
Abstract
An 18-year-old woman presented with galactorrhea and was thought to have a prolactinoma. As acromegaly signs and symptoms progressed, she proved to have an elevated GH nadir on oral glucose tolerance test but IGF1 was initially normal. The disease progressed and IGF-1 became elevated over time. She required two pituitary surgeries and radiation in addition to medical therapies for acromegaly. The case of discrepancy between IGF1 and GH levels is discussed as well as the management strategy for aggressive cases of acromegaly in which disease persists after pituitary surgery.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
Case Presentation
This is a woman who initially presented with galactorrhea at age 18 years and was found to have an elevated prolactin of 75 (normal <20 ng/mL). She reported progressive headaches, increase in nose size, increasing ring and shoe size, and swelling of fingers. On exam her vital signs were normal. She stood at 63 in. with a weight of 125 lb, blood pressure of 110/80, and heart rate of 72. Notable exam findings included an absence of the typical facial features of acromegaly except for a subtle increase in her nose and chin size compared to old photographs and a larger tongue. She had enlargement of feet and hands and thickened fingers. Visual field exam was normal, and the remainder of the exam was unremarkable. Biochemical testing revealed insulin growth factor 1 (IGF-1) was in the upper normal range at 624 (normal range 182–780 ng/mL) and a random growth hormone (GH) was 14 ng/mL. GH nadir on oral glucose tolerance test (OGTT) was high at 7.2 ng/mL, where normal OGTT nadir is <1 ng/mL. Morning cortisol and thyroid hormone levels were normal, and repeated prolactin (diluted) was again mildly elevated at 51.3 (0–20 ng/mL). Cosyntropin stimulation was normal, excluding adrenal insufficiency. Fasting glucose and hemoglobin AIc were normal. An MRI of the pituitary was ordered, which revealed a sellar mass approximately 1.4 cm in size without compression of the chiasm (Fig. 6.1).

Pituitary MRI post contrast
Given her elevated GH and a macroadenoma , she underwent transsphenoidal surgery by an experienced pituitary surgery. A GH-secreting adenoma was confirmed on pathology which also showed rare prolactin-secreting cells. Six weeks postoperatively, she felt well and reported decrease in ring size. Pituitary MRI six weeks after surgery revealed a small lateral superior remnant of tumor, which the neurosurgeon determined to be likely accessible with repeat surgery. Twelve weeks postoperatively, after the second surgery, her menses became regular and IGF-1 improved at 463 (normal range 182–780 ng/mL). Nadir GH by OGTT, while improved, remained high at 4.3 ng/mL, where normal OGTT nadir is <1 ng/mL.
My Management
-
1.
A repeat surgery was recommended and performed by an expert pituitary surgeon .
We considered repeat pituitary surgery versus medical therapy with somatostatin analog, cabergoline, or pegvisomant. Given her young age, uncertainty of ongoing access to medical insurance coverage for drugs, and the fact that the tumor remnant was deemed likely to be accessible with a second surgery, after discussion of options and risks and benefits of each, a second surgery was recommended and the patient agreed.
-
2.
A postoperative MRI and anterior pituitary function tests were obtained.
-
3.
Clinical assessment for signs and symptoms of acromegaly and biochemical testing for GH excess: OGTT for GH and IGF-1 levels were obtained at 6–12 weeks postoperatively.
-
4.
A colonoscopy was suggested.
Assessment and Diagnosis
The diagnosis of acromegaly is based on signs and symptoms of acromegaly and biochemical evidence of GH excess: a high GH and/or high IGF-1. The presence of pituitary tumor is visible on MRI in >90% of cases, and most of these, approximately 70% are macroadenomas [1]. This patient was originally thought to have a prolactinoma because her first presentation to her gynecologist was for galactorrhea. When she was seen by neuroendocrine, she was found to have subtle features of acromegaly, high GH but normal IGF-1, and ultimately pathology confirmed a GH-secreting macroadenoma.
