also produced minimal bone uptake at this time point (0.04 ± 0.02 %ID/g), and the activity concentrations in the spleen, kidneys, liver, and intestines were all <1.5 %ID/g at 6 h p.i. as well. Biodistribution data were also reported for [89Zr]Zr-L6-RGD; however, no comparisons were made against a 89Zr-labeled variant of DFO-RGD nor [89Zr]Zr-L6, experiments which could have made the study more meaningful within the context of this chapter.

(a) Fusarinine (L6) is a macrocyclic chelator comprised of three hydroxamic acids that can coordinate Zr in a hexadentate manner; (b) L6 can be attached to RGD through an amide linker
Both TAFC and L6 offer unique advantages with regard to the coordination of 89Zr. To wit, TAFC can bind [89Zr]Zr4+ at pH 1–5, a significant departure from the established protocols for other chelators such as DFO. In addition, the multiple amines of L6 open the door for simultaneously conjugating the chelator to as many as three vectors, making the creation of multimers possible. Ultimately, L6 and TAFC undoubtedly show some promise for the coordination of 89Zr. To date, however, the chelators have only been used for the construction of peptide-based radioconjugates, and the use of 89Zr with peptide-based vectors is of dubious merit. The performance of both chelators with radioimmunoconjugates certainly warrants exploration.
Hydroxamate-Functionalized Cyclams (L7)
The cyclen 1,4,7,10-tetraazamacrocyclododecane and cyclam 1,4,8,11-tetraazacyclotetradecane were separately derivatized with three and four pendant hydroxamate arms to afford chelators with six and eight oxygen donors, respectively [59]. The radiolabeling of these complexes with [89Zr]Zr4+ was performed in typical fashion via incubation at room temperature over 1 h at pH ~ 7.4–7.6. When the radiometal complexes were subjected with a 55 mM EDTA challenge and monitored over 144 h at 37 °C, L7 (Fig. 16)—which possesses long hydroxamate arms—was observed to be the most promising chelator and was selected for further comparison against DFO. The log D values of [89Zr]Zr-L7 and [89Zr]Zr-DFO are comparable at −3.4 vs. −3.1, respectively. Furthermore, both [89Zr]Zr-L7 and [89Zr]Zr-DFO performed similarly when challenged with EDTA, with 91 ± 2% and 87 ± 1% of the complexes remaining intact after 6 days of incubation. Furthermore, [89Zr]Zr-L7 (94% intact) proved more inert than [89Zr]Zr-DFO (53% intact) after 72 h incubation in rat plasma assays. An in vivo pharmacokinetic analysis of [89Zr]Zr-L7 and [89Zr]Zr-DFO reveals that the two complexes have similar serum half-lives and are primarily excreted via the renal system. However, the uptake in the bone produced by [89Zr]Zr-L7 (0.60 ± 0.19 %ID/g at 24 h p.i.) was markedly higher than that created by [89Zr]Zr-DFO at the same time point (0.05 ± 0.02 %ID/g). DFT in silico analysis validated the thermodynamic stability of [89Zr]Zr-L7, with the ligand providing a favorable coordination geometry with little steric strain.

Hydroxamate-functionalized cyclam (L7) is a macrocyclic construct modified with longer tri-hydroxamate arms. It provides the most stable coordinating properties for Zr4+ compared to other hydroxamate-modified cyclens and cyclams
L7 was functionalized with an N-hydroxy-succinimidyl ester to create a bifunctional chelator capable of being attached to antibodies like trastuzumab. Imaging and biodistribution studies in mice bearing both HER2-positive and HER2-negative tumors revealed that [89Zr]Zr-L7-trastuzumab and [89Zr]Zr-DFO-trastuzumab displayed generally similar in vivo behavior. The former, however, produced much higher activity concentrations in the bone than the latter. For example, at 96 h post-injection, the activity concentrations in the bone for [89Zr]Zr-L7-trastuzumab and [89Zr]Zr-DFO-trastuzumab were 19.0 ± 2.1 %ID/g and 2.8 ± 2.2 %ID/g, respectively.
