
Abstract
In his medical treatise De Medicina dating back two millenniums, Aulus Cornelius Celsus refers to the signs of inflammation as “redness and swelling with heat and pain.” Inflammation is a reaction of the body to foreign stimuli and has presumably evolved to restore homeostasis in response to infections, tissue damage, or toxins. In conditions when the basal homeostatic state cannot be restored, persistent inflammatory signals usually lead to a maladaptive state as we observe in diet-induced obesity. Inflammatory process typically involves an inducing factor, such as bacterial infection, which is recognized by sensory molecules, e.g., as toll-like receptors, which then leads to secretion of inflammatory mediators including cytokines, chemokines, and a subclass of eicosanoids called prostaglandins. Final stage of the acute inflammatory cycle involves a resolution stage aimed to return to the pre-inflammatory homeostatic boundaries (Serhan et al. 2007), and failure to do so might lead to chronic inflammation. As opposed to acute inflammatory response commonly observed, following tissue injury or bacterial or viral infections, metabolic syndrome, and obesity per se manifest in the form of a chronic low-grade inflammatory state also called para−/meta-inflammation (Medzhitov 2008; Hotamisligil 2017). The inducing factor(s) of this chronic low-grade inflammation is still not entirely clear; however common mediators are involved in the acute and chronic inflammation. In contrast to many other chronic inflammatory conditions where the response is localized to the site of action of the inducing factor (e.g., site of infection), obesity-associated inflammation is manifested at a systemic level incorporating the peripheral as well as central tissues. Low-grade chronic inflammation has been demonstrated in a variety of metabolic tissues, such as the white adipose (Xu et al. 2003), liver (Cai et al. 2005), skeletal muscle (8, 9), and pancreas (Ehses et al. 2008; Donath et al. 2010), and appears to play a causative role in metabolic dysregulations including insulin resistance during obesity. For example, treatment with an anti-inflammatory agent amlexanox, an inhibitor of the NF-κB kinases IKKε and TBK-1, reduces obesity in rodents and improves glucose homeostasis in mice (Reilly et al. 2013) and a subset of diabetics (Oral et al. 2017).
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Inflammation
In his medical treatise De Medicina dating back two millenniums, Aulus Cornelius Celsus refers to the signs of inflammation as “redness and swelling with heat and pain.” Inflammation is a reaction of the body to foreign stimuli and has presumably evolved to restore homeostasis in response to infections, tissue damage, or toxins. In conditions when the basal homeostatic state cannot be restored, persistent inflammatory signals usually lead to a maladaptive state as we observe in diet-induced obesity. Inflammatory process typically involves an inducing factor, such as bacterial infection, which is recognized by sensory molecules, e.g., as toll-like receptors, which then leads to secretion of inflammatory mediators including cytokines, chemokines, and a subclass of eicosanoids called prostaglandins. Final stage of the acute inflammatory cycle involves a resolution stage aimed to return to the pre-inflammatory homeostatic boundaries (Serhan et al. 2007), and failure to do so might lead to chronic inflammation. As opposed to acute inflammatory response commonly observed, following tissue injury or bacterial or viral infections, metabolic syndrome, and obesity per se manifest in the form of a chronic low-grade inflammatory state also called para−/meta-inflammation (Medzhitov 2008; Hotamisligil 2017). The inducing factor(s) of this chronic low-grade inflammation is still not entirely clear; however common mediators are involved in the acute and chronic inflammation. In contrast to many other chronic inflammatory conditions where the response is localized to the site of action of the inducing factor (e.g., site of infection), obesity-associated inflammation is manifested at a systemic level incorporating the peripheral as well as central tissues. Low-grade chronic inflammation has been demonstrated in a variety of metabolic tissues, such as the white adipose (Xu et al. 2003), liver (Cai et al. 2005), skeletal muscle (8, 9), and pancreas (Ehses et al. 2008; Donath et al. 2010), and appears to play a causative role in metabolic dysregulations including insulin resistance during obesity. For example, treatment with an anti-inflammatory agent amlexanox, an inhibitor of the NF-κB kinases IKKε and TBK-1, reduces obesity in rodents and improves glucose homeostasis in mice (Reilly et al. 2013) and a subset of diabetics (Oral et al. 2017).
Inflammation in the central nervous system (CNS) also parallels the excess calorie intake and obesity, and this chapter will focus on the key mediators, signaling components, and the various cell types involved in the inflammatory process in the CNS. We will describe genetic and pharmacological approaches utilized to target the inflammatory mediators or signaling pathways involved. A significant number of studies now suggest that obesity and the associated inflammatory state form a positive feedback loop where induction of inflammation exacerbates various abnormalities observed in obesity such as the impaired glucose metabolism (Okin and Medzhitov 2016). We further address the central inflammation in regard to neurons and glial cell populations with specific emphasis on the hypothalamus and regulation of energy homeostasis.
1.1 Hallmarks of Inflammation
Classical activators of inflammation, such as lipopolysaccharide (LPS or endotoxin), tumor necrosis factor alpha (TNFα), and interleukin-1 (IL-1), induce a series of physiological outcomes involving fever and anorexia through their central action. Fever is mediated through activation of cyclooxygenase expression, which is targeted – either directly or indirectly – by most commonly used antipyretic (fever reducing) agents including common nonsteroidal anti-inflammatory drugs (NSAID), such as salicylic acid (aspirin) and ibuprofen. Some inflammatory signals derived from the periphery act through their receptors in the brain; however their action is further amplified through local production of other inflammatory mediators. For example, peripheral administration of LPS (endotoxin) triggers hypothalamic expression of pro-inflammatory cytokines including TNFα, IL-6, and IL-1 (Hillhouse and Mosley 1993; Layé et al. 1994; Breder et al. 1994; Wong et al. 1997), and the LPS-induced anorexia is presumably an integrated response to all these factors, which activate an overlapping set of signaling cascades: These include the stress-activated kinase pathways including, but not limited to, c-Jun N-terminal kinase (JNK) and p38 kinase, canonical and noncanonical nuclear factor kappa B (NF-κB) pathways, and protein kinase R (PKR). Some of these pathways are also utilized by metabolic hormones and important for the regulation of energy balance. For example, sickness and leptin-induced anorexia are mediated in part by acute hypothalamic inflammation and require central NF-κB signaling (Jang et al. 2010). Although LPS also results in a systemic increase in TNFα concentrations, LPS-induced upregulation of central TNFα seems to follow a different mechanism and precedes the rise in circulating TNFα concentration (Sacoccio et al. 1998). Furthermore, there appears to be a functional redundancy on certain phenotypes induced by the central action of the pro-inflammatory cytokines. For example, most effects of systemic LPS administration, including anorexia, hypoglycemia, and activation of the hypothalamus-pituitary-adrenal axis, were intact in IL-1β knockout mice. However, the anorexic response to central administration of LPS still required central IL-1β production (Yao et al. 1999).
A classical hallmark of inflammation is upregulation of the hypothalamic-pituitary-adrenal (HPA) axis. Upon inflammatory challenge, plasma ACTH concentrations rise, which act on the adrenal glands and result in systemic elevated glucocorticoid levels. Glucocorticoids are anti-inflammatory agents and therefore act as a protective negative feedback loop to block persistent HPA activation (Besedovsky and del Rey 1996). Classical pro-inflammatory cytokines such as TNFα, IL-1, and IL-6 as well as LPS stimulate transcription of hypothalamic corticotropin-releasing hormone/factor (CRH/F), which acts on pituitary to induce ACTH release into the circulation. There is a certain degree of overlap in the physiology as well as the mechanism of action of leptin and the pro-inflammatory cytokines. For example, central IL-1 infusion into the VMH results in acute decreases in food intake and body weight in rats (Kent et al. 1994) and mice (Kent et al. 1996), and LPS administration upregulates the immediate early gene c-fos, which has been widely used as a marker for neuronal activation (Sagar and Sharp 1993), in the hypothalamus, the amygdala, and the nucleus tractus solitarius (NTS) (Elmquist et al. 1993). LPS, like leptin, induces the hypothalamic STAT3 phosphorylation (Hosoi et al. 2004), although this is a secondary response to LPS-induced IL-6 upregulation. In addition, LPS-induced anorexia and fever depend in part on circulating leptin (Sachot et al. 2004). Rats lacking leptin receptor signaling are defective in activating the HPA axis upon LPS exposure (Steiner et al. 2004). However, central action of leptin counteracts the fasting-induced activation of HPA axis, such that restoration of fasting-induced decrease in plasma leptin concentrations back to the fed levels blocks the rise in plasma corticosterone and ACTΗ levels observed upon food deprivation (Ahima et al. 1996).
As discussed below, NF-κB activation is a common hallmark of diet-induced obesity in the hypothalamus and peripheral metabolic tissues (Arkan et al. 2005; Wunderlich et al. 2008; Chiang et al. 2009). The canonical (classical) NF-κB signaling is comprised of a family of five transcription factors (p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1), and p100/52 (NF-κB2)), which are under normal conditions sequestered in the cytosol by inhibitor of κB (IκB). Phosphorylation of IκB by upstream IκB kinases (IKK) results in its proteasome-mediated degradation. Free NF-κB proteins then translocate to the nucleus to activate the transcription of target genes. Besides classical IKKs (IKKα, IKKβ), there are IKK-related kinases (IKKε, TBK1) that play a critical role in the obesity-induced inflammation. In the adipose tissue and liver of diet-induced obese mice, the activity of these noncanonical (alternate) IKKs increases (Chiang et al. 2009). IKKε knockout mice are protected from diet-induced inflammation and obesity mainly due to elevated energy expenditure (Chiang et al. 2009). Furthermore, treatment of diet-induced knockout mice with an IKKε/TBK1 inhibitor reverses diet-induced obesity and associated complications including glucose metabolism (Reilly et al. 2013). Obesity activates IKKε in the hypothalamus as well (Weissmann et al. 2014). Pharmacological or genetic inhibition of hypothalamic IKKε signaling in obese rodents leads to decreased NF-κB activity and ameliorates central insulin and leptin resistance (Weissmann et al. 2014).
IKKβ/NF-κB pathway plays a key role in neuronal stem cells (NSCs). For example, CNTF, a key factor involved in the maintenance of NSCs (Shimazaki et al. 2001), can suppress food intake and body weight even in leptin-resistant mice through a mechanism that involves neurogenesis in the hypothalamus (Kokoeva et al. 2005). Obese rodents have decreased numbers of hypothalamic NSCs, and inhibition of IKKβ/NF-κB increases their survival (Li et al. 2012). NSC-specific activation of IKKβ/NF-κB in adult mice results in decreased NSC number and increased food intake and weight gain (Li et al. 2012). One of the hallmarks of obesity is induction of a pathophysiology of aging at earlier ages in peripheral as well as central tissues. Hypothalamic markers of inflammation increase as mice get older. Most notably, ablating neuronal IKKβ expression or blocking IKKβ activity specifically in the mediobasal hypothalamus results in extended life-span accompanied by improved cognitive and motor functions, whereas activation of NF-κB induces an opposite phenotype and results in mice with shorter life-span (Zhang et al. 2013a). Most of these phenotypes could be mimicked by microglia-specific IKKβ inhibition as well, suggesting the importance of non-neuronal cells in hypothalamic inflammation and associated metabolic abnormalities.
Inflammatory ligands in the form of foreign pathogens or endogenous signals are typically recognized by pattern recognition receptors (toll-like receptors, TLRs) expressed on the cell surface or intracellular membranes such as ER. The ligands recognized by different TLRs have been characterized; for example, TLR4 acts as the receptor for LPS. TLR activation classically leads to NF-κB activation and induces the expression of interleukin-1 (IL-1) family of cytokines (IL-1 and IL-18), which are processed from their pro-interleukin precursors to mature forms by active caspase 1. The canonical pathway of caspase 1 activation involves inflammasomes (Martinon et al. 2002), large protein complexes assembled by innate immune receptors called nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs), absent in melanoma 2 (AIM2) and pyrin (Stutz et al. 2009; Broz and Dixit 2016). Glial cells in the CNS including microglia and astrocytes express TLRs (Lehnardt 2010), and inflammasome components are constitutively expressed in the brain (Yin et al. 2009), where they act as major regulators of central inflammation (Song et al. 2017). For example, the microglial IL-1β production observed in Alzheimer’s disease is mediated by amyloid beta recognized by the inflammasome NALP3 (also called NLRP3), which leads to caspase 1 activation and the processing of pro-IL-1β to active IL-1β (Halle et al. 2008). Inflammasome components are also detected in neurons (Kummer et al. 2007) and seem to play essential roles during conditions such as brain injury (de Rivero Vaccari et al. 2008), headache (Silverman et al. 2009; Karatas et al. 2013), and response to certain viral infections (Ramos et al. 2012). The overall clinical importance of inflammasomes extends beyond infections, and its dysregulation plays a key role in peripheral as well as central inflammation associated with metabolic syndrome including obesity. For example, NLRP3 ablation protected mice from age-associated central inflammation and astrogliosis, improved glucose homeostasis, attenuated bone loss and thymus dysfunction (Youm et al. 2013), and enhanced the obesity-associated defects in insulin signaling (Vandanmagsar et al. 2011). We next discuss in more detail the inflammation of the nervous system or “neuroinflammation.”
