
Abstract
Neoepitope peptides are newly formed antigens presented by major histocompatibility complex class I (MHC-I) on cell surfaces. The cells presenting neoepitope peptides are recognized and subsequently killed by cytotoxic T-cells. Immunopeptidomic approaches aim to characterize the peptide repertoire (including neoepitope) associated with the MHC-I molecules on the surface of tumor cells using proteomic technologies, providing critical information for designing effective immunotherapy strategies. We developed a novel constrained de novo sequencing algorithm to identify neo-epitope peptides from tandem mass spectra acquired in immunopeptidomic analyses. Our method incorporates prior probabilities to putative peptides according to position specific scoring matrices (PSSMs) representing the sequence preferences recognized by MHC-I molecules. We implemented a dynamic programming algorithm to determine the peptide sequences with an optimal posterior matching score for each given MS/MS spectrum. Similar to the de novo peptide sequencing, the dynamic programming algorithm allows an efficient searching in the entire peptide sequence space. On an LC-MS/MS dataset, we demonstrated the performance of our algorithm in detecting the neoepitope peptides bound by the HLA-C*0501 molecules that were superior to database search approaches and existing general purpose de novo peptide sequencing algorithms.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
The peptide epitopes presented by major histocompatibility complex class I (MHC-I) molecules on cell surfaces display a representative image of the collection of (endogenously synthesized or exogenous) proteins in the cell, allowing immune cells (e.g., the \(CD8^+\) cytotoxic T-cells) to monitor the biological activities occurring inside the cell, a process known as the immune surveillance [2, 7, 28]. A typical process of the peptide processing and presentation involves three steps: (1) the cytosolic proteins are first degraded into peptides by the proteasome; (2) the resulting peptides are loaded onto MHC-I molecules; and (3) the MHC-I/peptide complex is transported into the plasma membrane of the cell via endoplasmic reticulum (ER), while the extracellular domain of MHC-I, where the epitope peptide binds, is exported outside the membrane. In normal cells, the peptides presented by MHC-I will not induce immune responses. However, when abnormal processes (e.g., viral infection or tumorigenesis) occur inside cells, a fraction of MHC-I molecules may present peptides from foreign or novel proteins (e.g., due to somatic mutations in tumor cells), often referred to as the neoepitope peptides or neoantigens. Consequently, the cells presenting such peptides will likely to be recognized and subsequently killed by cytotoxic T-cells.
It is now well known that, during tumor development, maintenance and progression, tumor cells accumulate thousands of somatic mutations, many of these occurring in protein-coding regions of tumor genes [6, 22, 29]. Among them, missense or frameshift mutations have the potential to generate neoepitope peptides, which can be used as biomarkers for characterizing the states and subtypes of cancer, or can be selected as potential therapeutic cancer vaccines to induce robust and tumor-specific responses [7, 30]. Furthermore, neoepitope peptides were recently demonstrated as potential targets in cancer immunotherapies such as adoptive T-cell therapy [39].

An example of positional specific scoring matrix (PSSM) (shown as a frequency heatmap) derived from neoepitope peptides of 9 amino acid residues bound to HLA-C*0501. The third position is dominated by Asp while at the ninth position, Leu and Val are preferred.
In the past decade, clinical evidence has been accumulated on tumor-specific immune activities, leading to the implementation of successful strategies of cancer immunotherapy [9]. Because of the strong implications of neoepitope peptides in the design of effective cancer immunotherapy, different genomic and proteomic methods have been developed to identify neoepitope peptides presented by tumor cells from cancer patients. The genomic approaches start from exon and transcriptome sequencing of normal and tumor tissues in attempt to identify proteins over- or under-expressed tumor issues, as well as missense or frameshift mutations in tumor proteins [20, 25], and then use computational methods [1, 13, 40] to predict neoepitope candidate from these tumor proteins based on the immunogenicity of peptides, i.e., the likelihood of peptides being presented by MHC-I molecules in tumor cells and furthermore likely to provoke an immune response. Notably, the genomic approaches may not report accurate neoepitope peptides due to various limitations of the methods. First, some very low abundant proteins that may not be identified using transcriptome sequencing are often presented by the MHC-I molecules, and can provoke robust immune responses. Second, current immunogenicity prediction algorithms cannot yet accurately model the process of antigenic peptide processing and presentation by MHC-I, and thus may report many false positives and false negatives of neoepitope peptides. Most importantly, as multiple MHC-I molecules are encoded by the highly polymorphic human leukocyte antigen (HLA) genes (including three major types of HLA-I, HLA-II and HLA-III) in an individual patient, the peptide immunogenicity is indeed a private measure specific to this cancer patient, and thus cannot be modeled without sufficient neoepitope peptides already identified from the patient’s own sample [10].
