
Abstract
Genome editing using sequence-specific nucleases has been one of the fast-evolving technologies ever since the discovery of CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR-associated endonuclease) system. CRISPR locus present in the genomes of various bacteria and archaea offer a programmable defense mechanism against foreign agents like viruses. The understanding of this biological phenomenon leads to the development of genome editing tool for making specific modifications in the genomes of various organisms. The CRISPR/Cas9 technology basically depends upon two components: Cas9 enzyme and sgRNA (single-guide RNA). These two components are delivered into plant cells using different gene transfer methods such as Agrobacterium-mediated, biolistic (particle gun) approach, pre-assembled ribonucleoproteins followed by homology-based detection of target DNA by sgRNA, DNA cleavage by Cas9, and DNA repair by native cell machinery employing nonhomologous end joining. This usually leads to a frameshift and knockout mutation in the targeted gene. Providing a template DNA for homology-dependent repair can extend this technique to knock in mutations as well. This technology thus opens up a unique opportunity for directed alterations in chosen genes. Hence, CRISPR/Cas technology has huge potential as a precise and rapid plant breeding method. The absence of foreign DNA introduction (particularly in the case of gene knockout) is anticipated to attract fewer biosafety concerns as compared to GMOs in the regulatory frameworks coming up in different countries. This chapter reviews the CRISPR-Cas strategy by focusing on components of the tool kit and available variants, delivery into plant cells, and gene modification detection assays. A set of crop improvement-related studies, targeting genes of basic as well as applied significance, are listed to illustrate its current use.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
5.1 Introduction
Creation of genetic variation is the key element for crop improvement. As the world population is increasing, food security has become a challenging task with respect to limited land available for crop production (Ma et al. 2018). During the twentieth century, plant breeding has seen a tremendous change fueled by our increasing knowledge of genetic phenomena. Modern plant breeding is entering a new era with the emergence of various sequence-specific nucleases (SSNs). SSNs mediate efficient editing of genomes of various mammalian and plant species. These SSNs induce double-strand breaks (DSBs) at specific chromosomal sites followed by their repair through nonhomologous end-joining (NHEJ) pathway resulting in nucleotide insertions and deletions. If homologous donor templates are available at the site of DSB, homology-directed repair (HDR) can also occur (Symington and Gautier 2011). The different types of SSNs are briefly described below:
5.1.1 Zinc-Finger Nucleases (ZFNs)
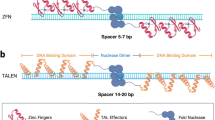
ZFNs are the hybrid proteins (nuclease domain is fused with DNA-binding domain) which provided a breakthrough for the manipulation of genomes of various crop species. ZFNs are regarded as the first generation (Bibikova et al. 2002) of the hybrid proteins to be used for genome editing purpose.
ZFNs are composed of zinc fingers which carry 30-amino-acid-long Cys2-His2 motif (folded into a ββα configuration and stabilized by a Zn2+ ion) constituting the DNA recognition and binding domain (Beerli and Barbas 2002). Each zinc-finger unit consists of three to four binding modules where each module recognizes a triplet nucleotide by inserting its α-helix into the major groove of the DNA double helix. Nuclease domain of Fok1 endonuclease (a type II restriction enzyme) when joined as a dimer to this zinc-finger unit further directs the cleavage at the identified site causing a double-strand break (DSB) (Carroll 2011; Weinthal et al. 2010). As tandem arrays of fingers can recognize extended contiguous sequences, at least three consecutive fingers are needed to provide an adequate binding affinity. As 18 bp of DNA sequence can confer specificity within billions of base pairs of DNA, designing arrays of six to eight ZFs with characterized recognition sites allows specific sequences to be targeted in the complex genome of different plant species. Since the availability of zinc-finger proteins as a site-specific nuclease to cleave target sites in the DNA, for many years, ZF protein technology was the only approach available to create custom site-specific DNA-binding protein and enzymes. Successful use of ZFNs for gene editing in model plants, Arabidopsis (Lloyd et al. 2005) and tobacco (Wright et al. 2005), was first reported in 2005, followed by its use for highly efficient gene targeting in tobacco (Townsend et al. 2009) and maize (Shukla et al. 2009). Several methodologies like “modular assembly” approach (Segal et al. 1999) and selection-based oligomerized pool engineering (OPEN)/context-dependent assembly (CoDA) (Maeder et al. 2008) have been developed for assembling longer arrays and constructing zinc fingers. Companies like Gandaq Ltd. and Sangamo BioSciences (Richmond, CA, USA) have eased the use of ZFs by accumulating an extensive proprietary inventory of ZFs and ZF pairs and making them commercially available. The complex interactions between each amino acid in zinc-finger module with each base pair of target limit the designing of zinc-finger modules (Kumar and Jain 2015).
5.1.2 Transcription Activator-Like Effector Nucleases (TALENs)
In TALENs, the cleavage domain of Fok1 endonuclease is fused in-frame to TALE protein (transcription activator-like effector), a major virulence factor secreted by the phytopathogenic bacteria of the genus Xanthomonas that causes disease in plants by activating transcription of a specific target gene (Bogdanove and Voytas 2011; Weber et al. 2011). The value of these proteins for genome engineering was realized in 2009, when the TALE-DNA-binding code was discovered (Boch et al. 2009; Moscou and Bogdanove 2009). Each TALE protein is composed of variable copies of 33–35 amino acid repeats with each amino acid repeat carrying repeat variable di-residue (RVDR) at positions 12 and 13, respectively, which determines pairing with a specific base of target DNA sequence (Streubel et al. 2012). Different methods like fast ligation-based automatable solid-phase high-throughput (FLASH) and golden gate cloning have been developed to assemble target-specific repeat arrays in correct order. This potential led the journal Nature Methods to declare targetable nucleases as “The method of the year 2011” (Baker 2012), and TALENs also constitute second generation of SSNs (Boch et al. 2009). Apart from the custom TALEN construction facility offered by different companies like Life Technologies, Cellectis Bioresearch, ToolGen, CoWin Biotech, and Transposagen Biopharmaceuticals, TALEN core facilities are also available at different academic institutes like University of California, San Francisco, University of Utah, and University of Wisconsin, Madison. TALENs have been used for genome editing in a number of crops like rice, tobacco, wheat, etc. (Shan et al. 2013 and Wang et al. 2014).
5.1.3 CRISPR/Cas System
CRISPR (clustered regularly interspaced short palindromic repeats) and Cas (CRISPR-associated enzyme) are the latest addition to the toolbox of genome engineering. It is a more versatile genome editing platform developed in the recent years (Wang et al. 2016b). Among the various genome editing approaches (ZFNs, TALENs, and CRISPR) (Beumer et al. 2008; Cui et al. 2011; Bassett et al. 2013), the CRISPR/Cas system is the method of choice because unlike ZFNs and TALENs, it does not involve intensive protein engineering (Mali et al. 2013; Gaj et al. 2013; Johnson et al. 2015) and requires only 22-nucleotide gRNA sequence for site-specific editing. Type II CRISPR/Cas system from Streptococcus pyogenes (Jinek et al. 2012) is an adaptive immune system in prokaryotes and provides defense against various viruses (Ishino et al. 1987) by degradation of foreign DNA in sequence-specific manner. This defense is acquired by integration of short fragments (spacers) of foreign agent between two adjacent repeats at the proximal end of CRISPR locus. In 2002, these “tandem repeats” were called “clustered regularly interspaced short palindromic repeats” (CRISPR) (Jansen et al. 2002; Horvath and Barrangou 2010). In 2005, the CRISPR spacer sequence was found to be highly homologous with exogenous sequences from bacterial plasmids and phages (Mojica et al. 2005; Pourcel et al. 2005). Due to the presence of homology between host and foreign DNA, CRISPR is able to cleave foreign DNA resulting in the development of CRISPR/Cas system as sequence-specific genome editing tool (Doudna and Charpentier 2014). During subsequent attack by viral/foreign DNA, the CRISPR arrays, including the spacers, are transcribed and are processed into small CRISPR RNAs (crRNAs), approximately 40 nt in length, which combine with trans-activating CRISPR RNAs (crRNAs) to activate and guide Cas9 nuclease (Barrangou et al. 2007), resulting in the cleavage of homologous double-stranded DNA sequences (protospacers) of foreign DNA. An essential requirement for cleavage is the presence of protospacer adjacent motif (PAM), i.e., NGG, downstream of target DNA (Jinek et al. 2012) but less frequently NAG (Hsu et al. 2013). The region presenting 12 base pairs upstream of PAM is the seed region, which must match between target DNA and RNA. This three-component system of crRNA, tracrRNA, and Cas protein was reduced to two-component system when the targeting specificity of crRNA was combined with structural properties of tracr in a chimeric sgRNA (Jinek et al. 2012). CRISPR/Cas system is an example of RNA-guided endonucleases (RGENs) which are based on the RNA (and not protein)-guided mode of DNA recognition. Ever since the first report published in 2013, stating the deployment of CRISPR/Cas system for gene targeting in mammalian cells appeared, extensive studies have followed up to explore the potential of this game-changing technology to target different traits in diverse crop species.
