
Abstract
Familial Mediterranean fever (FMF) is a monogenic autoinflammatory disease which has a high prevalence in the eastern Mediterranean. The typical FMF attack is characterized by fever and polyserositis in the form of abdominal or chest pain or arthritis along with high acute phase reactants. FMF is caused by different mutations in the MEFV gene coding for pyrin. The gain-of-function mutations in the MEFV gene are associated with an exaggerated inflammatory response via an increased production of interleukin-1.
An international group of experts has published the recommendations for the management of FMF. Colchicine is still the mainstay of FMF treatment, and its regular use prevents attacks, controls subclinical inflammation, and decreases the development of amyloidosis which is the most significant complication of FMF. However, a minor proportion of patients can be resistant to colchicine. If colchicine fails, anti-IL-1 therapy is a promising second-line therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Familial Mediterranean fever is the most common autoinflammatory disease (AID) over the world. FMF affects primarily the ethnic groups from the eastern Mediterranean basin such as Turks, Armenians, Arabs, and non-Ashkenazi Jews [1]. FMF is associated with mutations of the MEFV gene on chromosome 16p, which codes for the pyrin protein [2, 3]. Mutated pyrin causes an excessive inflammatory response via uncontrolled production of interleukin (IL)-1 [4]. Although the responsible gene for FMF has been identified, the molecular and genetic studies showed that the pathogenesis, inheritance, and penetrance of FMF are more complicated. It has been 20 years since the gene associated with FMF has been defined. However, the past 20 years have taught us that the chapter is not closed and that there is still much to be investigated.
Clinically FMF is characterized by recurrent and self-limited attacks of fever and polyserositis. The most significant complication of the chronic inflammation in FMF is secondary amyloidosis.
Colchicine is still the mainstay of FMF treatment. The regular use of colchicine can reduce the severity and frequency of inflammatory attacks and suppress chronic subclinical inflammation and prevent complications [5,6,7,8]. Recent advances in molecular studies have led new therapeutic options in FMF. If colchicine fails, anti-IL-1 therapy is a promising second-line therapy.
Genetics
The mutations in the MEFV gene , which are composed of 10 exons, were first described in 1997. The MEFV gene is located on chromosome 16 (16p 13.3) and encodes a protein consisting of 781 amino acids termed pyrin or marenostrin [2, 3, 9], which is a part of the NLRP3 inflammasome complex. Mutant pyrin causes an exaggerated inflammation with excessive production of IL-1 [4]. To date, there are about 300 known sequence variants of MEFV, and all reported mutations and the associated phenotypes are recorded in the INFEVERS database (http://fmf.igh.cnrs.fr/ISSAID/infevers/) [10]. However, some of them are not associated with the FMF phenotype and their significance is not clear. The most common pathogenic mutations worldwide are the p.M694V, M694I, M680I, and V726A. In 2009, Booty et al. showed that screening for the set of most common mutations appears sufficient to diagnose FMF in presence of clinical symptoms [11]. In 2012, Shinar et al. proposed recommendations for interpretation of genetic testing for FMF [12]. The consensus was set to test for 14 variants (the first 9 are defined as clearly pathogenic, and the other 5 are variants of uncertain significance): M694V, M694I, M680I, V726A, R761H, A744S, E167D, T267I, I692del, K695R, E148Q, P369S, F479L, and I591T. In 2015, the SHARE (Single Hub and Access point for pediatric Rheumatology in Europe) initiative has established evidence-based recommendations for genetic diagnosis of FMF [10]. These recommendations are presented in Table 9.1.
The MEFV gene mutations in exon 10 are associated with the most severe form of disease, and genetic variants on exon 2 and on exon 3 are associated with milder form of disease. M694V is the most common mutation, which has been associated with severe disease, early onset, and a higher risk of AA amyloidosis [10, 13]. According to the SHARE recommendations, the patients carrying two mutated alleles in position 680–694 on exon 10 must be considered at risk of having more severe disease [10]. The second most frequent mutation after M694V is the E148Q on exon 2, and its pathogenic role is still controversial. E148Q is a common variant in the general population. Ben-Chetrit et al. showed a similar frequency for E148Q mutation in patients and healthy controls [14]. Tchernitchko et al. demonstrated that E148Q allele frequency was comparable among patients and asymptomatic relatives, and they concluded E148Q as a benign polymorphism [15]. However, some reports suggest that patients with homozygous E148Q might have FMF phenotype [16, 17]. In 2012, Shinar et al. has defined E148Q as a variant of unknown significance in a consensus conference [12], and according to the SHARE recommendations, this fact is also highlighted [10].