In contrast, the immunopeptidomic approaches aim to directly analyze the peptide repertoire bound by the MHC-I molecules on the surface of tumor cells using proteomic technologies, and thus can overcome the limitations of genomic approaches. Because of its high throughput and sensitivity, liquid chromatography coupled tandem mass spectrometry (LC-MS/MS) has been routinely used in proteomics in an attempt to identify and quantify proteins in complex protein mixtures, and also becomes the technology of choice for the identification of neoepitope peptides eluted from MHC molecules [5]. From the MS/MS spectra acquired in an immunopeptidomic experiment, potential neoepitope peptides are identified often using a database search engines designed for peptide identification in proteomics (e.g. Sequest [12], Mascot [8] or MSGF+ [19]). However, the neoepitope peptides have some distinct features comparing to the peptides from general proteomic analysis. On one hand, neoepitope peptides bound to different classes of MHC-I molecules have relatively fixed length; for example, human HLA class I (HLA-I) recognizes peptides 8 to 12 amino acid residues in length [4]. On the other hand, unlike the peptides in proteomic experiments typically from tryptic digestion at specific basic amino acid residues, neoepitope peptides can be cleaved by proteasome at any arbitrary position in the target proteins. As a result, when MS/MS spectra from an immunopeptidomic study is searched against a target protein database (e.g, consisting of all human proteins), all non-tryptic peptides of the lengths within a range (8–12 residues) are considered; in the human protein database, there are \(\approx 10^7-10^8\) such peptides, much greater than the number of tryptic peptides (\(\approx 10^6\)). Furthermore, a recent study demonstrated that a surprisingly large fraction (about a third) of neoepitope peptides are generated by proteasome-catalyzed peptide splicing (PCPS) that cuts and pastes peptide sequences from different proteins [24]. If all concatenate peptides (with two subpeptides from the same or different proteins) are considered in the database search, the number of target peptides increases to \(\approx 10^{15}\), close to the total number of peptides 8–12 residues in length. Which poses great challenges to database search not only on the running time but also on potential false positives in peptide identification. Finally, strong sequence patterns are present in neoepitope peptides, largely because of the preferences in the binding affinity and specific structures of MHC-I molecules. The sequence pattern in neoepitope peptides recognized by a specific class of MHC-I molecule can be represented by a positional specific scoring matrices (PSSMs; see Fig. 1 as an example for HLA-C) [17], or more complex machine learning models for predicting peptide immunogenicity [1]. However, these sequence information are not used by current approaches for neoepitope peptide identification in proteomic experiments.
De novo peptide sequencing algorithms (such as Peaks [27], pepNovo [14], pepHMM [37] and most recently, uniNovo [16], Novor [26] and DeepNovo [32, 33]) represent a different approach to peptide identification in proteomics, that attempt to reconstruct the peptide sequence directly from an MS/MS spectrum. Comparing to database search algorithms, de novo sequencing algorithms explore the entire space of peptides, but are often more efficient because of the employment of a dynamic programming algorithm. From a Bayesian perspective, the database search approach can be viewed as a special case of de novo peptide sequencing, which assumes that only the proteins in the database can be present in the sample, and thus the peptides from these proteins have the prior probabilities of 1 while the other peptides have the prior probabilities of 0 [23]. Previous studies have showed that although the top peptide reported by the de novo sequencing algorithm for an MS/MS spectrum was sometimes incorrect, the correct one was usually the peptide in the database that received the highest score in de novo sequencing [14, 27], indicating that the incorporation of the protein database as prior knowledge significantly improves peptide identification.
In this paper, we present a novel constrained de novo sequencing algorithm for neoepitope peptide identification. The method can be viewed as a hybrid approach of the de novo sequencing and the database searching algorithms: it explores the entire space of peptide sequences 9–12 residues in length, but assigns a different prior probability to each putative peptide according to MHC-I specific PSSMs, such that the peptide with a motif with high immunogenicity incorporates a high prior probability into the posterior probability score of the peptide-spectrum matches (PSMs). Utilizing the sequential property of the PSSMs, we extended the dynamic programming (DP) algorithm for de novo peptide sequencing to determine the peptide sequences with the optimal posterior matching scores for each given MS/MS spectrum. Notably, similar to de novo peptide sequencing algorithms, the dynamic programming algorithm allows an efficient searching in the entire peptide sequence space, which, as shown above, is comparable to the size of the database consisting of all putative neoepitope peptides (including the concatenate peptides) derived from human proteins. We tested our algorithm in a LC-MS/MS dataset for detecting the neoepitope peptides bound by the HLA-C*0501 molecules [18]. Our method could detect about 19,017 neoepitope peptides of lengths between 9 to 12 residues with estimated false discovery rate below \(1\%\). In contrast, the database search approach (using MSGF+ against the human protein database) identified about 4,415 PSMs (1,804 unique peptides), in which 2,104 PSMs (764 unique peptides) have the length between 9 to 12 residues as putative neoepitope peptides. Out of the 2,104 PSMs, 1,269 were also identified by our method. A majority (791 out of 1,269) of the PSMs were exact matches, while most (360 out of 478) remaining PSMs contain only a swap of consecutive residues in peptide sequences. Finally, we tested a conventional de novo sequencing algorithm uniNovo [16] on the same dataset. It reported sequence tags on 1,863 MS/MS spectra, but with low sequence coverage (on average three amino acid residues per peptide), and thus cannot be used in neoepitope peptide sequencing. These results imply that the constrained de novo sequencing algorithm benefit from the prior probabilities (provided by the PSSMs) to distinguish the most likely neoepitope peptides from other peptides sharing similar sequences.