Unlike ZFs and TALENs, site-specific catalytic action of sgRNA-Cas9 complex is governed by the sequence of only 20 consecutive nucleotides that constitute chimeric gRNA. Accordingly, designing gRNA corresponding to the target DNA sequence and synthesis of this oligonucleotide is far simpler. Additionally, Cas9 nuclease does not require reengineering for each new target site. Shorter length of sgRNA sequence allows a more convenient delivery into the cells than the long and highly repetitive ZFN-/TALEN-encoding vectors. Off-targets due to non-specific binding of zinc-finger motifs have also been reported (Weinthal et al. 2010; Voytas 2013). Multiplexing sgRNA corresponding to different genes and simultaneous delivery/co-transformation is far more easily achievable relative to TALENs/ZFNs.
5.2 Components of CRISPR/Cas System as an Efficient Genome Editing Tool
To cleave the target DNA, Cas9 derived from S. pyogenes (SpCas9) recognizes PAM motif in the target site (Mojica et al. 2009), binds the target sequence as recognized by sgRNA which pairs with 19–22 bases complementary to DNA sequence upstream of the PAM, and cleaves the target DNA (Jinek et al. 2012). Different components of the CRISPR-Cas9 system are briefly discussed below:
5.2.1 Cas9 Enzyme (CRISPR-Associated Endonuclease)
Cas9 nuclease is maintained in an auto-inhibited conformation as observed during single-particle electron microscopy, and it becomes active only after gRNA gets loaded into it (Jinek et al. 2014). Upon recognition of PAM sequence, it further undergoes conformational change to form a central channel for RNA-DNA heteroduplex binding (Anders et al. 2014). Cas9 is a bilobed structure consisting of two major lobes, an N-terminal large REC lobe (recognition lobe) and a C-terminal small NUC lobe (nuclease lobe) (Nishimasu et al. 2014). REC lobe does not share structural similarity with other known proteins, indicating that it is a Cas9-specific functional domain. REC lobe further consists of three domains, i.e., α-helical segment, RuvC domain, and bridge helix domain. α-Helical segment consists of two sub-domains (RecI and RecII), out of which RecI is the largest sub-domain and is responsible for binding of gRNA by Watson-Crick base-pairing. The function of RecII domain is still unclear. Bridge helix domain is arginine rich and is responsible for linking REC lobe to NUC lobe. NUC lobe also possesses three domains, namely, helix-nuclease-helix (HNH) domain, RuvC endonuclease domain, and PAM-interacting (PI) domain. Helix-nuclease-helix is a nuclease domain which cleaves the DNA strand complementary to gRNA, three nucleotides upstream of PAM sequence. RuvC endonuclease is another nuclease domain which cleaves the DNA strand not complementary to gRNA. PAM-interacting (PI) domain is responsible for recognition of protospacer adjacent motif (PAM) on noncomplementary strand of DNA.
There are varieties of Cas nucleases available today, which can be utilized for various applications like high-throughput genome editing, knocking out the expression of gene, and transcriptional control with improved specificity and reduced off-target effects in various systems, ranging from fruit fly, yeast, bacteria, plants, etc. (Khatodia et al. 2016). Different variants of Cas9 nuclease are explained as follows:
5.2.1.1 SpCas9
SpCas9 is a wild-type or native Cas9, derived from Streptococcus pyogenes (size, 900–1600 amino acids). It cleaves dsDNA at a specific site resulting in the creating of double-strand break repair machinery, resulting in dsDNA cleavage, further activating nonhomologous end joining (NHEJ). If a donor template having homology to target locus is present, homology-directed pathway for repair (HDR) can also be followed for the introduction of specific changes (Hsu et al. 2013). It recognizes NGG as PAM sequence. It is successfully used in plant and mammalian cells.
5.2.1.2 Inactive Cas9 or dCas9
dCas9 is a nuclease-deficient but DNA-binding enzyme formed by the induction of mutations in H840A subunit in HNH domain and D10A subunit in RuvC domain (Qi et al. 2013). It is used for overexpressing (CRISPRa) or silencing of gene (CRISPRi) by fusion with effector domains. It is used as a visualization tool for the detection of repetitive and non-repetitive loci. dCas9 tagged with an affinity is being used for studying protein-genome interactions through chromatin immunoprecipitation assays (Fujita and Fujii 2014).
5.2.1.3 SaCas9
SaCas9 is derived from Staphylococcus aureus. It functions similar to SpCas9 but recognizes NNGRRT (where R = A or G) as PAM sequence. The size of SaCas9 is relatively smaller (1053 amino acids), which makes the process of delivery relatively easier (Ran et al. 2013). It is successfully used in mammalian cells.
5.2.1.4 Cas9 D10A
Cas9D10Ais formed by the induction of mutations in D10A subunit of SpCas9 (Shen et al. 2014). It cuts only one strand of DNA, thereby, generating single-strand breaks, which can be repaired and no indels are produced. This enzyme is used for minimizing off-target effects by employing double-nicking strategy with paired nickases targeting adjacent regions.
5.2.2 Target Site Identification and gRNA Designing
sgRNA sequence is composed of a constant tracrRNA and variable crRNA (Jinek et al. 2012). Within sgRNA, the guide sequence has an established length of 20 nucleotides followed by a three-base PAM sequence at its 3′ end. The potential of CRISPR-Cas9 system to target a specific sequence may be limited by the availability of PAM sites (Wei et al. 2015). The selection of a suitable target site is one of the most important determinants of efficiency of Cas9 and induction of on-target mutations. Aside from the manual selection of sgRNA target site, several web-based tools are available for genome-wide prediction of highly specific sgRNA and detection of off-targets in model plants and major crops (Bae et al. 2014; Heigwer et al. 2014; Xiao et al. 2014). Any undesired change or off-target cleavage could lead to cytotoxicity, apoptosis, and gross chromosomal rearrangements like inversion, deletion, or translocations (Lee et al. 2012; Park et al. 2014; Hendel et al. 2015).
sgRNA on-target cleavage efficiency is determined by DNA sequence profile, accessibility of the gene locus (packaging of chromosome), and nucleotide composition of DNA downstream of the spacer region. Different softwares provide unique scores for the activity (Cas9 on-target efficiency) and specificity (off-target prediction accuracy) of the target site cleavage by Cas9 and accordingly rank the suggested sgRNA sequences by their efficiency and specificity (Bolukbasi et al. 2016). Based on the mode of delivery chosen for CRISPR reagent, separate scores may be used, for example, for U6 promoter-based assays, Fusi/Doench score (Doench et al. 2016) and, for assays based on direct delivery of gRNA produced by T7-based IVT, Moreno-Mateos score may be used (Haeussler et al. 2016). For a large set of possible guides to pick from an additional ranking criteria, Wong score for U6 promoter-based guides (Wong et al. 2015) or GG rule (for T7-based IVT) may be followed. The softwares also calculate and provide a score for off-target mutation based on the distance of the mismatch to the PAM—Hsu Zhang score, MIT score (all potential off-targets may be summarized into guide specificity score that ranges from 0 to 100), and CFD (cutting frequency determination) score that distinguishes between validated and false-positive off-targets (Doench et al. 2016). Off-target scores are generally identified and reported for exonic regions (and not intergenic regions, introns, or UTRs). Further these softwares may provide information about restriction enzyme site within the predicted gRNA, primer sequence for cloning gRNA into vector and detecting on-target mutations (Periwal 2017). A few of the sgRNA design tools like CCTOP, COSMID, CRISPR optimal, Target Finder, and SAPTA have shown experimental validation of their prediction results, thus substantiating their effectiveness. Others like CROP-IT sgRNA scorer predict sgRNA based on the information obtained from previously tested engineered nucleases.