The inheritance of FMF is autosomal recessive inheritance. However, a substantial number of clinically FMF patients have only one MEVF mutation . Furthermore, 10–20% of patients who meet the diagnostic criteria for FMF have no demonstrable MEFV mutations [11, 13]. Because of the incomplete penetrance and the varying expression of FMF phenotype, it has been suggested that there are additional factors like modifier genes, epigenetics, or environmental factors affecting the expression of the disease. Marek et al. studied the clinical and genetic features of heterozygous FMF patients and performed haplotype studies. They showed that the heterozygous patients tend to have milder disease, but the disease could not be practically distinguished clinically from that in homozygous patients [18]. Federici et al. suggested a “dose effect” associated with the MEFV gene mutations . They demonstrated an increasing symptom frequency from patients carrying a single low-penetrance mutation to patients with high-penetrance mutations [19]. Lachmann et al. demonstrated that both basal and peak acute phase protein concentrations were higher in MEFV heterozygotes than in wild-type controls, demonstrating a “pro-inflammatory” condition among FMF carriers [20]. In addition, Kalyoncu et al. showed that acute rheumatic fever, arthralgia, and febrile episodes of more than four times per year were more frequent in asymptomatic heterozygous parents of children with FMF [21]. It has been shown that serum amyloid could increase the severity of the symptoms of FMF by activating NLRP3 inflammasome, resulting in excessive IL-1B secretion [22]. Supporting this observation, the presence of SAA1 allele was strongly associated with renal amyloidosis in FMF patients [23, 24].
The first study showing the effect of environment on FMF was in 1974. Schwabe et al. reported that the Armenian FMF patients who lived in the USA did not develop renal amyloidosis [25]. In the same line, Touitou et al. analyzed the features of 2482 FMF patients from 14 different countries, and they concluded that the country recruitment, rather than MEFV genotype, was the most important risk factor for renal amyloidosis in FMF [26]. Our group has shown that Turkish FMF children who live in Germany have a milder disease phenotype compared with the ones living in Turkey [16]. This observation was subsequently confirmed in the Eurofever registry [16].
Pathogenesis
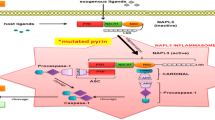
The MEFV gene encodes pyrin , 781 amino acid protein, expressed mainly in cells polymorph nuclear neutrophils, eosinophils, and monocyte series [27]. Pyrin is essential to form inflammasomes, multiprotein complexes playing a major role in both innate and adaptive immune systems [28]. Pyrin consists of three fundamental domains: N-terminal RING domain, B-box domain, and C-terminal coiled-coil domain. It has an additional C-terminal. B30.2/rfp/PRY/SPRY domain is where most of the major disease associated mutations are clustered [29]. Pyrin interacts with the inflammasome adaptor protein (ASC) and modulates caspase-1 and IL-1 activation [30]. Mutation of pyrin causes an exaggerated inflammation via excessive IL-1 production [4]. Until recently there were conflicting results on whether the mutations on MEFV gene were gain-of-function mutations [11, 31] or loss-of-function [32, 33] mutations. In 2011, with the generation of knock-in mice harboring the mouse pyrin protein fused to the human B30.2 domain containing FMF-associated mutations, large amounts of IL-1β secretion have been shown in an ASC and caspase-1-dependent, NLRP3-independent manner [34]. These data [34] and several later studies [35, 36] supported that FMF-associated mutations are gain of function and pyrin itself promotes ASC oligomerization and forms a caspase-1-activating complex.
