
Abstract
Essentially every organ in the human body, including the intestine, can be affected by fibrotic reactions. Under normal homeostatic conditions these reactions are self-limited and constitute an important reparative process aimed at the restoration of the functional integrity of injured tissues. However, under pathologic circumstances the homeostatic regulatory mechanisms evolve into an uncontrolled fibrotic process characterized by the accumulation of large amounts of fibrotic tissue, which disrupts normal organ architecture and ultimately leads to organ failure. Even though their etiology varies greatly, all fibrotic reactions share common features. It is universally accepted that myofibroblasts are the cells ultimately responsible for the pathologic fibrotic process. Myofibroblasts, expressing α-smooth muscle actin (α-SMA), comprise a distinctive population of mesenchymal cells. When activated, they markedly increase the production of fibrillar collagens (types I, III, V, and VI) and other extracellular matrix (ECM) macromolecules coupled with an increased inhibition of ECM-degradative enzymes which may result in the production of injurious scar tissue in the intestine. While abdominal adhesions may be caused by infection, inflammation or ischemia, surgical procedures are the primary cause. Unfortunately, adequate therapeutic solutions have proven elusive. The peritoneal surfaces, both visceral and parietal, are covered by a monolayer of mesothelial cells bound to a basement membrane. Because the mesothelial cells are weakly connected, the peritoneal surface is delicate and easily injured, resulting in a series of events, which can be broken down into coagulation cascade and inflammatory stages leading to a fibrous adhesion stage. TGF-β, IL-6 and likely other cytokines and growth factors play critical roles in adhesion formation by mediating the formation of myofibroblasts and stimulating the production of ECM. In the pathogenesis of fibrosis in inflammatory bowel disease (IBD), many factors need to be considered, including a much more sustained inflammatory response, a clear if still poorly understood genetic predisposition, the potential involvement of multiple mesenchymal cells, exposure of the mucosa to intestinal bacteria and the involvement of the immune system. In IBD, the normal wound healing process triggered by injury and inflammation fails and, instead of resolution, there is continued ECM production by myofibroblasts. Because of a protracted inflammatory response, one could imagine that anti-inflammatory therapy might be an effective approach. Unfortunately, this has not been the case, and it appears that once the damaging fibrotic reaction has been initiated in fibrosis-prone individuals, it is self-propagating. Thus, as in other fibrotic situations, the aberrant myofibroblast becomes the ultimate target. However, unlike adhesions in which the potential instigators can be anticipated and candidate drugs given over a fairly short time, in IBD the pathogenesis is much more protracted. There are a number of FDA—approved drugs capable of intercepting pathways potentially critical in the fibrotic reaction. TGF-β signaling is, of course, the primary target. However, because of the manifold activities of TGF-β, one or more downstream events in the signaling pathways must be judiciously selected so as not to elicit toxic responses. The same caution must be applied when dealing with other potential targets. Because of the inherent redundancy in signaling from multiple cytokines/growth factors involved in fibrotic reactions, it is likely that more than one drug must be administered simultaneously to obtain effective beneficial inhibition.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Fibrosis
- Myofibroblasts
- TGF-β
- Abdominal adhesions
- Inflammatory bowel disease
- Crohn’s disease
- Ulcerative colitis
22.1 Introduction
In order to place abdominal adhesions and luminal fibrosis in inflammatory bowel disease (IBD) on a more comprehensive platform to take advantage of what is known in other systems characterized by fibrotic reactions, we first discuss the pathophysiology of fibrosis in a general sense. Essentially every organ in the human body can be affected by fibrotic reactions. Under normal homeostatic conditions, these reactions are self-limited and constitute an important reparative process aimed at the restoration of the functional integrity of injured tissues through a complex sequence of events constituting tissue repair. However, under pathologic circumstances, the homeostatic regulatory mechanisms evolve into an unrestrained fibrotic process characterized by the progressive and uncontrolled accumulation of large amounts of connective tissue which disrupts the normal organ architecture and ultimately leads to organ failure [1,2,3]. These reactions can cause multi-system diseases such as Systemic Sclerosis (SSc) [4, 5], as well as fibrotic disorders affecting individual organs including those of the gastro-intestinal system. Despite considerable understanding of the pathogenesis of the fibrotic process attained recently [1,2,3], disease-modifying therapy for the fibrotic diseases is extremely limited. Even though etiologic agents vary greatly, the fibrotic diseases all share common molecular alterations that result in the exaggerated and uncontrolled accumulation of extracellular matrix (ECM) macromolecules in the affected tissues which may result in the replacement of functioning tissue such as alveoli in the lung, myocytes in the heart, or nephrons in the kidney either with non-functional fibrotic tissue or injurious scar tissue in the gut [6,7,8,9]. At the cellular level, it is universally accepted that myofibroblasts are the cells ultimately responsible for pathologic ECM synthesis in fibrotic disorders [10,11,12,13]. Myofibroblasts, expressing alpha smooth muscle actin (α-SMA), comprise a distinctive population of mesenchymal cells, which markedly increase the production of fibrillar collagens (types I, III, V, and VI) and other ECM macromolecules coupled with an increased inhibition of ECM-degradative enzymes [14,15,16]. Furthermore, such alterations in the ECM produce changes in the biomechanical properties of the affected tissues causing a progressive increase in tissue stiffness, a potentially potent pro-fibrotic stimulus [17,18,19,20].
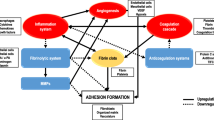
The origin of myofibroblasts, still a contentious issue, may vary depending upon the organ affected and the particular fibrotic reaction [11, 13, 21,22,23]. There are several potential sources including: [1] recruitment of fibroblast precursor cells (fibrocytes) from bone marrow, [2] trans-differentiation of various cell types including pericytes, adipocytes, and epithelial, mesothelial, and endothelial cells into a mesenchymal phenotype, [3] proliferation and activation of quiescent tissue- resident fibroblasts into a myofibroblast phenotype (see Fig. 22.1) and [4] sub-epithelial myofibroblasts. Although epithelial to mesenchymal transition (EMT), endothelial to mesenchymal transition (EndoMT), or pericyte to myofibroblast transition may play a role under specific circumstances [13, 21,22,23], the current preponderance of opinion is that the activation of tissue-resident fibroblasts is the major source of activated myofibroblasts. However, even though the trans-differentiation of various cell types may not be a predominant source of myofibroblasts during fibrotic disorders, alterations in the phenotype of the trans-differentiated cells may result in the production and secretion of pro-fibrotic factors, including TGF-β, which play an important role in the fibrotic process. Furthermore, these phenotypically-modified cells may produce numerous macromolecules which may enhance the fibrotic response such as the EDA form of fibronectin (FnEDA) and other ECM components including proteoglycans and several matricellular molecules [21,22,23,24,25,26].

Cellular origins and pathways leading to the formation of an activated myofibroblast. Injuries caused by a variety of putative causative agents such as bacteria, viruses, toxins, ROS, gadolinium-based contrast agents (GdBSAs) or radiation in genetically predisposed hosts result in inflammation. Activated inflammatory cells secrete cytokines and growth factors such as TGF-β and interleukin-6 causing trans-differentiation of resident and non-resident fibroblast cells as well as endothelial, epithelial and mesothelial cells into myofibroblasts. These cells produce excess amounts of structural macromolecules which contribute to fibrosis leading to alterations in tissue architecture causing pathological dysfunction. PAMPs Pathogen-Associated Molecular Patterns, TLR Toll-Like Receptor, TGF-β Transforming Growth Factor beta, ROS Reactive Oxygen Species, GdBSAs Gadolinium-Based Contrast Agents
22.2 Targeting Myofibroblasts
There are multiple potential levels that could be targeted for inhibition of fibrotic responses, such as elimination of the primary cause as in treatment of viral hepatitis for liver fibrosis, diminution of the immunologic and inflammatory responses in SSc and Idiopathic Pulmonary Fibrosis (IPF), and elimination of the untoward pro-fibrotic activities of myofibroblasts. Regrettably, owing to the lack of a comprehensive understanding of the etiologic mechanisms in the majority of the fibrotic disorders, opportunities for elimination of the originating cause of the fibrotic reaction are rare, and reduction of the immunologic and inflammatory responses has proven to be generally ineffective in abrogating pathologic fibrotic processes. Thus, modulation of the deleterious pro-fibrotic activity of myofibroblasts remains the most attractive therapeutic approach. This, in turn, requires a precise understanding of the molecular pathways most important in generating the excessive ECM by the myofibroblast as discussed recently [27] and briefly reviewed below.