There are certain points that must be kept in mind while selecting the target site and designing the gRNA for CRISPR-Cas experiment. To select a unique target sequence and minimize chances of off-target cleavage, a BLAST search of the whole genome sequence of the target specific for the relevant 22-nucleotide sequence may be performed (Chuai et al. 2016). CRISPR-Cas9 system discriminates efficiently against any potential off-target sites with perfect match in the 12-nucleotide-long PAM-proximal sequence (seed sequence), while mismatches in the PAM-distal sequence are actually tolerated. sgRNA with two extra target-independent GG nucleotides at the 5′ terminus have been found to confer significant specificity to the Cas9 action when compared with conventional sgRNA (Cho et al. 2014). Target sequence at the 3′ end of the coding region or intron should be avoided. While searching, PAM sequence should be looked upon in the complementary genomic sequence as well. Quadruplex-forming homopolymer or a stretch of repeat sequence in the target gene should be avoided. GC content should be >30% but less than 80%. The chosen gRNA should not lead to the formation of a stable secondary structure in the protospacer. It should be designed from the proximal region, i.e., 5′ end of the gene. The functional domains of the gene of interest may be considered, but targeting highly conserved region of a gene family should be avoided. More than three mismatches (especially in the seed region) between DNA target and gRNA may not be tolerated.
5.3 Modes of Delivery of CRISPR Components into Plants
RNA-guided endonucleases (RGENs) were proclaimed as “Breakthrough of the year 2015” by the Science journal for its broader application to numerous disciplines of life sciences. CRISPR-Cas9 system superseded the potential of previous GE tools (MegaN, ZFNs, and TALENs) on account of its easier preparation, affordable nature, and flexible usage (Kim and Kim 2014). Because of the presence of a rigid plant cell wall, delivery of GE reagents into the plant cells is a major barrier to the use of this technology for creating novel traits (Baltes et al. 2014). Therefore, introduction of CRISPR/Cas construct into plant cells is carried out through various direct (vectorless) and indirect (vector-mediated) gene transfer methods. The various direct methods for delivery of CRISPR/Cas construct into plant cells are electroporation, PEG (polyethylene glycol)-mediated transfection, and particle gun method. The indirect methods involve the introduction of construct through a vector or intermediate such as bacteria (e.g., Agrobacterium tumefaciens or A. rhizogenes) or plant virus systems [e.g., tobacco rattle virus (TRV), lentivirus, geminivirus, etc.].
5.3.1 Direct or Vectorless Methods
5.3.1.1 Electroporation or PEG-Mediated Method
Electroporation (using electrical impulse) or chemical transformation (using PEG) is usually carried out for testing the efficacy of various constructs for transient assays using protoplasts obtained after digestion of cell with cell wall-degrading enzymes like cellulose and pectinase. It is usually carried out for the validation of gRNAs. Protoplasts isolated from different crop species (rice, wheat, maize, lettuce, tobacco, tomato, Arabidopsis, etc.) have been used to evaluate gene editing reagents using CRISPR/Cas9-based systems (Liang et al. 2014; Shan et al. 2014; Cermak et al. 2015). Protoplasts can be used to determine target site mutagenesis efficiency and can be regenerated into plants (Woo et al. 2015).
5.3.1.2 Particle Gun Method
Particle gun method, also known as biolistic method, is a method of choice for the direct delivery of CRISPR/Cas construct into target cells of many crop plants using gold/tungsten microcarriers and uses the physical force to transfer the construct into plant cells. Transgene-free genome-edited crop plants have been produced in maize (Svitashev et al. 2016) and hexaploid wheat (Liang et al. 2017) through biolistic approach.
5.3.2 Indirect or Vector-Mediated Methods
5.3.2.1 Agrobacterium-Mediated Genome Editing
Agrobacterium tumefaciens-mediated genetic transformation is one of the most suitable and efficient methods for the production of stable transgenic plants. A number of crop plants have been genome-edited in the past few years using Agrobacterium tumefaciens. It is carried out by infecting various explants like callus tissues in Agrobacterium broth followed by the regeneration of T0 plants under in vitro conditions (Gao et al. 2015). Agrobacterium-mediated delivery of CRISPR/Cas9 construct into immature embryos of maize resulted in editing of various genes (Svitashev et al. 2016). But there are two major limitations of Agrobacterium-mediated genome editing: (i) it cannot be used for introducing ribonucleoprotein complex into target cells and (ii) there are greater chances of stable transformation leading to the integration of Ti plasmid backbone into the plant genome resulting in the production of transgenic plants.
5.3.2.2 Viral Vector-Mediated Genome Editing
Viruses are also used as vectors for the delivery of CRISPR/Cas constructs alone or along with donor DNA templates for carrying out programmable editing in plants. Viral vectors are one of the desired vectors for the introduction of genome editing reagents into target cells (Maggio and Goncalves 2015). They are also used for direct delivery of recombinant proteins into target cells after fusion to structural components of vector particles (Skipper and Mikkelson 2015). Lentiviral vectors are used for genetic modification of target cells due to integrase-dependent mechanisms which results in chromosomal insertion of transported foreign nucleic acids. However, for genome editing experiments, integrase mechanisms of lentivirus must be disabled to ensure episomal vector templates as substrates for homologous recombination or for transient designer nuclease expression. The first viral vector used for gene-silencing (VIGS) study was tobacco mosaic virus (TMV) (Kumagai et al. 1995). The process of inoculation becomes easier using geminiviruses (DNA viruses), since it requires only viral DNA. Geminiviruses have been modified for homology-directed recombination-mediated genome targeting and achieving desired modifications in crop plants (Gil-Humanes et al. 2017). Virus-based guide RNA delivery system for CRISPR-Cas9-mediated genome editing (VIGE) using geminivirus was carried out for targeting endogenous PDS3 (phytoene desaturase) and IspH (isopentenyl/dimethylallyl diphosphate synthase) genes, involved in non-mevalonate pathway of isoprenoid synthesis, using cabbage leaf curl virus because it can infect plants systemically resulting in albino phenotype of tobacco plants (Yin et al. 2015). TRV is also being used as a tool for genome editing because it has a bipartite genome consisting of two positive-sense single-guide RNAs, RNA1 and RNA2. RNA2 genome can be modified to carry exonic gene fragments (Kumar et al. 2003). Inoculation of virus is carried out through mechanical or agro-infiltration process. As there occurs no integration of viral genome into plant genome, the edited products are not transformed.
5.4 Detection of On-Target Modifications Following Genome Editing
The indel-forming nonhomologous end-joining (NHEJ) pathway repairs double-strand breaks of various crop plants under in vivo conditions (Jinek et al. 2013), resulting in mutation following genome editing. NHEJ is an error-prone DNA repair mechanism, which results in small insertions and/or deletions (indels) at the site of the break. As a result of indels, there occurs frameshift mutation and production of termination codon which can result in knockout of the function of the gene due to the production of truncated polypeptides (Perez et al. 2008; Santiago et al. 2008; Sung et al. 2013; Ramleet et al. 2015). Therefore, after the introduction of CRISPR/Cas construct or gRNA-Cas ribonucleoprotein complex into the genome of host plant, the next step is to characterize the mutations caused by that particular construct. If we target a diploid crop plant (e.g., rice), there are four potential outcomes: no mutation, a heterozygous mutation (only one allele is mutated), a biallelic mutation (both alleles are mutated, but the sequence of each allele is distinct), or a homozygous mutation (same mutation on both alleles) (Zischewski et al. 2016). The techniques employed to identify various kinds of mutations are mismatch cleavage assays [T7E1 assay, Surveyor™ nuclease assay (Transgenomic, Gaithersburg, MD, USA)], PCR-RE assay (Beumer et al. 2008), quantitative PCR high-resolution melting (qPCR-HRM) curve analysis technique (Yu et al. 2014) and sequencing.