2 Method
Constrained de novo Peptide Sequencing. Given an MS/MS spectrum M, the constrained de novo peptide sequencing problem is to find the peptide sequence T within a range of length (\(l_{min} \le |T| \le l_{max}\)) that maximizes a posterior matching score S:
where P(T) represents the prior probability of the peptide T, and P(M|T) represents the matching probability, i.e., the probability of observing the MS/MS spectra from the peptide T. For peptides with a fixed length l, their prior probabilities are defined by a PSSM \(p_{ij}\) (\(\sum _i p_{ij} = 1\)) for residue i at the position j (\(j=1, 2, ..., l\)) in the peptide; thus, for the peptide \(T=t_1t_2...t_l\), \(P(T)=\prod ^l_{j=1} p_{t_j j}\). The matching probability P(M|T) is modeled by the independent fragmentation at each peptide bond: \(P(M|T)=\prod ^l_j=1 P(f_{M, j})\), where \(P(f_{M, j})\) stands for the probability of observing \(f_{M, j}\), the occurrence pattern of the set of fragment ions, including the b-ion, y-ion and the neutral loss ions, derived from the fragmentation between the precursor (\(t_1t_2...t_j\)) and the suffix (\(t_{j+1}t_{j+2}...t_l\)) peptide in M. Notably, \(f_{M, j}\) is dependent only on \(m_j\), the j-th prefix mass of the prefix peptide \(t_1t_2...t_j\), but is not dependent on the peptide sequences. Therefore,
where \(P(F(m_j))\) represents probability of observing the set of fragment ion \(F(m_j)\) associated with the prefix mass \(m_j\) in M. These probabilities can be learned from a training set of identified MS/MS spectra [14], in which the peaks are assigned. Alternatively, as adopted here, \(P(F(m_j))\) is assigned empirically based on the logarithm transformed ion intensities of the matched b- or y-ions (within a mass tolerance). Let S(j, m) be the maximum posterior matching score between an MS/MS spectrum and any peptide of length j with a total mass of m, which can be computed by using a dynamic programming algorithm,
where k is an amino acid in the alphabet A. Note that the multiplication of probabilities in Eq. 3 can be transformed into the summation of the logarithms of probabilities. Finally, the optimal potential matching score of a peptide with a fixed length l, implicated as the number of columns in the PSSM, matching a given spectrum M, is \(S(M; l, m_{pr})\), in which \(m_{pr}\) is the precursor mass of M. The algorithm can be applied to each putative peptide length between \(l_{min}\) and \(l_{max}\) with a corresponding PSSM, and the peptides will be reported in the order of their posterior matching scores. The dynamic programming algorithm is executed in \(O(l \cdot m_{pr})\) time using \(O(l \cdot m_{pr})\) space (where the fragment ion masses are binned according to the mass resolution), but can be further accelerated by heuristics as described below. Note that the prefix mass scoring has been previously proposed as a useful tool for de novo peptide sequencing [14], database searching [19] and spectrum alignment to identify mutations and post-translation modifications (PTMs) [31]. The dynamic programming algorithm presented here can be view as matching a predefined PSSM against a vector of prefix mass scores (probabilities) in order to find the optimal matches between a peptide and a subset of prefix masses.
Accelerating the Dynamic Programming Algorithm. For an input MS/MS spectrum of the precursor mass \(m_{pr}\) and a PSSM with a specific neoepitope peptide length l, the above algorithm explores all potential prefix masses between 0 and \(m_{pr}\) for each prefix peptide of the length from 0 to l. However, there are only a limited number of prefix masses corresponding to prefix peptides of a fixed length, indicating that the matrix of S(j, m) computed in Eq. 3 has many zeroes, especially when for small j. To compute only the non-zero elements in S(j, m), we exploited a branch-and-bound approach to explore the peptide space, while retaining only the best scored sub-peptide among those with the same prefix mass.

A schematic illustration of the exploration of the peptide sequence space in the constrained de novo algorithm (see text for details).
The sequencing algorithm maintains a pool of putative prefix peptides, each associated with a posterior matching score. The pool starts with N (\(N=|A|=20\) representing the number of amino acid masses) prefix peptides of length 1 (Fig. 2) with posterior matching scores of \(S(1, m(k))=p_{1k}\cdot P(F(m(k)))\) (where m(k) is the mass of the amino acid k). At each following iteration j, for \(j= 2, ..., l\), every prefix peptide in the pool generates N new prefix peptides, one for every amino acid, by appending a new amino acid to the end of each existing peptide (of length \(j-1\)) in the pool.