22.3 The Transforming Growth Factor-β (TGF-β) Pathways
The TGF-β family of growth factors plays wide-ranging roles in numerous physiological processes including embryogenesis, cellular differentiation, immunologic system development, inflammatory response, and wound repair [28,29,30,31]. Furthermore, numerous studies have shown that the three TGF-β isoforms are potent inducers of myofibroblasts either through activation of quiescent fibroblasts, or through the phenotypic conversion of various cell types into activated myofibroblasts [32,33,34]. Owing to their potent pro-fibrotic activities, they have been implicated in the pathogenesis of various fibrotic human diseases [35,36,37,38,39]. The intracellular transduction pathways following TGF-β binding to its cognate receptors are complex and involve both Smad-mediated pathways referred to as canonical and non-Smad pathways referred to as non-canonical [40, 41]. These pathways are diagrammatically illustrated in Fig. 22.2 along with other relevant cytokine/growth factor pathways. In the canonical pathway, the ligand-bound TGF-β receptor II (TβRII) recruits and phosphorylates a TGF-β receptor I (TβRI) which is known as activin-like kinases (ALKs), with ALK-5 being the most important in the context of the fibrotic process. Signaling from the phosphorylated TβRI to the nucleus occurs through the receptor activated RSmads, Smad2 and Smad3, which are phosphorylated by TβR1. The phosphorylated Smad2/Smad3 then bind to the co-Smad, Smad4, forming a complex that translocates across the nuclear membrane. Within the nucleus, the Smad complex, in association with various transcription factors, co-activators and co-repressors, modulates the expression of target genes [39, 40]. Non-canonical TGF-β-initiated signaling cascades are independent of RSmads. These non-canonical pathways can be activated in a cell-specific and context-dependent manner and mediate important TGF-β pro-fibrotic effects [41, 42]. For example, TGF-β stimulation leads to the activation of PI3K, which, in turn, activates two important pro-fibrotic pathways: p21 activated kinase (PAK2)-Abelson kinase (c-Abl) and Akt-mTOR1 [43]. Downstream targets of c-Abl include several mediators involved in the fibrotic response. Activated c-Abl phosphorylates protein kinase C-δ (PKC-δ), a potent pro-fibrotic mediator was recently shown to up-regulate collagen gene transcription in SSc dermal fibroblasts [44]. Phosphorylated PKC-δ has also been shown to in turn phosphorylate the transcription factor Fli-1, reversing its inhibitory effect on collagen gene expression [45]. Another important non-Smad signaling pathway is through activation of Jun-N-terminal kinase (JNK) resulting in the activation of c-Jun, a critical pro-fibrotic transcription factor [41, 42]. Besides serine/threonine phosphorylation, TβRII can also be phosphorylated on tyrosine residues in response to TGF-β [46, 47] leading to activation of Erk1/2 MAPK which play an important role by regulating myofibroblast formation as well as ECM synthesis [48, 49].

Critical TGF-β, Il-6 and Growth Factor/Cytokine signaling pathways important in fibrogenesis. Illustrated is the canonical pathway originating from a representative dimeric receptor. Following TGF-β binding, the TGFβRll receptor recruits a TGFβRl, either activin-like kinase-1 or activin-like kinase-5 and activates it by phosphorylation. Activin-like kinase 5 (Alk 5) then specifically phosphorylates receptor-regulated Smad2 and Smad3 which then complex with Co-Smad4 resulting in their transport to the nucleus where they interact with various co-activators or co-repressors to regulate transcription of critical pro-fibrotic genes, here represented by connective tissue growth factor (CTGF) fibronectin isoform EDA (FnEDA) and Col1a1 collagen genes. Also shown is Il-6 binding to its receptor with activation of Ras, Raf and MEK1/2 and ultimately Erk1/2. On the far left and right are illustrated are the signaling pathways for (PDGF), Wnt and Hedgehog. Each of these pathways is activated by ligand-binding to specific receptors, but the subsequent signaling transmission mechanisms between these pathways differ dramatically (see text). For clarity of presentation these pathways have been abbreviated with only the essential features presented. This figure has been modified from two produced by Protein Lounge. TGF-β Transforming Growth Factor beta, TGFβR1 & II Transforming Growth Factor beta Receptor 1 & II, LTBP Latent TGF-binding protein, Smad Sma and Mad related family of signal transducers, ERK Extracellular signal-regulated Kinase, TF Transcription Factor, Il-6 Interleukin -6, GP130 Glycoprotein 130, SARA Smad Anchor for Receptor Activation, PDGF Platelet Derived Growth Factor, SHP2 Tyrosine phosphatase, GRB2 Growth factor receptor-bound 2, SHC Src homolog 2, Ras Fas family of genes, Raf Raf kinases, MEK 1/2 MAPK/ERK pathway, Wnt Signal transduction pathway, Shh Sonic hedgehog, Ptch Protein patched homologue 1, Smo G-protein coupled receptor, Src Family of protein tyrosine kinases, c-Abl Abelson tyrosine kinase
One of the important aspects of TGF-β action is the stimulation of other mediators having fibrogenic potential. These include, but are not limited to, connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF) and epidermal growth factor (EGF/Erb-B) ligands. For example, it has been demonstrated that TGF-β-induced ErbB activation was achieved by up-regulation of ErbB ligands through autocrine signaling from the PDGF-receptor (PDGFR) via MEK/ERK [50]. While the molecular changes initiated by TGF-β are complex, and challenging, approaches with therapeutic agents can be carefully designed to take advantage of specific points within critical pathways to abrogate their deleterious pro-fibrotic effects.
22.4 Other Molecular Pathways Involved in the Fibrotic Process
Although the TGF-β family of growth factors plays the most important role in fibrosis, there are numerous other molecular pathways that also participate depending on the specific trigger initiating the fibrotic process and the tissues or organs involved. Although the diverse mechanisms mediated by these pathways result in an extremely intricate picture, the detailed understanding of their components and of their interactions may provide novel therapeutic targets to modify the devastating effects of fibrotic diseases. Some of these pathways are illustrated in Fig. 22.2 and will be briefly reviewed in the following sections.
Endothelin-1 Endothelin-1 (ET-1), a polypeptide with potent vasoconstrictive activity and a major factor in the pathophysiology of pulmonary arterial hypertension [51, 52], may also have pro-fibrotic activity and play a role in organ fibrosis [53,54,55]. ET-1 not only can increase the production of ECM macromolecules such as collagen types I and III, but also has been shown to inhibit production of matrix metalloproteinase-1 in normal human fibroblasts [56]. Increased levels of ET-1 have been found in several fibrotic diseases and in experimental pulmonary fibrosis [54, 56, 57]. Additionally, ET-1 may have a potential role in the generation of myofibroblasts through EMT or EndoMT, effects possibly mediated through the endothelin A receptor [58, 59] and through the synergistic stimulation of TGF-β induced EndoMT [60]. These findings, collectively, strongly suggest that ET-1 may play an important role in the pathophysiology of human fibrotic diseases and can be targeted with currently available therapeutics.
22.4.1 Connective Tissue Growth Factor (CTGF/CCN2)
CTGF, also known as CCN2, a growth factor with multiple effects, is now regarded as an important effector in both normal and pathologic fibrotic responses [61,62,63]. CTGF provokes a potent pro-fibrotic response when added to cultured fibroblasts and elevated CTGF levels have been found in a variety of experimental fibrotic models in mice while suppression of CTGF reduced bleomycin-induced lung fibrosis [61,62,63,64,65]. TGF-β stimulates CTGF synthesis in a variety of cell types, and CTGF can act as a downstream mediator to enhance the pro-fibrotic activity of TGF-β by stimulating the production of ECM components including type I collagen and fibronectin [61, 62]. Importantly, imatinib mesylate, an inhibitor of c-Abl, blocked activation of the Smad1 pathway when normal fibroblasts were stimulated with TGF-β and inhibited stimulation of CTGF expression in SSc fibroblasts [49]. Therefore, at least in some circumstances, c-Abl appears to be required for Smad1 activation. Furthermore, CTGF expression can be stimulated through the RhoA/Rock pathway [66]. Thus, CTGF is a targetable pro-fibrotic mediator.
22.4.2 Platelet Derived Growth Factor
The platelet derived growth factor (PDGF) family consists of four different polypeptides (PDGF-A, -B, -C, -D) which form disulphide-bonded dimers such as PDGF-AA and PDGF-BB as well as PDGF-AB heterodimers. Two structurally related tyrosine kinase receptors, PDGFRα and PDGFRβ bind PDGF ligands which leads to receptor homo- or hetero-dimerization and autophosphorylation of specific tyrosine residues within the receptor cytoplasmic domain [67,68,69]. This receptor activation initiates multiple signal translocation pathways including PI3K, Ras-MAPK, Src family kinases and phospholipase Cγ(PLCγ) resulting in important cellular responses including proliferation, chemotaxis and actin reorganization. It is now clear that PDGF can be involved in fibrotic reactions affecting multiple organs, including pulmonary, renal and hepatic fibrosis as well as SSc [70]. Fibroblasts can be regarded as both a major source and target since they secrete PDGF-A as well as express PDGFRα on their cell surface [71,72,73]. Thus, a PDGF-A/PDGFRα signaling loop can stimulate fibroblasts to synthesize ECM and release pro-fibrotic mediators. Since PDGF-B is released primarily by macrophages and hepatic stellate cells, this suggests a major role for PDGF-B/PDGFRβ signaling in liver fibrosis [74, 75]. PDGF signaling becomes activated upon tissue injury to promote wound closure and is turned off when the repair processes are completed [76]. However, excessive scar formation and tissue fibrosis can result, if PDGF signaling is not terminated.