1.2 Central Nervous System (CNS) Components of the Inflammatory Process
1.2.1 Neurons Are Not Alone in the CNS
In order to understand the CNS inflammation, it is important first to get a broad understanding of the cellular architecture of the CNS as term neuroinflammation extends beyond the neurons. Majority of the cells in the CNS are non-neuronal cells called glia. There are three glial cell population: astrocytes, microglia, and oligodendrocytes. The main function of these cells is to maintain the homeostasis in the nervous system. Astrocytes send projections (called astrocytic end feet) to meet the blood capillaries and have traditionally been viewed as a gate between the circulation and the rest of the CNS. For example, the classical view of CNS glucose utilization has been the import of glucose from the bloodstream by the astrocytes to be first oxidized to lactic acid, which is then taken up by neurons and used as the energy source. This indirect route of glucose utilization, called the astrocyte-neuron lactate shuttle, was reported to be key to the central regulation of glucose metabolism (Lam et al. 2005). Likewise, astrocytes can oxidize fatty acids to ketones, which could then be used by neurons as energy source (Le Foll and Levin 2016). Although recent findings suggest that neurons might play a more direct role in nutrient utilization (Lundgaard et al. 2015), astrocytes play an indispensible role in the central regulation of energy homeostasis: They express the receptors for key metabolic factors including leptin, insulin, and ghrelin (Kim et al. 2014; García-Cáceres et al. 2016; Frago and Chowen 2017). Astrocytic insulin signaling is required for brain glucose uptake and systemic glucose homeostasis (García-Cáceres et al. 2016). By regulating extracellular adenosine concentration, astrocytes regulate AgRP neuronal activity and actively participate in the regulation of feeding (Yang et al. 2015). Leptin induces astrocyte proliferation in the hypothalamus (Rottkamp et al. 2015), and removal of leptin receptor from astrocytes attenuates the response to leptin-induced anorexia while potentiating ghrelin-induced food intake (Kim et al. 2014).
Microglia are resident macrophages of the CNS derived from erythromyeloid precursors in the yolk sac (Kierdorf et al. 2013). Their function includes maintenance as well as the plasticity of synaptic circuitry, synaptic transmission, neuronal surveillance, and neurogenesis (Walton et al. 2006; Paolicelli et al. 2011; Pascual et al. 2012; Wake et al. 2013). Rodents are estimated to have between 3 and 4 million microglia. Their distribution is not uniform throughout the brain, with more microglia detected in the hippocampus, olfactory telencephalon, basal ganglia, and substantia nigra than regions such as the cerebellum and brain stem (Lawson et al. 1990). Contrary to differentiated adult neurons, microglia can proliferate. Earlier studies have suggested that a source of resident microglia was monocytes that are recruited from the periphery, which in turn could differentiate into resident microglia (Lawson et al. 1992). However, recent studies show that postnatal hematopoiesis makes a marginal contribution to steady-state resident microglial cell population (Ginhoux et al. 2010). Microglia development and survival depend on a receptor tyrosine kinase called colony-stimulating factor 1 receptor (CSF1R) (Ginhoux et al. 2010; Erblich et al. 2011) whose inhibition leads to complete microglial depletion in adult mice. However, in adult mice, nestin-positive latent microglial progenitors are capable of renewing the CNS microglia pool (Elmore et al. 2014). The third glial population is comprised of the oligodendrocytes (ODs). These cells mainly function to provide electrical insulation (myelination) to neurons. Besides this fundamental role, ODs produce a variety of neurotrophic and growth factors such as BDNF and IGF-1 to promote neuronal survival (Wilkins et al. 2001, 2003; Du and Dreyfus 2002). Another glial population is NG2 glia, which are also called polydendrocytes or oligodendrocyte precursors (Nishiyama et al. 2009). These cells are characterized by their expression of a single membrane-spanning chondroitin sulfate proteoglycan, called NG2, and are able to differentiate into myelinating oligodendrocytes and engage in tissue repair (Hughes et al. 2013). Accordingly, NG2 glia gain a hypertrophic characteristic upon demyelinating insults (Keirstead et al. 1998; Di Bello et al. 1999; Levine and Reynolds 1999). In the hypothalamus, particularly in the circumventricular organ the median eminence (ME), NG2 glia play a crucial role in sensing circulating leptin (Djogo et al. 2016). Leptin receptor-positive neurons have their dendritic projections in the ME in close proximity of NG2 glia. Following depletion of NG2 glia, the ARC neurons lose their response to leptin, which in turn triggers obesity (Djogo et al. 2016). This finding has more imminent consequences for humans as NG2 depletion upon cranial X-ray irradiation was also proposed to contribute to weight gain in humans (Djogo et al. 2016).
The resident astrocytes and microglia are the innate immune cells of the CNS, and there exists a dynamic interaction between them including during central inflammation. Activated microglia can induce a subpopulation of astrocytes by secreting inflammatory mediators including IL1α, TNF, and C1q, which in turn blunts the neuroprotective capacity of the astrocytes leading to neuronal death (Liddelow et al. 2017). Contrary to the periphery, central innate immune response cannot initiate adaptive immunity: In the peripheral innate immune cells (e.g., dendritic cells), foreign antigens are typically presented by MHC class II molecules on the cell surface to T cells to trigger an adaptive immune response. However, this process is not adapted by the glia. This is consistent with the anti-inflammatory environment of the brain parenchyma as well as the physical limitations induced by the blood-brain barrier (BBB) that would block migration/communication of innate and adaptive immune cell components. However, it is worth to mention that the notion that brains are devoid of adaptive immunity is not absolute. For example, dendritic cell-like properties have been reported in meninges and choroid plexus in healthy mice (Anandasabapathy et al. 2011), and a rare expression pattern of MHCs exists in neurons (Neumann et al. 1995). Surprisingly, recent evidence suggests that inflammatory signals regulate peripheral adaptive immunity through pathways including their central actions. For example, hypothalamic responses to TNFα result in increased sympathetic tone to white adipose tissue. As a result, increased lipolysis produces long-chain fatty acids, which mediate the accumulation of the cells of adaptive immunity, the lymphocytes, in spleen and adipose tissue (Kim et al. 2015).
1.2.2 Hypothalamic Inflammation and Energy Homeostasis
As explained above, obesity manifests itself as a chronic low-grade inflammatory state. Increase in the adipose tissue expression and plasma levels of the pro-inflammatory cytokine TNFα in obesity was first reported in 1993 in rodents (Hotamisligil et al. 1993). TNFα inhibits insulin signaling by inducing an inhibitory phosphorylation on insulin receptor substrate 1 (IRS1) (Hotamisligil et al. 1994, 1996), an insulin receptor-interacting protein. Accordingly, TNFα knockout mice are protected from obesity-induced insulin resistance (Uysal et al. 1997). Likewise, macrophage-specific ablation of c-Jun N-terminal kinase (JNK) blocks pro-inflammatory macrophage polarization, decreases their adipose tissue infiltration, and confers protection from obesity-induced insulin resistance (Han et al. 2013). Markers of inflammation in obesity are observed in several peripheral tissues including the liver, adipose tissue, and muscle (Schenk et al. 2008). Hypothalamus is no exception; high-fat feeding results in increased activation of JNK and NF-κB pathways in the hypothalamus (De Souza et al. 2005); results in elevated expression of pro-inflammatory mediators in the hypothalamus, including IL-1β and TNFα; and attenuates insulin, leptin, and ghrelin (Naznin et al. 2015) signaling. Hypothalamic PTP1B expression, a negative regulator of insulin and leptin receptor signaling, increases in diet-induced obesity; and this response can be mimicked by TNFα administration to lean healthy mice (Bence et al. 2006; Zabolotny et al. 2008). Central injection of neutralizing antibodies targeted against TLR4 or TNFα in obese rats reduces markers of hypothalamic inflammation and improves hepatic steatosis and gluconeogenesis through parasympathetic output to the liver (Milanski et al. 2012). Furthermore, reducing hypothalamic inflammation by neuronal-, glial-, or hypothalamus-specific deletion of IKKβ in mice confers protection from diet-induced obesity (Zhang et al. 2008; Valdearcos et al. 2017).
The hypothalamic inflammation observed in obesity is temporally different than peripheral inflammation. While peripheral inflammation is thought to develop secondary to diet-induced adiposity, rapid changes in the hypothalamic inflammatory signaling cascades could be observed within only 1 day following high-fat diet consumption. For example, rodents display elevated markers of inflammation (Thaler et al. 2012) and insulin resistance in the hypothalamus-brain circuitry (Ono et al. 2008) within 24 h of exposure to high-fat diet, much earlier than accumulation of adiposity. HFD-induced proliferation and activation of glia in the hypothalamus seem to be important for the accompanying peripheral inflammation such that blocking central cell proliferation attenuates not only the central but also the peripheral inflammation (André et al. 2017). The central inflammation observed upon high-fat diet consumption, at least in part, depends on the direct effect of saturated fatty acids on the hypothalamus rather than the total calories consumed, and central infusion of palmitate, a saturated fatty acid, to lean rats mimics the hypothalamic insulin resistance and IKKβ activation observed in rats exposed to high-fat diet (Posey et al. 2009). Hypothalamic inflammation could be stimulated by administration of saturated fatty acids by intragastric gavage for 3 days; however, the same treatment does not induce systemic inflammation. In parallel with their central effects in vivo, saturated fatty acids induce endoplasmic reticulum (ER) stress in neuronal cultures (Mayer and Belsham 2010; Choi et al. 2010). However, their pro-inflammatory effects were attenuated in cultured hypothalamic neurons, suggesting a role for non-neuronal cells in the fatty acid-induced central inflammation (Choi et al. 2010). Consumption of high-calorie diet results in accumulation of saturated fatty acids in the hypothalamus (but not the cerebral cortex) and activates the microglia in the ARC (Valdearcos et al. 2014). Diet-induced hypothalamic gliosis, activation of the glial population, is also reported in humans (Thaler et al. 2012). At least in high-fat diet-fed rodents, some of the hypothalamic gliosis is also attributed to monocyte-derived non-microglial myeloid cells recruited by resident microglia (Berkseth et al. 2014). Furthermore, the detected inflammation was restricted to hypothalamic microglia, but not the astrocytes, although both cell population accumulate in the mediobasal hypothalamus following high-fat diet consumption. Accordingly, saturated fatty acids induce secretion of inflammatory mediators, including TNFα, IL-6, and monocyte chemoattractant protein-1 (also called CCL2), from primary microglia but not primary astrocytes (Valdearcos et al. 2014), and hypothalamic infiltration of astrocytes following high-fat diet does not seem to blunt leptin responsiveness (Balland and Cowley 2017). Astrocytes also express some components of the inflammasome such as NLRP2 (Minkiewicz et al. 2013) but not others (e.g., NLRP3) (Gustin et al. 2015), and microglia appear to be the main inflammatory glial population involved in CNS (Gustin et al. 2015). The hypothalamic microglial inflammation is accompanied by elevated markers of neuronal injury and decreased cognitive function, and this was reported in both rodents and humans (Thaler et al. 2012; Valdearcos et al. 2014; Puig et al. 2015). Saturated fatty acids induce hypothalamic inflammation mainly acting through TLR4 (Milanski et al. 2009), and in line with above findings, activation of microglial TLR4 by LPS induces neurodegeneration (Lehnardt et al. 2003). Upon microglial depletion, the response of the CNS to LPS challenge is compromised for certain pro-inflammatory markers including IL-1β and TNFα (Elmore et al. 2014). Microglia also express TLR2, which was proposed to acutely mediate the sickness-induced anorexia and POMC activation (Jin et al. 2016), at least in part through microglia-derived TNFα release (Yi et al. 2017). Although the inflammatory pathways might contribute to the regulation of energy balance by anorectic hormones including leptin (Jang et al. 2010), these effects seem to be acute, and blocking TNFα signaling in the mediobasal hypothalamus results in attenuation of diet-induced weight gain (Yi et al. 2017). Depleting microglia by CSF1R antagonism augmented leptin signaling and suppressed food intake, suggesting a negative role of microglial activation on central leptin action (Valdearcos et al. 2014). When mice are fed with HFD supplemented with the CSF1R antagonist, the food intake and weight gain on the mice are attenuated (Valdearcos et al. 2017). Likewise, blocking microglial activation by specific depletion of IKKβ from microglia confers resistance to high-fat diet-induced weight gain in part by reducing food intake (Valdearcos et al. 2017). When hypothalamic gliosis is induced by specifically activating the microglial NF-κB pathway, the transgenic mice had increased food intake and weight gain and reduced energy expenditure (Valdearcos et al. 2017). Considering that microglial depletion in adult mice does not result in overt behavioral phenotypes or cognitive abnormalities (Elmore et al. 2014), CSF1R antagonism might be a promising anti-obesity mechanism. Astrocyte activation is also observed upon acute HFD challenge. Surprisingly, blocking HFD-induced astrocyte activation by astrocyte-specific inhibition of NF-κB results in increased food intake, although this response is very acute and does not persist upon continuous HFD feeding (Buckman et al. 2015). In obesity as well as aging, elevated TGFβ production by the astrocytes impairs glucose homeostasis by acting on POMC neurons and inducing an inflammatory response (Yan et al. 2014). Diet-induced hypothalamic inflammation and gliosis are not permanent in rodents and could largely be reversed upon exercise (Ropelle et al. 2010; Yi et al. 2012a) or weight loss (Berkseth et al. 2014). In humans, at least one study showed that hypothalamic inflammation in obese human MBH was not reversed following gastric bypass-induced weight loss and elevated insulin sensitivity (Kreutzer et al. 2017). More research is certainly warranted to dissect the potential differences between humans and rodents in regard to the kinetics as well as the reversible nature of diet-induced hypothalamic inflammation.