Twenty years after the identification of the gene, we recently have understood the function of pyrin. Xu et al. have also demonstrated that the modification and inactivation of Rho GTPases by different pathogenic bacteria toxins induce the activation of the pyrin inflammasome [36]. They suggested that pyrin senses a downstream event in the actin cytoskeleton pathway rather than directly recognizing Rho modification [36]. Park et al. has enlightened these mechanisms further. They demonstrated that pyrin is phosphorylated by RhoA-activated serine-threonine kinases (PKN1 and PKN2) and binds to regulatory 14-3-3 protein leading to inactivation of the pyrin inflammasome formation. In the presence of several bacterial toxins and FMF causing MEFV mutations, RhoA is inactivated resulting in a lowered threshold for activation of the pyrin inflammasome [37]. Gao et al. have observed that site-specific dephosphorylation and microtubule dynamics influenced activation of pyrin inflammasome. Targeting drugs, including colchicine, blocked activation of the pyrin inflammasome by inhibiting oligomerization of pyrin with ASC. It did not affect pyrin dephosphorylation and 14-3-3 dissociation [38]. Moreover, Gorp et al. have showed that pyrin inflammasome activation led by microtubules formation is not effective in FMF patients that harbor mutations in B30.2/SPRY domain [39]. Although recent studies have elucidated more about the mechanism of pyrin, there are still many questions that we need to answer about the pathogenesis of FMF.
Clinical Manifestations
Clinical episode usually starts during childhood or adolescence, 90% of them having had an onset of the disease by age 20 [1, 40, 41]. FMF attacks can last for 12–72 h. Attacks are characterized by fever and serositis along with an increase in acute phase reactants. The serositis may manifest as peritoneal, pleural, joint, or skin inflammation, sometimes in combination. Abdominal pain with fever is the most frequent presentation. Pain can be generalized or focused in a quadrant, sometimes mimicking acute appendicitis, and the range is from mild to severe [1, 41, 42]. Pleural pain is generally unilateral. Rarely, a small effusion, friction rub, or atelectasis may be present [42]. Joint manifestations, especially arthralgia which is more common than arthritis, can sometimes be the first sign of the disease in children [43]. FMF arthritis is usually monoarticular; however in children, it may have involvement of several joints symmetrically or asymmetrically, with pain and large effusions [41, 44, 45]. The aspirate will be sterile but may have leukocyte counts as high as 100,000/mm3. Three clinical phenotypes have been suggested for FMF: type 1, which is usually associated with recurrent short episodes of inflammation and serositis; type 2, characterized by the evidence of reactive amyloid-associated (AA) amyloidosis, the most common complication of FMF, as the first clinical manifestation of the disease in an otherwise asymptomatic individual; and type 3, known as the “silent” homozygous or compound heterozygote state, in which two MEFV mutations are found without signs or symptoms of FMF nor of AA amyloidosis [46]. Twenty percent of patients have the so-called exertional leg pain, muscle pain in the lower extremities after physical exercise [47]. Colchicine-induced myopathy is a very rare side effect [47, 48]. The only cutaneous finding in FMF is the erysipeloid erythematous rash on the dorsum of the foot, ankle, or lower leg [41, 44, 49, 50].
Pericarditis is a rare condition [51]. In prepubescent boys, unilateral acute scrotal pain episodes may rarely occur [41, 52]. In patients with FMF, certain rheumatic diseases such as Behçet disease [53, 54], polyarteritis nodosa [53, 55], microscopic polyarteritis [56], and glomerulonephritis [57, 58] are more frequent as compared to the healthy population. The increase in these rheumatic diseases has been suggested to be due to the inflammatory milieu [21]. Neurological symptoms are rare; however headache may rarely occur in pediatric patients.
Laboratory Investigations
Acute phase reactants such as ESR, C-reactive protein, and serum amyloid A (SAA) increase during FMF attacks [44]. Elevation of acute phase serum proteins in between attacks is accepted to reflect ongoing inflammation and susceptibility to develop systemic amyloidosis, which is the most serious sequela of FMF [59,60,61]. Systemic amyloidosis presents with SAA deposition mainly in the kidney but also in many organs such as the gastrointestinal tract, spleen, kidneys, adrenals, thyroid, and lungs [1]. There are some predisposing risk factors for amyloidosis such as a positive family history of this complication, male sex, the α/α genotype at the SAA1 locus, and poor compliance with colchicine therapy. Many studies confirm that M694V mutation is more common in patients with amyloidosis, arthritis, and erysipeloid erythema [43, 62,63,64,65]. Microalbuminuria is an early indicator of impaired renal function. It is recommended to do periodic urinalysis which is an important part of continuing care for FMF patients. Amyloidosis can be confirmed by biopsy of the kidney or rectum, if proteinuria is detected [66].