22.4.3 Wnt-Signaling
While the Wnt proteins, consisting of a multi-gene family of secreted glycoproteins, play crucial roles in embryogenesis, numerous studies now have shown that the Wnt/β catenin pathway is involved in several pro-fibrotic processes including myofibroblast activation via Smad-dependent autocrine TGF-β signaling [77,78,79,80,81]. Besides its structural role, β-catenin plays a critical role in canonical Wnt signaling. In the absence of Wnt signals, β-catenin is phosphorylated by a complex consisting of adenomatosis polyposis coli (APC), axin, glycogen synthase kinase-3β (GSK-3β) and casein kinase which promotes subsequent ubiquitin-mediated β-catenin degradation. When secreted, Wnt proteins bind to cell surface Frizzled receptors (FZD) and lower density lipoprotein receptor-related protein co-receptor (LRP516) and the degradation complex is disrupted resulting in the stabilization of β-catenin which translocates to the nucleus where it binds to T-cell factor/lymphoid enhancer-binding factor (Tcf/Lef) to induce target gene transcription [82, 83]. Aberrant canonical Wnt signaling has been implicated in SSc [80] as well as in pulmonary, renal, dermal and liver fibrosis, in addition to scarring following myocardial infarction and fibrosis accompanying muscular dystrophy [84,85,86]. Under homeostatic conditions Wnt signaling must be tightly controlled. Indeed, an array of potent negative regulators has been identified, among which the Dickkopf proteins (DHK 1–4) play key roles. Dhk-1 is the best studied of them and it functions as a natural antagonist of Wnt signaling [87, 88].
22.4.4 Hedgehog Signaling
Three different mammalian orthologs of the Drosophila melanogaster hedgehog (Hh) morphogen have been identified in humans. They are highly hydrophobic secreted peptides called sonic hedgehog (SHh), Indian hedgehog, and Desert Hedgehog with SHh being the most important in the present context [89]. Patched (Ptc) a twelve-pass transmembrane protein binds Hh ligands, but in the absence of ligand Ptc interacts with and inhibits Smoothened (Smo), a seven-pass transmembrane protein [90]. However, binding of SHh to Ptc induces conformational changes that prevent Ptc from inhibiting Smo, which, initiates signaling events resulting in stabilization of Gli family zinc finger transcription factors and in enhanced expression of Hh target genes [91]. While Hh signaling is critical during embryonic development, inappropriate activation in adults has been implicated in the pathogenesis of various diseases, including malignancies [92, 93]. In SSc cultured fibroblasts, overexpression of SHh causes accumulation of Gli-2 and increased expression of Hh target genes [94]. An extensive immunofluorescence analysis of affected SSc skin demonstrated intense staining in dermal fibroblasts and endothelial cells. Other results from this study found that TGF-β increased expression of SHh and that SHh induced strong stimulation of fibroblast to myofibroblast transition in normal dermal fibroblasts comparable to that caused by TGF-β [94]. Overexpression of SHh in the skin of mice induced fibrosis and mice lacking one allele of the inhibitor receptor Ptc 1 gene were more sensitive to experimentally-induced fibrosis [95].
22.4.5 Notch Signaling
Also first discovered in Drosophila, Notch signaling is initiated by binding of members of two ligand families, Jagged and Delta-like to Notch receptors, which results in cleavage of these receptors by the secretase complex and release of the active Notch intracellular domain (NICD) [95, 96]. Translocation of the NCID into the nucleus activates transcription of multiple target genes such as Hairy/Enhancer of Split (Hes) [97]. As with Hedgehog, Notch signaling is crucial during embryonic development, and is highly regulated in the adult. There is accumulating evidence for the participation of Notch signaling in fibrotic diseases, although the molecular mechanisms involved in fibroblast activation and enhanced ECM production need further clarification [98,99,100].
22.4.6 Matrix Stiffness and Rho-Associated Kinases
While activation of myofibroblasts and stimulation of ECM production by TGF-β and other cytokines are complex and incompletely understood, recent studies have focused on the role of actin cytoskeleton reorganization. There is increasing interest in the mechanical properties of the ECM, particularly of the effect of tissue stiffness on the biosynthetic activities of resident fibroblasts/myofibroblasts [18,19,20, 101, 102]. Although the exact mechanisms whereby increased matrix stiffness stimulates production of polymerized α-SMA remains to be elucidated, this effect can result in nuclear translocation of MKL-1 (MRTF-A), a transcription factor that plays a critical role in the stimulation of expression of fibrotic genes [101]. Furthermore, matrix stiffening can promote RhoA production and activation, resulting in increased ROCK activity and enhanced fibroblast contractility. In addition, there may be cross-talk between the MAP kinase ERK 1/2, a potential downstream target of ROCK, and TGF-β [103]. All these findings suggest that as the fibrotic process proceeds and tissues become increasingly stiff, a vicious cycle gets established in which the matrix stiffness itself promotes further ECM production.
22.5 Pathophysiology of Fibrotic Abdominal Adhesions
While peritoneal adhesions may be caused by infection, inflammation or ischemia, surgical procedures are primarily responsible and can cause pelvic pain, bowel obstruction and infertility [104,105,106,107]. Although modern advances in surgical technique, including laparoscopy, have led to a decrease in their incidence, abdominal adhesions still pose a very significant medical as well as economic problem. Unfortunately, similar to other fibrotic reactions, adequate therapeutic solutions have proven elusive. This section briefly reviews the present state of affairs, including consideration of existing therapies and then places abdominal adhesions in the context of fibrotic reactions in general. Within this framework, an argument is developed suggesting that drugs which target signaling pathways known to be instrumental in the pathogenesis of many other fibrotic reactions be tested for their efficacy either in preventing or ameliorating intestinal adhesions.
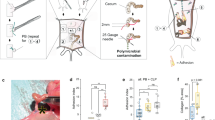
The visceral peritoneum covering the gut and other viscera accounts for about 80% of the total peritoneal surface, while the parietal peritoneum lining the walls of the abdominal cavity accounts for the remaining 20%. These surfaces are covered by a monolayer of mesothelial cells bound to a basement membrane which, in turn, rests on a bed of connective tissue containing fibroblasts, macrophages and other cell types [108]. This layer also contains a rich capillary network and lymphatics. Because the mesothelial cells are weakly connected, the peritoneal surface is delicate and easily injured. Injury to the peritoneum exposing the basement membrane causes a local inflammatory response resulting in deposition of a fibrin-rich exudate as part of the haemostatic process. While a fibrinous exudate is essential for normal repair, if it is not resolved in a timely fashion, it can provide a matrix for invading fibroblasts and blood vessels leading ultimately to adhesion formation.
Activation of the coagulation cascade results in the formation of thrombin from prothrombin, which converts fibrinogen into fibrin monomers and which polymerize forming a fibrin clot, a reaction which can be inhibited by anti-thrombin. An essential feature of the restoration of normal tissue architecture is the degradation of fibrin by the proteolytic enzyme, plasmin, which is formed from the inactive precursor, plasminogen, by tissue plasminogen activator (tPA) or urokinase plasminogen activator (uPA) with tPA being the most important in the present context. A very significant inhibitor of tPA and uPA is plasminogen activator inhibitor (PAI-1) which is produced by a variety of cells and whose production is enhanced by a number of factors including surgery, inflammation, IL-1 and tumor necrosis factor (TNF). The control of these competing reactions is not well-understood at the present time.
22.6 Stages in Abdominal Adhesion Formation
Successful treatment of abdominal adhesions is bedeviled by several confounding factors, not the least of which is the complex pathogenesis. A variety of causes can be responsible for initiation of adhesion formation which then likely proceeds through a common pathway. This process has been broken down into several stages, each one of which has been considered as a therapeutic target.
22.6.1 Coagulation Stage
As discussed above, the coagulation cascade is a critical factor in adhesion pathogenesis [109, 110]. This sequence of reactions involves a number of protein factors which facilitate or inhibit the ultimate formation of a fibrin clot. While, in many cases, the formation of a clot is essential to limit injury, resolution of the clot, in a timely manner, is necessary to prevent adhesion formation. Thus, the balance between fibrin clot formation and its lysis is critical. Much attention has been devoted to the measurement of factors such as thrombin that promote the clot formation as well as those like plasminogen activator that resolve it. These studies provide a rational basis for enhancing clot lysis as a therapeutic strategy. However, in practice, this has proven difficult.
22.6.2 Inflammatory Stage
Although this stage can overlap with the coagulation stage, it is clear that a major inflammatory response is initiated during abdominal adhesion formation. This stage is characterized by an influx of multiple cell types and production of a variety of cytokines and growth factors is and is elicited by a number of inciting events. This has led to inhibition of inflammation as a therapeutic approach to adhesion prevention which, by and large, has proven to be unsuccessful, although there are some reports of a positive response [111].
22.6.3 Fibrous Adhesion Stage
The final stage in the adhesion process is formation of a connective tissue scar, which is of critical importance since it is this fibrous tissue that causes the most severe complications. The exact inciting events in this final stage have received insufficient attention. However, based upon many studies in multiple organ systems, abdominal adhesion formation shares many attributes with fibrotic reactions found elsewhere in the body. Unfortunately, in none of these fibrotic reactions have any biomarkers been identified which can be used to detect the early stages of pro-fibrotic pathology. Much more work is needed in the molecular characterization of early adhesion formation and the composition of the scar itself in order to formulate novel therapeutic approaches.
22.7 Focus on Fibrous Adhesion Formation
22.7.1 The Myofibroblast
It is now well-known and accepted that although the causative mechanisms of fibrotic disorders are diverse and vary widely, they all share important cellular and molecular features which provide a fundamental framework for therapeutic approaches. At the cellular level, as discussed above, it is universally appreciated that a particular cell with unique characteristics, the myofibroblast, is responsible in all instances for the formation of the connective tissue scar that disrupts normal architecture and function [112]. The accumulation of myofibroblasts and the uncontrolled persistence of their elevated biosynthetic functions are crucial determinants of the extent and rate of progression of fibrotic reactions and of their clinical course, prognosis and response to therapy. While the origins of myofibroblasts in other organs may differ, in the case of abdominal adhesions, there is a unique cell type, namely the mesothelial surface cells, which likely mediate adhesion formation. These cells can undergo mesothelial/mesenchymal transition and trans-differentiate into myofibroblasts which can be facilitated by TGF-β [113, 114]. As previously discussed, both epithelial and endothelial cells can be induced by TGF-β and other cytokines to trans-differentiate into myofibroblasts [23, 115,116,117,118]. In this transition, the cells loose expression of cadherins, rearrange their cytoskeleton, change morphology, gain expression of α-SMA and produce substantial amounts of ECM [119, 120] .