5.4.1 T7E1 Assay
T7 endonuclease 1 (T7E1), an enzyme, identified from bacteriophage (Center and Richardson 1970), cleaves the cruciform DNA at 5′ end and provides a cost-effective and easy method for the detection of CRISPR/Cas-induced indel mutations. It recognizes structural abnormalities in dsDNA and cleaves the heteroduplexed DNA at specific sites (Dickie et al. 1987). It is the most commonly used methods for the detection of mutations created through CRISPR-Cas9-mediated genome editing. During genome editing through CRISPR technology, CRISPR/Cas constructs are introduced into plant cells using Agrobacterium-mediated or biolistic approaches resulting in the generation of targeted mutations. After few days, amplification of the genomic DNA surrounding the target region is carried out through PCR. The obtained amplicon is denatured and recomplexed by heating and subsequent slow cooling. During cleavage by Cas9 enzyme, if nonhomologous end joining occurs between the cut ends of DNA, the formation of heteroduplex occurs between PCR products of different lengths (e.g., mutant and WT amplicons), leading to DNA distortion that is recognized and cleaved by T7E1. The frequency of mutation is determined by the banding patterns of the cleaved products between control and experimental samples. However, the performance of the assay may be impacted by the length and identity of base pair mismatches, flanking sequence, secondary structure, and relative abundance of mutant sequences (Mashal et al. 1995; Vouillot et al. 2015). The sensitivity of T7E1 assay ranges between 0.5 and 5% (Kim et al. 2013). This method is suitable for detecting indels; however, SNPs cannot be identified by this method; rather it also tends to miss small indels in many cases (Qiu et al. 2004).
5.4.2 Surveyor™ Assays
Surveyor™ nuclease is an endonuclease derived from celery and is a site-specific enzyme. The sensitivity of surveyor assay is less (3%) as compared to T7E1 assay (Qiu et al. 2004) but is suitable for the detection of SNPs and small indels. The mechanism of action of surveyor assay relies on cutting both the DNA strands downstream of mismatch.
Therefore, the choice of mismatch cleavage using either T7E1 or Surveyor™ nuclease depends on which types of mutations are expected after genome editing. There are standard protocols which are provided with Surveyor™ nuclease assay kits. The mismatch cleavage assay usually underestimates the mutation frequency due to the preferential cleavage properties of each enzyme.
5.4.3 Quantitative PCR High-Resolution Melting(qPCR-HRM) Curve Analysis
During this method, amplification of DNA sequence surrounding genomic target is carried out through real-time PCR followed by incorporation of fluorescent dye and determining the melt curve analysis of amplicons (Dahlem et al. 2012). The principle behind HRMA method is during denaturation step of real-time PCR; intercalating dyes are released from dsDNA resulting in the loss of fluorescence. The data are collected over smaller temperature increments of 0.2 °C, followed by signal normalization and analysis as compared to the melt curves analyzed in typical quantitative PCR (qPCR) experiment. Melting temperature shifts and the shape of the melting curves can both provide useful information: homozygous allelic variants may cause a temperature shift in the melt curve compared to the wild-type homoduplex, whereas heteroduplexes representing heterozygous mutations change the shape of the melt curve due to the presence of mismatches (Taylor et al. 2010). HRMA is a sensitive and highly effective method as it does not involve direct handling of PCR sample and is a high-throughput screening method. It is a nondestructive method, i.e., it does not cause any harm to amplicons; therefore, they can further be analyzed by sequencing or heteroduplex mobility assay. This method is able to differentiate between homozygous wild type and homozygous mutants (Zischewski et al. 2016). The estimated detection limit in a ~100 bp amplicon is at least 2%, i.e., one mutant among 50 wild-type genomes for indels larger than 4 bp (Dahlem et al. 2012). Larger indels could not be detected by this method.
5.4.4 Polymerase Chain Reaction-Restriction Enzyme (PCR-RE) Assay
During CRISPR-Cas9-mediated genome editing, Cas9 nuclease cleaves double-stranded DNA three-base pair upstream of PAM sequence. If the site of any restriction endonuclease is present in the region upstream of PAM sequence, then the Cas9 nuclease will cut within a restriction enzyme site. The amplification of target locus and ~300–1000 bp of flanking material is achieved using 20–30-nucleotide-long fragments with Tm = 60 °C (Shan et al. 2014) followed by their digestion with appropriate restriction enzyme. The amplicons with no mutation (wild type) will be cleaved because no disruption of restriction site occurs, whereas the uncleaved bands will be amplicons of mutated region where there occurs loss of restriction enzyme site. The frequency of mutation can be observed from the uncleaved bands. PCR-RE assay can detect SNPs, small indels, and large indels (Zischewski et al. 2016). Subcloning of the uncleaved fragments followed by their sequencing can be used for detailed characterization of induced mutation.
5.4.5 Sequencing
CRISPR/Cas9-mediated mutations can be detected by amplification of the target sequence using specific primers followed by Sanger’s sequencing and next-generation sequencing (NGS), which provides a direct proof for the type of mutation. This procedure could be laborious and time consuming. Analysis of large number of PCR amplicons sequenced through NGS can be carried through CRISPR-Genome Analyzer (CRISPR-GA) software (Güell et al. 2014). Large molecules can be identified through single-molecule real-time (SMRT) sequencing (Hendel et al. 2014), with an average read length of 8.5 kb.
5.5 Recent Advances in Genome Editing
Ever since the first report of the use of CRISPR/Cas9 to edit plant genome in 2013, there came many reports on the proof of concept, optimization of workflow in different plant species, advances in web-based tools to design sgRNA and identify off-target sites and incessant evolution of this technology in terms of identification/generation of SpCas9 variants to knock down a gene, identification of direct delivery agents, amenability of CRISPR/Cas system to interference (CRISPRi) or activation (CRISPRa), and use of choice-based editing enzymes to precisely edit a specific base in the gene sequence. Certainly, CRISPR/Cas9 has triggered a revolution in the laboratories which are using this technology for innovative applications in biology. Some of the recent advances of genome editing are briefly discussed below:
5.5.1 Cas9 Variants
5.5.1.1 Cpf1
Cpf1 is also an RNA-guided endonuclease of type II CRISPR system derived from Prevotella and Francisella bacteria. The Cpf1 protein has a RuvC-like endonuclease domain that is similar to the RuvC domain of Cas9. Furthermore, Cpf1 does not have a HNH endonuclease domain, and the N-terminus of Cpf1 does not have the alpha-helical recognition lobe of Cas9, and the size is smaller as compared to Cas9. It recognizes 5′-YTN-3′ as PAM sequence (where Y = pyrimidine and N = nucleobase) (Fonfara et al. 2016). Recently, CRISPR/Cpf1 system was used for targeting two genes, OsPDS and OsBEL, resulting in specific and heritable mutations in rice (Xu et al. 2016).
5.5.1.2 Cas13
Cas13 is sequence-specific RNA endonuclease. It is used for downregulating the transcripts of specific genes due to the presence of HEPN (higher eukaryotes and prokaryotes nucleotide binding) domain which is associated with RNase activity of single-stranded RNA. The most active orthologue of Cas13 enzyme is Cas13a isolated from Leptotrichia wadei which is being used in mammalian cells for knockdown of targeted RNA (Abudayyeh et al. 2017).