After appending an amino acid k to an existing prefix peptide with mass \(m'\), the mass of the resulting prefix peptide (i.e., the prefix mass m) is used to compute P(F(m)), and then the posterior matching score of the new prefix peptide is computed by \(S(j,m)=S(j-1,m') \cdot p_{jk} \cdot P(F(m))\), where \(S(j-1,m')\) is the posterior matching score associated with the existing prefix peptide of length \(j-1\). At each step, the precursor mass m should match at least one of b- and y-ions; otherwise, the precursor peptide is labeled with one miscleavage, which is tracked on each iteration of an algorithm: if a prefix peptide contains too many miscleavages, it is eliminated from further extension. Once the posterior matching score of a prefix peptide is obtained, it will be compared with other peptides in the pool with the same prefix mass, and the k (default = 5) best scoring peptides are retained. After each step, at most \(N \times m_{pr}\) prefix peptides are retained in the pool. The algorithm is illustrated in Fig. 2. We note that, although the worst-case running time of the de novo sequencing algorithm is still \(O(l \cdot m_{pr})\) for each spectrum, in practice, it runs much faster as many un-realistic prefix masses were not evaluated, especially for small l.
In the final step (with prefix peptides of the expected length l), all peptides with masses matching the precursor mass are re-assessed by using a global scoring scheme (see below), and are reported in the order of their global scores. Note that for each input MS/MS spectrum, the constrained de novo algorithm was conducted four times, with an input PSSM for peptides of length 9, 10, 11 and 12, respectively.
Pre-processing of MS/MS Spectra. Prior to constrained de novo sequencing algorithm, several pre-processing steps were conducted on the MS/MS spectra, including: (1) peaks with an intensity of 0 were removed; (2) the precursor peak was removed; (3) any converted mass greater than precursor mass was removed; (4) Isotopic masses of precursor masses were removed; (5) the intensities of all peaks were logarithm-transformed.
Construction of PSSMs. Peptides of length 9–12 were extracted from the IEDB [35] database http://www.iedb.org/, and separated by length. A total of 892 peptides of length 9, 191 peptides of length 10, 110 peptides of length 11, and two peptides of length 12 were considered. Four PSSMs were created, one for each peptide length, in which the amino acid frequency in every position in the PSSM was computed based on these peptide sequences and the pseudo-count of 1 was incorporated to ensure there were no frequencies of 0.
Re-Assessment of Peptide-Spectrum Matches (PSMs) by Global Scoring. The global score of a PSM is a probability measure, based on a combination of the prior probability based on the input PSSM, and how well it’s theoretical fragmentation of the peptide matches to the experimental spectrum. It is calculated using Eq. (1), where P(T) is the probability of the peptide given the PSSM, normalized to the length of the peptide, and P(M|T) is the probability of observing MS/MS M from peptide T based off of the theoretical fragmentation of T. P(M|T) is calculated by
where \(e_i\) is a normalized intensity of the experimental spectrum E, \(a_i\) is the mass accuracy (in ppm) between experimental mass i and theoretical fragmentation mass i (or W if there is no matching mass between the two), from the mass accuracy vector A, W is the lowest allowable mass accuracy between an experimental and theoretical mass, and k is the number of peaks in the experimental spectrum M.

Score distributions of PSMs reported by the constrained de novo sequencing algorithm and the decoy PSMs from the reverse peptides.
False Discovery Rate Estimation. After the global scores were computed for all PSMs, it was necessary to determine a score threshold to validate whether a peptide match was reliably identified from an MS/MS spectrum by our constrained de novo sequencing algorithm. Note that it is possible for multiple similar peptide sequences to score high enough to indicate that any of them could be the correctly identified neoepitope peptide producing the corresponding MS/MS spectrum. In this case, the de novo sequencing algorithm reports all of them. As shown in the results section, in practice, usually only a few peptides( ) are reported for each spectrum.
) are reported for each spectrum.
To obtain an appropriate score threshold, we adopted similar strategy to the target-decoy search in database searching [11] to estimate the false discovery rate (FDR) of PSMs. We generated a decoy peptide database consisting of about 40 million randomly selected and reversed peptides with lengths of 9–12 residue from the proteins in the Uniprot database. Additionally, a second database was created for the reversed peptides found by the constrained de novo sequencing algorithm. For each spectrum in our analysis, up to 10 peptides matching the spectrum precursor mass within the mass resolution (35 ppm) were selected from both databases as decoys. The top scoring peptides among these decoy peptides were used to form the decoy PSMs, whose global scores were computed. The score distributions are depicted in Fig. 3, containing the scores from both decoy PSMs and the PSMs reported by the constrained de novo sequencing algorithm. We then used the following formula to estimate the FDR at a certain score threshold t: \(FDR_t = N_{decoy}/N_{cons}\), where \(N_{decoy}\) and \(N_{cons}\) represent the numbers of decoy and positive (from the sequencing algorithm) PSMs with global scores above t, respectively. We then estimated that PSMs with higher than 0.0058 have FDR lower than 1%.
Datasets. The dataset was obtained from ProteomeXChange [36] (accession number: PXD006455). The experiments were conducted on two common HLA-C: HLA-C*05:01 and HLA-C*07:02. These HLA class I molecules were isolated from the cell surface of C*05 and C*07 transfected 721.221 cells, and sequenced bound peptides by mass spectrometry. As observed in the original article [18], HLA-C*05:01 has higher expression level and more diversified binding peptides. In our testing, we chose the binding peptides of HLA-C*05:01 (with length between 9 to 12 residues) to demonstrate the performance of our method. In total, there are 339,513 spectra acquired in a total 25 fractions of LC-MS/MS analysis using the Q Exactive HF-X MS (Thermo Fisher Scientific) [36].