Prior to becoming a myofibroblast, the precursor cell undergoes a transition to an intermediate stage (proto-myofibroblast) characterized by increased actin-myosin stress fibers and prominent focal adhesion structures [121, 122], which is characterized by the expression of an isoform of the protein fibronectin containing the EDA domain (FnEDA) . It is expressed at the cell surface during embryonic development, but is not normally found in adults, which distinguished it from the plasma form which is secreted by the liver into the blood stream. Expression of this cellular form of fibronectin (FnEDA) characterizes the intermediate stage of myofibroblast formation and results from the splicing of a unique exon into the primary mRNA transcript [123]. This exon encodes for an extra 91 amino acids in the final FnEDA molecule and is only synthesized in adults as a consequence of tissue damage, inflammation and wound healing [124]. Thus, the proto-myofibroblast synthesizes FnEDA but not α-SMA [125]. Clearly, FnEDA represents a potential diagnostic and therapeutic target.
22.7.2 Critical Cytokines, Signaling Pathways and Therapeutic Strategies
While the underlying etiology of a particular fibrotic reaction is frequently unknown, certain signaling pathways activated by several cytokines and growth factors undoubtedly play key roles in the pathogenesis with the TGF-β family playing a critical and predominant role by mediating the formation of myofibroblasts and stimulating the productions of ECM [126]. TGF-β is secreted into the ECM as a large latent complex which can undergo several alternative proteolytic or conformational activating events prior to binding to its cognate receptor. While the intracellular transduction pathways following TGF-β receptor binding can directly stimulate molecular effectors involved in the pro-fibrotic response, another important aspect of TGF-β action is the stimulation of other mediators having pro-fibrotic potential. In addition, a number of other cytokines and growth factors have been shown to be involved in adhesion formation, each of which may involve signaling via unique pathways and which can be targeted therapeutically.
Because of the multiplicity of signaling pathways elicited by TGF-β, multiple potential therapeutic targets exist. However, judicious choices must be made since TGF-β has numerous, and diverse critical functions. Thus, inhibiting all of its manifold activities by blocking its cell surface receptor, for example, is not desirable since toxic effects may ensue. Importantly, the use of this approach has been largely unsuccessful in past clinical trials [127,128,129]. Therapeutic strategies must be developed which limit the effect, as much as possible, to inhibiting signaling reactions crucial to, and only to, the fibrotic response. This implies that the most desirable targets are those that are essential for the response, but are as far “down- stream” in the relevant signaling pathways as possible. Furthermore, recent studies have shown that the c-Able-PKCδ pathway participates in the process of endothelial-mesenchymal transition. Additionally, the Akt-mTOR pathway plays an important role in various cell processes including regulation of cell proliferation and metabolism as well as being involved in some epithelial/mesenchymal transitions. These pathways are potential therapeutic targets.
It is likely that there are several cytokines in addition to TGF-β that may participate in adhesion formation. Of these, IL-6 is of considerable importance. The diverse functions of IL-6 are mediated by several protein components which include a receptor that is specific for IL-6 (IL-6R) and gp130 which together form a heterodimer complex and activate two pathways: the JAK/Stat-pathway and the Ras-MAPK (mitogen-activated protein kinase) pathway [130]. Most importantly, IL-6, in addition to TGF-β, was also found to be elevated in peritoneal fluid during abdominal surgeries. In a model of repetitive peritoneal inflammation, IL-6 was found to be capable of mediating a peritoneal fibrotic process [131]. Studies have shown that IL-6 can promote EMT in colorectal cell lines [132]. These data, as well as recent evidence from our laboratory [114], demonstrated that MMT can be induced by IL-6 and thus may represent an important cellular mechanism which can mediate the formation of abdominal adhesions.
22.7.3 Role of Hypoxia
It has been shown that hypoxia may play a role in abdominal adhesion formation by several mechanisms based largely on experimental model systems. Hypoxia can decrease tPA and increase PAI expression thereby inhibiting fibrin lysis. The PAI-1 gene promoter contains hypoxia response elements HRE-1 and HRE-2 which bind the oxygen-regulated transcription factor HIF-1α resulting in increased PAI-1 expression. HIF-1α also increases the production of vascular endothelial growth factor (VEGF), which plays a critical role in angiogenesis and formation of blood vessels in adhesions. In addition, hypoxia can increase production of TGF-β1 by human mesothelial cells and peritoneal fibroblasts while TGF-β1 can stimulate VEGF and CTGF expression. Hypoxia also increased expression of TIMP-1, thereby potentially causing a decrease in matrix degradation.
22.7.4 Material Barriers
Currently, the most frequent approach in the prevention of adhesion formation is use of material barriers [111, 133]. While a number of such materials are available, including both liquid and solid based ones, no completely acceptable one has been developed for several reasons. These include toxicity, difficulty in handling membrane films, and potentially limited efficacy. However, several of these are currently in use including Seprafilm® which we describe as a typical example. Seprafilm® is a transparent, resorbable membrane composed of sodium hyaluronic acid and carboxymethylcellulose. It degrades in 7 days under physiologic conditions, is safe, and provides some effectiveness in preventing postoperative adhesions after abdominal surgery. However, while Seprafilm® covers the treated tissue, it does not protect remote areas and thus allows adhesion formation at distant sites, and, in addition, it is highly fragile. A sprayable form of Seprafilm® is also currently being tested.
22.7.5 Pharmaceutical Approaches
Attempts have been made to modify adhesion formation by use of pharmaceuticals mainly through use of anti-inflammatory and anti-coagulant agents. For example, these have included systemic and local application of steroids, cyclooxygenase inhibitors, heparin and tissue plasminogen activator (t-PA). In some instances, barrier membranes have been used in combination with an agent e.g., heparin. However, the results have been mixed and no clear-cut beneficial result has been obtained. On a more promising note, efforts have been made to inhibit the effect of the pro-inflammatory peptide substance P by blocking its major receptor, neurokinin receptor (NK1R) which is believed to play a role in adhesion formation, possibly by lowering expression of metalloproteinases. In animal adhesion models, administration of NK1R antagonists increased metalloproteinase activity, increased fibrinolysis and significantly lowered adhesion formation [111, 133].
22.8 Preventing Adhesions By Blocking Connective Tissue Formation
While abdominal adhesions present a significant and recurring medical problem, unfortunately, there are no really effective therapeutic measures available either for prevention or cure. It is noteworthy that TGF-β1 and IL-6 were found to be elevated in the peritoneal fluid of patients during/after abdominal surgery and that the levels of the cytokines appeared to be related to the severity of abdominal adhesion formation [134, 135]. In contrast to crohn’s disease (CD) and IBD, abdominal adhesion formation is unusual because of the potential major role of mesothelial cells. Because of their cellular environment, mesothelial cells are bathed in peritoneal fluid containing high concentrations of both TGF-β and IL-6 and have the potential to undergo MMT providing a major source of myofibroblasts responsible for adhesion formation. The control of MMT is achieved primarily by three families of transcription factors: zinc finger Snail (SNAl1, SNAl2), basic helix-loop helix (Twisted 1) and ZEB (ZEB1, ZEB2) whose expression can be increased by both TGF-β and IL-6. Furthermore, both cytokines can elicit the phosphorylation/activation of a critical intracellular effector molecule, Erk1/2, required for the MMT of peritoneal mesothelial cells. MEK 1/2 is responsible for Erk1/2 phosphorylation and specific inhibition of MEK 1/2 prevents Erk1/2 phosphorylation and MMT. This implies that MEK inhibitors may be good therapeutic candidates for adhesion prevention, e.g., MEK inhibitors such as trametinib and selumetinib have been tested in clinical trials for melanoma [136].
22.8.1 Intestinal Fibrosis in Inflammatory Bowel Disease (IBD)
The pathophysiology of fibrotic reactions such as CD and IBD is considerably more complex in comparison to that of abdominal adhesions which occur secondary to surgery. Although much remains to be determined in the pathogenesis of adhesions with respect to genetic predisposition, firm identification of the source of myofibroblasts and detailed understanding of critical signaling pathways in IBD, the pathophysiology is more multifaceted because potential inciting events are more numerous. Additional considerations include a much more sustained inflammatory response, a clear, if still poorly understood, genetic pre-disposition, the potential involvement of multiple mesenchymal cells/myofibroblasts, exposure of the mucosa to intestinal bacteria and the involvement of the immune system.
22.8.2 Genetic Basis in IBD
While there may be familial pre-disposition in the formation of adhesions, the evidence remains anecdotal and there is no firm supporting evidence. In contrast, significant evidence supports the concept that pre-disposition to development of CD is polygenic with 163 loci contributing to either susceptibility or disease severity including fibrostenosis [137,138,139,140]. However attempts to link genetic loci to clinical phenotype remain problematic with the strongest association linked to susceptibility being to NOD2 (polymorphisms with disturbed surveillance of bacterial microflora), IL23 receptor (polymorphisms linked to regulation of adaptive immunity) and ATG16L1 and IRGM (deficit in autophagy) [140]. Individuals with NOD2/CARD15 mutations have a tenfold greater risk of aggressive disease and different genetic variations correlate with distinct phenotypes such as fibrostenosis [141, 142]. Interestingly, deep sequencing of loci identified by genome-wide association study (GWAS) have identified matrix metalloproteinases (MMP-1, MMP-2, MMP-3, MMP-9) and their tissue inhibitors (TIMP-1 and TIMP-2) as prognostic indicators for diagnostic and surgical recurrence in CD [143].