Neurons are rather resistant to saturated fatty acid-induced inflammation compared to glia (Choi et al. 2010). There is relatively higher expression of IKKβ and IκBα in the hypothalamus compared to peripheral tissues including the liver, skeletal muscle, adipose tissue, and kidney (Zhang et al. 2008). In diet-induced obese mice, IKKβ-NF-κB signaling gets activated in the neurons located in the mediobasal hypothalamus (MBH) (Zhang et al. 2008). Neuronal activation of IKKβ in MBH neurons stimulates food intake and weight gain, while a dominant negative IKKβ reverses both parameters (Zhang et al. 2008). AgRP-specific IKKβ knockout mice also eat less and gain less weight on HFD (Zhang et al. 2008), and activating IKKβ specifically in AgRP neurons impairs glucose homeostasis without changing body weight (Tsaousidou et al. 2014). Activation of the inflammatory signals and ER stress are mechanistically coupled at least through NF-κB signaling (Zhang et al. 2008). Active IKKβ triggers ER stress, which in turn leads to further activation of IKKβ, resulting in a positive feedback loop to attenuate central insulin and leptin signaling leading to positive energy balance (Zhang et al. 2008). Besides overnutrition, inhibition of hypothalamic autophagy also leads to IKKβ activation and hypothalamic inflammation, increased food intake, and decreased energy expenditure (Meng and Cai 2011). Increased hypothalamic IKKβ-NFκB signaling decreases the number of hypothalamic neural stem cells, results in dysregulated neurogenesis (Li et al. 2012), inhibits gonadotropin-releasing hormone (GnRH) expression (Zhang et al. 2013a), and triggers hypertension (Purkayastha et al. 2011a). For example, increased MBH-specific IKKβ activation increases blood pressure, and POMC-specific IKKβ KO mice are protected from obesity-induced hypertension (Purkayastha et al. 2011a). Consequently, neuronal IKKβ KO mice exhibit extended life-span (Zhang et al. 2013a).
Prominent role of NF-κB signaling in the regulation of energy balance is further emphasized by studies utilizing glucocorticoids (GC). GCs are potent anti-inflammatory molecules, secreted from the adrenal glands typically in response to stress-induced activation of the HPA axis. GCs are steroids and act through the intracellular glucocorticoid receptor (GR). GR directly interacts with p65 (Rel A) (Ray and Prefontaine 1994) and inhibits NF-κB signaling by increasing the nuclear export rate of p65 (Nelson et al. 2003) and increasing IkB expression level (Deroo and Archer 2001). Targeting a GR agonist, dexamethasone (Dexa), to glucagon-like peptide-1 (GLP-1) expressing cells by using a GLP-1/Dexa conjugate results in decreased hypothalamic and systemic inflammation, improves glucose homeostasis, and decreases body weight in DIO mice (Quarta et al. 2017). GLP-1 receptor (GLP1R) is mostly expressed by neurons in human and rodent brain. It is also expressed by the immune cells in the circulation. Accordingly, the metabolic improvements observed upon GLP-1/Dexa treatment are mediated by both central and peripheral GLP1R-expressing cells (Quarta et al. 2017).
JNKs are typically activated by high-fat feeding (Prada et al. 2005), and genetic studies have indicated the ablation of peripheral JNK activity to be protective from diet-induced metabolic abnormalities. JNK1 global KO mice, but not JNK2 KO mice, are protected from diet-induced obesity (Hirosumi et al. 2002). Adipose tissue-specific ablation of JNK1 does not alter body weight or adiposity but confers significant protection from hepatic insulin resistance (Sabio et al. 2008). Similar protection from peripheral insulin resistance is observed upon deletion of JNK1 and JNK2 from macrophages (Han et al. 2013) or skeletal muscle (Sabio et al. 2010b). Although liver-specific JNK1 KO mice were reported to exhibit hepatic insulin resistance, glucose intolerance, and steatosis (Sabio et al. 2009), deletion of both JNK1 and JNK2 from hepatocytes significantly improves systemic glucose homeostasis and insulin signaling (Vernia et al. 2014).
In the CNS, JNKs are essential for proper brain development (Kuan et al. 1999; Chang et al. 2003; Amura et al. 2005). JNK2 and JNK3 deletion confers protection from various forms of neurodegeneration including cerebral ischemic hypoxia (Kuan et al. 2003; Hunot et al. 2004). In JNK1 KO mice, glucocorticoid-induced feeding was exacerbated (Unger et al. 2010); however the animals exhibit increased central insulin sensitivity (Unger et al. 2010). Accordingly, neuron-specific ablation of JNK1 results in increased central as well as peripheral insulin sensitivity, reduced hepatic steatosis, and decreased body weight in diet-induced obese mice, without altering central leptin sensitivity (Kleinridders et al. 2009; Sabio et al. 2010a; Belgardt et al. 2010). Most of these effects were due to decreased food intake and elevated HPT axis activity (Sabio et al. 2010a). Accordingly, disrupting HPT axis by blocking thyroxine production attenuated the protective effect of neuronal JNK1 depletion from diet-induced weight gain (Sabio et al. 2010a). Triiodothyronine treatment induced hypothalamic JNK1 activity, which in turn led to hepatic steatosis (Martínez-Sánchez et al. 2017). AgRP-specific JNK1 activation blunted leptin signaling specifically in AgRP neurons, increased AgRP neuronal firing, and consequently induced weight gain (Tsaousidou et al. 2014) without significant alterations in glucose homeostasis.
Role of JNK3 in feeding and body weight regulation is different than the other isoforms. HFD activated hypothalamic JNK3 phosphorylation, and embryonic deletion of JNK3 induces food intake, significantly exacerbates weight gain, increases adipose tissue inflammation, and impairs systemic glucose homeostasis specifically on HFD (Vernia et al. 2016). These phenotypes were largely mimicked when JNK3 was ablated from leptin receptor-positive cells or specifically from AgRP neurons (Vernia et al. 2016). However, POMC-specific JNK3 KO mice had normal feeding and glucose homeostasis (Vernia et al. 2016). Despite these findings based on genetic studies, central or peripheral administration of a JNK2/3-specific inhibitor suppressed food intake and resulted in robust weight loss (Gao et al. 2017).
TLR4 acts as the endogenous receptor for LPS and saturated fatty acids and plays a critical role in the lipid-induced insulin resistance in the periphery (Shi et al. 2006). In the CNS, TLR4 is expressed predominantly in microglia (Chakravarty and Herkenham 2005) and plays a critical role for fatty acid-induced hypothalamic inflammation (Milanski et al. 2009). Adult neuronal progenitor cells also express TLR4, and its absence promotes neuronal differentiation (Rolls et al. 2007). Hypothalamic TLR4 signaling has also been implicated in the etiology of metabolic syndrome. HFD-induced obesity triggers neuronal expression of a chemokine, CX3CL1, in the hypothalamus and mediates the recruitment of peripheral monocytes and induction of hypothalamic inflammation (Morari et al. 2014). TLR4 was proposed to also interact with resistin, an adipokine that negatively regulates insulin signaling (Benomar et al. 2013), such that the central effects of resisting are blocked in TLR4 knockout mice (Benomar et al. 2016). Hypothalamic TLR4 also appears to have protective effects such that TLR4 blocks excess apoptotic neuronal death upon HFD feeding (Moraes et al. 2009). Acting in the paraventricular nucleus of the hypothalamus, TLR4 signaling mediates the obesity-induced increase hypertension (Dange et al. 2015; Masson et al. 2015). MyD88 knockout mice are resistant to LPS- or IL-1β-induced anorexia but not weight loss (Ogimoto et al. 2006; Yamawaki et al. 2010). Neuron-specific ablation of MyD88 blocked high-fat diet-induced ARC IKKβ activation (but not JNK1 activation) and conferred partial protection from fatty acid-induced leptin resistance and weight gain (Kleinridders et al. 2009).
Diet-induced obesity and associated inflammation have been shown to alter the structure and permeability of the BBB (Mauro et al. 2014; Varatharaj and Galea 2017). Certain regions of the CNS involved in the regulation of energy homeostasis lie outside the BBB; these include, among other regions, the ME and area postrema (Maolood and Meister 2009; Riediger 2012). The barriers between the CNS-resident cells and circulation have a fundamental role in the regulation of energy homeostasis during normal physiology as well as pathophysiological conditions including inflammation. Permeability of BBB is not rigid especially at the blood-hypothalamus barrier. A specialized and modified ependymal microglial population called tanycytes line the ventricles and form a barrier between the cerebrospinal fluid and circulation (Langlet et al. 2013b). Tanycytes are critically important in the sensing of circulating factors, such as leptin by hypothalamic neurons (Balland et al. 2014), while actively participating in neurogenesis (Lee et al. 2012). During fasting, decreased blood glucose concentration triggers the release of a permeability factor (vascular endothelial growth factor-A, VEGF-A) from tanycytes (Langlet et al. 2013a). Tanycytes are glucose-sensitive cells; their stimulation by glucose evokes ATP-mediated Ca+2 responses, including ATP release (Frayling et al. 2011). VEGF-A results in increased access of the ARC to circulating factors during fasting compared to other regions. However, astrocytes also express VEGF-A and contribute to pathologically increased BBB permeability and lymphocyte infiltration into the brain parenchyma during neuroinflammatory diseases (Argaw et al. 2012). IL-1β administration increases BBB permeability, probably by altering the tight junction profile of the endothelium, and its effect is amplified by other inflammatory mediators such as TNFα (Quagliarello et al. 1991; Nadeau and Rivest 1999; Blamire et al. 2000; Beard et al. 2014). Upon exposure to HFD, there is elevated degeneration in the endothelium of the hypothalamic BBB vasculature, which leads to infiltration and population of the ARC by IgG-type antibodies, and a pathological increase in blood vessel length and density in the ARC (Yi et al. 2012b, c). Notably, similar findings have been observed in the hypothalami of diabetic patients (Yi et al. 2012b).
While it is not going to be extensively discussed in this chapter, it is important to note that central inflammation is not restricted to the hypothalamus and extends to other brain regions. Diet-induced weight gain increases inflammatory markers in the cortex, amygdala, cerebellum, and brain stem and is overall associated with anxiety, depression, cognitive defects, and hypertension (Russo et al. 2011; Wu et al. 2012; Speretta et al. 2016; Guillemot-Legris et al. 2016; Carlin et al. 2016; Almeida-Suhett et al. 2017; Spencer et al. 2017). Hippocampus is another site where neuroinflammation is associated with high-fat diet, where increased TLR4 expression is detected (Dutheil et al. 2016). Hippocampal inflammation acts as a causative factor for high-fat diet-induced anxiety such that blocking the activation of inflammasome attenuates the anxious phenotype in rodents. Obesity and inflammatory cytokines are associated with a decline in cognitive functions (Yehuda et al. 2005; Gunstad et al. 2006; Nguyen et al. 2014) and impaired neurogenesis in the hippocampus (Lindqvist et al. 2006; Park et al. 2010; Chesnokova et al. 2016). The observed cognitive decline appears to require microglial activation associated with decreased BDNF levels (Pistell et al. 2010). Anti-inflammatory manipulations including pharmacological interventions or exercise increase cognitive function in rodents and humans (Jeon et al. 2012; Kang et al. 2016; Veronese et al. 2017; Pérez-Domínguez et al. 2017; Wang et al. 2017). For example, treadmill exercise decreases hippocampal microgliosis in obese mice. Furthermore, blocking hippocampal IL-1β signaling reverses the defects in synaptic plasticity and cognitive decline (Erion et al. 2014).
Genetic and pharmacological studies targeting different receptors or intracellular signaling cascades of the inflammatory molecules have revealed that, as opposed to their acute effects, inflammatory molecules in the CNS contribute to a positive energy balance, and their inhibition contributes to weight loss and improves systemic glucose metabolism. Diet-induced obesity and aging display similar signs of neuroinflammation, neurodegeneration, and cognitive decline, some of which could be blocked or in some cases reversed by noninvasive interventions in lifestyle including a healthy diet and exercise. However it remains a reality that obesity has reached epidemic proportions throughout the world, which increases the demand on pharmacological interventions to combat obesity and associated metabolic abnormalities. Studies obtained in rodents and in part in humans collectively suggest that targeting central inflammation might present promising approaches.