Database Searching. We used MSGF+ [19] here as the database searching engine. The parameters for the MSGF+ are set as following to match the experimental conditions of the LC-MS/MS analyses: (1) instrument type: high-resolution LTQ; (2) the enzyme type: unspecific cleavage; (3) precursor mass tolerance: 35 ppm; (4) isotope error range: −1, 2; (5) modifications: oxidation as variable and carboamidomethyl as fixed; (6) maximum charge is 7 and minimum charge is 1. The FDR is estimated by using a target-decoy search approach (TDA) [11].
3 Results
Constrained de novo Sequencing. We implemented the constrained de novo sequencing algorithm in C. It spends a total of 8,910 min on a Linux computer (Intel(R) Xeon(R) CPU E5-2670 0 @ 2.60 GHz) as single thread to process 339,513 input MS/MS spectra in the HLC-C peptidomic dataset, i.e., about 1.6 s per MS/MS spectrum. Among the entire set of spectra, the sequencing algorithm reported one or more peptide sequences for 136,249 (40.14%) spectra, resulting a total of 2,775,977 peptide-spectrum matches (PSMs), i.e., 20 PSMs (peptides) per spectra. Among them, 81,888 PSMs over 28,759 spectra (i.e., 2.85 PSMs per spectra) received a global matching score above 0.0058 (corresponding to about \(1\%\) FDR; see Methods), corresponding to 57,449 unique peptides, are retained for further analysis.

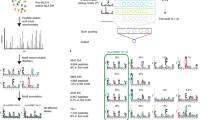
The length distributions of the top-ranked peptides reported by the constrained de novo sequencing algorithm (A); and the sequence logos representing the position specific frequency pattern among the top-ranked peptides with different lengths (B).

(A) The comparison of PSMs and identified unique peptides (in parentheses) reported by database searching and constrained de novo sequencing. (B) Number of amino acids difference in overlapped IDs from database search and constrained de novo. (C) The prior probability and matching scores of the PSMs reported by the constrained de novo sequencing and database search approach. The PSMs are depicted in different colors: orange for those detected by both approaches, red for those detected by database searching only, and black for those detected by de novo sequencing only while blue for those reported by de novo sequencing and also have at least 50% sequence similarity to human proteins (Color figure online)
The top-ranked peptides of the 28,759 spectra corresponds to 19,017 unique peptides. The length distribution of these peptides is illustrated in Fig. 4. A majority (13,648, \(71.76\%\)) of them are 9 residues in length, which is consistent with previous observations [18] and the IEDB database [35], in which 892 out of 1,195 (74.64%) HLA-C*0501 bounded peptides are 9 residues in length. Figure 4B shows the sequence logo [34] generated by using the identified peptides by the de novo sequencing method. Specifically, 13,648 peptides have 9 residues, 2,904 have 10 residues, 1,647 have 11 residues, and 818 have 12 residues. Those sequences were used to generate the sequence logos in Fig. 4. For peptides of length 9, the sequence logo showed that the positions of P2, P3 and P9 have strong amino acid preferences: P2 is enriched by Ala, P9 is enriched by Leu/Ile, and P3 is dominated by Asp. For peptides of other lengths, Asp is predominant at multiple positions, especially in the peptides N-termini, while Leu/Ile are predominant in peptides C-termini.
If all the sequences are retained as long as the global matching score is above the threshold, our method reported 57,449 unique peptide sequences. To be noted, we kept the all the de novo sequences here, because in many cases multiple peptide sequences containing swapped consecutive amino acids are reported, possibly due to missing fragment peaks to distinguish them in the MS/MS spectra. For those cases, the constrained de novo peptide sequencing algorithm will report very similar peptides with nearly identical global matching scores.
Comparison with Database Searching Results. MSGF+ is employed to identify peptides by searching against the human proteome database. The computation takes 1,102 min on a Linux computer (Intel(R) Xeon(R) CPU E5-2670 0 @ 2.60 GHz). It reported 4,415 PSMs given 5% false discovery rateFootnote 1. Among these PSMs, 2,104 are identified as peptides of lengths between 9 to 12 residues (corresponding to 764 unique peptide sequences), which are putative HLA-C*0501 bounded neoepitope peptides. We compared the peptides identified by our constrained de novo sequencing algorithm with those identified by the database searching method in a Venn diagram shown in Fig. 5A. A total of 1,269 spectra are identified by both the database searching and the de novo sequencing method, among which 791 spectra were identified as identical peptidesFootnote 2 by both methods: for 360 spectra, the peptides identified by the de novo sequencing method differ only in no more than two amino acid residues from the peptides identified by the database searching (where most of cases are two consecutive residues swaps); and for the remaining 118 spectra, the two identified peptides by these two methods differ in more than two residues, but share over 50% sequence similarity.