5.5.2 DNA-Free Direct Genome Editing Using Ribonucleoproteins
The expressed sgRNA and Cas9 can be made to enter into the plant cell either with the help of an agent (plasmid-carrying gene construct into the target cell by means of a bacteria like A. tumefaciens and A. rhizogenes or a virus like tobacco rattle virus and geminivirus) or by direct delivery of RNA (in the form of in vitro transcripts (IVTs), Cas9 mRNA) and protein via protoplast transfection or microprojectile bombardment or in the form of a pre-assembled ribonucleoprotein complex. Agrobacterium-mediated genetic transformation, PEG-mediated protoplast transformation, and particle bombardment have been extensively used as conventional delivery methods for CRISPR/Cas9 reagents (Jiang et al. 2013; Shan et al. 2013, 2014). However, its wider applicability is limited by off-target effects, unwanted and random integration of DNA segments from plasmid vectors into the host genome (Kim et al. 2014; Koo et al. 2015).
Unlike the plasmid-based methods, ribonucleoprotein (RNP) complex has come up as a very efficient method for the direct delivery of active components of CRISPR machinery into a plant cell. These RGEN components may be prepared separately and assembled in vitro and subsequently introduced into the plant genome (classical chemical transfection) for the desired genetic modifications. In vector delivery method, there is a possibility of integration of remnants of the constructs into the plant genome, but RNP completely rules out any such possibility. RNPs act immediately upon delivery with highest mutation frequency 1 day after delivery into the cell. sgRNA and Cas9 protein have a decreased functional time within the cell as they rapidly get degraded in vivo (by the action of endogenous proteases) after editing and, therefore, leaving no trace of foreign DNA in the plant genome. Accordingly, the chances of cleavage at off-target sites are greatly reduced (Kim et al. 2014). Woo et al. (2015) first demonstrated the use of this next-generation plasmid-independent CRISPR/Cas9 genome editing approach in plants by transfecting the protoplasts of Arabidopsis, tobacco, lettuce, and rice and obtaining targeted mutagenesis up to 46% in regenerated protoplasts. Liang et al. (2017) succeeded in editing two genes TaGW2 and TaGASR7 by delivering CRISPR/Cas9 RNPs via particle bombardment. The application of this direct delivery mode has also been tested in other food crops like maize (Svitashev et al. 2016) and soybean (Kim et al. 2017) and horticultural crops like apple and grapes for increasing resistance to powdery mildew and fire blight disease (Malnoy et al. 2016). To adopt this next-generation, RGEN RNP tool to develop vector-free plants, different options may be exploited depending upon the amenability of different species to these methods. These methods include PEG-mediated protoplast transformation using microinjection mesoporous silica nanoparticles (MSNs) and cell-penetrating peptides (Martin-Ortigosa and Wang 2014; Jensen et al. 2014; Masani et al. 2014; Woo et al. 2015).
RNP has a much broader spectrum of applications to offer than mere gene disruption. It dramatically reduces off-target mutations without compromising on-target efficiency. It offers high specificity, paving way for precision crop breeding and developing DNA-free genetically edited crop plants (Kanchiswamy 2016). Though the frequency of mutagenesis obtained via RNP delivery or use of RNA is low, chances of off-target mutagenesis are considerably reduced. RNP delivery also enables HR-mediated precise gene editing as seen in the case of endogenous ALS2 where a 127-base single-stranded repair template for HR was co-delivered (Maize, Mark Cigan group). Truly, RNP has marked the evolution of next generation of GE tools. As mutations induced by these protein complexes do not fall under the current regulatory legislations, they may be qualified as non-GMOs, promoted for commercialization, public acceptance, and practical cereal improvement (Wolter and Puchta 2017).
5.5.3 CRISPR-Cas9-Mediated Base Editing
Homology-directed repair (HDR) or targeting-induced local lesions in genome (TILLING) has been used for a long time for generating point mutations in crops. Genome editing in plants via NHEJ or HDR pathway involves double-strand break repair. HDR has been found to be relatively inefficient due to the competition by NHEJ, the dominant pathway to repair DSB in plants. Moreover, the delivery of DNA repair template into the cell is also quite challenging.
Targeted base editing offers itself as a very promising technique that may enable precise and efficient base replacement in the target locus (Lu and Zhu 2017) without the need for a foreign DNA donor or dsDNA cleavage. It may offer a direct and irreversible conversion of one target base into another in a programmable manner (Komor et al. 2016). Since this approach does not involve a donor DNA template, it avoids the chances of random integration of donor DNA in the genome of the edited plants. The first generation of experimental optimization has been conducted for converting blue fluorescent protein (BFP) into a green fluorescent protein (GFP) by fusing rat cytidine deaminase APOBEC1 with a Cas9 variant which may be a nickase or a dead Cas9. CRISPR-Cas9 nickase-cytidine deaminase fusion protein enabled editing of a single base in the plant genome when directed by sgRNA. Zong et al. (2017) used CRISPR-Cas9 nickase-cytidine deaminase fusion protein to achieve targeted conversion of cytosine to uracil/thymine from position P3 to P9 within the protospacer region of OsCDC48, OsNRT1.1B, and OsSPL14 in rice, TaLox2 in wheat, and ZmCENH3 in maize with a mutagenesis frequency up to 43.48%. Relative to gene disruption or replacement strategy, indel frequency was found to be greatly reduced. Accordingly, targeted nucleotide substitution within the desired locus may be made without the need for double-strand break (NHEJ) or the introduction of any foreign DNA (HDR). Lu and Zhu (2017) too tested the applicability of this strategy in rice for two agronomically important genes—NRT1.1B (nitrogen transporter) and SLR1 (DELLA proteins).
5.6 Applications of Genome Editing for Crop Improvement
CRISPR/Cas9 RGEN system has completely revolutionized the genome editing platform. It may be used for sequence-specific integration of more than one gene for pyramiding useful traits in elite cultivars that may help develop improved varieties in a shorter time. It may be utilized for the creation and use of novel allelic variants, a new source of genetic variation, and broaden the spectrum of existing genetic variability for breeding crops. Hybridizing the transgenic plant carrying CRISPR-Cas9 system with elite cultivar may provide non-transgenic genome modification in plants which may address ethical concerns raised against commercialization of transgenic plants. Further, the introduction of desired genetic/epigenetic variation via CRISPR-Cas9 system may provide a promising platform to engineer gene networks in their native context to improve quantitative traits of agronomic importance like increased yield, enhanced tolerance to diseases, insect pests, and abiotic stress. Successful genome editing has been carried out in different crops like wheat, rice, maize, sorghum, barley, and Brassica (Table 5.1). Plant diseases caused by microorganisms are the major factor that reduces quality and yield of crops. Powdery mildew is one of the most common plant diseases caused by fungal species. Wang et al. (2014) were successful in inducing selective mutation in one of the three homeoalleles of mildew resistance locus (MLO) in wheat with mutation frequency of 5.9%. Resistance to powdery mildew through CRISPR/Cas technology was also achieved by Zhang et al. (2017) by knocking out of TaEDR1 gene. The quality of rice was improved by knocking out the expression of starch-branching enzymes (OsSBEI and OsSBEII), genes for reducing the content of amylopectin and increasing amylose content in rice (Sun et al. 2017). Table 5.1 gives a comprehensive elaboration of the use of CRISPR-Cas strategy for improving important traits in different crop species.
5.7 Regulatory Considerations
The next wave of genetically edited crops will soon make its way from laboratory into the market. But before that, these products should be able to stand the test of field trials, proceed through multiple regulatory loops, and win over the concerns of the masses assuring general public acceptance. USDA and APHIS designate a crop as genetically modified (GM) if it is produced using a plant pathogen or if it contains a foreign gene. These GMOs are subjected to stringent regulatory procedures before commercialization. The modified plants generated by CRISPR-Cas9 system are by and large indistinguishable from naturally occurring mutant crops or that developed by conventional breeding approaches. Regulatory authorities around the world are currently considering how to handle the plants that lack a transgene yet carry targeted mutations such as those created by NHEJ (Kuzma and Kokotovich 2011). USDA does not regulate plants if they contain targeted mutagenesis occurring by self-repair mechanism. GE canola; non-browning, acrylamide-safe potatoes; and GE mushrooms have already been approved by the USDA and waived off from GMO regulations (Waltz 2016; Haun et al. 2014).