2 Endoplasmic Reticulum Stress and Unfolded Protein Response
Endoplasmic reticulum (ER) is a multifaceted membranous structure extending from the outer nuclear membrane to the cytoplasm and is involved in several biological processes including protein folding, secretion, lipid biosynthesis, calcium storage, and posttranslational modifications such as glycosylation and lipidation. About one third of the human proteome including the secreted, ER-resident, and membrane proteins go through ER. Depending on the tissue, representation of the secreted and membrane proteins in the total proteome greatly varies. For example, the majority of pancreatic proteome is composed of secretory proteins, which however constitute a small percentage of the skeletal muscle proteome. The intracellular expression profile of the chaperones has evolved to match the nature of the proteome of the corresponding tissue: While pancreas and liver, tissues with higher secreted and/or membrane protein pools, have higher ER chaperone expression, tissues such as skeletal muscle and skin, where the soluble proteins constitute the majority of the proteome, have higher expression of cytosolic chaperones. Accordingly, of more than 300 proteins in the human chaperone proteome (chaperome), 48 are ER-specific proteins (Brehme et al. 2014). Together with the ER-associated degradation (ERAD) pathways, the ER chaperones coordinate the folding capacity of the ER to regulate ER protein homeostasis. When the ER protein load exceeds the folding capacity, a condition called ER stress, a series of signaling pathways collectively called the unfolded protein response – UPR – is activated. The primary function of UPR is to restore the ER homeostasis by informing the cytosol and nucleus about the excess protein load to activate a series of protective mechanisms. Among these responses are global shutdown of the protein synthesis while specifically activating the translation of chaperone proteins, elevated membrane synthesis to increase the ER volume to remodel the secretory apparatus, and degradation of unfolded proteins through either ubiquitin-proteasome pathway (ER-associated degradation) or lysosomes (autophagy). While UPR might achieve in reinstating the ER homeostasis, persistent ER stress leads to apoptotic cell death.
We discuss below the signaling components of UPR and the results of genetic studies designed to study the role of individual UPR components in metabolism. Before that, we find it helpful to briefly summarize some of the chemical tools employed to study the biology of ER stress. Some of the commonly used chemical agents known to induce ER stress are tunicamycin, thapsigargin, and brefeldin A (BFA). Tunicamycin is an antifungal and antibiotic nucleoside, which acts as an inhibitor of N-acetylglucosamine transferases to block N-linked glycosylation (Takatsuki and Tamura 1971). Resulting defective protein folding triggers ER stress. Thapsigargin is an inhibitor of the ER Ca2+ importer called sarco−/endoplasmic reticulum Ca2+ ATPase (SERCA). Upon thapsigargin treatment, cytosolic Ca2+ concentration rises, while ER Ca2+ concentration gets depleted. There is a significant difference between the cytosolic and ER Ca2+ concentrations (300 μM in the ER vs. 5–50 μM in the cytosol) (Pozzan et al. 1994). Ca2+ is important for protein-protein interactions and ER chaperone function, thereby affecting protein synthesis, folding, and posttranslational modifications (Corbett et al. 1999). BFA is an antiviral lactone, which blocks ER-Golgi transport (Helms and Rothman 1992), resulting in accumulation of the polypeptides in the ER, and triggers ER stress. Another ER stress inducer, homocysteine, is a nonprotein cysteine analog and triggers the expression of ER stress-responsive genes, including GRP78, and decreases the expression of antioxidant enzymes, without inducing heat shock response (Outinen et al. 1998). There is also a set of other compounds, called chemical chaperones, which help stabilize the native conformation of proteins, which are now extensively used in research to alleviate ER stress (Welch and Brown 1996). 4-phenyl butyric acid (4PBA) and tauroursodeoxycholic acid (TUDCA) are two chemical chaperones commonly used in biomedical research. Both compounds are pleiotropic agents: While 4PBA also acts as a broad-spectrum HDAC inhibitor (Bora-Tatar et al. 2009), TUDCA is a bile acid that acts as a ligand for the G-protein-coupled receptor TGR5 (also called G-protein-coupled bile acid receptor 1), which is expressed in BAT, microglia, and other tissues (Kawamata et al. 2003; Yanguas-Casás et al. 2017), and acts as an anti-inflammatory molecule.
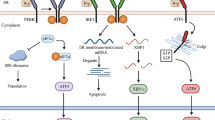
UPR is classically composed of three arms that are each initiated by distinct ER membrane proteins: PERK (protein kinase RNA (PKR)-like ER kinase) (Harding et al. 1999), IRE1 (inositol- requiring protein-1) (Morl et al. 1993; Cox et al. 1993), and ATF6 (activating transcription factor 6) (Haze et al. 1999). These proteins function as stress sensors that detect the ER protein load and induce adaptive responses aimed to bring the ER folding capacity back to homeostatic boundaries.
PERK’s lumenal domain is a stress sensor, whereas its cytosolic domain has enzymatic activity for its substrate eIF2α (eukaryotic initiation factor 2 subunit α). The other known target of PERK is a transcription factor nuclear erythroid-related factor 2 (Nrf2), the master regulator of the antioxidant response that is involved in the PERK-mediated cell survival in stressed cells (Cullinan et al. 2003). The enzyme(s) that dephosphorylate and inactivate PERK are currently unknown. eIF2α dephosphorylation is accomplished by the phosphatase PP1 through its association with either the growth arrest and DNA damage-inducible protein GADD34 (also called PPP1R15A) (Novoa et al. 2001; Lee et al. 2009; Rojas et al. 2015) or PPP1R15B (Jousse et al. 2003). GADD34 itself is an ER stress-inducible gene acting in a negative feedback loop, whereas PPP1R15B is a constitutive repressor of eIF2α phosphorylation.
PERK is one of four eIF2α kinases (other being GNC2, PKR, HRI) that halt translation in response to different stimuli (Donnelly et al. 2013). Besides ER stress, eIF2α phosphorylation could be triggered by amino acid starvation, double-stranded RNA accumulation, oxidants, and heme depletion. Amino acid deficiency (as well as UV light) is sensed by GCN2 (general control nonderepressible 2, also called EIF2AK4). PKR (protein kinase R, also known as RNA-activated or double-stranded RNA-activated protein kinase, EIF2AK2) responds to viral infections, and HRI (heme-regulated eIF2α kinase, EIF2AK1) senses heme deficiency. Depletion of all four eIF2α kinases in mouse embryonic fibroblasts results in complete loss of eIF2α phosphorylation in response to various stress stimulations (Taniuchi et al. 2016). All these stimuli merge on eIF2α and regulate the same downstream pathways and collectively constitute the integrated stress response (ISR) (Pakos-Zebrucka et al. 2016). In unstressed cells, PERK appears as a monomer bound to the major ER chaperone GRP78 (78 kDa glucose-regulated protein also known as binding immunoglobulin protein-BiP). GRP78 is the primary chaperone in the ER lumen that interacts with the translocating nascent polypeptides. Accumulation of unfolded proteins in the ER lumen triggers the dissociation of the GRP78-PERK complex, which results in PERK homodimerization followed by trans-autophosphorylation. Therefore, GRP78 dissociation couples the ER protein load (or ER stress) to PERK dimerization and activation. PERK phosphorylates eIF2α at Ser51 (Harding et al. 1999), which results in the GDP-bound, inactive form of eIF2α. While eIF5 inactivates eIF2α by hydrolyzing GTP to GDP, eIF2B acts as the guanine nucleotide exchange factor for eIF2α. Phosphorylation status and duration of eIF2α have significant impact in mammalian physiology. An activator of eIF2B called ISRIB, e.g., acts as a chemical inhibitor of the ISR and blocks eIF2α phosphorylation, and has been implicated in increased cognitive function in rodents (Sidrauski et al. 2013). GTP-bound eIF2α interacts with the initiator methionyl-tRNA, which recognizes the start codon for initiation of translation. Therefore, eIF2α phosphorylation links ER proteostasis to the regulation of global protein synthesis. Translational attenuation, however, is not the only function of eIF2α. Induction of ER stress by an inhibitor of glycosylation (tunicamycin) in PERK knockout cells results in accumulation of endogenous peroxides (Harding et al. 2003). A list of mRNAs whose transcription is linked to eIF2α phosphorylation has been characterized and includes genes regulating amino acid sufficiency and resistance to oxidative stress (Harding et al. 2003). Accordingly, there are selective mRNAs whose translation actually increases upon eIF2α phosphorylation (Harding et al. 2000). For example, ATF4 (activating transcription factor 4) mRNA translation is elevated following eIF2α phosphorylation by PERK (Harding et al. 2000), which then induces the expression of genes involved in amino acid and cholesterol metabolism, glutathione biosynthesis, resistance to oxidative stress, and the proapoptotic transcription factor CHOP (Harding et al. 2000, 2003; Fusakio et al. 2016).
The mechanism of translational control of the mRNAs whose translation increases by ER stress represents a prominent example of adaptation. ATF4, e.g., has two upstream open reading frames (uORF), uORF1 and uORF2, which are followed by the ATF4 coding sequence. Under ER stress or other conditions that result in phosphorylated (thus inactive) eIF2α, ribosomes do not have sufficient time to reassemble at the uORF2, which would otherwise be translated. uORF2 is inhibitory and translated in unstressed cells where active eIF2α is abundant (Vattem and Wek 2004; Lu et al. 2004). Therefore, uORF1 has a stimulatory effect on the reinitiation of translation at AUGs downstream of uORF2.
UPR activation also leads to activation of the NF-κB pathway. Phospho-eIF2α-induced NF-κB activation involves two possible mechanisms: Phosphorylated eIF2α triggers the release of the I-κB from NF-κB (Jiang et al. 2003); when bound, I-κB sequesters NF-κB in the cytosol and blocks its nuclear localization. An alternative mechanism is that the amount of I-κB decreases due to the translational inhibition imposed by phospho-eIF2α (Deng et al. 2004). ATF6 branch (discussed below) and the IRE1-IKK-TRAF2 complex can also induce NF-κB activity (Hu et al. 2006; Yamazaki et al. 2009). ER stress and NF-κB connection is of particular importance in the context of metabolism. For example, ER stress-induced NF-κB triggers hypothalamic leptin resistance (Zhang et al. 2008). High-fat diet feeding results in elevated ER stress in the hypothalami of obese mice. Resulting activation of NF-κB upregulates the transcription of SOCS3 (Zhang et al. 2008), which is a negative regulator of the leptin-induced STAT3 phosphorylation.
IRE1 has a lumenal domain, which interacts with GRP78 and can also recognize the unfolded proteins, and a cytosolic kinase domain (Morl et al. 1993; Cox et al. 1993). As described below, the best characterized function of IRE1 involves the splicing of the mRNA of a transcription factor (HAC1 in yeast and XBP1 in metazoans) (Calfon et al. 2002), and the IRE1-HAC1/XBP1 arm constitutes the most ancient UPR pathway. Dissociation of GRP78 triggers oligomerization of the monomeric IRE1s, but is not sufficient for IRE1 activation. IRE1 oligomers form a luminal surface which allows IRE1 to directly sense unfolded proteins (Kimata et al. 2007), which in turn is thought to trigger a conformational change in the protein allowing IRE1 dimers to trans-autophosphorylate each other. Other than itself, IRE1 does not have any other known substrates as far as its kinase activity is concerned. IRE1 activation is prone to regulation by other factors as well. For example, the cytosolic non-receptor ABL tyrosine kinases localize to the ER membrane during ER stress and potentiate IRE1’s RNase activity (Morita et al. 2017).