The PSMs reported by both the database searching and the de novo sequencing algorithm, and those reported by only one of these methods were investigated in the context of their prior probabilities and matching scores (Fig. 5C). The PSMs reported by both methods receive generally higher matching scores and comparable prior probabilities. 825 out of 835 PSMs reported only by the database searching method received a global matching score below the threshold 0.0058 used for selecting de novo sequencing results. The remaining ten PSMs received prior probabilities less than 0.1 (on average, prior probability is 0.05), indicating they are less likely neoepitope peptides. On the other hand, among the top-ranked 27,476 PSMs reported only by the de novo sequencing algorithm, 23,857 have the prior probabilities above 0.1. We further analyzed the 18,905 unique peptides from these 27,476 top-ranked PSMs. When searching against the human protein database containing 21,006 sequences from Uniprot [3] using Rapsearch2 [41], 14,658 (77.53%) peptides have 50% or higher sequence similarity with some peptides from human proteins, while 7,737 (40.93%) peptides differ at most two amino acids (i.e, a swap of two consecutive residues), including 1,910 (10.10%) identical peptides. Notably, although these identified peptides are more likely the true neoepitope peptides, some of the rest peptides may also be neoepitope peptides, e.g., those generated by novel gene splicing and fusion events, or PCPS [24].
Comparison with Current de novo Sequencing Methods. We attempted to compare our method with the most recently developed de novo sequencing method uniNovo [16] on the HLA-C peptidomic dataset. The parameters of uniNovo are chosen in consistence with the experimental settings: (1) the ion tolerance: 0.3 Da; (2) precursor ion tolerance: 100 ppm (3) fragmentation method: HCD; (4) no enzyme specificity is selected; (5) five peptide sequences per spectrum are reported; (6) minimum length of peptides: 9; and (7) minimum accuracy: 0.8. A total of 1,863 spectra are identified by uniNovo under these parameters. Most of the sequencing results are non-conclusive: only 3–6 (on average 3.1) amino acid residues were reported in these peptides, and the gaps between the residues were reported as mass intervals (e.g., a typical output of uniNovo is [406.2043]D[204.10266]QI). Because of the non-conclusive peptide sequences in uniNovo report, we did not further compare it with the results from our constrained de novo sequencing algorithms. We also compared our method with another up-to-date and user-friendly de novo sequencing software, Novor [26]. We used the default parameters of the software for comparison. In total, Novor reported 337,717 peptide-spectrum matches (PSMs), with only one top peptide for each spectrum. We note that, as Novor inherently considers only trypsin-digested peptides in the de novo sequencing algorithm, and most neo-epitope peptides do not have K/R at their C-termini, we limited our comparison on those top-scored tryptic-like peptides (with K/R at their C-termini) reported by our constrained de novo sequencing algorithm under 1% FDR. Only 2,259 spectra were identified as tryptic-like peptides by our method, the peptide sequences reported by both methods on these spectra are, however, quite different, with an average hamming distance of 5.9. When compared to the MS-GF+ results, the peptides reported by Novor have average 4.54 hamming distance, while our de novo results have average 3.84 hamming distance. This comparison suggests that the prior information (i.e., the PSSM) employed in the constrained sequencing algorithm helps to identify the peptide sequences that are more likely neoepitope peptide than a generic de novo sequencing algorithm without using this prior information.
4 Discussion
The constrained de novo sequencing method was designed specifically for characterizing neoepitope sequences from their MS/MS spectra acquired in immunopeptidomic experiments. The algorithm does not rely on a database of potential neoepitope peptides, and thus can identify peptides that are not contiguous subsequences of proteins in a database, including those resulting from novel insertion, deletion, splicing or gene fusion events, or those containing mutations (e.g., in tumor cells) or those generated by proteasome-catalyzed peptide splicing (PCPS) [24]. The dynamic programming algorithm adopted here allows for efficient searching in the entire space of peptide sequences within a range of desirable lengths (e.g., 9–12 residues). The results showed that, when peptides can be obtained by both methods, the peptide sequence reported by the de novo sequencing method often match with that from database searching, with at most one swap between two consecutive amino acid residues. Notably, unlike existing de novo sequencing algorithms (e.g., uniNovo) often reporting many putative sequence tags each with relatively low sequence coverage of target peptide, the constrained de novo sequencing method report one or a few complete peptide sequence with desirable length. As a result, it is straightforward to search for the occurrence of peptide sequences in a protein database, even for those generated by PCPS (e.g., concatenated from two subpeptides in different proteins).
The results on the testing dataset showed that many MS/MS spectra that were not identified by the database searching approach were identified as putative neoepitope peptides by the constrained de novo sequencing algorithm. This is probably due to the fact that the constrained de novo sequencing method benefits from the incorporation of PSSMs as prior probabilities, which prefers the peptides with high immunogenicities (i.e., likely to be presented by MHC-I). This is consistent with the typical experimental setting in immunopeptidomics, where peptides bound to a target MHC-I protein (e.g., HLA-C for the dataset used here) are enriched before the LC-MS/MS analyses. Hence, we anticipate a majority of MS/MS spectra result from the those peptides and thus can be identified using the constrained de novo sequencing method. On the other hand, other peptides (not bound to the target MHC-I molecule) are not of interests in immunopeptidomics, and thus it is not a concern if the de novo sequencing method cannot identify them.