Non-hydroxamates
DOTA (L8), DOTP (L9), and DOTAM (L10)
Tetraazamacrocycles such as 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, L8, Fig. 17a) and variants of DOTA with phosphate- (DOTP, L9, Fig. 17b) and amine- (DOTAM, L10, Fig. 17c) derivatized arms have also been investigated for their ability to coordinate zirconium-89 [60]. Unlike acyclic ligands, these macrocycles required heating at 90 °C at 45 min to sufficiently encapsulate the radiometal. This need for heating puts these chelators at a disadvantage in the context of biomolecular vectors, because antibodies and proteins generally cannot tolerate high temperatures. Furthermore, differences in the complexation of [89Zr]Zr4+ have been observed when using [89Zr]Zr-oxalate and [89Zr]Zr-Cl4 as the starting material, with the former providing lower yields, possibly as a result of the competition between the ligand and the oxalates in solution. Competition assays with excess EDTA revealed that the complexes observe the following decreasing ordering of stability: [89Zr]Zr-L8 > [89Zr]Zr-L9 > [89Zr]Zr-L10 = [89Zr]Zr-DFO. Challenging [89Zr]Zr-L8, [89Zr]Zr-L9, and [89Zr]Zr-L10 with excess Fe(III) and Ga(III) did not yield any appreciable loss of [89Zr]Zr4+ from the macrocycles, while similar metal competition experiments with [89Zr]Zr-DFO resulted in the partial dissociation of the radiometal from DFO.

(a) DOTA (L8), (b) DOTP (L9), and (C) DOTAM (L10) are tetraazamacrocycles with carboxylic acid-, phosphate-, and amine-bearing pendant arms, respectively, that have been explored for the coordination of zirconium-89
The in vivo behavior of [89Zr]Zr-L8, [89Zr]Zr-L9, [89Zr]Zr-L10, and [89Zr]Zr-DFO were compared via biodistribution experiments. High levels of [89Zr]Zr-L10 were observed and retained in the liver and spleen over the course of 3 days, primarily due to the aggregation of the complex and its precipitation with serum proteins. Similar biodistribution profiles—i.e. accumulation in the blood, liver, and bone—were observed for both [89Zr]Zr-L8 and [89Zr]Zr-DFO within the first hour p.i., with the latter retained in the kidneys at 4 h p.i.. Compared to these two complexes, [89Zr]Zr-L9 exhibited higher accretion in the blood, liver, kidneys, and bone at 72 h p.i. Notably, [89Zr]Zr-L8 produced lower activity concentrations in the bone (0.03 ± 0.009 %ID/g) than both [89Zr]Zr-L9 (2.6 ± 0.1 %ID/g) and [89Zr]Zr-DFO (0.08 ± 0.01 %ID/g). Pandya et al. rationalized that the higher uptake of [89Zr]Zr-L9 in the bone may be due to transchelation and the influence of the ligand’s four phosphate moieties (which could theoretically cause the deposition of the intact complex onto bone tissue). This has been observed for other metals complexed by phosphate-containing macrocycles. Moreover, the authors found that L8 appears to provide superior kinetic inertness and in vivo stability against transchelation compared to other reported Zr chelators, including TAFC, L1, and L2. In retrospect, the main limitation to L8 is the requirement for elevated temperatures when radiolabeling, which can be circumvented by exploring other synthetic methodologies such as “click” chemistry.
2-Hydroxyisophthalamide (L11-L12)
Four 2-hydroxylisophthalamide (IAM) moieties were fused to form the octadentate ligands L11 (Fig. 18a) and L12 (Fig. 18b) [61]. IAMs possess phenolic and carbonyl oxygen donor groups that preferentially bind hard metals and were chosen for these chelators based on their similarities to lanthanide-sequestration moieties produced by bacteria. The coordination of zirconium-89 by L11 and L12 was performed in water at a pH~7.0–7.5. Because of L11’s rigidity, it was incubated with zirconium-89 for 2 h at a relatively high temperature, 95 °C. L12 is less rigid but was still heated at 50 °C for 1 h to induce the coordination of zirconium-89. The Log P values for natZr-L11 and natZr-L12 are −2.97 ± 0.02 and − 1.45 ± 0.06, making both more hydrophilic than [89Zr]Zr-DFO.