Active IRE1 has endoribonuclease activity toward X-box-binding protein-1 (XBP1) mRNA (Yoshida et al. 2001; Calfon et al. 2002). Unprocessed XBP1 encodes a protein that represses UPR genes. Excision of a 26 nucleotide intron results in a frameshift in the XBP1 reading frame. Upon removal of this intron, cleaved XBP1 mRNA is ligated by RtcB (Lu et al. 2014), and the resulting shorter mRNA encodes an active transcription factor of UPR called XBP1 spliced (XBP1s). XBP1 transcription is upregulated by UPR (through ATF6) as well (Yoshida et al. 2001), resulting in increased unspliced form of XBP1, which serves as an inhibitor of UPR, thereby forming a negative feedback loop (Yoshida et al. 2006). XBP1s are involved in the transcriptional regulation of genes regulating ER biogenesis and ER-associated degradation. Apparently, these two key processes that have to be carried out in order to relieve ER stress reveal the vital role of XBP1 in UPR. For example, XBP1s upregulate the expression of several ER-resident chaperones and increase the biosynthesis of the predominant ER phospholipid phosphatidylcholine to enable ER expansion (Sriburi et al. 2004). XBP1 mediates the upregulation of ER degradation-enhancing α-mannosidase-like protein, which is required for the degradation of misfolded glycoprotein substrates (Yoshida et al. 2003). XBP1s play a fundamental role in overall mammalian development and physiology. Global deletion of XBP1 results in embryonic lethality from anemia (Reimold et al. 2000). In the liver, XBP1 is required for recovery from ER stress such that liver-specific XBP1 KO mice display liver injury and fibrosis following ER stress induction (Olivares and Henkel 2015). Hepatic XBP1 positively regulates the expression of lipogenic genes, and liver-specific XBP1 KO mice have decreased plasma levels of cholesterol and triglyceride (TG) and hepatic lipid biosynthesis (Lee et al. 2008). In the liver, there is a postprandial transient upregulation of UPR, which is important for the remodeling of hepatic metabolic flux in the transition from fasting to refeeding, and liver-specific XBP1s overexpression can mimic this metabolic switch (Deng et al. 2013). Deletion of IRE1α from hepatocytes decreases very low-density lipoprotein (VLDL) assembly and secretion, without altering TG synthesis or de novo lipogenesis (Wang et al. 2012). Adipocyte-specific XBP1 KO females have defective milk production resulting in decreased litter growth (Gregor et al. 2013). Depletion of XBP1 from the pancreas and the hypothalamus by RIP-Cre (rat insulin promoter)-mediated recombination triggers glucose intolerance due to β-cell failure and decreased insulin secretion (Lee et al. 2011). XBP1 deletion results in constitutive activation of IRE1, which is capable of cleaving prohormone convertase 1 and 2, and carboxypeptidase E mRNAs in pancreatic β cells (Lee et al. 2011). Likewise, deletion of IRE1α from RIP-positive cells leads to glucose intolerance and attenuates the HFD-induced β-cell proliferation commonly observed in obesity, but does not affect body weight or food intake of lean or HFD-induced obese mice (Xu et al. 2014). In the brain XBP1 regulates memory formation (Martínez et al. 2016), regulates neurite growth (Hayashi et al. 2007), and confers neuroprotection from degenerative diseases (Valdés et al. 2014). However, neuron-specific XBP1 KO mice are not different than wild-type mice in neuronal loss or survival under conditions associated with prion protein misfolding (Hetz et al. 2008). Furthermore, the neuronal XBP1 KOs have comparable body weights to wild-type mice (Hetz et al. 2008; Martínez et al. 2016).
XBP1 plays an important role in innate and adaptive immunity: It is required for the development and survival of dendritic cells, differentiation of lymphocytes (Reimold et al. 2001; Iwakoshi et al. 2003), and TLR2/4-mediated activation of pro-inflammatory cytokines by macrophages (Martinon et al. 2010). Eosinophil differentiation also requires XBP1s activity (Bettigole et al. 2015). Despite its well-characterized function in peripheral immune system, the role of XBP1 in microglia and CNS inflammation is not uncovered.
Another important function of IRE1 – that is implicated in linking ER stress to the metabolic diseases – was revealed by David Ron and coworkers, where they showed that IRE1 can activate c-Jun N-terminal kinases (JNK) (Urano et al. 2000). Tumor necrosis factor receptor (TNFR)-associated factor 2 (TRAF2), an adaptor protein, is recruited by phospho-IRE1 so that TRAF2 associates with the JNK kinases that ultimately result in JNK phosphorylation and activation (Urano et al. 2000). Tumor necrosis factor (TNF) receptor family is known to elicit its activity partly through JNK activation (Smith et al. 1994), and this pathway depends on intact TRAF2 (Lee et al. 1997). JNK could phosphorylate insulin receptor substrate 1 (IRS1) at Ser307, which results in the blockage of insulin-mediated IRS1 activation (Aguirre et al. 2000). Upon high-fat feeding, certain peripheral tissues such as the liver develop insulin resistance, at least in part, due to the high-fat diet-induced activation of ER stress, which in turn results in inhibition of JNK-mediated insulin signaling (Ozcan et al. 2004). Ser307Ala mutation eliminates phosphorylation of IRS-1 by JNK and removes the inhibitory effect of TNFα on insulin-stimulated tyrosine phosphorylation of IRS-1 (Aguirre et al. 2000).
IRE1 is also engaged in an RNA degradation pathway called IRE1-dependent decay of mRNA (RIDD) (Maurel et al. 2014), which is not limited to mRNA cleavage and extends to pre-microRNA processing (Upton et al. 2012). Substrates for IRE1’s RNase activity are comprised of nuclear, cytosolic, ER, and extracellular targets (Maurel et al. 2014). IRE1β, one of the two IRE1 isoforms, e.g., regulates selective degradation of secretory pathway protein mRNAs (Nakamura et al. 2011).
ATF6 is an ER transmembrane protein that has a stress-sensing lumenal domain and a cytoplasmic basic leucine zipper domain that acts as a transcription factor upon dissociation from the rest of the molecules (Haze et al. 1999). In unstressed cells, ATF6 exists bound to GRP78. During ER stress, GRP78 dissociates from ATF6, which triggers ATF6 to localize to Golgi, where it is processed to release its cytoplasmic DNA-binding domain (Shen et al. 2002). ATF6 is processed by Site 1 and Site 2 proteases in Golgi in a similar manner as the processing of sterol regulatory element-binding proteins (SREBPs) (Brown and Goldstein 1997; Ye et al. 2000). ATF6 has two isoforms, ATF6α and ATF6β. Knocking out both gene results in embryonic lethality, while single knockouts develop normally (Yamamoto et al. 2007). Another ER transmembrane protein that is activated in a similar manner to ATF6 is CREBH (cyclic AMP response element-binding protein hepatocyte), which is a liver transcription factor. CREBH has a cytoplasmic domain, which upon cleavage of CREBH in Golgi transits to the nucleus to activate genes involved in acute inflammatory response (Zhang et al. 2006). Because ER stress activates cleavage of CREBH in Golgi, CREBH acts as a mediator of ER stress-induced inflammatory response (Zhang et al. 2006). Activated ATF6 increases phosphatidylcholine synthesis independently of XBP1, suggesting a redundancy between ATF6 and XBP1 (Lee et al. 2003; Yamamoto et al. 2007) for certain genes including the ones responsible for ER stress-activated lipid biosynthesis and ER expansion (Bommiasamy et al. 2009). In hypothalamic cultures obtained from ATF6α KO mice, ER stress-induced regulation of various genes including CHOP-, GRP78-, and ERAD-associated genes is defective, suggesting that hypothalamic XBP1s cannot compensate for the absence of ATF6α (Lu et al. 2016). ATF6 and XBP1s can physically interact, and a dominant negative truncated version of XBP1 can suppress the activity of XBP1s or AFT6 (Lee et al. 2003). ATF6 activates the transcription of XBP1 and ER chaperones including GRP78 (Yoshida et al. 2001). ATF6 participates in the hepatic control of glucose homeostasis. In fasting, gluconeogenesis is triggered in the liver partly through the activity of CREB-regulated transcription coactivator 2 (CRTC2). Induction of ER stress in hepatocytes stimulates the dephosphorylation and nuclear entry of CRTC2. ATF6 recruits CRTC2 to ER stress-responsive genes, which results in increased expression of ER quality control genes and decreased hepatic glucose output (Wang et al. 2009). In the brain, ATF6 activation confers neuroprotection (Naranjo et al. 2016), and astroglial activation is attenuated in ATF6 knockout mice upon ischemic brain injury (Yoshikawa et al. 2015).
As discussed above, UPR has evolved to maintain the ER homeostasis acting in a cell-autonomous manner. Additionally, there are other proteostatic signaling routes evolved to enable communication between the nucleus and the cytosolic proteome (i.e., heat shock response) (Li et al. 2017) or the mitochondria (mitochondrial UPR) (Fiorese and Haynes 2017). However, there is an evolutionarily conserved yet incompletely understood intertissue signaling mechanism, also called the transcellular chaperone signaling (van Oosten-Hawle et al. 2013), described at least in metazoans that enable non-autonomous protein homeostasis by communication between different tissues (van Oosten-Hawle and Morimoto 2014). As UPR has evolved to allow communication between ER and other parts of the cellular structures, such as the nucleus, this trans-chaperone signaling enables the individual cells/tissues to communicate with each other. While it is beyond the scope of this chapter, it is worth to note that the regulation of ER homeostasis including ER stress within individual tissues should not be evaluated independently of its effect on other tissues. While induction of ER stress and impairment of UPR at individual tissues have detrimental metabolic consequences, some tissues respond to organelle stress, including ER stress, by secreting factors that improve the overall physiology. For example, during ER stress in the liver, hepatocytes secrete FGF21 (Schaap et al. 2013), which confers broad metabolic improvements including ameliorating ER stress and inducing weight loss (Kharitonenkov et al. 2005; Coskun et al. 2008). Likewise, neuronal overexpression of XBP1 in C. elegans activates UPR in non-neuronal cells, confers stress resistance, and increases longevity (Taylor and Dillin 2013).
2.1 ER Stress and the Regulation of Energy Balance
Regulation of metabolism in mammals requires a coordination between multiple tissues that is mediated by secreted factors, hormones and others, and neuronal inputs. The past decades in the metabolism field have uncovered numerous secreted factors, most notably leptin, and the dysregulation in the secretion, processing, recognition, or signaling of these factors play a fundamental role in the etiology of metabolic disorders. The canonical secretory pathway in mammals involves the translocation of nascent polypeptides into the ER, their regulated processing, posttranslational modifications, packing, and secretion. The hypothalamus is a heterogeneous population of chemically distinct neurons, with at least 50 distinct cell types in the ARC and ME identified based on their transcriptome profile (Campbell et al. 2017). Even within defined classes of neuronal populations, such as AgRP and POMC neurons, there are various subtypes. For example, there are at least three different subsets of POMC neurons in the MBH (Campbell et al. 2017). Likewise, among the AgRP neurons, which are exclusively expressed in the ARC, the leptin receptor-positive subset does not project to intra-hypothalamic sites (Betley et al. 2013). Therefore, the secretory profile of these neuronal populations, and the relative role of UPR in the respective neuronal subsets, is likely not uniform. The wide abundance and variety of the hypothalamic neuropeptides, which are synthesized as inactive precursors that require processing, demand a well-orchestrated ER quality control machinery. The physiological relevance and requirement of UPR in hypothalamic feeding centers are evident by a study on the transcriptional profile of AgRP and POMC neurons under satiated and fasted states showing UPR activation in AgRP neurons upon fasting (Henry et al. 2015). Numerous target genes for XBP1s including ER chaperones were upregulated by fasting in AgRP neurons. Genes involved in ER protein translocation and Golgi trafficking were also upregulated by fasting (Henry et al. 2015). ATF4 and ATF6 transcriptions also increased in AgRP neurons, although ATF6 nuclear localization was not altered by fasting. AgRP-specific ERAD-related transcripts were also upregulated by food deprivation. Besides UPR, fasting activated Nrf2 oxidative stress pathway in AgRP neurons. Notably, most of these fasting-induced changes in UPR pathway were specific to AgRP neurons, and no significant regulation was detected in POMC cells (Henry et al. 2015). However, upon short-term refeeding of fasted mice, POMC-specific XBP1s, ATF4, and ATF6 expression increased (Williams et al. 2014). Changes in AgRP neurons are probably related to fasting-induced AgRP neuronal activation and increased AgRP biosynthesis and secretion. For example, hypothalamic ATF4 regulates AgRP expression by stimulating FOXO1 (Deng et al. 2017), a transcription factor that positively regulates AgRP expression (Kitamura et al. 2006; Kim et al. 2006; Ren et al. 2012). However, whether and/or to what extent UPR contributes to the orexigenic response elicited by AgRP neurons is not known.
Overexpression of ATF4 in the MBH marginally induces food intake and weight gain and results in an attenuated hepatic insulin signaling, while a dominant negative ATF4 improves insulin signaling and glucose metabolism (Zhang et al. 2013b). Adverse effects of ATF4 overexpression could be reversed by hepatic vagotomy or by knocking down hypothalamic S6K expression (Zhang et al. 2013b). As explained in more detail in Chap. 8, while acute activation of hypothalamic S6K activity appears to block hepatic insulin signaling, overexpression of constitutively active S6K in the MBH blocks weight gain and protects against adverse consequences of HFD (Ono et al. 2008; Blouet et al. 2008). AgRP ATF4 KO mice are protected from weight gain on regular chow and HFD due to decreased food intake and increased energy expenditure (Deng et al. 2017). Deletion of ATF4 from POMC neurons also result in a similar phenotype with mice resistant to HFD-induced weight gain (Xiao et al. 2017a, b). Effect of ATF4 on POMC neurons was largely dependent on the negative regulation of ATG5 by ATF4 such that mice lacking both genes in POMC neurons had reduced energy expenditure on HFD and gained more weight (Xiao et al. 2017a, b). ATG5 is required for the regulation of autophagy, and POMC-specific ATG5 KO mice have defective autophagy in POMC neurons, although their body weight is not affected (Malhotra et al. 2015). Deletion of ATF4 led to the ATG5-dependent autophagy and increased αMSH production, which in turn led to increased energy expenditure (Xiao et al. 2017a, b).