22.8.3 Cells Responsible for IBD Fibrostenosis
As in in other respects, the cellular fibrotic reaction in IBD is considerably more complex than that generally occurring in abdominal adhesion formation. Much of this complexity has to do with inciting events and the extent of tissue involvement. As discussed above, the majority of abdominal adhesions are owing to injury at the serosal surface during surgical procedures. However, in IBD, a multiplicity of factors interact in a complex, and in, as yet, poorly understood fashion, to initiate and prolong the fibrotic response. These factors include genetic predisposition, active participation of a diverse number of cell types including those of the immune system, an extended inflammatory response, exposure to bacterial products from the microbiome, and potential involvement of a number of cytokines and growth factors.
The production of ECM, particularly that of collagens I and III, is a fundamental factor in fibrostenosis formation. In IBD, as in other fibrotic diseases, myofibroblasts play a critical role in the overall pathophysiological mechanism and there are numerous potential sources of them. Unique to the intestine are the cells of Cajal in the submucosa and muscularis [144]. Another major source is the subepithelial myofibroblasts (SEMFs). Interestingly, there is substantial evidence of cross-talk between SEMFs and the epithelium [145]. Conditioned medium from cultured epithelial cells treated with pro-inflammatory cytokines enhanced SEMF migration and production of collagen and MMPs [146]. SEMFs exposed to pro-inflammatory cytokines can themselves then express interleukins IL-6 and IL-8 as well as granulocyte and macrophage stimulatory factors and pro-fibrotic cytokines IL-17A and TGF-β [147, 148]. The SEMFs also express Toll-like receptors (TLR-2, TLR-4 and TLR-5) which enables them to respond to lipopolysaccharide and flagellin and further cytokine production [149, 150]. Another aspect of the involvement of SEMFs in fibrosis pathogenesis is that they may act as non-professional antigen-presenting cells since they constitutively express class II major histocompatibility complex to promote CD4+ T cell differentiation [151].
Other potential sources of myofibroblasts in IBD include stromal fibroblasts and pericytes which, upon activation, can proliferate, express α-SMA and produce ECM [152, 153]. Pericytes are found in capillaries and small blood vessels surrounding endothelial cells and during inflammation they differentiate into cells producing large amounts of ECM [153]. Another source of myofibroblast-like cells is bone marrow-derived fibrocytes which travel to the tissues through the blood stream. Multipotent bone marrow mesenchymal cells differentiate into fibrocytes which express hematopoietic markers such as CD45, CD11, CD13 and CD6, but they also express collagen and α-SMA [153]. Increased amounts of such cells have been found in the blood and tissues of CD patients.
22.8.4 Smooth Muscle Cells
An unusual feature of the fibrotic response in the intestine is the involvement of smooth muscle cells which are found in the muscularis externa and which represent the largest mesenchymal component in the intestine. This is particularly critical in CD because the full thickness of the intestinal wall is frequently involved. There is marked thickening of the muscularis with cellular hyperplasia, hypertrophy and ECM deposition which contributes to stricture formation [154]. The smooth muscle cells in the muscularis externa and the muscularis mucosae may also be involved in both CD and UC. Smooth muscle cells, in addition to producing ECM, also produce significant amounts of cytokines and growth factors including TGF-β, Il-6, IGF-1, PDGF, and CTGF [155].
22.8.5 Pathogenesis of Fibrosis in IBD
In both CD and ulcerative colitis (UC), injury to the mucosal epithelium exposes the underlying tissues to inflammatory mediators derived from the intestinal microbiome. Such exposure can generate a damaging immune response in susceptible individuals. Such events generate a prolonged and recurrent inflammatory reaction which, in turn, can initiate and extend fibrotic reactions characterized by the activation of myofibroblasts resulting in stricture formation. Such fibro-stenosis occurs much more frequently in CD (30–50%) in which the full thickness of the intestine is affected as opposed to much less extent in UC (~5%) where usually only the mucosa and submucosa are primarily affected [156]. While inflammation is clearly necessary for the initiation of the fibrotic reaction; however, once started, it can progress in the absence of continued inflammation.
22.9 ECM Homeostasis in IBD
The net concentration of ECM is governed by its rate of production and by the rate of turnover of its components. Such turnover is regulated largely by the activity of matrix metalloproteinases (MMPs) of which there are 23 human types. MMPs are zinc and calcium proteases produced by a variety of cells including epithelial, myofibroblasts, and macrophages The MMPs include collagenases, gelatinases, stromelysins and matrilysins and thus comprise a broad level of substrate specificity [157,158,159]. Increased expression of MMPs such a collagenase MMP8 and gelatinase MMP-9 have been found adjacent to intestinal ulcerations in CD. The activity of these proteases is controlled by four tissue inhibitors of proteases (TIMPs). Cytokines and growth factors such as TGF-β and TNF-α can regulate MMP and TIMP expression. TGF-β down regulates MMP-1 and MMP-3 in intestinal myofibroblasts and enhances TIMP-1 expression, while, contrarily, TNF-α decreases MMP-2 activity and increases TIMP-1 expression [158]. The balance between MMP activity and TIMP inhibitory regulation is disturbed in the intestine of patients with fibrostenotic CD [157,158,159,160]. The T-cell derived cytokine, IL-21, that is increased in IBD, can increase expression of MMPs by intestinal myofibroblasts thereby contributing to mucosal ulcer formation [161]. In active CD, increased expression of MMP-1, MMP-3 and TIMP-1 has been found in the intestinal muscularis [162]. Thus, it is likely that these substances play a role in the pathophysiology of CD; however, the mechanism(s) which govern their expression is largely unknown.
22.10 Role of Immune Responses in IBD Fibrotic Reactions
A major difference in the pathogenesis of the fibrotic reaction between IBD and abdominal adhesions secondary to surgery is the pronounced role of the immune system in IBD and its comparatively small role in abdominal adhesion generation. Injury to the intestinal epithelium permits entry of bacteria into the mucosa and the generation of activating molecules including DAMPs and PAMPs which stimulate pro-inflammatory cytokines and chemokines by macrophages which recruit innate immune cells [163]. In a complex subsequent series of events, activated (M1) macrophages secrete interleukins (IL-1, IL-2 IL-23), TNF-α and reactive oxygen species which activate myofibroblasts [163]. Activation of TLRs can also stimulate myofibroblast proliferation and collagen production [164]. In chronic inflammation, macrophages shift to a M2 phenotype and release anti-inflammatory and pro-fibrotic mediators including IL-4, IL-13, IL-10 and TGF-β [163]. A correlation between collagen accumulation and elevated IL-13 and TIMP-1 expression by mononuclear cells has been reported in CD tissue samples [165]. Not surprisingly, the adaptive immune responses are complex. In a Th1 immunity response, interferon (INF)γ was found in early stages of CD and is considered as an anti-fibrotic cytokine blocking TGF-β and CTGF expression, but INF-γ therapy of fibrotic reactions, for the most part, has not proven successful [166,167,168]. Th17 immune responses associated with fibrosis have also been reported in CD. IL-17A was elevated in stricturing CD tissue with increased collagen, MMP-3, MMP-12 and TIMP-1 [169]. IL-17 may have other effects in fibrosis by regulating expression of TGF-β and CTGF in myofibroblasts and upregulating collagen production [170].
22.11 Potential Anti-Fibrotic Therapy in IBD
Because the pathophysiology of abdominal adhesion formation may be less diverse in comparison to IBD, potential therapeutic targets are more easily selected as discussed above. Table 22.1 illustrates the greater complexity of IBD versus abdominal adhesions. In IBD there are multiple potential levels at which therapy could be applied. For example, there are several endogenous anti-fibrotic mediators with peroxisome proliferator-activated receptor (PPAR)-γ being the most promising. It has been found that this ligand-activated nuclear receptor, that is widely expressed in various cell types, has been found to decrease inflammatory responses in the intestine and other tissues [171]. PPAR-γ inhibits pro-fibrotic signaling by TGF-β and Wnt-β-catenin [172, 173], blocks TGF-β pro-fibrotic effects such as collagen and fibronectin production [174] and suppresses TGF-β-induced EMT [175]. Since ECM accumulation is governed by the comparative rates of production and degradation, this implies that increased degradation by MMPs might be an effective approach to minimize ECM formation. There is considerable in vitro information with respect to the control of the activity of multiple MMPs [158, 176]. Regrettably however, because of severe limitations in present knowledge regarding the ability to up-regulate MMP activity in a controlled manner in vivo, this approach is not presently feasible.