One way to escape GMO legislation and produce non-transgenic genome-edited plants is to switch from DNA to RNA or proteins, particularly the use of ribonucleoprotein complexes (Wolter and Puchta 2017). In addition to these GMO-based legislative regulations, the unresolved patent claims are delaying the widespread use of CRISPR/Cas9 technology. The rapid resolution of license negotiation and the establishment of licensing free structure shall give a tremendous boost to this breakthrough technology to help in solving the problem of food shortage and environmental protection.
5.8 Conclusion
CRISPR/Cas is a versatile, precise, and efficient tool that can be used to target any region of the genome of an organism by changing the gRNA sequence. It will help in annotating large amounts of sequence information which will further help in discovering the genomes of different crop plants by overcoming the limitations of searching plant populations with adequate variation and conventional crossing methods. The simple knockout strategy holds significant promise for creating targeted allelic variation for crop improvement. Base editing and homology-dependent insertion of new DNA further expand these possibilities. Ribonucleoprotein-based methodologies are likely to reduce the possibilities of apprehended off-target genome modifications. The CRISPR/Cas system thus needs to be integrated in the crop improvement programs as a quicker, safer, and accurate methodology to take advantage of the presently available vast genome sequence data to develop crop varieties for the future needs of the farmers and consumers.
References
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, Lander ES, Voytas DF, Ting AY, Zhang F (2017) RNA targeting with CRISPR–Cas13. Nature 550:280–284. https://doi.org/10.1038/nature24049
Anders C, Niewoehner O, Duerst A, Jinek M (2014) Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513:569–573
Bae S, Park J, Kim JS (2014) Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30:1473–1475
Baker M, (2012) Gene-editing nucleases. Nature Methods 9(1):23–26
Baltes NJ, Gil-Humanes J, Cermak T, Atkins PA, Voytas DF (2014) DNA replicons for plant genome engineering. Plant Cell 26:151–163
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. https://doi.org/10.1126/science.1138140
Bassett AR, Tibbit C, Ponting CP, Liu JL (2013) Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep 4(1):220–228
Beerli RR, Barbas CF III (2002) Engineering polydactyl zincfinger transcription factors. Nat Biotechnol 20:135–141
Beumer KJ, Trautman JK, Bozas A, Liu JL, Rutter J, Gall JG, Carroll D (2008) Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc Natl Acad Sci 105(50):19821–19826
Bibikova M, Golic M, Golic KG, Carroll D (2002) Targetedchromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161:1169–1175
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U (2009) Breaking the codeof DNA binding specificity of TAL-type III effectors. Science 326:1509–1512
Bogdanove AJ, Voytas DF (2011) TAL effectors: customizable proteins for DNA targeting. Science 333(6051):1843–1846. https://doi.org/10.1126/science.1204094
Bolukbasi MF, Gupta A, Wolfe SA (2016) Creating and evaluating accurate CRISPR-cas9 scalpels for genomic surgery. Nat Methods 13:41–50
Braatz J, Harloff HJ, Mascher M, Stein N, Himmelbach A, Jung C (2017) CRISPR-Cas9 targeted mutagenesis leads to simultaneous modification of different homoeologousgene copies in polyploid oilseed rape (Brassica napus). Plant Physiol 174(2):935–942
Carroll D (2011) Genome engineering with zinc-finger nucleases. Genetics 188(4):773–782
Center MS, Richardson CC (1970) An endonuclease induced afer infection of Escherichia coli with bacteriophage T7. II. Specifcityof the enzyme toward single- and double-stranded deoxyribonucleic acid. J Biol Chem 245:6292–6299
Cermak T, Baltes NJ, Cegan R, Zhang Y, Voytas DF (2015) High-frequency precise modification of the tomato genome. Genome Biology 16:232 https://doi.org/10.1186/s13059-015-0796-9
Char SN, Neelakandan AK, Nahampun H, Frame B, Main M, Spalding MH, Becraft PW, Meyers BC, Walbot V, Wang K, Yang B (2017) An Agrobacterium-delivered CRISPR/Cas9system for high-frequency targeted mutagenesis in maize. Plant Biotechnol J 15:257–268
Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS (2014) Analysis of off-target effects of CRISPR/Cas-derived RNAguided endonucleases and nickases. Genome Res 24:132–141. https://doi.org/10.1101/gr.162339.113
Chuai G-H, Wang Q-L, Liu Q (2016) Insilico meets in vivo: towards computational CRISPR-based sgRNA design. Trends Biotechnol. https://doi.org/10.1016/j.tibitech.2016.06.008
Cui X, Ji D, Fisher DA, Wu Y, Briner DM, Weinstein EJ (2011) Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol 29:64–67
Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, Weis AM, Voytas DF, Grunwald DJ (2012) Simple methods for generating and detectinglocus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet 8(8):e1002861
Dickie P, McFadden G, Morgan AR (1987) The site-specifc cleavage of synthetic Holliday junction analogs and related branchedDNA structures by bacteriophage T7 endonuclease I. J Biol Chem 262:14826–14836
Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF et al (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34:184–191
Doudna JA, Charpentier E (2014) Genome editing- the new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. https://doi.org/10.1126/Science.1258096
Feng C, Su H, Bai H, Wang R, Liu Y, Guo X, Liu C, Zhang J, Yuan J, Birchler JA, Han F (2018) High efficiency genome editing using a dmc1 promoter-controlledCRISPR/Cas9 system in maize. Plant Bitechnol J. https://doi.org/10.1111/pbi.12920
Fonfara I, Richter H, Bratovič M, Le Rhun A, Charpentier E (2016) The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532:517–521. https://doi.org/10.1038/nature17945
Fujita T, Fujii H (2014) Identification of proteins associated with an IFNγ-responsive promoter by a retroviral expression system for enChIP using CRISPR. PLoS One. https://doi.org/10.1371/journal.pone.0103084
Gaj T, Gersbach CA, Barbas CF (2013) ZFN, TALEN and CRISPR/Cas –based methods for genome engineering. Trends Biotechnol 31:397–405. https://doi.org/10.1016/j-tbiotech.2013.04.004
Gao J, Wang G, Ma S, Xie X, Wu X, Zhang X, Wu Y, Zhao P, Xia Q (2015) CRISPR/Cas9-mediated targeted mutagenesis in Nicotianatabacum. Plant Mol Biol 87:99–110
Gil-Humanes J, Wang Y, Liang Z, Shan Q, Ozuna CV, Sanchez-Leon S, Baltes NJ, Starker C, Barro F, Gao C, Voytas DF (2017) High efficiency gene targeting in hexaploid wheat using DNA replicons and CRISPR/Cas9. Plant J 89:1251–1262
Güell M, Yang L, Church GM (2014) Genome editing assessment using CRISPR genomeanalyzer (CRISPR-GA). Bioinformatics 30(20):2968–2970
Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud J-B, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, Joly J-S, Concordet J-P (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17:148
Haun W, Coffman A, Clasen BM, Demorest ZL, Lowy A, Ray E, Retterath A, Stoddard T, Juillerat A, Cedrone F, Mathis L, Voytas DF, Zhang F (2014) Improved soybean oil quality by targeted mutagenesis of the fatty acid desaturase 2 gene family. Plant Biotechnol J 12:934–940
Heigwer F, Kerr G, Boutros M (2014) E-CRISP: fast CRISPR target site identification. Nat Methods 122:122–123
Hendel A, Kildebeck EJ, Fine EJ, Clark JT, Punjya N, Sebastiano V, Bao G, Porteus MH (2014) Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep 7(1):293–305