Four 2-hydroxylisophthalamide (IAM) chelating units were combined to form the octadentate ligands (a) L11 and (b) L12
Stability studies in the presence of 50 mM DTPA suggested that [89Zr]Zr-L11 is more inert to transchelation than both [89Zr]Zr-L12 and [89Zr]Zr-DFO, though parallel experiments in serum somewhat curiously revealed that [89Zr]Zr-DFO (100% intact after 7 days) is more stable than [89Zr]-Zr-L11 (~75.1% intact) or [89Zr]-Zr-L12 (~17% intact) in serum over 7 days. In vivo biodistribution experiments revealed that [89Zr]Zr-L12 had a higher retention in the blood after 1 day than either [89Zr]Zr-L11 or [89Zr]Zr-DFO, with uptake values of 0.01 ± 0.006 %ID/g, 0.003 ± 0.000 %ID/g, and 0.001 ± 0.001 %ID/g, respectively. Furthermore, both [89Zr]Zr-L11 (0.38 ± 0.04 %ID/g) and [89Zr]Zr-L12 (1.52 ± 0.15 %ID/g) were observed to accumulate in the liver to greater degrees than [89Zr]Zr-DFO (0.081 ± 0.012 %ID/g). Finally, and perhaps most importantly, [89Zr]Zr-DFO was observed to produce lower activity concentrations in the bone (0.078 ± 0.014 %ID/g) after 3 days than either of the two IAM-based chelators (0.1 ± 0.01 %ID/g for [89Zr]Zr-L11 and 0.68 ± 0.33 %ID/g for [89Zr]Zr-L12). These disappointing in vitro and in vivo results seem to disqualify both L11 and L12 as potential chelators of zirconium-89.
The Future
Over the last decade, advances in the production of zirconium-89 as well as the development of chelators to stably sequester the radiometal have fueled the advent of 89Zr-based PET imaging. Perhaps not surprisingly, this rise in 89Zr-based PET has run parallel to a growth in the importance of—and appreciation for—the role of antibodies as vectors for diagnostic and theranostic nuclear imaging. Today, DFO remains the “gold standard” chelator for 89Zr and has been employed in all clinical trials to date. Yet the stability of the [89Zr]Zr-DFO complex in preclinical models has motivated a number of investigations aimed at developing new chelators for the radiometal. A wide range of promising alternatives have been developed, though as we embark on these discoveries, we should be sure to evaluate both the chelator’s toxicity and—even more importantly—its potential to change the pharmacokinetic properties of its targeting vector. After all, we should not lose sight of the big picture: ultimately, our overarching goal is to use 89Zr to track targeting vectors in order to accurately stage disease and predict and monitor the response of patients to therapy.
The Bottom Line
In this chapter, we have tried to provide an overview of the production, chemistry, and radiochemistry of zirconium-89 as well as several techniques to consider when working with the radiometal. While a variety of novel chelators have been developed for zirconium-89, only a handful have matched or exceeded DFO’s ability to coordinate the radiometal. We understand that this chapter—like all those in this textbook—has provided a great deal of information, so in the end, we sincerely hope the readers keep the following points foremost in their mind.
-
Zirconium-89 is a residualizing, positron-emitting radionuclide whose physical half-life provides an excellent match for the pharmacokinetic profiles of many antibodies.
-
The current “gold standard” chelator for zirconium-89 in both the laboratory and clinic is desferrioxamine (DFO).
-
Over time, [89Zr]Zr-DFO is susceptible to transchelation in the presence of endogenous proteins, producing free [89Zr]Zr4+ that can subsequently deposit in the bones. As a result, the imaging of osseous lesions can become difficult, and the radiosensitive bone marrow can be exposed to nontrivial radiation doses.
-
Zirconium(IV) is a hard metal, and thus hard donor atoms (e.g. oxygens) are necessary for its coordination.
-
The ideal chelator for Zr4+ should be octadentate in order to saturate the metal’s preference for a coordination number of 8. For example, several of the most successful “second-generation” chelators for zirconium-89—e.g. DFO★, DFO-HOPO, and L1—possess four hydroxamate groups that collectively offer eight donor atoms.