Why was her IGF-1 normal if she had acromegaly, and what are the causes for the discrepancy between IGF-1 and GH? Possible reasons for a lower IGF-1 include exogenous estrogen, liver disease, starvation/malnourishment, and poorly controlled DM [2]. None of these applied in her case. She may have been restricting calories but was not known to have anorexia. Another possibility includes a range of problems with IGF-1 assays [3]. In this case, the normal range for her age was up to 780 ng/mL, and this may have been inappropriately high, placing her fairly high level of 624 within the normal range. GH may be elevated in anorexia, normal puberty , pregnancy, diabetes mellitus and hypoglycemia, liver disease, and exogenous estrogen [4]. These conditions should be considered if IGF-1 is normal in the setting of high GH or the clinical diagnosis is in question; but none of these explained the high GH in this case of isolated GH excess, and ultimately pathology confirmed acromegaly.
She was only 18 years at her first presentation of acromegaly. The mean age at diagnosis is typically in the fourth decade, and most patients have had already had the disease for 7–10 years prior to diagnosis [1]. Younger patients are more likely to have more aggressive disease and more likely to have germ-line aryl hydrocarbon receptor-interacting protein (AIP) mutations even in the absence of known family history [5]. Gigantism occurs when the GH excess begins before the pubertal closure of the epiphyses. At 63 inches, she was not tall relative to her mid-parental height because menarche and puberty had been completed before the onset of the acromegaly, and therefore she presumably had closure of her epiphyses prior to the onset of acromegaly.
At the time of diagnosis of acromegaly, assessment should include evaluations of comorbidities associated with acromegaly. These include evaluation of glucose and hemoglobin AIC to screen for diabetes or glucose intolerance, a test for sleep apnea if there are suggestive symptoms, an echocardiogram if there are cardiac symptoms, and a screening colonoscopy to evaluate for colonic polyps and colon cancer [6]. In addition, testing anterior hormonal function to assess for co-secretion or deficiencies is indicated at baseline and after surgical therapy [7]. Visual field testing is appropriate in patients with MRI evidence of tumors which contact or invade the optic chiasm and preconception in reproductive age patients with macroadenomas [6].
Management
Pituitary surgery remains a first-line therapy for acromegaly unless contraindicated, refused, inaccessible, or if there is no compression of chiasm and patients are unlikely to be surgically cured; in these cases primary medical therapy is appropriate [6].
Medical therapy includes cabergoline, somatostatin analogs (SSAs) , and GH receptor antagonist [8]. Cabergoline is more likely to be effective as monotherapy if the IGF-1 is less than double normal and typically has minimal effect on tumor reduction [9]. Dopamine agonists such as cabergoline are generally well tolerated but can increase impulsive behavior rarely and must be avoided in patients with psychotic disorders as they may stimulate psychosis in such patients [10, 11]. The first-generation somatostatin analogs include lanreotide and octreotide, target somatostatin subtype receptors (SSTR) 2 and 5, and are available as long-acting monthly injections, which decrease GH excess and control tumor in 20–70% of patients. Side effects may include diarrhea, nausea, abdominal pain, gallstones, diabetes, injection site pain, and rarely hypoglycemia , bradycardia, and hair loss. Pasireotide , a second-generation somatostatin analog , binds to SSTR 1, 2, 3, and 5 and has slightly greater efficacy in controlling GH excess and also confers tumor control in responsive patients with a side effect profile similar to a first-generation SSA but with increased risk of diabetes and hyperglycemia. Pegvisomant , a growth hormone receptor antagonist, does not typically decrease tumor but effectively controls IGF-1 in most patients with minimal side effects: rarely causing increase in liver enzymes, headaches, hives, or lipohypertrophy at injection sites. If monotherapy is not effective, then therapies can be combined: SSA plus pegvisomant, SSA plus cabergoline, and cabergoline plus SSA.
If medical therapies are not tolerated or inaccessible, radiation is an option. Gamma knife or proton beam can be given as a single dose unless the tumor is very near the optic chiasm, in which case single-dose radiotherapy cannot be used, and traditional fractionated radiation therapy can be given over many weeks. Since radiotherapy takes 2–20 years to produce a remission [12], medical therapy must be used until then to control GH excess.