Deletion of XBP1 from POMC neurons does not significantly alter body weight on regular chow or HFD, although the mice show a tendency for elevated weight gain (https://tez.yok.gov.tr/UlusalTezMerkezi/TezGoster?key=1zw6GvYMe-q3Hf6HR-3USykM7dHpyqsbqQ-p1MsCgPMp7KeLv7nO_Vnsn0mL6Afc). While suppressing caloric intake on lean mice, HFD-fed POMC-specific XBP1 KOs tend to eat more (https://tez.yok.gov.tr/UlusalTezMerkezi/TezGoster?key=1zw6GvYMe-q3Hf6HR-3USykM7dHpyqsbqQ-p1MsCgPMp7KeLv7nO_Vnsn0mL6Afc). Increased food intake of obese mice is however compensated by increased energy expenditure, and the obese KO mice have decreased fat percentage and decreased respiratory exchange ratios, indicative of elevated fat oxidation, than their POMC-Cre control counterparts (https://tez.yok.gov.tr/UlusalTezMerkezi/TezGoster?key=1zw6GvYMe-q3Hf6HR-3USykM7dHpyqsbqQ-p1MsCgPMp7KeLv7nO_Vnsn0mL6Afc). These results should, however, be evaluated taking into account the phenotype of the POMC-Cre mice itself and the significant difference in the co-localization of POMC-Cre-expressing cells and POMC neurons in adult mice (Padilla et al. 2012). Genetic depletion of IRE1α expression from POMC neurons does not alter body weight on regular diet, but induces food intake and weight gain with increased adiposity on HFD (Yao et al. 2017). Obese POMC-IRE1α KO mice also display decreased energy expenditure, decreased cold tolerance, and impaired glucose tolerance (Yao et al. 2017). Additionally, these KO mice had elevated expression of negative regulators of leptin receptor signaling including PTP1B and SOCS3, which might contribute to their hyperphagic phenotype (Yao et al. 2017). Overexpression of XBP1s in POMC neurons results in hypophagic mice with increased energy expenditure and confers resistance to obesity (Williams et al. 2014). Interestingly, POMC-specific XBP1 overexpression also led to increased hepatic XBP1s expression and increased the BAT- and beige-specific genes in the adipose tissue. Considering that hypothalamic induction of ER stress attenuates POMC processing and αMSH production (Cakir et al. 2013), it is worth investigating whether the metabolic improvements observed in mice with POMC-specific XBP1s overexpression are secondary to improved POMC processing.
ER stress and UPR play a fundamental role in metabolic regulation. Studies conducted in rodents showed that HFD-induced obesity leads to ER stress in multiple peripheral systems including the liver and adipose tissue. Furthermore, there is a functional interaction between ER stress and inflammation. For example, ER stress-induced inflammation in the liver couples obesity to insulin resistance (Ozcan et al. 2004), and deletion of IRE1α from the cells of myeloid lineage, including the macrophages but not the lymphocytes (T and B cells), confers almost complete protection from HFD-induced obesity and other associated metabolic abnormalities (Shan et al. 2017). The role of central ER stress and its effect on energy metabolism have been an area of active research during the past decade. Findings from our laboratory and others have indicated that development of obesity is accompanied by elevated markers of ER stress in the hypothalamus, predominantly in the MBH. Elevated ER stress in turn triggers central inflammation, resulting a positive feedback loop that is ultimately detrimental to the CNS circuitry regulating food intake and energy expenditure. ER stress has a more direct, cell-autonomous effect on the neuroendocrine hypothalamic population of neurons encoding bioactive neuropeptides, such as POMC (Cakir et al. 2013).
Initial evidence on the relationship between HFD-feeding and hypothalamic ER stress came from in vitro studies as well as results obtained from rodents using pharmacological and genetic tools. Treatment of leptin-responsive cells with inducers of ER stress, such as homocysteine, tunicamycin, and thapsigargin, induces the expression of negative regulators of leptin receptor signaling including SOCS3 and PTP1B and reduces leptin-induced STAT3 phosphorylation (Hosoi et al. 2008; Cakir et al. 2013). These results are reproduced when ER stress was induced in the hypothalamus of lean rodents (Hosoi et al. 2008; Cakir et al. 2013). Homocysteine treatment induced XBP1 splicing in the brain (Hosoi et al. 2010), also blocked the leptin-induced ERK phosphorylation, and did not affect the phospho-JNK levels (Hosoi et al. 2008). Furthermore, 4PBA treatment reversed the ER stress-induced attenuation in leptin receptor signaling. Central infusion of thapsigargin also blocked leptin and insulin signaling in the hypothalamus and had an orexigenic effect.
Diet-induced obesity correlates with increased markers of hypothalamic ER stress. IRE1 and PERK phosphorylation, XBP1 splicing, and expression of CHOP and GRP78 increase in HFD-fed rodent hypothalamus (Zhang et al. 2008; Won et al. 2009; Cakir et al. 2013). It is possible that the diet composition plays a fundamental role in the induction of hypothalamic ER stress. For example, central ceramide infusion induces hypothalamic ER stress and causes weight gain (Contreras et al. 2014). Ceramide-induced ER stress and weight gain can be reversed by VMH-specific overexpression of GRP78, while a dominant negative GRP78 induces ER stress and weight gain (Contreras et al. 2014). Overexpression of GRP78 in the VMH of Zucker rats decreased ER stress, suppresses weight gain, and increases BAT-mediated thermogenesis without altering food intake (Contreras et al. 2014). Effect of GRP78 and ER stress in the VMH is likely independent of leptin signaling and involves the sympathetic output to the adipose tissue and involves browning of WAT- and UCP1-induced thermogenesis (Contreras et al. 2017). Saturated fatty acids, such as palmitate, activate ER stress in hypothalamic neurons and attenuate leptin and insulin signaling (Kleinridders et al. 2009; Mayer and Belsham 2010; Diaz et al. 2015). As discussed above, TLR4 acts as a receptor for saturated fatty acids, and deletion of the TLR adaptor protein MyD88 from neurons confers protection from diet-induced obesity (Kleinridders et al. 2009). Central infusion of palmitate induces inflammatory cytokine expression and ER stress through activation of toll-like receptor 4 (TLR4) (Milanski et al. 2009; Kanczkowski et al. 2013), and central inhibition or genetic inactivation of TLR4 confers protection from diet-induced obesity in rodents (Milanski et al. 2009). Likewise, knockout of the TLR4 adapter protein MyD88 prevents excess weight gain in mice on high-fat diet (Kanczkowski et al. 2013). TLR4 signaling can also activate IRE1-XBP1 axis in macrophages while suppressing the ATF4 pathway (Woo et al. 2009). Distinct from the ER-related XBP1 targets, TLR4-mediated XBP1 activation triggers the production of pro-inflammatory cytokines (Martinon et al. 2010). In rodent and human CNS, XBP1 expression is highest in microglia (Zhang et al. 2014; Bennett et al. 2016), and whether a similar link exists between fatty acids and IRE1-XBP1 pathway in CNS immune cells, such as in microglia, is worth investigating.
As discussed above, diet-induced obese mice have elevated hypothalamic IKKβ-NF-κB axis, which in turn activated the inflammatory gene expression in the hypothalamus, but also act as a factor in a positive feedback loop where its constitutive activation promoted ER stress (Zhang et al. 2008): HFD-fed mice have elevated levels of phosphorylated PERK and eIF2α in the hypothalamus (Zhang et al. 2008). Induction of central ER stress in lean mice by tunicamycin led to the activation of NF-κB, suggesting a causative relationship between ER stress and central inflammation. Accordingly, overexpression of constitutively active IKKβ in the MBH of lean mice was sufficient to induce ER stress, while neuronal deletion of IKKβ decreased PERK phosphorylation. Activated IKKβ-NF-κB signaling induced the expression of SOCS3, which is a negative regulator of insulin and leptin signaling. While activation of ER stress increased NF-κB activity, relieving ER stress with chemical chaperone TUDCA alleviated the HFD-induced hypothalamic NF-κB activation (Zhang et al. 2008). In summary, hypothalamic IKKβ and ER stress can activate each other, forming a positive feedback loop leading to dysregulation of central energy balance. DIO rats also display increased hypothalamic PERK and eIF2α phosphorylation (Cakir et al. 2013), and these responses seem to be ARC specific, as DIO rats do not suffer ER stress in the PVN (Cakir et al. 2013). Accordingly, PVN retains its leptin sensitivity in DIO rodents (Perello et al. 2010). While induction of ER stress in lean animals induces markers of leptin resistance, including SOCS3 and PTP1B, and suppressed energy expenditure (Zhang et al. 2008; Cakir et al. 2013), central infusion of TUDCA in obese rats did not alter STAT3 phosphorylation but acutely suppressed food intake and triggered increased oxygen consumption (Cakir et al. 2013).
Central ER stress does not only alter energy metabolism but also adversely affect other metabolic parameters. For example, thapsigargin-induced hypothalamic ER stress results in glucose tolerance, peripheral insulin resistance, and increases blood pressure, mimicking the phenotype in DIO rodents (Purkayastha et al. 2011b; Cakir et al. 2013). These responses could be reversed by genetic inhibition of IKK signaling in the ARC or partially reversed by treatment of DIO mice with TUDCA without altering food intake or body weight (Purkayastha et al. 2011b; Horwath et al. 2017). 4PBA treatment of rats resulted in attenuation of LPS-induced heart rate thought to be mediated, at least in part, through TLR4 action in the PVN (Masson et al. 2015). In obese mice that undergo vertical sleeve gastrectomy, hypothalamic markers of ER stress were decreased, which was accompanied by a significant decrease in mean arterial pressure (McGavigan et al. 2017).
The physical interaction between the ER and mitochondria is also affected by ER stress. Mitofusin 1 and mitofusin 2 (MTF1/2) are proteins located on the ER membrane and the outer surface of the mitochondria tethering the two organelles. MTF deficiency in the liver results in ER stress and causes glucose intolerance and defective insulin signaling in the liver and muscle, a defect that can be rescued with TUDCA treatment (Sebastián et al. 2012). AgRP-specific MTF1 or MTF2 KO mice are protected from HFD-induced weight gain (Dietrich et al. 2013). On the contrary, ablation of MTF2 in POMC neurons results in elevated ER stress and promotes food intake and weight gain (Schneeberger et al. 2013), while POMC-specific MTF1 KOs have dysfunctional glucose metabolism without altered hypothalamic ER stress (Ramírez et al. 2017). As we continue to discuss in the following section, several studies have indicated that ER stress leads to a profound defect in the processing of prohormones including POMC.
2.2 Hypothalamic ER Stress and Neuropeptides
ER stress-induced abnormalities in energy as well as glucose metabolism are causally related to altered hypothalamic POMC processing. POMC-specific MTF2 KO mice develop glucose intolerance prior to development of their obese phenotype largely due to increased hepatic gluconeogenesis (Schneeberger et al. 2015). These mice had reduced hypothalamic αMSH levels that could be rescued with central TUDCA treatment (Schneeberger et al. 2015). In addition, acute HFD exposure also increased the hypothalamic ER stress, decreased αΜSH levels, and impaired glucose homeostasis, which could be rescued by central αMSH replenishment (Schneeberger et al. 2015). MTF2 deletion in POMC neurons was proposed to decrease ER-mitochondrial contacts in the POMC neurons of obese mice; however the observed decreased ER-mitochondrial physical interaction is likely not a direct effect of the MTF2 deletion but rather secondary to the obese phenotype of the KO mice. MTF2 depletion in mouse embryonic fibroblasts results in increased ER-mitochondrial tethering (Filadi et al. 2015). AgRP-specific MTF1 or MTF2 KOs did not have differences in mitochondria-ER contacts, compared to control mice, and did not display altered markers of ER stress (Dietrich et al. 2013). POMC-MTF2 KO mice are resistant to anorectic action of exogenous leptin (Schneeberger et al. 2013). As discussed below, these mice have a decreased expression of POMC mRNA but an accumulation in the POMC protein. Although the expression of the POMC-processing enzymes PC2, PAM, and CPE was also elevated in the KO mice, α-MSH level was significantly lower, which accounts for the obese phenotype of the mice (Schneeberger et al. 2013). However, upon chronic central infusion of 4PBA or TUDCA, their food intake decreased, αΜSH levels increased, and the mice lost their excess adiposity, and their body weight returned to the range observed in their chow-fed counterparts (Schneeberger et al. 2013). Accordingly, overexpression of MTF2 in the ARC protects against HFD-induced weight gain and decreases markers of ER stress (Schneeberger et al. 2013). The adverse effect of HFD on hypothalamic MTF expression might be, at least in part, dependent on SFAs. HFD feeding or palmitate alone decreases the expression of mitofusin 2 (MTF2) the ARC (Diaz et al. 2015).