In IBD, the normal wound healing process triggered by injury and inflammation fails and instead of resolution, there is continued myofibroblast activity. Because the inflammatory response is protracted, one could imagine that anti-inflammatory therapy might be an effective approach. Unfortunately, this has not been the case, and it appears that once the damaging fibrotic reaction has been initiated in fibrosis-prone individuals, it is self-propagating. Thus, as in other fibrotic situations, the aberrant myofibroblast becomes the ultimate target. However, unlike abdominal adhesions in which the potential effector cells (mesothelial cells) can be anticipated and candidate drugs given over a fairly short time, in IBD, the pathogenesis is much more protracted. There a number of FDA-approved drugs capable of blocking pathways essential to fibrotic reactions such as trametinib [177] (Fig. 22.2). TGF-β is, of course, the primary target; however, total blockade has not worked because of adverse toxic responses because of its manifold activities. The same caution must be exercised when dealing with other potential targets (Fig. 22.2) [6]. Because of the inherent redundancy in signaling from multiple cytokines/growth factors involved in the activation of genes responsible for ECM synthesis, it is likely that more than one drug must be administered simultaneously to obtain effective beneficial inhibition of pro-fibrotic pathways.
22.12 Conclusions
Although abdominal adhesions and IBD have varying etiologies, they share many features of fibrotic reactions found in other organs and tissues of the body. These include the synthesis of excess extracellular matrix composed of collagen and other macromolecules by myofibroblasts. These fibrotic reactions are driven by various cytokines/growth factors with TGF-β predominating. Despite considerable recently obtained understanding of the pathogenesis of the fibrotic process, disease-modifying therapy for these diseases is extremely limited. However, one potentially productive way forward will be to simultaneously use several FDA-approved drugs which can act together to effectively block multiple pathways preventing the activation of pro-fibrotic genes. In addition, biomarkers need to be identified which can signal the early onset of disease which may permit more effective use of currently available therapeutics.
References
Rosenbloom J, Castro SV, Jimenez SA. Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Ann Intern Med. 2010;152(3):159–66.
Rockey DC, Bell PD, Hill JA. Fibrosis--a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138–49.
Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210.
Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117(3):557–67.
Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis--a lethal component of systemic sclerosis. Nat Rev Rheumatol. 2014;10(7):390–402.
Rosenbloom J, Mendoza FA, Jimenez SA. Strategies for anti-fibrotic therapies. Biochim Biophys Acta. 2013;1832(7):1088–103.
Denton CP. Systemic sclerosis: from pathogenesis to targeted therapy. Clin Exp Rheumatol. 2015;33(4 Suppl 92):S3–7.
Karsdal MA, Manon-Jensen T, Genovese F, Kristensen JH, Nielsen MJ, Sand JM, et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2015;308(10):G807–30.
Thannickal VJ, Henke CA, Horowitz JC, Noble PW, Roman J, Sime PJ, et al. Matrix biology of idiopathic pulmonary fibrosis: a workshop report of the national heart, lung, and blood institute. Am J Pathol. 2014;184(6):1643–51.
Gabbiani G. The myofibroblast: a key cell for wound healing and fibrocontractive diseases. Prog Clin Biol Res. 1981;54:183–94.
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170(6):1807–16.
Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180(4):1340–55.
McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39(4):666–71.
Kirk TZ, Mark ME, Chua CC, Chua BH, Mayes MD. Myofibroblasts from scleroderma skin synthesize elevated levels of collagen and tissue inhibitor of metalloproteinase (TIMP-1) with two forms of TIMP-1. J Biol Chem. 1995;270(7):3423–8.
Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. 2014;5:123.
Gilbane AJ, Denton CP, Holmes AM. Scleroderma pathogenesis: a pivotal role for fibroblasts as effector cells. Arthritis Res Ther. 2013;15(3):215.
Laurent GJ, Chambers RC, Hill MR, McAnulty RJ. Regulation of matrix turnover: fibroblasts, forces, factors and fibrosis. Biochem Soc Trans. 2007;35(Pt 4):647–51.
Wells RG, Discher DE. Matrix elasticity, cytoskeletal tension, and TGF-beta: the insoluble and soluble meet. Sci Signal. 2008;1(10):pe13.
Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep. 2009;11(2):120–6.
Parker MW, Rossi D, Peterson M, Smith K, Sikstrom K, White ES, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124(4):1622–35.
Postlethwaite AE, Shigemitsu H, Kanangat S. Cellular origins of fibroblasts: possible implications for organ fibrosis in systemic sclerosis. Curr Opin Rheumatol. 2004;16(6):733–8.
Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97.
Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to Mesenchymal Transition (EndoMT) in the pathogenesis of human fibrotic diseases. J Clin Med. 2016;5(4):E45.
Iozzo RV, Schaefer L. Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11–55.
Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biol. 2014;37:1–14.
Resovi A, Pinessi D, Chiorino G, Taraboletti G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014;37:83–91.
Kramann R, DiRocco DP, Humphreys BD. Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J Pathol. 2013;231(3):273–89.
Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83(12):4167–71.
Sporn MB, Roberts AB. Transforming growth factor-beta. Multiple actions and potential clinical applications. JAMA. 1989;262(7):938–41.
Moses HL, Roberts AB, Derynck R. The discovery and early days of TGF-beta: a historical perspective. Cold Spring Harb Perspect Biol. 2016;8(7):a021865.
Fujio K, Komai T, Inoue M, Morita K, Okamura T, Yamamoto K. Revisiting the regulatory roles of the TGF-beta family of cytokines. Autoimmun Rev. 2016;15(9):917–22.
Goumans MJ, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009;19(1):116–27.
Medici D, Potenta S, Kalluri R. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J. 2011;437(3):515–20.
van Meeteren LA, ten Dijke P. Regulation of endothelial cell plasticity by TGF-beta. Cell Tissue Res. 2012;347(1):177–86.
Jimenez SA, Castro SV, Piera-Velazquez S. Role of growth factors in the pathogenesis of tissue fibrosis in systemic sclerosis. Curr Rheumatol Rev. 2010;6(4):283–94.
Lafyatis R. Transforming growth factor beta--at the Centre of systemic sclerosis. Nat Rev Rheumatol. 2014;10(12):706–19.
Pohlers D, Brenmoehl J, Loffler I, Muller CK, Leipner C, Schultze-Mosgau S, et al. TGF-beta and fibrosis in different organs – molecular pathway imprints. Biochim Biophys Acta. 2009;1792(8):746–56.
Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-beta signaling in fibrosis. Growth Factors. 2011;29(5):196–202.
Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–38.
ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29(5):265–73.
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–84.
Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118(Pt 16):3573–84.
Wilkes MC, Leof EB. Transforming growth factor beta activation of c-Abl is independent of receptor internalization and regulated by phosphatidylinositol 3-kinase and PAK2 in mesenchymal cultures. J Biol Chem. 2006;281(38):27846–54.
Jimenez SA, Gaidarova S, Saitta B, Sandorfi N, Herrich DJ, Rosenbloom JC, et al. Role of protein kinase C-delta in the regulation of collagen gene expression in scleroderma fibroblasts. J Clin Invest. 2001;108(9):1395–403.
Bujor AM, Asano Y, Haines P, Lafyatis R, Trojanowska M. The c-Abl tyrosine kinase controls protein kinase Cdelta-induced Fli-1 phosphorylation in human dermal fibroblasts. Arthritis Rheum. 2011;63(6):1729–37.
Lawler S, Feng XH, Chen RH, Maruoka EM, Turck CW, Griswold-Prenner I, et al. The type II transforming growth factor-beta receptor autophosphorylates not only on serine and threonine but also on tyrosine residues. J Biol Chem. 1997;272(23):14850–9.
Galliher AJ, Schiemann WP. Src phosphorylates Tyr284 in TGF-beta type II receptor and regulates TGF-beta stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 2007;67(8):3752–8.
Caraci F, Gili E, Calafiore M, Failla M, La Rosa C, Crimi N, et al. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res. 2008;57(4):274–82.
Pannu J, Asano Y, Nakerakanti S, Smith E, Jablonska S, Blaszczyk M, et al. Smad1 pathway is activated in systemic sclerosis fibroblasts and is targeted by imatinib mesylate. Arthritis Rheum. 2008;58(8):2528–37.
Andrianifahanana M, Wilkes MC, Gupta SK, Rahimi RA, Repellin CE, Edens M, et al. Profibrotic TGFbeta responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases. FASEB J. 2013;27(11):4444–54.
Kawanabe Y, Nauli SM. Endothelin. Cell Mol Life Sci. 2011;68(2):195–203.
Thorin E, Clozel M. The cardiovascular physiology and pharmacology of endothelin-1. Adv Pharmacol. 2010;60:1–26.
Shi-Wen X, Denton CP, Dashwood MR, Holmes AM, Bou-Gharios G, Pearson JD, et al. Fibroblast matrix gene expression and connective tissue remodeling: role of endothelin-1. J Invest Dermatol. 2001;116(3):417–25.
Jing J, Dou TT, Yang JQ, Chen XB, Cao HL, Min M, et al. Role of endothelin-1 in the skin fibrosis of systemic sclerosis. Eur Cytokine Netw. 2015;26(1):10–4.
Xu SW, Howat SL, Renzoni EA, Holmes A, Pearson JD, Dashwood MR, et al. Endothelin-1 induces expression of matrix-associated genes in lung fibroblasts through MEK/ERK. J Biol Chem. 2004;279(22):23098–103.
Park SH, Saleh D, Giaid A, Michel RP. Increased endothelin-1 in bleomycin-induced pulmonary fibrosis and the effect of an endothelin receptor antagonist. Am J Respir Crit Care Med. 1997;156(2 Pt 1):600–8.
Ross B, D’Orleans-Juste P, Giaid A. Potential role of endothelin-1 in pulmonary fibrosis: from the bench to the clinic. Am J Respir Cell Mol Biol. 2010;42(1):16–20.
Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121(22):2407–18.