Hendel A, Fine EJ, Bao G, Porteus MH (2015) Quantifying on- and off-target genomeediting. Trends Biotechnol 33(2):132–140
Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F (2013) DNA targeting specificity ofRNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433
Jansen R, Embden JD, Gaastra W, Schouls LM (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43(6):1565–1575
Jensen SP, Febres VJ, Moore GA (2014) Cell penetrating peptides as an alternative transformation method in citrus. J Citrus Pathol 1:10–15
Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP (2013) Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res 41(20):e188
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J (2013) RNA-programmed genome editing in human cells. eLife 2:e00471. https://doi.org/10.7554/eLife.0047
Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343:1247997
Johnson RA, Gurevich V, Filler S, Samach A, Levy AA (2015) Comparative assessments of CRISPR-Cas nucleases cleavageefficiency in planta. Plant Mol Biol 87:143–156
Kanchiswamy CN (2016) DNA-free genome editing methods for targeted crop improvement. Plant Cell Rep 35:1469–1474
Kapusi E, Corcuera-Gómez M, Melnik S, Stoger E (2017) Heritable genomic fragment deletions and small indels in the putative ENGase gene induced by CRISPR/Cas9 in barley. Front Plant Sci 8. https://doi.org/10.3389/fpls.2017.00540
Khatodia S, Bhatotia K, Passricha N, Khurana SMP, Tuteja N (2016) The CRISPR/Casgeneome editing tool: application in improvement of crops. Front Plant Sci 7. https://doi.org/10.3389/fpls.2016.00506
Kim H, Kim JS (2014) A guide to genome engineering with programmable nucleases. Nat Rev Genet 15:321–334. https://doi.org/10.1038/nrg3686
Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E (2013) A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 31(3):251–258
Kim S, Kim D, Cho SW, Kim J, Kim JS (2014) Highly efficient RNA guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012–1019. https://doi.org/10.1101/gr.171322.113
Kim H, Kim ST, Riju J, Kang B-C, Kim J-S, Kim S-G (2017) CRISPR/Cpf-1 mediated DNA-free plant genome editing. Nat Commun 8:14406
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533:420–424
Koo T, Lee J, Kim JS (2015) Measuring and reducing off-target activities of programmable nucleases including CRISPR-Cas9. Mol Cells 38:475–481. https://doi.org/10.14348/molcells.2015.0103
Kumagai MH, Donson J, della-Cioppa G, Harvey D, Hanley K, andGrill LK (1995) Cytoplasmic inhibition of carotenoid biosynthesiswith virus-derived RNA. Proc Natl Acad Sci USA 92:1679–1683
Kumar V, Jain M (2015) The CRISPR-Cas system for plant genome editing: advances and opportunities. J Exp Bot 66:47–57
Kumar D, Anandalakshmi SP, Marathe R, Schiff M, Liu Y (2003) Virus-induced gene silencing. Methods Mol Biol 236:287–294
Kuzma J, Kokotovich A (2011) Renegotiating GM crop regulation. EMBO Rep 12(9):883–888
Lee HJ, Kweon J, Kim E, Kim S, Kim JS (2012) Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res 22:539–548. https://doi.org/10.1101/gr.129635.111
Li J, Zhang X, Sun Y, Zhang J, Du W, Guo X, Li S, Zhao Y, Xia L (2018) Efficient allelic replacement in rice by gene editing: a case study of the NRT1.1B gene. J Integr Plant Biol. https://doi.org/10.1111/jipb.12650
Liang Z, Zhang K, Chen K, Gao C (2014) Targeted mutagenesis in Zea mays using TALENs and CRISPR/Cas system. J Genet Genom 41:63–68
Liang Z, Chen K, Li T, Zhang Y, Wang Y, Zhao Q, Liu J, Zhang H, Liu C, Ran Y, Gao C (2017) Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nat Commum 8:14261
Lloyd A, Plaisier CL, Carroll D, Drews GN (2005) Targeted mutagenesis using zinc-finger nucleases in Arabidopsis. Proc Natl Acad Sci USA 102(6):2232–2237
Lu Y, Zhu JK (2017) Precise editing of a target base in the rice genome using a modified CRISPR/Cas9 system. Mol Plant 10:523–525
Ma X, Mau M, Sharbel TF (2018) Genome editing for global food security. Trends Biotechnol 36(2):123–127
Macovei A, Sevilla NR, Cantos C, Jonson G, Loedin IS, Cermak T, Voytas D, Choi IR, Mohanty PC (2018) Novel alleles of rice eIF4G generated by CRISPR/Cas9-targeted mutagenesis confer resistance to rice tungro spherical virus (RTSV). Plant Biotechnol J. https://doi.org/10.1111/pbi.12927
Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, Unger-Wallace E, Sander JD, Muller-Lerch F, Fu F, Pearlberg J, Gobel C, Dassie JP, Pruett-Miller SM, Porteus MH, Sgroi DC, Iafrate AJ, Dobbs D, McCray PB Jr, Cathomen T, Voytas DF, Joung JK (2008) Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31:294–301
Maggio I, Goncalves MA (2015) Genome editing at the cross-roads of delivery, specificity and fidelity. Trends Biotechnol 33(5):280–291
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339(6121):823–826
Malnoy M, Viola R, Jung M-H, Koo O-J, Kim S, Kim J-S, Velasco R, Kanchiswamy CN (2016) DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoprotein. Front Plant Sci 7. https://doi.org/10.3389/fpls.2016.01904
Martin-Ortigosa S, Wang K (2014) Proteolistics: a biolistics method for intracellular delivery of proteins. Transgenic Res 23:743–756
Masani MY, Noll GA, Parveez GK, Sambanthamurthi R, Prufer D (2014) Efficient transformation of oil palm protoplasts by PEG mediated transfection and DNA microinjection. PLoS One 9:e96831. https://doi.org/10.1371/journal.pone.0096831
Mashal RD, Koontz J, Sklar J (1995) Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases. Nat Genet 9:177–183
Miao J, Guo D, Zhang J, Huang Q, Qin G, Zhang X, Wan J, Gu H, Qu LJ (2013) Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res 23:1233–1236
Mojica F, Garcia-Martinez J, Soria E (2005) Intervening sequences ofregularly spaced prokaryotic repeats derive from foreign geneticelements. J Mol Evol 60:174–182
Mojica FJ, Díez-Villaseñor C, García-Martínez J, Almendros C (2009) Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–740
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 11:326
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156(5):935–949
Park CY, Kim J, Kweon J, Son JS, Lee JS, Yoo JE, Cho SR, Kim JH, Kim JS, Kim DW (2014) Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc Natl Acad Sci USA 111:9253–9258. https://doi.org/10.1073/pnas.1323941111
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee Y-L, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC, June CH (2008) Establishment of HIV-1 resistance in CD4+ T cells by genomeediting using zinc-finger nucleases. Nat Biotechnol 26(7):808–816
Periwal V (2017) A comprehensive overview of computational resources to aid in precision genome editing with engineered nucleases. Brief Bioinform 18:698–711
Pourcel C, Salvignol G, Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 51:653–663
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152(5):1173–1183
Qiu P, Shandilya H, Alessio JMD, Connor KO, Durocher J, Gerard GF (2004) Mutation detection using surveyor™ nuclease. BioTechniques 36:702–707
Ramleet MK, Yan T, Cheung AM, Chuah CT, Li S (2015) High throughput genotyping of CRISPR/Cas9 mediated mutants using fluorescent PCR capillary gel electrophoresis. Sci Rep 5:15581
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protocol 8(11):2281–2308
Sánchez-León S, Gil-Humanes J, Ozuna CV, Giménez MJ, Sousa C, Voytas DF, Barro F (2018) Low-gluten, nontransgenic wheat engineered with CRISPR/Cas9. Plant Biotechnol J 16(4):902–910
Santiago Y, Chan E, Liu P-Q, Orlando S, Zhang L, Urnov FD, Holmes MC, Guschin D, Waite A, Miller JC, Rebar EJ, Gregory PD, Klug A, Collingwood TN (2008) Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci U S A 105(15):5809–5814