In HFD-induced obese rodents, there is a gradual decrease in the amount of POMC processing evidenced by accumulation of POMC but decreased amounts of its processing products ACTH and αMSH in the MBH (Enriori et al. 2007; Cakir et al. 2013; Schneeberger et al. 2013). Furthermore, although leptin could stimulate, albeit at a compromised level, the expression of POMC mRNA and hypothalamic ACTH levels, αMSH level, is completely insensitive to leptin administration (Cakir et al. 2013). Decreased αMSH levels in DIO rodents were not due to increased αMSH degradation (Wallingford et al. 2009; Cakir et al. 2013; Schneeberger et al. 2013). These findings suggested that there was a defect in the step leading from the POMC precursor to the production of αMSH. While there was no significant difference in the mRNA levels of POMC-processing enzymes between lean and DIO animals, there was a significant reduction in the protein levels of PC2 in DIO ARC. Reduced PC2 expression and αMSH levels could be induced in lean rat hypothalamus simply by central infusion of thapsigargin or tunicamycin, which suggested that ER stress negatively regulated PC2 protein levels (Cakir et al. 2013), which in turn leads to decreased αMSH levels. The thapsigargin- or tunicamycin-induced defects in POMC processing could be partially rescued by chemical chaperones PBA or TUDCA (Cakir et al. 2013). As explained in detail in Chap. 5, PC2 is critical in the processing of ACTH to αMSH. Decreased PC2 and αMSH levels could also be induced in different cell lines upon induction of ER stress in a way that could be rescued by chemical chaperones. Furthermore, central TUDCA infusion rescued the defect in the POMC processing in DIO rats and increased the αMSH levels. However, the same treatment did not alter the level of hypothalamic NPY, suggesting that DIO-induced ER stress affects the processing of hypothalamic neuropeptides differently (Cakir et al. 2013). Salubrinal is an agent that selectively inhibits the dephosphorylation of p-eIF2α (Boyce et al. 2005) and enhances the FFA-induced activation of the PERK pathway, but not the ATF6 and IRE1 branches (Ladrière et al. 2010). Blocking the dephosphorylation of eIF2α with salubrinal increased PC2 and reversed the effect of thapsigargin on PC2 levels, suggesting that PERK-eIF2α branch of the UPR could protect the decrease in PC2 during ER stress.
A recent study has further revealed the overall importance of ER homeostasis on POMC processing in regard to energy balance. Proper trafficking and processing of POMC require a protein complex between suppressor enhancer of lin-like 1 (Sel1L) and hydroxymethylglutaryl reductase degradation protein (Hrd1, also called synoviolin). Sel1L-Hrd1 is an evolutionarily conserved arm of ERAD, and their genetic deletion results in embryonic lethality in mice (Yagishita et al. 2005; Francisco et al. 2010). Both genes are expressed in the hypothalamus and in POMC neurons (Kim et al. 2017). Feeding and leptin positively regulate their expression. Deletion of Sel1L from POMC neurons results in loss of response to leptin’s anorectic effect, hyperphagia, and age-associated obesity. Sel1L deficiency did not induce central ER stress or inflammation (Kim et al. 2017). Although POMC mRNA level was unaltered in POMC-specific Sel1L deficiency, there was a significant accumulation of the POMC protein. Hrd1 is an E3 ligase and together with Sel1L targets POMC to proteasomal degradation. Consequently, in the absence of this quality control system, the nascent POMC polypeptide cannot be properly targeted for further processing resulting in aggregation and accumulation in the ER (Kim et al. 2017). It is possible that diet-induced obesity also results in a functional dysregulation of the ERAD system in POMC neurons, which might contribute to the accumulation of POMC observed in obese rodents (Cakir et al. 2013). Finally, while it is beyond the scope of this chapter, it is worth noting that the effect of hypothalamic ER stress extends beyond regulation of energy balance. Central ER stress affects the polyA tail length of mRNA for certain neuropeptides including vasopressin and oxytocin, resulting in decreased mature mRNA levels (Morishita et al. 2011), which could be reversed by TUDCA treatment (Morishita et al. 2011). Furthermore, ERAD deficiency also leads to aggregation and accumulation of proAVP, the precursor of the precursor for the antidiuretic hormone arginine vasopressin, resulting in central diabetes insipidus (Shi et al. 2017) (Fig. 4.1).

Propose changes in POMC processing and biosynthesis of α-MSH in the obese state. While the levels of processing enzyme PC2 adjust under fasting (i.e., low leptin, first panel) and fed (i.e., basal leptin, second panel), in the DIO (i.e., high leptin, third panel), there is a decrease in PC2 and an accumulation of POMC compared to the fed control. Compared to the basal leptin condition, fasting causes diminished α-MSH by a leptin-mediated reduction in POMC mRNA along with reduced POMC processing due to lower PC1 and PC2 levels. In the DIO condition, endoplasmic reticulum stress triggers an unfolded response causing an accumulation of POMC in the ER partly caused by an unfolded response and a possible degradation of PC2 through the ERAD system, conditions that ultimately cause less α-MSH production. (Image from our prior publication (Cakir et al. 2013))
3 Conclusion
Across species in all kingdoms of life, maintenance of homeostasis is key for survival. Both at the cellular and the organismal level, various signaling routes have evolved and are activated in response to internal or environmental stimuli to keep the homeostasis within certain boundaries. This chapter has focused on two of these adaptive mechanisms, inflammation and ER stress/UPR.
Infection, tissue damage, or stress could trigger inflammation, which in turn increases defense mechanisms to cope with the infection (immune response) or tissue repair and adaptation to the stress stimuli. As outlined above, the inflammatory state associated with obesity is different than classical inflammatory response; it is a chronic but rather low-grade inflammatory state, which, unlike the infection-induced inflammation, is not coupled to induction of an immune response. Furthermore, the central vs. the peripheral inflammation associated with high-calorie intake are different. Central markers of inflammation can be detected following consumption of high-calorie diets prior to any weight gain, while peripheral inflammation, tissue macrophage infiltration, and polarization proceeds gain of adiposity. The associated cell types in the periphery vs. the CNS are also different, and the anti-inflammatory nature of the brain parenchyma is not suitable for survival of immune cell components in the brain. CNS has its own macrophage pool, the microglia, and we are only beginning to understand the role of the glial cell populations in the regulation of energy balance and overall metabolism.
ER stress is classically triggered by an imbalance between the ER’s folding capacity and the steady-state unfolded protein pool in the ER lumen. There are numerous physiological or pathological triggers of ER stress, and nutrient deprivation, redox status, infection, and ER calcium levels are among some of them. The dysfunction of the ER is communicated to the nucleus and the translational machinery through a set of signaling routes called the UPR. Much like other stress response pathways, UPR initially functions to restore the homeostasis, in this case, of the ER. The temporal aspect of the stress-activated pathways including the UPR is of significant importance; prolonged UPR can result in detrimental consequences as induction of apoptosis. This makes sense from an organismal point of view to eliminate overly stressed/damaged cells which cannot be rescued. However, a chronic ER stress detected in an entire tissue as observed in obesity results in a maladaptive state which triggers a shift in homeostatic set points. This, in turn, results in maladaptive states, such as insulin and leptin resistance, which contributes to the etiology of obesity and diabetes.
Both ER stress and inflammation are intimately connected for a number of reasons. First of all, mediators of inflammation and ER stress utilize, at least in part, common downstream signaling pathways (e.g., NF-κB activation). This is one of the reasons that ER stress and inflammation can trigger each other even at a cell autonomous level. Second, signaling components of the inflammatory process as well as ER stress are also shared by several metabolic routes, resulting in a causative relationship between these processes and the regulation of energy metabolism. Third, either process is activated chronically at a moderate level during obesity. Fourth, nutrients, most prominently saturated fatty acids, can induce central ER stress as well as an inflammatory response. Fifth, both obesity-associated ER stress and elevated inflammation appear to be reversible, at least in rodents.
Various studies conducted in rodents as well as results from humans suggest that obesity is accompanied by elevated ER stress and inflammation in the CNS. Our knowledge on central ER stress in regard to the possible role of UPR in glia is rather limited, and most of the metabolic studies have so far focused on ER stress in neurons. Central inflammation encompasses neurons as well as glia, and astrogliosis and microgliosis have detrimental consequences in the hypothalamic circuits regulating energy balance. What triggers hypothalamic ER stress or inflammation in the first place is not fully uncovered; however nutrient-related inputs, such as saturated fatty acids, appear to play a causative role. Induction of hypothalamic ER stress contributes to a positive energy balance, and this is mediated at least in part due to the direct impact on altered prohormone processing as in the case for POMC. Inflammatory mediators are classically accompanied by an anorexic response; however prolonged activation of central inflammatory pathways results in elevated food intake and/or increased weight gain. This is a major distinction in the effect of central inflammation when considered in an acute vs. chronic setting. Genetic and pharmacological evidence suggest that amelioration of the hypothalamic ER stress and inflammation that accompanies obesity result in weight loss, improved glucose metabolism, and improved cardiovascular functions. It is finally important to note the similarity between obesity and the aging process in regard to their association to central inflammation and ER stress. The results indicating that suppression of neuronal inflammation confers longevity in mice point out the importance of CNS inflammation on overall mammalian physiology.
It is vital to the understanding of biology that the cells or organisms are more than a collection of individual components (organelles or tissues) put together. As Aristotle said, “the whole is more than the sum of its parts.” As such, there is a sophisticated communication system evolved to maintain the homeostasis in multicellular organisms. Components of the inflammatory process and the UPR should be evaluated from this perspective at the organismal level. Cell autonomous stress response pathways function to restore the homeostatic boundaries; however they have non-cell autonomous consequences (e.g., ER stress-induced hepatic FGF21 secretion) that translate into whole animal physiology. While cellular components of the UPR and inflammation are rather well-characterized, how these processes affect homeostatic brain centers including the hypothalamus and the brain stem is only beginning to be uncovered. A comprehensive understanding of the causes of the CNS inflammation and ER stress and how these processes interact with the metabolic regulatory routes will help us devise novel therapeutic opportunities for the treatment of metabolic diseases such as obesity.
Questions
-
1.
What are the differences between central and peripheral inflammation? How does innate vs. acquired immunity play a role in CNS inflammation?
-
2.
What are the causes and consequences of acute vs. chronic inflammation on energy balance?
-
3.
What is the relationship between ER stress and inflammation? What are the signaling pathways that connect them?
-
4.
What are the signaling routes that communicate between the ER and the nucleus? How conserved are they?
-
5.
How does hypothalamic ER stress interfere with the regulation of energy homeostasis?
References
Aguirre, V., Uchida, T., Yenush, L., et al. (2000). The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). The Journal of Biological Chemistry, 275, 9047–9054.
Ahima, R. S., Prabakaran, D., Mantzoros, C., et al. (1996). Role of leptin in the neuroendocrine response to fasting. Nature, 382, 250–252.
Almeida-Suhett, C. P., Graham, A., Chen, Y., & Deuster, P. (2017). Behavioral changes in male mice fed a high-fat diet are associated with IL-1β expression in specific brain regions. Physiology & Behavior, 169, 130–140.
Amura, C. R., Marek, L., Winn, R. A., & Heasley, L. E. (2005). Inhibited neurogenesis in JNK1-deficient embryonic stem cells. Molecular and Cellular Biology, 25, 10791–10802.
Anandasabapathy, N., Victora, G. D., Meredith, M., et al. (2011). Flt3L controls the development of radiosensitive dendritic cells in the meninges and choroid plexus of the steady-state mouse brain. The Journal of Experimental Medicine, 208, 1695–1705.
André, C., Guzman-Quevedo, O., Rey, C., et al. (2017). Inhibiting microglia expansion prevents diet-induced hypothalamic and peripheral inflammation. Diabetes, 66, 908–919.
Argaw, A. T., Asp, L., Zhang, J., et al. (2012). Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. The Journal of Clinical Investigation, 122, 2454–2468.
Arkan, M. C., Hevener, A. L., Greten, F. R., et al. (2005). IKK-beta links inflammation to obesity-induced insulin resistance. Nature Medicine, 11, 191–198.
Balland, E., & Cowley, M. A. (2017). Short-term high fat diet increases the presence of astrocytes in the hypothalamus of C57BL6 mice without altering leptin sensitivity. Journal of Neuroendocrinology. https://doi.org/10.1111/jne.12504.
Balland, E., Dam, J., Langlet, F., et al. (2014). Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metabolism, 19, 293–301.
Beard, R. S., Jr., Haines, R. J., Wu, K. Y., et al. (2014). Non-muscle Mlck is required for β-catenin- and FoxO1-dependent downregulation of Cldn5 in IL-1β-mediated barrier dysfunction in brain endothelial cells. Journal of Cell Science, 127, 1840–1853.
Belgardt, B. F., Mauer, J., Wunderlich, F. T., et al. (2010). Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proceedings of the National Academy of Sciences of the United States of America, 107, 6028–6033.