Diagnosis
Since FMF usually requires lifelong treatment, it is crucial to establish a timely, correct diagnosis . The diagnosis of FMF relies mainly on clinical findings, and molecular analysis of the MEFV gene provides genetic confirmation [46]. The presence of short (12–72 h), recurrent (three or more) febrile episodes and abdominal, chest, joint, or skin manifestations with no discernible infectious cause suggest a clinical diagnosis of FMF [67, 68]. The supportive factors can be positive family history, onset before the age of 20, ethnicity, and favorable response to colchicine therapy. There are some sets of classification and diagnostic criteria for FMF. The first set of criteria was created for adults by the experts in Tel Hashomer Hospital [1]. Livneh et al. [67] revised the criteria in 1997, excluding some manifestations of the Tel Hashomer criteria especially amyloidosis. However, there were clear differences between adult and pediatric FMF cases such as the fever-only attacks in some children and inability of some pediatric patients to express the severity and exact location of the pain. Although the Tel Hashomer criteria were very successful in diagnosing the patients, the specificity was low (54.6%) in children [69] In 2009, Turkish pediatricians [69] defined criteria for children with FMF as well (Table 9.2) [68]. Among Turkish children, the criteria (two out of five criteria for diagnosis) reached a sensitivity and specificity of 88.8% and 92.2%, respectively [68]. There are more than 50 mutations described in MEFV [70]; however a number of sequence variants are not pathogenic. The exchange of valine or isoleucine for methionine at position 694 (M694V and M694I), the substitution of alanine for valine at position 726 (V726A) and the substitution of isoleucine for methionine at residue 680 (M680I) are the most common mutations among patients. Exon 2 of MEFV includes a number of missense substitutions, most of which are variants of unknown significance; the most well known is the substitution of glutamine for glutamic acid at residue 148 (E148Q). Despite complete sequencing of the coding region of MEFV, it is more likely 30 % of patients with clinical signs of FMF just have one demonstrable mutation [11, 18, 71]. Recently mutations in exon 2 of MEFV (S242R and E244K) have been associated with neutrophilic dermatoses, inherited in a dominant fashion [72].
Treatment
An international group of experts published recommendations for the management of FMF in 2016 [73]. These recommendations are presented in Table 9.3. According to these recommendations, the main goal of treatment should be complete control of unprovoked attacks and minimizing subclinical inflammation between attacks. Colchicine has been the mainstay of FMF treatment since 1972 [74]. Colchicine can reduce the frequency and severity of attacks and suppress subclinical inflammation between attacks and improve quality of life [5,6,7]. Furthermore, it prevents the development of secondary amyloidosis in patients with FMF [8]. Colchicine is generally safe and well tolerated in children [75]. Colchicine is known to prevent microtubule elongation by binding to tubulin monomers and inhibit polymer formation, which is necessary for pyrin inflammasome assembly [76]. It is recommended that colchicine should be started as soon as the patient is clinically diagnosed. Physicians should follow up these asymptomatic individuals regularly [73]. If the asymptomatic patient has M694V homozygous mutations, which is more commonly associated with the secondary amyloidosis, the physician may start colchicine treatment, especially in countries where amyloidosis is frequent [12]. Colchicine treatment is generally started at the subtherapeutic dose of 0.5 mg/day (or 0.6 mg/day depending on the available drug formulation) and monitored according to disease activity and the patient’s tolerance. Higher doses up to 2 mg/day can be used to control ongoing disease activity and amyloidosis [73]. The major side effects of colchicine are gastrointestinal, diarrhea, and transient elevation of transaminases.
A minority group of FMF patients do not respond to colchicine or are intolerant to the drug because of its side effects. Since the mutation in the pyrin protein has been clearly associated with increased IL-1 production, anti-IL-1 treatment has emerged as a promising second-line therapy in patients with resistant disease [30, 77]. Several studies have reported successful results in colchicine-resistant patients with IL-1-blocking agents [78,79,80]. IL-1 blockade can also reverse proteinuria in patients with secondary amyloidosis [77, 79, 80]. However, there is no evidence for using anti-IL-1 treatment without colchicine to prevent amyloidosis. Thus, a maximal tolerated dose of colchicine is recommended with anti-IL-1 treatment [73]. Anti-TNF treatment can be successful, especially in patients with FMF and chronic arthritis and sacroiliitis [81].