-
The hydrophilicity of a chelator plays a role in the whole body clearance of the radiometal-bearing complex.
-
The radiolabeling of antibodies with zirconium-89 must be conducted at or close to neutral pH to preserve the structural integrity of the mAb. As a result, the pKa of the chelator should be within this range. Heating conditions should range from room temperature to 37 °C.
-
Zirconium-89 with high effective specific activity should be employed to achieve near quantitative radiolabeling yields and facilitate the creation of 89Zr-labeled mAbs with high specific activities.
References
Deri MA, Zeglis BM, Francesconi LC, Lewis JS. PET imaging with (8)(9)Zr: from radiochemistry to the clinic. Nucl Med Biol. 2013;40(1):3–14.
Abou DS, Ku T, Smith-Jones PM. In vivo biodistribution and accumulation of 89Zr in mice. Nucl Med Biol. 2011;38(5):675–81.
Ulaner GA, Hyman DM, Ross DS, Corben A, Chandarlapaty S, Goldfarb S, et al. Detection of HER2-positive metastases in patients with HER2-negative primary breast cancer using 89Zr-trastuzumab PET/CT. J Nucl Med. 2016;57(10):1523–8.
Greenwood NN, Earnshaw A. Chemistry of the elements. 2nd ed. Oxford: Elsevier Butterworth-Heinemann; 1997.
Kozak CM, Mountford P. Zirconium & hafnium. Inorganic & coordination chemistry. In: King RB, editor. Encyclopedia of inorganic chemistry. 2nd ed. Chichester/West Sussex: Wiley; 2006.
Intorre BI, Martell AE. Zirconium complexes in aqueous solution. I. Reaction with multidentate ligands1. J Am Chem Soc. 1960;82(2):358–64.
Intorre BJ, Martell AE. Aqueous zirconium complexes. II. Mixed chelates. J Am Chem Soc. 1961;83(17):3618–23.
Ekberg C, Kallvenius G, Albinsson Y, Brown PL. Studies on the hydrolytic behavior of Zr(IV). J Solution Chem. 2004;33(1):47–79.
Conti M, Eriksson L. Physics of pure and non-pure positron emitters for PET: a review and a discussion. EJNMMI Physics. 2016;3:8.
Moses WW. Fundamental limits of spatial resolution in PET. Nucl Instr Methods Phys Res A. 2011;648(Suppl 1):S236–S40.
Disselhorst JA, Brom M, Laverman P, Slump CH, Boerman OC, Oyen WJ, et al. Image-quality assessment for several positron emitters using the NEMA NU 4-2008 standards in the Siemens Inveon small-animal PET scanner. J Nucl Med. 2010;51(4):610–7.
Link JM, Krohn KA, Eary JF, Kishore R, Lewellen TK, Johnson MW, et al. Zr-89 for antibody labeling and positron emission tomography. J Labelled Compd Radiopharm. 1986;23(10–12):1297–8.
Eary JF, Link JM, Kishore R, Johnson MW, Badger CC, Richter KY, et al. Production of positron emitting Zr89 for antibody imaging by PET. J Nucl Med. 1986;27(6):983.
Dejesus OT, Nickles RJ. Production and purification of Zr-89, a potential PET antibody label. Appl Radiat Isot. 1990;41(8):789–90.
Meijs WE, Herscheid JDM, Haisma HJ, Wijbrandts R, Vanlangevelde F, Vanleuffen PJ, et al. Production of highly pure no-carrier added Zr-89 for the labeling of antibodies with a positron emitter. Appl Radiat Isot. 1994;45(12):1143–7.
Holland JP, Sheh Y, Lewis JS. Standardized methods for the production of high specific-activity zirconium-89. Nucl Med Biol. 2009;36(7):729–39.
Wooten A, Madrid E, Schweitzer G, Lawrence L, Mebrahtu E, Lewis B, et al. Routine production of 89Zr using an automated module. Appl Sci. 2013;3(3):593.
Queern SL, Aweda TA, Massicano AVF, Clanton NA, El Sayed R, Sader JA, et al. Production of Zr-89 using sputtered yttrium coin targets 89Zr using sputtered yttrium coin targets. Nucl Med Biol. 2017;50:11–6.