The PSSMs adopted in this study were constructed by using known peptide sequences bound to a target MHC-I protein (HLA-C). The PSSMs for some desirable lengths are not informative as there are only very few known peptides of the respective length (e.g., only two known sequences have 12 residues in lengths). The PSSMs for some other classes of MHC-I may be even less characterized in current literature. We expect more accurate PSSMs can be derived after more neoepitope peptides become available with the advances of immunopeptidomic analyses, which can further improve the constrained de novo sequencing as presented here. Moreover, it is anticipated the preferences of MHC-I can be different in different patient because of the presence of many alleles of MHC-I encoding genes in human population. Therefore, specific PSSMs may be needed to be constructed for different MHC-I alleles so that appropriate PSSMs can be selected (based on HLA typing from the patient’s genomic sequencing data [15, 38]) for neoepitope peptide analyses of an individual patient.
The method presented here can also be applied to sequencing of other types of neoepitope peptides. For example, even though the attention has been most focused on the peptides presented by MHC-I that stimulates the cytotoxic killer T-cell responses, the peptides presented by MHC-II that are important for CD4+ helper T-cell responses [21] can also be characterized using a similar approach. The MHC-II presented peptides are typically longer in length and more variable, and thus more data are required to derive useful prior PSSM models.
Notes
- 1.
We used a FDR threshold of 5% to be consistent with the original article [18]. When a more common FDR threshold 0.01 is used, much fewer (1,280) MS/MS spectra were identified, among which only 97 were identified as peptides with lengths between 9 and 12.
- 2.
Note that, here only the top-ranked peptides reported by the de novo sequencing algorithm were considered, and ILE and LEU are considered as identical amino acids in this comparison.
References
Bhattacharya, R., Sivakumar, A., Tokheim, C., Guthrie, V.B., Anagnostou, V., Velculescu, V.E., Karchin, R.: Evaluation of machine learning methods to predict peptide binding to MHC class I proteins. bioRxiv, p. 154757 (2017)
Blum, J.S., Wearsch, P.A., Cresswell, P.: Pathways of antigen processing. Annu. Rev. Immunol. 31, 443–473 (2013)
Boutet, E., Lieberherr, D., Tognolli, M., Schneider, M., Bairoch, A.: UniProtKB/Swiss-Prot: the manually annotated section of the UniProt knowledgebase. Plant Bioinf.: Methods Protoc. 406, 89–112 (2007)
Bouvier, M., Wiley, D.C.: Importance of peptide amino and carboxyl termini to the stability of MHC class I molecules. Science 265(5170), 398–402 (1994)
Caron, E., Kowalewski, D.J., Koh, C.C., Sturm, T., Schuster, H., Aebersold, R.: Analysis of major histocompatibility complex (MHC) immunopeptidomes using mass spectrometry. Mol. Cell. Proteomics 14(12), 3105–3117 (2015)
Chalmers, Z.R., Connelly, C.F., Fabrizio, D., Gay, L., Ali, S.M., Ennis, R., Schrock, A., Campbell, B., Shlien, A., Chmielecki, J., et al.: Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9(1), 34 (2017)
Comber, J.D., Philip, R.: MHC class I antigen presentation and implications for developing a new generation of therapeutic vaccines. Ther. Adv. Vaccines 2(3), 77–89 (2014)
Cottrell, J.S., London, U.: Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20(18), 3551–3567 (1999)
Dustin, M.L.: Cancer immunotherapy: killers on sterols. Nature 531(7596), 583–584 (2016)
Editorial, N.B.: The problem with neoantigen prediction. Nat. Biotech. 35(2), 97 (2017)
Elias, J.E., Gygi, S.P.: Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4(3), 207–214 (2007)
Eng, J.K., McCormack, A.L., Yates, J.R.: An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5(11), 976–989 (1994)
Flower, D.R.: Towards in silico prediction of immunogenic epitopes. TRENDS Immunol. 24(12), 667–674 (2003)
Frank, A., Pevzner, P.: PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77(4), 964–973 (2005)
Gabriel, C., Fürst, D., Faé, I., Wenda, S., Zollikofer, C., Mytilineos, J., Fischer, G.: HLA typing by next-generation sequencing-getting closer to reality. HLA 83(2), 65–75 (2014)
Jeong, K., Kim, S., Pevzner, P.A.: UniNovo: a universal tool for de novo peptide sequencing. Bioinformatics 29(16), 1953–1962 (2013)
Jones, D.T.: Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292(2), 195–202 (1999)
Kaur, G., Gras, S., Mobbs, J.I., Vivian, J.P., Cortes, A., Barber, T., Kuttikkatte, S.B., Jensen, L.T., Attfield, K.E., Dendrou, C.A., et al.: Structural and regulatory diversity shape HLA-C protein expression levels. Nat. Commun. 8 (2017)