The Autoinflammatory Disease Activity Index (AIDAI) , which is a patient-based symptom diary, is used to monitor disease activity in FMF and other autoinflammatory diseases. AIDAI contains 13 symptoms, and it should be scored daily by patients or by parents as yes or no (Fig. 9.1). AIDAI score is very easy to use, and it provides to assess disease activity and response to therapy [82].

Autoinflammatory Diseases Activity Index diary . Notes: each line refers to a day in a month (Adapted from Ref. [82])
The research in the past 20 years has taught us a lot about the clinical and pathogenic characteristics of FMF. However much remains to be investigated. Why do some patients have more severe disease? Why do some heterozygotes display the phenotype? What is the phenotype-genotype correlation in regard to the many variants that have been identified? Are all pyrin mutations associated with a FMF phenotype ? What is the effect of environment? These are questions that will keep us working on the field in the years to come.
References
Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med. 1967;43(2):227–53.
The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90(4):797–807.
French FMFC. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17(1):25–31.
Berkun Y, Ben-Chetrit E. Pyrin and cryopyrin--similar domain sequence but opposite inflammatory consequence. Clin Exp Rheumatol. 2007;25(4 Suppl 45):S6–8.
Livneh A, Langevitz P. Diagnostic and treatment concerns in familial Mediterranean fever. Baillieres Best Pract Res Clin Rheumatol. 2000;14(3):477–98.
Majeed HA, Rawashdeh M, el-Shanti H, Qubain H, Khuri-Bulos N, Shahin HM. Familial Mediterranean fever in children: the expanded clinical profile. QJM. 1999;92(6):309–18.
Zemer D, Livneh A, Danon YL, Pras M, Sohar E. Long-term colchicine treatment in children with familial Mediterranean fever. Arthritis Rheum. 1991;34(8):973–7.
Zemer D, Pras M, Sohar E, Modan M, Cabili S, Gafni J. Colchicine in the prevention and treatment of the amyloidosis of familial Mediterranean fever. N Engl J Med. 1986;314(16):1001–5.
Stoffels M, Szperl A, Simon A, Netea MG, Plantinga TS, van Deuren M, et al. MEFV mutations affecting pyrin amino acid 577 cause autosomal dominant autoinflammatory disease. Ann Rheum Dis. 2014;73(2):455–61.
Giancane G, Ter Haar NM, Wulffraat N, Vastert SJ, Barron K, Hentgen V, et al. Evidence-based recommendations for genetic diagnosis of familial Mediterranean fever. Ann Rheum Dis. 2015;74(4):635–41.
Booty MG, Chae JJ, Masters SL, Remmers EF, Barham B, Le JM, et al. Familial Mediterranean fever with a single MEFV mutation: where is the second hit? Arthritis Rheum. 2009;60(6):1851–61.
Shinar Y, Obici L, Aksentijevich I, Bennetts B, Austrup F, Ceccherini I, et al. Guidelines for the genetic diagnosis of hereditary recurrent fevers. Ann Rheum Dis. 2012;71(10):1599–605.
Touitou I. The spectrum of Familial Mediterranean Fever (FMF) mutations. Eur J Hum Genet. 2001;9(7):473–83.
Ben-Chetrit E, Lerer I, Malamud E, Domingo C, Abeliovich D. The E148Q mutation in the MEFV gene: is it a disease-causing mutation or a sequence variant? Hum Mutat. 2000;15(4):385–6.
Tchernitchko D, Legendre M, Cazeneuve C, Delahaye A, Niel F, Amselem S. The E148Q MEFV allele is not implicated in the development of familial Mediterranean fever. Hum Mutat. 2003;22(4):339–40.
Ozen S, Demirkaya E, Amaryan G, Kone-Paut I, Polat A, Woo P, et al. Results from a multicentre international registry of familial Mediterranean fever: impact of environment on the expression of a monogenic disease in children. Ann Rheum Dis. 2014;73(4):662–7.
Yilmaz E, Ozen S, Balci B, Duzova A, Topaloglu R, Besbas N, et al. Mutation frequency of Familial Mediterranean Fever and evidence for a high carrier rate in the Turkish population. Eur J Hum Genet. 2001;9(7):553–5.