Pandey MK, Engelbrecht HP, Byrne JP, Packard AB, DeGrado TR. Production of 89Zr via the 89Y(p,n)89Zr reaction in aqueous solution: effect of solution composition on in-target chemistry. Nucl Med Biol. 2014;41(4):309–16.
Verel I, Visser GW, Boellaard R, Stigter-van Walsum M, Snow GB, van Dongen GA. 89Zr immuno-PET: comprehensive procedures for the production of 89Zr-labeled monoclonal antibodies. J Nucl Med. 2003;44(8):1271–81.
Guerard F, Lee YS, Tripier R, Szajek LP, Deschamps JR, Brechbiel MW. Investigation of Zr(IV) and 89Zr(IV) complexation with hydroxamates: progress towards designing a better chelator than desferrioxamine B for immuno-PET imaging. Chem Commun (Camb). 2013;49(10):1002–4.
Hoard JL, Glen GL, Silverton JV. The configuration of Zr(C2O4)4-4 and the stereochemistry of discrete eight-coordination. J Am Chem Soc. 1961;83(20):4293–5.
Lin M, Mukhopadhyay U, Waligorski GJ, Balatoni JA, González-Lepera C. Semi-automated production of 89Zr-oxalate/89Zr-chloride and the potential of 89Zr-chloride in radiopharmaceutical compounding. Appl Radiat Isot. 2016;107(Supplement C):317–22.
Bickel H, Gaeumann E, Keller-Schierlein W, Prelog V, Vischer E, Wettstein A, Zaehner H. On iron-containing growth factors, sideramines, and their antagonists, the iron-containing antibiotics, sideromycins. Experientia. 1960;16:129–33. [Article in German]
Bergeron RJ, McManis JS. Reagents for the stepwise functionalization of spermine. J Org Chem. 1988;53(13):3108–11.
Perk LR, Vosjan MJ, Visser GW, Budde M, Jurek P, Kiefer GE, et al. P-Isothiocyanatobenzyl-desferrioxamine: a new bifunctional chelate for facile radiolabeling of monoclonal antibodies with zirconium-89 for immuno-PET imaging. Eur J Nucl Med Mol Imaging. 2010;37(2):250–9.
Vosjan MJ, Perk LR, Visser GW, Budde M, Jurek P, Kiefer GE, et al. Conjugation and radiolabeling of monoclonal antibodies with zirconium-89 for PET imaging using the bifunctional chelate p-isothiocyanatobenzyl-desferrioxamine. Nat Protoc. 2010;5(4):739–43.
Marquez BV, Ikotun OF, Zheleznyak A, Wright B, Hari-Raj A, Pierce RA, et al. Evaluation of 89Zr-pertuzumab in breast cancer xenografts. Mol Pharm. 2014;11(11):3988–95.
Meijs WE, Haisma HJ, Klok RP, van Gog FB, Kievit E, Pinedo HM, et al. Zirconium-labeled monoclonal antibodies and their distribution in tumor-bearing nude mice. J Nucl Med. 1997;38(1):112–8.
Tinianow JN, Gill HS, Ogasawara A, Flores JE, Vanderbilt AN, Luis E, et al. Site-specifically 89Zr-labeled monoclonal antibodies for ImmunoPET. Nucl Med Biol. 2010;37(3):289–97.
Houghton JL, Zeglis BM, Abdel-Atti D, Aggeler R, Sawada R, Agnew BJ, et al. Site-specifically labeled CA19.9-targeted immunoconjugates for the PET, NIRF, and multimodal PET/NIRF imaging of pancreatic cancer. Proc Natl Acad Sci U S A. 2015;112(52):15850–5.
Tavare R, McCracken MN, Zettlitz KA, Salazar FB, Olafsen T, Witte ON, et al. Immuno-PET of murine T cell reconstitution postadoptive stem cell transplantation using anti-CD4 and anti-CD8 cys-diabodies. J Nucl Med. 2015;56(8):1258–64.
Meijs WE, Herscheid JDM, Haisma HJ, Pinedo HM. Evaluation of desferal as a bifunctional chelating agent for labeling antibodies with Zr-89. Int J Rad Appl Instrum A. 1992;43(12):1443–7.