Kim, S., Pevzner, P.A.: MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5 (2014)
Kvistborg, P., Clynes, R., Song, W., Yuan, J.: Immune monitoring technology primer: whole exome sequencing for neoantigen discovery and precision oncology. J. Immunother. Cancer 4(1), 22 (2016)
Laidlaw, B.J., Craft, J.E., Kaech, S.M.: The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat. Rev. Immunol. 16(2), 102–111 (2016)
Le Gallo, M., Rudd, M.L., Urick, M.E., Hansen, N.F., Zhang, S., Lozy, F., Sgroi, D.C., Vidal Bel, A., Matias-Guiu, X., Broaddus, R.R., et al.: Somatic mutation profiles of clear cell endometrial tumors revealed by whole exome and targeted gene sequencing. Cancer 123, 3261–3268 (2017)
Li, Y.F., Arnold, R.J., Radivojac, P., Tang, H.: Protein identification problem from a Bayesian point of view. Stat. Interface 5(1), 21 (2012)
Liepe, J., Marino, F., Sidney, J., Jeko, A., Bunting, D.E., Sette, A., Kloetzel, P.M., Stumpf, M.P., Heck, A.J., Mishto, M.: A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 354(6310), 354–358 (2016)
Linnemann, C., Van Buuren, M.M., Bies, L., Verdegaal, E.M., Schotte, R., Calis, J.J., Behjati, S., Velds, A., Hilkmann, H., El Atmioui, D., et al.: High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 21(1), 81–85 (2015)
Ma, B.: Novor: real-time peptide de novo sequencing software. J. Am. Soc. Mass Spectrom. 26(11), 1885–1894 (2015)
Ma, B., Zhang, K., Hendrie, C., Liang, C., Li, M., Doherty-Kirby, A., Lajoie, G.: PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17(20), 2337–2342 (2003)
Neefjes, J., Jongsma, M.L., Paul, P., Bakke, O.: Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 11(12), 823–836 (2011)
Nik-Zainal, S., Davies, H., Staaf, J., Ramakrishna, M., Glodzik, D., Zou, X., Martincorena, I., Alexandrov, L.B., Martin, S., Wedge, D.C., et al.: Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534(7605), 47–54 (2016)
Schumacher, T.N., Schreiber, R.D.: Neoantigens in cancer immunotherapy. Science 348(6230), 69–74 (2015)
Tanner, S., Shu, H., Frank, A., Wang, L.-C., Zandi, E., Mumby, M., Pevzner, P.A., Bafna, V.: InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 77(14), 4626–4639 (2005)
Tran, N.H., Levine, Z., Xin, L., Shan, B., Li, M.: Protein identification with deep learning: from abc to xyz. arXiv preprint (2017). arXiv:1710.02765
Tran, N.H., Zhang, X., Xin, L., Shan, B., Li, M.: De novo peptide sequencing by deep learning. Proc. Natl. Acad. Sci. 114(31), 8247–8252 (2017)
Vacic, V., Iakoucheva, L.M., Radivojac, P.: Two sample logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics 22(12), 1536–1537 (2006)
Vita, R., Overton, J.A., Greenbaum, J.A., Ponomarenko, J., Clark, J.D., Cantrell, J.R., Wheeler, D.K., Gabbard, J.L., Hix, D., Sette, A., et al.: The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 43(D1), D405–D412 (2014)
Vizcaíno, J.A., Deutsch, E.W., Wang, R., Csordas, A., Reisinger, F., Rios, D., Dianes, J.A., Sun, Z., Farrah, T., Bandeira, N., et al.: Proteomexchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32(3), 223–226 (2014)
Wan, Y., Yang, A., Chen, T.: PepHMM: a hidden markov model based scoring function for mass spectrometry database search. Anal. Chem. 78(2), 432–437 (2006)
Xie, C., Yeo, Z.X., Wong, M., Piper, J., Long, T., Kirkness, E.F., Biggs, W.H., Bloom, K., Spellman, S., Vierra-Green, C., et al.: Fast and accurate HLA typing from short-read next-generation sequence data with xHLA. Proc. Natl. Acad. Sci. 201707945 (2017)
Yarchoan, M., Johnson III, B.A., Lutz, E.R., Laheru, D.A., Jaffee, E.M.: Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 17(4), 209–222 (2017)
Zhang, L., Udaka, K., Mamitsuka, H., Zhu, S.: Toward more accurate pan-specific MHC-peptide binding prediction: a review of current methods and tools. Briefings Bioinf. 13(3), 350–364 (2011)
Zhao, Y., Tang, H., Ye, Y.: RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 28(1), 125–126 (2011)
Acknowledgements
This work was supported by the NIH grant 1R01AI108888 and the Indiana University Precision Health Initiative (IU-PHI).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this paper

Cite this paper
Li, S., DeCourcy, A., Tang, H. (2018). Constrained De Novo Sequencing of neo-Epitope Peptides Using Tandem Mass Spectrometry. In: Raphael, B. (eds) Research in Computational Molecular Biology. RECOMB 2018. Lecture Notes in Computer Science(), vol 10812. Springer, Cham. https://doi.org/10.1007/978-3-319-89929-9_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-89929-9_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-89928-2
Online ISBN: 978-3-319-89929-9
eBook Packages: Computer ScienceComputer Science (R0)