Marek-Yagel D, Berkun Y, Padeh S, Abu A, Reznik-Wolf H, Livneh A, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum. 2009;60(6):1862–6.
Federici S, Calcagno G, Finetti M, Gallizzi R, Meini A, Vitale A, et al. Clinical impact of MEFV mutations in children with periodic fever in a prevalent western European Caucasian population. Ann Rheum Dis. 2012;71(12):1961–5.
Lachmann HJ, Sengul B, Yavuzsen TU, Booth DR, Booth SE, Bybee A, et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology (Oxford). 2006;45(6):746–50.
Kalyoncu M, Acar BC, Cakar N, Bakkaloglu A, Ozturk S, Dereli E, et al. Are carriers for MEFV mutations “healthy”? Clin Exp Rheumatol. 2006;24(5 Suppl 42):S120–2.
Niemi K, Teirila L, Lappalainen J, Rajamaki K, Baumann MH, Oorni K, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011;186(11):6119–28.
Atoyan S, Hayrapetyan H, Sarkisian T, Ben-Chetrit E. MEFV and SAA1 genotype associations with clinical features of familial Mediterranean fever and amyloidosis in Armenia. Clin Exp Rheumatol. 2016;34(6 Suppl 102):72–6.
Cazeneuve C, Ajrapetyan H, Papin S, Roudot-Thoraval F, Genevieve D, Mndjoyan E, et al. Identification of MEFV-independent modifying genetic factors for familial Mediterranean fever. Am J Hum Genet. 2000;67(5):1136–43.
Schwabe AD, Peters RS. Familial Mediterranean Fever in Armenians. Analysis of 100 cases. Medicine (Baltimore). 1974;53(6):453–62.
Touitou I, Sarkisian T, Medlej-Hashim M, Tunca M, Livneh A, Cattan D, et al. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum. 2007;56(5):1706–12.
Centola M, Wood G, Frucht DM, Galon J, Aringer M, Farrell C, et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95(10):3223–31.
de Torre-Minguela C, Mesa Del Castillo P, Pelegrin P. The NLRP3 and pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol. 2017;8:43.
Weinert C, Morger D, Djekic A, Grutter MG, Mittl PR. Crystal structure of TRIM20 C-terminal coiled-coil/B30.2 fragment: implications for the recognition of higher order oligomers. Sci Rep. 2015;5:10819.
Park H, Bourla AB, Kastner DL, Colbert RA, Siegel RM. Lighting the fires within: the cell biology of autoinflammatory diseases. Nat Rev Immunol. 2012;12(8):570–80.
Yu JW, Fernandes-Alnemri T, Datta P, Wu J, Juliana C, Solorzano L, et al. Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol Cell. 2007;28(2):214–27.
Papin S, Cuenin S, Agostini L, Martinon F, Werner S, Beer HD, et al. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ. 2007;14(8):1457–66.
Hesker PR, Nguyen M, Kovarova M, Ting JP, Koller BH. Genetic loss of murine pyrin, the Familial Mediterranean Fever protein, increases interleukin-1beta levels. PLoS One. 2012;7(11):e51105.
Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34(5):755–68.
Gavrilin MA, Abdelaziz DH, Mostafa M, Abdulrahman BA, Grandhi J, Akhter A, et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J Immunol. 2012;188(7):3469–77.
Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513(7517):237–41.
Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol. 2016;17(8):914–21.
Gao W, Yang J, Liu W, Wang Y, Shao F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc Natl Acad Sci U S A. 2016;113(33):E4857–66.
Van Gorp H, Saavedra PH, de Vasconcelos NM, Van Opdenbosch N, Vande Walle L, Matusiak M, et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc Natl Acad Sci U S A. 2016;113(50):14384–9.
Ben-Chetrit E, Levy M. Familial Mediterranean fever. Lancet. 1998;351(9103):659–64.
Gedalia A, Adar A, Gorodischer R. Familial Mediterranean fever in children. J Rheumatol Suppl. 1992;35:1–9.
Brauman A, Gilboa Y. Recurrent pulmonary atelectasis as a manifestation of familial Mediterranean fever. Arch Intern Med. 1987;147(2):378–9.
Brik R, Shinawi M, Kasinetz L, Gershoni-Baruch R. The musculoskeletal manifestations of familial Mediterranean fever in children genetically diagnosed with the disease. Arthritis Rheum. 2001;44(6):1416–9.