Zeglis BM, Lewis JS. The bioconjugation and radiosynthesis of 89Zr-DFO-labeled antibodies. J Vis Exp. 2015;96.
Kuda-Wedagedara ANW, Workinger JL, Nexo E, Doyle RP, Viola-Villegas N. (89)Zr-cobalamin PET tracer: synthesis, cellular uptake, and use for tumor imaging. ACS Omega. 2017;2(10):6314–20.
Bauman A, Valverde IE, Fischer CA, Vomstein S, Mindt TL. Development of 68Ga- and 89Zr-labeled exendin-4 as potential radiotracers for the imaging of insulinomas by PET. J Nucl Med. 2015;56(10):1569–74.
Ferris TJ, Charoenphun P, Meszaros LK, Mullen GE, Blower PJ, Went MJ. Synthesis and characterisation of zirconium complexes for cell tracking with Zr-89 by positron emission tomography. Dalton Trans. 2014;43(39):14851–7.
Charoenphun P, Meszaros LK, Chuamsaamarkkee K, Sharif-Paghaleh E, Ballinger JR, Ferris TJ, et al. [(89)Zr]oxinate4 for long-term in vivo cell tracking by positron emission tomography. Eur J Nucl Med Mol Imaging. 2015;42(2):278–87.
Asiedu KO, Koyasu S, Szajek LP, Choyke PL, Sato N. Bone marrow cell trafficking analyzed by (89)Zr-oxine positron emission tomography in a murine transplantation model. Clin Cancer Res. 2017;23(11):2759–68.
Bansal A, Pandey MK, Demirhan YE, Nesbitt JJ, Crespo-Diaz RJ, Terzic A, et al. Novel (89)Zr cell labeling approach for PET-based cell trafficking studies. EJNMMI Res. 2015;5:19.
Zhang P, Yue Y, Pan D, Yang R, Xu Y, Wang L, et al. Pharmacokinetics study of Zr-89-labeled melanin nanoparticle in iron-overload mice. Nucl Med Biol. 2016;43(9):529–33.
Boros E, Bowen AM, Josephson L, Vasdev N, Holland JP. Chelate-free metal ion binding and heat-induced radiolabeling of iron oxide nanoparticles. Chem Sci. 2015;6(1):225–36.
Abou DS, Thorek DL, Ramos NN, Pinkse MW, Wolterbeek HT, Carlin SD, et al. (89)Zr-labeled paramagnetic octreotide-liposomes for PET-MR imaging of cancer. Pharm Res. 2013;30(3):878–88.
Cheng L, Kamkaew A, Shen S, Valdovinos HF, Sun H, Hernandez R, et al. Facile preparation of multifunctional WS2 /WOx nanodots for chelator-free (89) Zr-labeling and in vivo PET imaging. Small. 2016;12(41):5750–8.
Chen F, Goel S, Valdovinos HF, Luo H, Hernandez R, Barnhart TE, et al. In vivo integrity and biological fate of chelator-free zirconium-89-labeled mesoporous silica nanoparticles. ACS Nano. 2015;9(8):7950–9.
Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson SM, Lewis JS. 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J Nucl Med. 2010;51(8):1293–300.
Vugts DJ, Klaver C, Sewing C, Poot AJ, Adamzek K, Huegli S, et al. Comparison of the octadentate bifunctional chelator DFO*-pPhe-NCS and the clinically used hexadentate bifunctional chelator DFO-pPhe-NCS for (89)Zr-immuno-PET. Eur J Nucl Med Mol Imaging. 2017;44(2):286–95.
Patra M, Bauman A, Mari C, Fischer CA, Blacque O, Haussinger D, Gasser G, Mindt TL. An octadentate bifunctional chelating agent for the development of stable zirconium-89 based molecular imaging probes. Chem Commun (Camb). 2014;50:11523–5.
White DL, Durbin PW, Jeung N, Raymond KN. Specific sequestering agents for the actinides. 16. Synthesis and initial biological testing of polydentate oxohydroxypyridinecarboxylate ligands. J Med Chem. 1988;31(1):11–8.