Kim KK, Chapman HA. Endothelin-1 as initiator of epithelial-mesenchymal transition: potential new role for endothelin-1 during pulmonary fibrosis. Am J Respir Cell Mol Biol. 2007;37(1):1–2.
Cipriani P, Di Benedetto P, Ruscitti P, Capece D, Zazzeroni F, Liakouli V, et al. The endothelial-mesenchymal transition in systemic sclerosis is induced by endothelin-1 and transforming growth factor-beta and may be blocked by macitentan, a dual endothelin-1 receptor antagonist. J Rheumatol. 2015;42(10):1808–16.
Grotendorst GR. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 1997;8(3):171–9.
Leask A, Abraham DJ. The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem Cell Biol. 2003;81(6):355–63.
Igarashi A, Nashiro K, Kikuchi K, Sato S, Ihn H, Grotendorst GR, et al. Significant correlation between connective tissue growth factor gene expression and skin sclerosis in tissue sections from patients with systemic sclerosis. J Invest Dermatol. 1995;105(2):280–4.
Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19(2):133–44.
Ponticos M, Holmes AM, Shi-wen X, Leoni P, Khan K, Rajkumar VS, et al. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 2009;60(7):2142–55.
Ruperez M, Rodrigues-Diez R, Blanco-Colio LM, Sanchez-Lopez E, Rodriguez-Vita J, Esteban V, et al. HMG-CoA reductase inhibitors decrease angiotensin II-induced vascular fibrosis: role of RhoA/ROCK and MAPK pathways. Hypertension. 2007;50(2):377–83.
Betsholtz C. Biology of platelet-derived growth factors in development. Birth Defects Res C Embryo Today. 2003;69(4):272–85.
Farooqi AA, Waseem S, Riaz AM, Dilawar BA, Mukhtar S, Minhaj S, et al. PDGF: the nuts and bolts of signalling toolbox. Tumour Biol. 2011;32(6):1057–70.
Alvarez RH, Kantarjian HM, Cortes JE. Biology of platelet-derived growth factor and its involvement in disease. Mayo Clin Proc. 2006;81(9):1241–57.
Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15(4):255–73.
Tallquist M, Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004;15(4):205–13.
Yamakage A, Kikuchi K, Smith EA, LeRoy EC, Trojanowska M. Selective upregulation of platelet-derived growth factor alpha receptors by transforming growth factor beta in scleroderma fibroblasts. J Exp Med. 1992;175(5):1227–34.
Olson LE, Soriano P. Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell. 2009;16(2):303–13.
Czochra P, Klopcic B, Meyer E, Herkel J, Garcia-Lazaro JF, Thieringer F, et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol. 2006;45(3):419–28.
Ogawa S, Ochi T, Shimada H, Inagaki K, Fujita I, Nii A, et al. Anti-PDGF-B monoclonal antibody reduces liver fibrosis development. Hepatol Res. 2010;40(11):1128–41.
Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79(4):1283–316.
Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205.
Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13(12):767–79.
Bergmann C, Distler JH. Canonical Wnt signaling in systemic sclerosis. Lab Invest. 2016;96(2):151–5.
Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, et al. Wnt/beta-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012;64(8):2734–45.
Beyer C, Schramm A, Akhmetshina A, Dees C, Kireva T, Gelse K, et al. Beta-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann Rheum Dis. 2012;71(5):761–7.
Huang H, He X. Wnt/beta-catenin signaling: new (and old) players and new insights. Curr Opin Cell Biol. 2008;20(2):119–25.
Nusse R. Wnt signaling in disease and in development. Cell Res. 2005;15(1):28–32.
He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol. 2009;20(4):765–76.
Konigshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W, et al. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS One. 2008;3(5):e2142.
Trensz F, Haroun S, Cloutier A, Richter MV, Grenier G. A muscle resident cell population promotes fibrosis in hindlimb skeletal muscles of mdx mice through the Wnt canonical pathway. Am J Physiol Cell Physiol. 2010;299(5):C939–47.
Pinzone JJ, Hall BM, Thudi NK, Vonau M, Qiang YW, Rosol TJ, et al. The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood. 2009;113(3):517–25.
Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/arrow. Nat Cell Biol. 2001;3(7):683–6.
Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75(7):1417–30.
Rohatgi R, Milenkovic L, Corcoran RB, Scott MP. Hedgehog signal transduction by smoothened: pharmacologic evidence for a 2-step activation process. Proc Natl Acad Sci U S A. 2009;106(9):3196–201.
Rohatgi R, Scott MP. Patching the gaps in hedgehog signalling. Nat Cell Biol. 2007;9(9):1005–9.
Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, et al. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–2.
Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–6.
Horn A, Palumbo K, Cordazzo C, Dees C, Akhmetshina A, Tomcik M, et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Rheum. 2012;64(8):2724–33.
Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Dev Cell. 2009;16(5):633–47.
D’Souza B, Miyamoto A, Weinmaster G. The many facets of Notch ligands. Oncogene. 2008;27(38):5148–67.
Borggrefe T, Liefke R. Fine-tuning of the intracellular canonical Notch signaling pathway. Cell Cycle. 2012;11(2):264–76.
Louvi A, Artavanis-Tsakonas S. Notch and disease: a growing field. Semin Cell Dev Biol. 2012;23(4):473–80.
Dees C, Tomcik M, Zerr P, Akhmetshina A, Horn A, Palumbo K, et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann Rheum Dis. 2011;70(7):1304–10.
Kavian N, Servettaz A, Weill B, Batteux F. New insights into the mechanism of notch signalling in fibrosis. Open Rheumatol J. 2012;6:96–102.
Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47(3):340–8.
Kessler D, Dethlefsen S, Haase I, Plomann M, Hirche F, Krieg T, et al. Fibroblasts in mechanically stressed collagen lattices assume a “synthetic” phenotype. J Biol Chem. 2001;276(39):36575–85.
Hayashida T, Decaestecker M, Schnaper HW. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. FASEB J. 2003;17(11):1576–8.
Al-Jaroudi D, Tulandi T. Adhesion prevention in gynecologic surgery. Obstet Gynecol Surv. 2004;59(5):360–7.
Boland GM, Weigel RJ. Formation and prevention of postoperative abdominal adhesions. J Surg Res. 2006;132(1):3–12.
Ellis H. The clinical significance of adhesions: focus on intestinal obstruction. Eur J Surg Suppl. 1997;577:5–9.
Ozel H, Avsar FM, Topaloglu S, Sahin M. Induction and assessment methods used in experimental adhesion studies. Wound Repair Regen. 2005;13(4):358–64.
Beyene RT, Kavalukas SL, Barbul A. Intra-abdominal adhesions: anatomy, physiology, pathophysiology, and treatment. Curr Probl Surg. 2015;52(7):271–319.
Arung W, Meurisse M, Detry O. Pathophysiology and prevention of postoperative peritoneal adhesions. World J Gastroenterol. 2011;17(41):4545–53.
Moris D, Chakedis J, Rahnemai-Azar AA, Wilson A, Hennessy MM, Athanasiou A, et al. Postoperative abdominal adhesions: clinical significance and advances in prevention and management. J Gastrointest Surg. 2017;21(10):1713–22.
Pados G, Venetis CA, Almaloglou K, Tarlatzis BC. Prevention of intra-peritoneal adhesions in gynaecological surgery: theory and evidence. Reprod Biomed Online. 2010;21(3):290–303.
Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200(4):500–3.
Strippoli R, Moreno-Vicente R, Battistelli C, Cicchini C, Noce V, Amicone L, et al. Molecular mechanisms underlying peritoneal EMT and fibrosis. Stem Cells Int. 2016;2016:3543678.
Jin X, Ren S, Macarak E, Rosenbloom J. Pathobiological mechanisms of peritoneal adhesions: the mesenchymal transition of rat peritoneal mesothelial cells induced by TGF-beta1 and IL-6 requires activation of Erk1/2 and Smad2 linker region phosphorylation. Matrix Biol. 2016;51:55–64.
Haensel D, Dai X. Epithelial-to-mesenchymal transition in cutaneous wound healing: where we are and where we are heading. Dev Dyn. 2017;247(3):473–80.
Sanchez-Duffhues G, Garcia de Vinuesa A, Ten Dijke P. Endothelial to mesenchymal transition in cardiovascular diseases: developmental signalling pathways gone awry. Dev Dyn. 2017;247(3):492–508.
Voon DC, Huang RY, Jackson RA, Thiery JP. The EMT spectrum and therapeutic opportunities. Mol Oncol. 2017;11(7):878–91.
Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7(344):re8.
Flier SN, Tanjore H, Kokkotou EG, Sugimoto H, Zeisberg M, Kalluri R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J Biol Chem. 2010;285(26):20202–12.
Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172(7):973–81.
Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27(5):549–50.
Singer II, Kawka DW, Kazazis DM, Clark RA. In vivo co-distribution of fibronectin and actin fibers in granulation tissue: immunofluorescence and electron microscope studies of the fibronexus at the myofibroblast surface. J Cell Biol. 1984;98(6):2091–106.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–63.
Brown LF, Dubin D, Lavigne L, Logan B, Dvorak HF, Van de Water L. Macrophages and fibroblasts express embryonic fibronectins during cutaneous wound healing. Am J Pathol. 1993;142(3):793–801.
Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142(3):873–81.
Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 2000;257(1):180–9.
Colak S, Ten Dijke P. Targeting TGF-beta signaling in cancer. Trends Cancer. 2017;3(1):56–71.
Tolcher AW, Berlin JD, Cosaert J, Kauh J, Chan E, Piha-Paul SA, et al. A phase 1 study of anti-TGFbeta receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2017;79(4):673–80.