Samuels J, Aksentijevich I, Torosyan Y, Centola M, Deng Z, Sood R, et al. Familial Mediterranean fever at the millennium. Clinical spectrum, ancient mutations, and a survey of 100 American referrals to the National Institutes of Health. Medicine. 1998;77(4):268–97.
Ince E, Cakar N, Tekin M, Kendirli T, Ozkaya N, Akar N, et al. Arthritis in children with familial Mediterranean fever. Rheumatol Int. 2002;21(6):213–7.
Soriano A, Manna R. Familial Mediterranean fever: new phenotypes. Autoimmun Rev. 2012;12(1):31–7.
Langevitz P, Zemer D, Livneh A, Shemer J, Pras M. Protracted febrile myalgia in patients with familial Mediterranean fever. J Rheumatol. 1994;21(9):1708–9.
Majeed HA, Al-Qudah AK, Qubain H, Shahin HM. The clinical patterns of myalgia in children with familial Mediterranean fever. Semin Arthritis Rheum. 2000;30(2):138–43.
Azizi E, Fisher BK. Cutaneous manifestations of familial Mediterranean fever. Arch Dermatol. 1976;112(3):364–6.
Barzilai A, Langevitz P, Goldberg I, Kopolovic J, Livneh A, Pras M, et al. Erysipelas-like erythema of familial Mediterranean fever: clinicopathologic correlation. J Am Acad Dermatol. 2000;42(5 Pt 1):791–5.
Kees S, Langevitz P, Zemer D, Padeh S, Pras M, Livneh A. Attacks of pericarditis as a manifestation of familial Mediterranean fever (FMF). QJM. 1997;90(10):643–7.
Eshel G, Vinograd I, Barr J, Zemer D. Acute scrotal pain complicating familial Mediterranean fever in children. Br J Surg. 1994;81(6):894–6.
Ben-Chetrit E, Cohen R, Chajek-Shaul T. Familial mediterranean fever and Behcet’s disease--are they associated? J Rheumatol. 2002;29(3):530–4.
Schwartz T, Langevitz P, Zemer D, Gazit E, Pras M, Livneh A. Behcet's disease in Familial Mediterranean fever: characterization of the association between the two diseases. Semin Arthritis Rheum. 2000;29(5):286–95.
Ozdogan H, Arisoy N, Kasapcapur O, Sever L, Caliskan S, Tuzuner N, et al. Vasculitis in familial Mediterranean fever. J Rheumatol. 1997;24(2):323–7.
Ozen S, Ben-Chetrit E, Bakkaloglu A, Gur H, Tinaztepe K, Calguneri M, et al. Polyarteritis nodosa in patients with Familial Mediterranean Fever (FMF): a concomitant disease or a feature of FMF? Semin Arthritis Rheum. 2001;30(4):281–7.
Akpolat T, Akpolat I, Karagoz F, Yilmaz E, Kandemir B, Ozen S. Familial Mediterranean fever and glomerulonephritis and review of the literature. Rheumatol Int. 2004;24(1):43–5.
Said R, Hamzeh Y, Said S, Tarawneh M, al-Khateeb M. Spectrum of renal involvement in familial Mediterranean fever. Kidney Int. 1992;41(2):414–9.
Duzova A, Bakkaloglu A, Besbas N, Topaloglu R, Ozen S, Ozaltin F, et al. Role of A-SAA in monitoring subclinical inflammation and in colchicine dosage in familial Mediterranean fever. Clin Exp Rheumatol. 2003;21(4):509–14.
Korkmaz C, Ozdogan H, Kasapcopur O, Yazici H. Acute phase response in familial Mediterranean fever. Ann Rheum Dis. 2002;61(1):79–81.
Tunca M, Kirkali G, Soyturk M, Akar S, Pepys MB, Hawkins PN. Acute phase response and evolution of familial Mediterranean fever. Lancet. 1999;353(9162):1415.
Cazeneuve C, Sarkisian T, Pecheux C, Dervichian M, Nedelec B, Reinert P, et al. MEFV-gene analysis in armenian patients with Familial Mediterranean fever: diagnostic value and unfavorable renal prognosis of the M694V homozygous genotype-genetic and therapeutic implications. Am J Hum Genet. 1999;65(1):88–97.