Allott L, Da Pieve C, Meyers J, Spinks T, Ciobota DM, Kramer-Marek G, et al. Evaluation of DFO-HOPO as an octadentate chelator for zirconium-89. Chem Commun (Camb). 2017;53(61):8529–32.
Deri MA, Ponnala S, Zeglis BM, Pohl G, Dannenberg JJ, Lewis JS, et al. Alternative chelator for (8)(9)Zr radiopharmaceuticals: radiolabeling and evaluation of 3,4,3-(LI-1,2-HOPO). J Med Chem. 2014;57(11):4849–60.
Deri MA, Ponnala S, Kozlowski P, Burton-Pye BP, Cicek HT, Hu C, et al. P-SCN-Bn-HOPO: a superior bifunctional chelator for (89)Zr immunoPET. Bioconjug Chem. 2015;26(12):2579–91.
Tinianow JN, Pandya DN, Pailloux SL, Ogasawara A, Vanderbilt AN, Gill HS, et al. Evaluation of a 3-hydroxypyridin-2-one (2,3-HOPO) based macrocyclic chelator for (89)Zr(4+) and its use for immunopet imaging of HER2 positive model of ovarian carcinoma in mice. Theranostics. 2016;6(4):511–21.
Rousseau J, Zhang Z, Dias GM, Zhang C, Colpo N, Benard F, et al. Design, synthesis and evaluation of novel bifunctional tetrahydroxamate chelators for PET imaging of (89)Zr-labeled antibodies. Bioorg Med Chem Lett. 2017;27(4):708–12.
Ma MT, Cullinane C, Imberti C, Baguna Torres J, Terry SY, Roselt P, et al. New tris(hydroxypyridinone) bifunctional chelators containing isothiocyanate groups provide a versatile platform for rapid one-step labeling and PET imaging with (68)Ga(3.). Bioconjug Chem. 2016;27(2):309–18.
Ma MT, Meszaros LK, Paterson BM, Berry DJ, Cooper MS, Ma Y, et al. Tripodal tris(hydroxypyridinone) ligands for immunoconjugate PET imaging with (89)Zr(4+): comparison with desferrioxamine-B. Dalton Trans. 2015;44(11):4884–900.
Knetsch PA, Zhai C, Rangger C, Blatzer M, Haas H, Kaeopookum P, et al. [(68)Ga]FSC-(RGD)3 a trimeric RGD peptide for imaging alphavbeta3 integrin expression based on a novel siderophore derived chelating scaffold-synthesis and evaluation. Nucl Med Biol. 2015;42(2):115–22.
Zhai C, Summer D, Rangger C, Franssen GM, Laverman P, Haas H, et al. Novel bifunctional cyclic chelator for (89)Zr labeling-radiolabeling and targeting properties of RGD conjugates. Mol Pharm. 2015;12(6):2142–50.
Boros E, Holland JP, Kenton N, Rotile N, Caravan P. Macrocycle-based hydroxamate ligands for complexation and immunoconjugation of (89)zirconium for positron emission tomography (PET) imaging. ChemPlusChem. 2016;81(3):274–81.
Pandya DN, Bhatt N, Yuan H, Day CS, Ehrmann BM, Wright M, et al. Zirconium tetraazamacrocycle complexes display extraordinary stability and provide a new strategy for zirconium-89-based radiopharmaceutical development. Chem Sci. 2017;8(3):2309–14.
Bhatt NB, Pandya DN, Xu J, Tatum D, Magda D, Wadas TJ. Evaluation of macrocyclic hydroxyisophthalamide ligands as chelators for zirconium-89. PLoS One. 2017;12(6):e0178767.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Marquez-Nostra, B.V., Viola, N. (2019). The Radiopharmaceutical Chemistry of Zirconium-89. In: Lewis, J., Windhorst, A., Zeglis, B. (eds) Radiopharmaceutical Chemistry. Springer, Cham. https://doi.org/10.1007/978-3-319-98947-1_21
Download citation
DOI: https://doi.org/10.1007/978-3-319-98947-1_21
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-98946-4
Online ISBN: 978-3-319-98947-1
eBook Packages: MedicineMedicine (R0)