Outcome
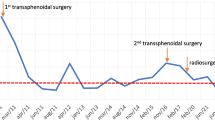
The first surgery was not effective but was uncomplicated and improved GH and IGF-1 levels and symptoms. The second surgery was not curative but associated with further improvement of GH. Patient could not tolerate the SSA due to nausea and weight loss . She could not tolerate cabergoline due to fatigue and mood changes. She had radiation therapy and was observed off medications since her IGF-1 was normal, and she did not have symptoms of acromegaly. However, for several years without medical insurance, she was lost to follow-up. Three years after radiotherapy, her IGF-1 began to rise, and signs and symptoms of acromegaly progressed including thickening of the fingers, joint pain, headaches, sweating, and subtle progression in facial features of acromegaly (Table 6.1). She was then treated with pegvisomant with normal IGF-1 levels. Annual MRI of the pituitary post-radiation demonstrated stability of the tumor. Anterior pituitary function tests were obtained at 6 and 12 months and yearly following radiation, and all remained normal except for the finding of central hypothyroidism 5 years after radiation.
Clinical Pearls and Pitfalls
-
Galactorrhea was the presenting sign and prolactin was high which made the diagnosis initially seem to be a prolactinoma. However, in such patients with hyperprolactinemia and adenoma, particularly with any signs or symptoms of acromegaly, tests for GH excess should be obtained given that there can be co-secretion of prolactin and GH.
-
When IGF-1 and GH show discrepant values, reasons for false positive and false negatives should be reviewed, and levels should be repeated in a reliable assay. If either value is confirmed, levels should be interpreted based on the clinical correlation and treated if there are symptoms or signs of acromegaly.
-
Repeat surgery can be considered if remnant tumor is surgically accessible.
-
Classic acromegaly appearance may not be obvious in early disease; enlargement of hands and feet may be only sign without facial features of acromegaly in some patients.
-
Galactorrhea may be a sign of acromegaly in women.
-
When acromegaly persists after surgery, medications should be administered to control GH and IGF-1 and tumor remnant; if a medication is ineffective alone, medical therapies can be combined.
-
If medical therapy is not effective, not tolerated, or inaccessible, radiotherapy should be considered in patients in whom surgery has been unsuccessful.
-
Following radiotherapy to the pituitary, long-term follow-up of anterior pituitary function should be evaluated at 6 months, 12 months, and yearly after the radiation therapy is completed since hormonal loss can occur for many years or even up to decades after the radiation therapy.
-
This case was notable in that the original IGF-I was normal at diagnosis when she had isolated elevation of growth hormone ; over time, as the disease progressed off therapy, her IGF-I level became elevated.
References
Melmed S. Acromegaly. N Engl J Med. 2006;355:2558–73.
Freda PU. Monitoring of acromegaly: what should be performed when GH and IGF-1 levels are discrepant? Clin Endocrinol (Oxf). 2009;71:166–70.
Frystyk J, Freda P, Clemmons DR. The current status of IGF-I assays—a 2009 update. Growth Horm IGF Res. 2010;20:8–18.
Freda PU. Current concepts in the biochemical assessment of the patient with acromegaly. Growth Horm IGF Res. 2003;13:171–84.
Cazabat L, et al. Germline inactivating mutations of the aryl hydrocarbon receptor-interacting protein gene in a large cohort of sporadic acromegaly: mutations are found in a subset of young patients with macroadenomas. Eur J Endocrinol. 2007;157:1–8.
Katznelson L, et al. Acromegaly: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99:3933–51.
Fleseriu M, et al. Hormonal replacement in hypopituitarism in adults: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101:3888–921.
Dineen R, Stewart PM, Sherlock M. Acromegaly. QJM. 2017;110(7):411–20. pii: hcw00.
Abs R, et al. Cabergoline in the treatment of acromegaly: a study in 64 patients. J Clin Endocrinol Metab. 1998;83:374–8.
Chang SC, Chen CH, Lu ML. Cabergoline-induced psychotic exacerbation in schizophrenic patients. Gen Hosp Psychiatry. 2008;30:378–80.
Barake M, et al. Investigation of impulsivity in patients on dopamine agonist therapy for hyperprolactinemia: a pilot study. Pituitary. 2014;17:150–6.
Shih HA, Loeffler JS. Radiation therapy in acromegaly. Rev Endocr Metab Disord. 2008;9:59–65.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Nachtigall, L.B. (2018). Acromegaly: Diagnosis and Management in Patients Who Present with Discrepancy Between IGF-1 and GH. In: Nachtigall, L. (eds) Pituitary Tumors. Springer, Cham. https://doi.org/10.1007/978-3-319-90909-7_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-90909-7_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-90907-3
Online ISBN: 978-3-319-90909-7
eBook Packages: MedicineMedicine (R0)