Castellone MD, Laukkanen MO. TGF-beta1, WNT, and SHH signaling in tumor progression and in fibrotic diseases. Front Biosci (Schol Ed). 2017;9:31–45.
Costa-Pereira AP. Regulation of IL-6-type cytokine responses by MAPKs. Biochem Soc Trans. 2014;42(1):59–62.
Fielding CA, Jones GW, McLoughlin RM, McLeod L, Hammond VJ, Uceda J, et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity. 2014;40(1):40–50.
Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124(4):1853–67.
Ward BC, Panitch A. Abdominal adhesions: current and novel therapies. J Surg Res. 2011;165(1):91–111.
Falk P, Bergstrom M, Palmgren I, Holmdahl L, Breimer ME, Ivarsson ML. Studies of TGF-beta(1-3) in serosal fluid during abdominal surgery and their effect on in vitro human mesothelial cell proliferation. J Surg Res. 2009;154(2):312–6.
Cheong YC, Shelton JB, Laird SM, Richmond M, Kudesia G, Li TC, et al. IL-1, IL-6 and TNF-alpha concentrations in the peritoneal fluid of women with pelvic adhesions. Hum Reprod. 2002;17(1):69–75.
Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. MEK and the inhibitors: from bench to bedside. J Hematol Oncol. 2013;6:27.
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40(8):955–62.
Brant SR. Promises, delivery, and challenges of inflammatory bowel disease risk gene discovery. Clin Gastroenterol Hepatol. 2013;11(1):22–6.
Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 2011;140(6):1704–12.
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–24.
Burke JP, Mulsow JJ, O’Keane C, Docherty NG, Watson RW, O’Connell PR. Fibrogenesis in Crohn’s disease. Am J Gastroenterol. 2007;102(2):439–48.
Hugot JP. Genetic origin of IBD. Inflamm Bowel Dis. 2004;10(Suppl 1):S11–5.
Meijer MJ, Mieremet-Ooms MA, Sier CF, van Hogezand RA, Lamers CB, Hommes DW, et al. Matrix metalloproteinases and their tissue inhibitors as prognostic indicators for diagnostic and surgical recurrence in Crohn’s disease. Inflamm Bowel Dis. 2009;15(1):84–92.
Mostafa RM, Moustafa YM, Hamdy H. Interstitial cells of Cajal, the Maestro in health and disease. World J Gastroenterol. 2010;16(26):3239–48.
Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. II. Intestinal subepithelial myofibroblasts. Am J Phys. 1999;277(2 Pt 1):C183–201.
Drygiannakis I, Valatas V, Sfakianaki O, Bourikas L, Manousou P, Kambas K, et al. Proinflammatory cytokines induce crosstalk between colonic epithelial cells and subepithelial myofibroblasts: implication in intestinal fibrosis. J Crohns Colitis. 2013;7(4):286–300.
Okuno T, Andoh A, Bamba S, Araki Y, Fujiyama Y, Fujiyama M, et al. Interleukin-1beta and tumor necrosis factor-alpha induce chemokine and matrix metalloproteinase gene expression in human colonic subepithelial myofibroblasts. Scand J Gastroenterol. 2002;37(3):317–24.
Rogler G, Gelbmann CM, Vogl D, Brunner M, Scholmerich J, Falk W, et al. Differential activation of cytokine secretion in primary human colonic fibroblast/myofibroblast cultures. Scand J Gastroenterol. 2001;36(4):389–98.
Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124(7):1866–78.
Zawahir S, Li G, Banerjee A, Shiu J, Blanchard TG, Okogbule-Wonodi AC. Inflammatory and immune activation in intestinal myofibroblasts is developmentally regulated. J Interf Cytokine Res. 2015;35(8):634–40.
Saada JI, Pinchuk IV, Barrera CA, Adegboyega PA, Suarez G, Mifflin RC, et al. Subepithelial myofibroblasts are novel nonprofessional APCs in the human colonic mucosa. J Immunol. 2006;177(9):5968–79.
Rieder F, Fiocchi C. Intestinal fibrosis in IBD--a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009;6(4):228–35.
Speca S, Giusti I, Rieder F, Latella G. Cellular and molecular mechanisms of intestinal fibrosis. World J Gastroenterol. 2012;18(28):3635–61.
Pinchuk IV, Mifflin RC, Saada JI, Powell DW. Intestinal mesenchymal cells. Curr Gastroenterol Rep. 2010;12(5):310–8.
Flynn RS, Murthy KS, Grider JR, Kellum JM, Kuemmerle JF. Endogenous IGF-I and alphaVbeta3 integrin ligands regulate increased smooth muscle hyperplasia in stricturing Crohn’s disease. Gastroenterology. 2010;138(1):285–93.
Rieder F, de Bruyn JR, Pham BT, Katsanos K, Annese V, Higgins PD, et al. Results of the 4th scientific workshop of the ECCO (Group II): markers of intestinal fibrosis in inflammatory bowel disease. J Crohns Colitis. 2014;8(10):1166–78.
Makitalo L, Sipponen T, Karkkainen P, Kolho KL, Saarialho-Kere U. Changes in matrix metalloproteinase (MMP) and tissue inhibitors of metalloproteinases (TIMP) expression profile in Crohn’s disease after immunosuppressive treatment correlate with histological score and calprotectin values. Int J Color Dis. 2009;24(10):1157–67.
McKaig BC, McWilliams D, Watson SA, Mahida YR. Expression and regulation of tissue inhibitor of metalloproteinase-1 and matrix metalloproteinases by intestinal myofibroblasts in inflammatory bowel disease. Am J Pathol. 2003;162(4):1355–60.
Di Sabatino A, Jackson CL, Pickard KM, Buckley M, Rovedatti L, Leakey NA, et al. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut. 2009;58(6):777–89.
Murphy G, Nagase H. Progress in matrix metalloproteinase research. Mol Asp Med. 2008;29(5):290–308.
Monteleone G, Caruso R, Fina D, Peluso I, Gioia V, Stolfi C, et al. Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut. 2006;55(12):1774–80.
Warnaar N, Hofker HS, Maathuis MH, Niesing J, Bruggink AH, Dijkstra G, et al. Matrix metalloproteinases as profibrotic factors in terminal ileum in Crohn’s disease. Inflamm Bowel Dis. 2006;12(9):863–9.
Lech M, Anders HJ. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta. 2013;1832(7):989–97.
Fiocchi C, Lund PK. Themes in fibrosis and gastrointestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2011;300(5):G677–83.
Bailey JR, Bland PW, Tarlton JF, Peters I, Moorghen M, Sylvester PA, et al. IL-13 promotes collagen accumulation in Crohn’s disease fibrosis by down-regulation of fibroblast MMP synthesis: a role for innate lymphoid cells? PLoS One. 2012;7(12):e52332.
Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut. 2009;58(8):1152–67.
Higashi K, Inagaki Y, Fujimori K, Nakao A, Kaneko H, Nakatsuka I. Interferon-gamma interferes with transforming growth factor-beta signaling through direct interaction of YB-1 with Smad3. J Biol Chem. 2003;278(44):43470–9.
Raghu G, Brown KK, Bradford WZ, Starko K, Noble PW, Schwartz DA, et al. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004;350(2):125–33.
Biancheri P, Pender SL, Ammoscato F, Giuffrida P, Sampietro G, Ardizzone S, et al. The role of interleukin 17 in Crohn’s disease-associated intestinal fibrosis. Fibrogenesis Tissue Repair. 2013;6(1):13.
Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143(3):765–76 e3.
Speca S, Dubuquoy L, Desreumaux P. Peroxisome proliferator-activated receptor gamma in the colon: inflammation and innate antimicrobial immunity. J Clin Gastroenterol. 2014;48(Suppl 1):S23–7.
Lu D, Carson DA. Repression of beta-catenin signaling by PPAR gamma ligands. Eur J Pharmacol. 2010;636(1–3):198–202.
Zhao C, Chen W, Yang L, Chen L, Stimpson SA, Diehl AM. PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. 2006;350(2):385–91.
Ghosh AK, Bhattacharyya S, Lakos G, Chen SJ, Mori Y, Varga J. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2004;50(4):1305–18.
Tan X, Dagher H, Hutton CA, Bourke JE. Effects of PPAR gamma ligands on TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells. Respir Res. 2010;11:21.
de Bruyn M, Vandooren J, Ugarte-Berzal E, Arijs I, Vermeire S, Opdenakker G. The molecular biology of matrix metalloproteinases and tissue inhibitors of metalloproteinases in inflammatory bowel diseases. Crit Rev Biochem Mol Biol. 2016;51(5):295–358.
Macarak Edward J, Lotto Christine E, Deepika K, Xiaoling J, Wermuth Peter J, Anna-Karin O, Matthew M, Joel R. Trametinib prevents mesothelial-mesenchymal transition and ameliorates abdominal adhesion.formation. J Surg Res. 2018;227:198–210. https://www.sciencedirect.com/science/article/pii/S0022480418300982?via%3Dihub
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Macarak, E., Rosenbloom, J. (2018). The Pathogenesis of Intraabdominal Adhesions: Similarities and Differences to Luminal Fibrosis. In: Rieder, F. (eds) Fibrostenotic Inflammatory Bowel Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-90578-5_22
Download citation
DOI: https://doi.org/10.1007/978-3-319-90578-5_22
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-90577-8
Online ISBN: 978-3-319-90578-5
eBook Packages: MedicineMedicine (R0)