Gershoni-Baruch R, Brik R, Zacks N, Shinawi M, Lidar M, Livneh A. The contribution of genotypes at the MEFV and SAA1 loci to amyloidosis and disease severity in patients with familial Mediterranean fever. Arthritis Rheum. 2003;48(4):1149–55.
Kone Paut I, Dubuc M, Sportouch J, Minodier P, Garnier JM, Touitou I. Phenotype-genotype correlation in 91 patients with familial Mediterranean fever reveals a high frequency of cutaneomucous features. Rheumatology. 2000;39(11):1275–9.
Shinar Y, Livneh A, Langevitz P, Zaks N, Aksentijevich I, Koziol DE, et al. Genotype-phenotype assessment of common genotypes among patients with familial Mediterranean fever. J Rheumatol. 2000;27(7):1703–7.
Blum A, Sohar E. The diagnosis of amyloidosis. Ancillary procedures. Lancet. 1962;1(7232):721–4.
Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40(10):1879–85.
Yalcinkaya F, Ozen S, Ozcakar ZB, Aktay N, Cakar N, Duzova A, et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology. 2009;48(4):395–8.
Sonmez HE, Batu ED, Ozen S. Familial Mediterranean fever: current perspectives. J Inflamm Res. 2016;9:13–20.
Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, et al. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat. 2004;24(3):194–8.
Ozen S. Changing concepts in familial Mediterranean fever: is it possible to have an autosomal-recessive disease with only one mutation? Arthritis Rheum. 2009;60(6):1575–7.
Moghaddas F, Llamas R, De Nardo D, Martinez-Banaclocha H, Martinez-Garcia JJ, Mesa-Del-Castillo P, et al. A novel Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis mutation further defines 14-3-3 binding of pyrin and distinction to Familial Mediterranean Fever. Ann Rheum Dis. 2017;76:2085.
Ozen S, Demirkaya E, Erer B, Livneh A, Ben-Chetrit E, Giancane G, et al. EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis. 2016;75(4):644–51.
Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med. 1972;287(25):1302.
Padeh S, Gerstein M, Berkun Y. Colchicine is a safe drug in children with familial Mediterranean fever. J Pediatr. 2012;161(6):1142–6.
Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, et al. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428(6979):198–202.
Ozen S, Bilginer Y, Aktay Ayaz N, Calguneri M. Anti-interleukin 1 treatment for patients with familial Mediterranean fever resistant to colchicine. J Rheumatol. 2011;38(3):516–8.
Grattagliano I, Bonfrate L, Ruggiero V, Scaccianoce G, Palasciano G, Portincasa P. Novel therapeutics for the treatment of familial Mediterranean fever: from colchicine to biologics. Clin Pharmacol Ther. 2014;95(1):89–97.
van der Hilst J, Moutschen M, Messiaen PE, Lauwerys BR, Vanderschueren S. Efficacy of anti-IL-1 treatment in familial Mediterranean fever: a systematic review of the literature. Biologics. 2016;10:75–80.
Hashkes PJ, Spalding SJ, Giannini EH, Huang B, Johnson A, Park G, et al. Rilonacept for colchicine-resistant or -intolerant familial Mediterranean fever: a randomized trial. Ann Intern Med. 2012;157(8):533–41.
Bilgen SA, Kilic L, Akdogan A, Kiraz S, Kalyoncu U, Karadag O, et al. Effects of anti-tumor necrosis factor agents for familial mediterranean fever patients with chronic arthritis and/or sacroiliitis who were resistant to colchicine treatment. J Clin Rheumatol. 2011;17(7):358–62.
Piram M, Kone-Paut I, Lachmann HJ, Frenkel J, Ozen S, Kuemmerle-Deschner J, et al. Validation of the auto-inflammatory diseases activity index (AIDAI) for hereditary recurrent fever syndromes. Ann Rheum Dis. 2014;73(12):2168–73.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Demir, S., Günel, İ.E., Özen, S. (2019). Familial Mediterranean Fever. In: Efthimiou, P. (eds) Auto-Inflammatory Syndromes. Springer, Cham. https://doi.org/10.1007/978-3-319-96929-9_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-96929-9_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-96928-2
Online ISBN: 978-3-319-96929-9
eBook Packages: MedicineMedicine (R0)