Segal DJ, Dreier B, Beerli RR, Barbas CF (1999) Towards controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 50-GNN-30 DNA target sequences. Proc Natl Acad Sci USA 96(6):2758–2763
Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, Gao C (2013) Targeted genome modification of crop plants using CRISPR/Cas system. Nat Biotechnol 31(8):686–688
Shan Q, Wang Y, Li J, Gao C (2014) Genome editing in rice and wheat using the CRISPR/Cas system. Nat Protoc 9:2395
Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, Skarnes WC (2014) Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11:399–402
Shi J, Gao H, Wang H, Lafitte HR, Archibald RL, Yang M, Hakimi SM, Mo H, Habben JE (2017) ARGOS8 variants generated byCRISPR-Cas9 improve maize grain yield under field droughtstress conditions. Plant Biotechnol J 15:207–216
Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, Worden SE, Mitchell JC, Arnold NL, Gopalan S, Meng X, Choi VM, Rock JM, Wu YY, Katibah GE, Zhifang G, McCaskill D, Simpson MA, Blakeslee B, Greenwalt SA, Butler HJ, Hinkley SJ, Zhang L, Rebar EJ, Gregory PD, Urnov FD (2009) Precise genome modification in the cropspeciesZea mays using zinc-finger nucleases. Nature 459:437–441
Skipper KA, Mikkelson JG (2015) Delivering the goods for genome engineering and editing. Hum Gene Ther 26:486. https://doi.org/10.1089/hum.2015.063
Streubel J, Blücher C, Landgraf A, Boch J (2012) TAL effector RVD specificities and efficiencies. Nat Biotechnol 30:593–595
Sun Y, Jiao G, Li J, Guo X, Du W, Du J, Francis F, Zhao Y, Xia L (2017) Generation of high-amylose rice through CRISPR/Cas9-mediated targeted mutagenesis of starch branching enzymes. Front Plant Sci 8:298
Sung YH, Baek I-J, Kim DH, Jeon J, Lee J, Lee K, Jeong D, Kim J-S, Lee H-W (2013) Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 31(1):23–24
Svitashev S, Schwartz C, Lenderts B, Young JK, Cigan AM (2016) Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nat Commun 7:13274
Symington LS, Gautier J (2011) Double-strand break end resection and repair pathway choice. Annu Rev Genet 45:247–271
Tang L, Mao B, Li Y, Lv Q, Zhang L, Chen C, He H, Wang W, Zeng X, Shao Y, Pan Y, Hu Y, Peng Y, Fu X, Li H, Xia X, Zhao B (2017) Knockout of OsNramp5 using the CRISPR/Cas9 system produces low Cd-accumulating indica rice without compromising yield. Sci Rep 7:14438. https://doi.org/10.1038/s41598-017-14832-9
Taylor S, Scott R, Kurtz R, Fisher C, Patel V, Bizouarn F (2010) A practical guide to high resolution melt analysis genotyping. Bio-Rad Laboratories, Inc, Hercules
Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK, Voytas DF (2009) High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature 459:442–445
Upadhyay SK, Kumar J, Alok A, Tuli R (2013) RNA-guided genome editing for target gene mutations in wheat. Gene Genomes Genet 3(12):2233–2238
Vouillot L, Télie A, Pollet N (2015) Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered byengineered nucleases. Genes Genomes Genet 5:407–415
Voytas DF (2013) Plant genome engineering with sequence-specific nucleases. Annu Rev Plant Biol 64:30.1–30.24
Waltz E (2016) Gene-edited CRISPR mushroom escapes US regulation. Nature 532:293
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qui J-L (2014) Simaltaneous editing of three homoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32:947. https://doi.org/10.1038/nbt.2969
Wang F, Wang C, Liu P, Lei C, Hao W, Gao Y, Liu YG, Zhao K (2016a) Enhanced rice blast resistance by CRISPR/Cas9-targeted mutagenesis of the ERF transcription factor gene OsERF922. PLoS One. https://doi.org/10.1371/journal.pone.0154027
Wang H, Russa ML, Qi LS (2016b) CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem 85:227–264
Wang W, Akhunova A, Chao S, Akhunov E (2016c) Optimizing multiplex CRISPR/Cas9 based genome editing for wheat. Biorxiv. https://doi.org/10.1101/051342
Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S (2011) A modular cloning system for standardized assembly ofmultigene constructs. PLoS One 6(2):e16765.; PMID:21364738. https://doi.org/10.1371/journal.pone.0016765
Wei Y, Terns RM, Terns MP (2015) Cas9 function and host genome sampling in type II-A CRISPR–Cas adaptation. Genes Dev 29(4):356–361
Weinthal D, Tovkach A, Zeevi V, Tzfira T (2010) Genome editing in plant cells byzinc finger nucleases. Trends Plant Sci 15:308–321
Wolter F, Puchta H (2017) Knocking out consumers concerns and regulator’s rules: efficient use of CRISPR/Cas9 ribonucleoprotein complexes for genome editing in cereals. Genome Biol 18:43
Wong N, Liu W, Wang X (2015) WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol 16:218
Woo JW, Kim J, Kwon S II, Corvalan C, Cho SW, Kim H, Kim S-G, Kim S-T, Choe S, Kim J-S (2015) DNA-free genome editing in plants with pre-assembled CRISPR-Cas9 ribonucleoproteins. Nat Biotechnol 33:1162–1164
Wright DA, Townsend JA, Winfrey RJ Jr, Irwin PA, Rajagopal J, Lonosky PM, Hall BD, Jondle MD, Voytas DF (2005) High-frequency homologous recombination in plants mediated by zinc-finger nucleases. Plant J 44(4):693–705
Xiao A, Cheng Z, Kong L, Zhu Z, Lin S, Gao G, Zhang B (2014) CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 30:1180–1182
Xu R, Li H, Qin R, Wang L, Li L, Wei P, Yang J (2014) Gene targeting using the Agrobacteriumtumefacians-mediated CRISPR/Cas system in rice. Rice 7:5
Xu R-F, Li H, Qin R-Y, Li J, Qiu C-H, Yang Y-C, Ma H, Li L, Wei P-C, Yang J-B (2015) Generation of inheritable and “transgene clean” targeted genome-modified rice in latergenerations using the CRISPR/Cas9system. Sci Rep 5:11491 https://doi.org/10.1038/srep 11491
Xu R, Qin R, Li H, Li D, Li L, Wei P, Yang J (2016) Generation of targeted mutant rice using CRISPR/Cpf1 system. Plant Biotechnol J. https://doi.org/10.1111/pbi.1266
Yin K, Han T, Liu G, Chen T, Wang Y, Yu AYL, Liu Y (2015) A geminivirus-based guide RNA delivery system for CRISPR/Cas9 mediatedplant genome editing. Sci Rep 5:14926
Yu C, Zhang Y, Yao S, Wei Y (2014) A PCR based protocol for detecting indel mutationsinduced by TALENs and CRISPR/Cas9 in zebrafish. PLoS One 9(6):e98282
Zhang H, Zhang J, Wei P, Zhang B, Gou F, Feng Z, Mao Y, Yang L, Zhang H, Xu N, Zhu JK (2014) The CRISPR/Cas9 system produces specific andhomozygous targeted gene editing in rice in onegeneration. Plant Biotechnol J 12:797–807
Zhang Y, Liang Z, Zong Y, Wang Y, Liu J, Chen K, Qiu J-L, Gao C (2016) Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat Commun 7:12617
Zhang Y, Bai Y, Wu G, Zou S, Chen Y, Gao C, Tang D (2017) Simultaneous modification of three homeologs of TaEDR1 by genome editing enhances powdery mildew resistance in wheat. Plant J 91:714. https://doi.org/10.1111/tpj.13599
Zischewski J, Fisher R, Bortesi L (2016) Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol Adv 35:95–104
Zong Y, Wang Y, Li C, Zhang R, Chen K, Ran Y, Qiu JL, Wang D, Gao C (2017) Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion. Nat Biotechnol 35:438–440
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Manchanda, P., Suneja, Y. (2018). Genome Editing for Crop Improvement: Status and Prospects. In: Gosal, S., Wani, S. (eds) Biotechnologies of Crop Improvement, Volume 3. Springer, Cham. https://doi.org/10.1007/978-3-319-94746-4_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-94746-4_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-94745-7
Online ISBN: 978-3-319-94746-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)