Bence, K. K., Delibegovic, M., Xue, B., et al. (2006). Neuronal PTP1B regulates body weight, adiposity and leptin action. Nature Medicine, 12, 917–924.
Bennett, M. L., Bennett, F. C., Liddelow, S. A., et al. (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America, 113, E1738–E1746.
Benomar, Y., Gertler, A., De Lacy, P., et al. (2013). Central resistin overexposure induces insulin resistance through toll-like receptor 4. Diabetes, 62, 102–114.
Benomar, Y., Amine, H., Crépin, D., et al. (2016). Central Resistin/TLR4 impairs adiponectin signaling, contributing to insulin and FGF21 resistance. Diabetes, 65, 913–926.
Berkseth, K. E., Guyenet, S. J., Melhorn, S. J., et al. (2014). Hypothalamic gliosis associated with high-fat diet feeding is reversible in mice: A combined immunohistochemical and magnetic resonance imaging study. Endocrinology, 155, 2858–2867.
Besedovsky, H. O., & del Rey, A. (1996). Immune-neuro-endocrine interactions: Facts and hypotheses. Endocrine Reviews, 17, 64–102.
Betley, J. N., Cao, Z. F. H., Ritola, K. D., & Sternson, S. M. (2013). Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell, 155, 1337–1350.
Bettigole, S. E., Lis, R., Adoro, S., et al. (2015). The transcription factor XBP1 is selectively required for eosinophil differentiation. Nature Immunology, 16, 829–837.
Blamire, A. M., Anthony, D. C., Rajagopalan, B., et al. (2000). Interleukin-1beta -induced changes in blood-brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: A magnetic resonance study. The Journal of Neuroscience, 20, 8153–8159.
Blouet, C., Ono, H., & Schwartz, G. J. (2008). Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metabolism, 8, 459–467.
Bommiasamy, H., Back, S. H., Fagone, P., et al. (2009). ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. Journal of Cell Science, 122, 1626–1636.
Bora-Tatar, G., Dayangaç-Erden, D., Demir, A. S., et al. (2009). Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorganic & Medicinal Chemistry, 17, 5219–5228.
Boyce, M., Bryant, K. F., Jousse, C., et al. (2005). A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science, 307, 935–939.
Breder, C. D., Hazuka, C., Ghayur, T., et al. (1994). Regional induction of tumor necrosis factor alpha expression in the mouse brain after systemic lipopolysaccharide administration. Proceedings of the National Academy of Sciences of the United States of America, 91, 11393–11397.
Brehme, M., Voisine, C., Rolland, T., et al. (2014). A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Reports, 9, 1135–1150.
Brown, M. S., & Goldstein, J. L. (1997). The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell, 89, 331–340.
Broz, P., & Dixit, V. M. (2016). Inflammasomes: Mechanism of assembly, regulation and signalling. Nature Reviews. Immunology, 16, 407–420.
Buckman, L. B., Thompson, M. M., Lippert, R. N., et al. (2015). Evidence for a novel functional role of astrocytes in the acute homeostatic response to high-fat diet intake in mice. Molecular Metabolism, 4, 58–63.
Cai, D., Yuan, M., Frantz, D. F., et al. (2005). Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nature Medicine, 11, 183–190.
Cakir, I., Cyr, N. E., Perello, M., et al. (2013). Obesity induces hypothalamic endoplasmic reticulum stress and impairs proopiomelanocortin (POMC) post-translational processing. The Journal of Biological Chemistry, 288, 17675–17688.
Calfon, M., Zeng, H., Urano, F., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature, 415, 92–96.
Campbell, J. N., Macosko, E. Z., Fenselau, H., et al. (2017). A molecular census of arcuate hypothalamus and median eminence cell types. Nature Neuroscience, 20, 484–496.
Carlin, J. L., Grissom, N., Ying, Z., et al. (2016). Voluntary exercise blocks Western diet-induced gene expression of the chemokines CXCL10 and CCL2 in the prefrontal cortex. Brain, Behavior, and Immunity, 58, 82–90.
Chakravarty, S., & Herkenham, M. (2005). Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. The Journal of Neuroscience, 25, 1788–1796.
Chang, L., Jones, Y., Ellisman, M. H., et al. (2003). JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Developmental Cell, 4, 521–533.
Chesnokova, V., Pechnick, R. N., & Wawrowsky, K. (2016). Chronic peripheral inflammation, hippocampal neurogenesis, and behavior. Brain, Behavior, and Immunity, 58, 1–8.
Chiang, S.-H., Bazuine, M., Lumeng, C. N., et al. (2009). The protein kinase IKKepsilon regulates energy balance in obese mice. Cell, 138, 961–975.
Choi, S. J., Kim, F., Schwartz, M. W., & Wisse, B. E. (2010). Cultured hypothalamic neurons are resistant to inflammation and insulin resistance induced by saturated fatty acids. American Journal of Physiology. Endocrinology and Metabolism, 298, E1122–E1130.
Contreras, C., González-García, I., Martínez-Sánchez, N., et al. (2014). Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Reports, 9, 366–377.
Contreras, C., González-García, I., Seoane-Collazo, P., et al. (2017). Reduction of hypothalamic endoplasmic reticulum stress activates browning of white fat and ameliorates obesity. Diabetes, 66, 87–99.
Corbett, E. F., Oikawa, K., Francois, P., et al. (1999). Ca2+ regulation of interactions between endoplasmic reticulum chaperones. The Journal of Biological Chemistry, 274, 6203–6211.
Coskun, T., Bina, H. A., Schneider, M. A., et al. (2008). Fibroblast growth factor 21 corrects obesity in mice. Endocrinology, 149, 6018–6027.
Cox, J. S., Shamu, C. E., & Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell, 73, 1197–1206.
Cullinan, S. B., Zhang, D., Hannink, M., et al. (2003). Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Molecular and Cellular Biology, 23, 7198–7209.
Dange, R. B., Agarwal, D., Teruyama, R., & Francis, J. (2015). Toll-like receptor 4 inhibition within the paraventricular nucleus attenuates blood pressure and inflammatory response in a genetic model of hypertension. Journal of Neuroinflammation, 12, 31.
de Rivero Vaccari, J. P., Lotocki, G., Marcillo, A. E., et al. (2008). A molecular platform in neurons regulates inflammation after spinal cord injury. The Journal of Neuroscience, 28, 3404–3414.
De Souza, C. T., Araujo, E. P., Bordin, S., et al. (2005). Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology, 146, 4192–4199.
Deng, J., Lu, P. D., Zhang, Y., et al. (2004). Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Molecular and Cellular Biology, 24, 10161–10168.
Deng, Y., Wang, Z. V., Tao, C., et al. (2013). The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. The Journal of Clinical Investigation, 123, 455–468.
Deng, J., Yuan, F., Guo, Y., et al. (2017). Deletion of ATF4 in AgRP neurons promotes fat loss mainly via increasing energy expenditure. Diabetes, 66, 640–650.
Deroo, B. J., & Archer, T. K. (2001). Glucocorticoid receptor activation of the I kappa B alpha promoter within chromatin. Molecular Biology of the Cell, 12, 3365–3374.
Di Bello, I. C., Dawson, M. R., Levine, J. M., & Reynolds, R. (1999). Generation of oligodendroglial progenitors in acute inflammatory demyelinating lesions of the rat brain stem is associated with demyelination rather than inflammation. Journal of Neurocytology, 28, 365–381.
Diaz, B., Fuentes-Mera, L., Tovar, A., et al. (2015). Saturated lipids decrease mitofusin 2 leading to endoplasmic reticulum stress activation and insulin resistance in hypothalamic cells. Brain Research, 1627, 80–89.
Dietrich, M. O., Liu, Z.-W., & Horvath, T. L. (2013). Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell, 155, 188–199.
Djogo, T., Robins, S. C., Schneider, S., et al. (2016). Adult NG2-glia are required for median eminence-mediated leptin sensing and body weight control. Cell Metabolism, 23, 797–810.
Donath, M. Y., Böni-Schnetzler, M., Ellingsgaard, H., et al. (2010). Cytokine production by islets in health and diabetes: Cellular origin, regulation and function. Trends in Endocrinology and Metabolism, 21, 261–267.
Donnelly, N., Gorman, A. M., Gupta, S., & Samali, A. (2013). The eIF2α kinases: Their structures and functions. Cellular and Molecular Life Sciences, 70, 3493–3511.
Du, Y., & Dreyfus, C. F. (2002). Oligodendrocytes as providers of growth factors. Journal of Neuroscience Research, 68, 647–654.
Dutheil, S., Ota, K. T., Wohleb, E. S., et al. (2016). High-fat diet induced anxiety and anhedonia: Impact on brain homeostasis and inflammation. Neuropsychopharmacology, 41, 1874–1887.
Ehses, J. A., Böni-Schnetzler, M., Faulenbach, M., & Donath, M. Y. (2008). Macrophages, cytokines and beta-cell death in type 2 diabetes. Biochemical Society Transactions, 36, 340–342.
Elmore, M. R. P., Najafi, A. R., Koike, M. A., et al. (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82, 380–397.
Elmquist, J. K., Ackermann, M. R., Register, K. B., et al. (1993). Induction of Fos-like immunoreactivity in the rat brain following Pasteurella multocida endotoxin administration. Endocrinology, 133, 3054–3057.
Enriori, P. J., Evans, A. E., Sinnayah, P., et al. (2007). Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metabolism, 5, 181–194.
Erblich, B., Zhu, L., Etgen, A. M., et al. (2011). Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One, 6, e26317.
Erion, J. R., Wosiski-Kuhn, M., Dey, A., et al. (2014). Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. The Journal of Neuroscience, 34, 2618–2631.
Filadi, R., Greotti, E., Turacchio, G., et al. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proceedings of the National Academy of Sciences of the United States of America, 112, E2174–E2181.
Fiorese, C. J., & Haynes, C. M. (2017). Integrating the UPR(mt) into the mitochondrial maintenance network. Critical Reviews in Biochemistry and Molecular Biology, 52, 304–313.
Frago, L. M., & Chowen, J. A. (2017). Involvement of astrocytes in mediating the central effects of ghrelin. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms18030536.
Francisco, A. B., Singh, R., Li, S., et al. (2010). Deficiency of suppressor enhancer Lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. The Journal of Biological Chemistry, 285, 13694–13703.
Frayling, C., Britton, R., & Dale, N. (2011). ATP-mediated glucosensing by hypothalamic tanycytes. The Journal of Physiology, 589, 2275–2286.
Fusakio, M. E., Willy, J. A., Wang, Y., et al. (2016). Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Molecular Biology of the Cell, 27, 1536–1551.
Gao, S., Howard, S., & LoGrasso, P. V. (2017). Pharmacological inhibition of c-Jun N-terminal kinase reduces food intake and sensitizes leptin’s anorectic signaling actions. Scientific Reports, 7, 41795.
García-Cáceres, C., Quarta, C., Varela, L., et al. (2016). Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell, 166, 867–880.
Ginhoux, F., Greter, M., Leboeuf, M., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330, 841–845.
Gregor, M. F., Misch, E. S., Yang, L., et al. (2013). The role of adipocyte XBP1 in metabolic regulation during lactation. Cell Reports, 3, 1430–1439.
Guillemot-Legris, O., Masquelier, J., Everard, A., et al. (2016). High-fat diet feeding differentially affects the development of inflammation in the central nervous system. Journal of Neuroinflammation, 13, 206.
Gunstad, J., Paul, R. H., Cohen, R. A., et al. (2006). Obesity is associated with memory deficits in young and middle-aged adults. Eating and Weight Disorders, 11, e15–e19.
Gustin, A., Kirchmeyer, M., Koncina, E., et al. (2015). NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One, 10, e0130624.
Halle, A., Hornung, V., Petzold, G. C., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nature Immunology, 9, 857–865.
Han, M. S., Jung, D. Y., Morel, C., et al. (2013). JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science, 339, 218–222.
Harding, H. P., Zhang, Y., & Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature, 397, 271–274.
Harding, H. P., Novoa, I., Zhang, Y., et al. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular Cell, 6, 1099–1108.
Harding, H. P., Zhang, Y., Zeng, H., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular Cell, 11, 619–633.
Hayashi, A., Kasahara, T., Iwamoto, K., et al. (2007). The role of brain-derived neurotrophic factor (BDNF)-induced XBP1 splicing during brain development. The Journal of Biological Chemistry, 282, 34525–34534.
Haze, K., Yoshida, H., Yanagi, H., et al. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular Biology of the Cell, 10, 3787–3799.
Helms, J. B., & Rothman, J. E. (1992). Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature, 360, 352–354.
Henry, F. E., Sugino, K., Tozer, A., et al. (2015). Cell type-specific transcriptomics of hypothalamic energy-sensing neuron responses to weight-loss. eLife. https://doi.org/10.7554/eLife.09800.
Hetz, C., Lee, A.-H., Gonzalez-Romero, D., et al. (2008). Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proceedings of the National Academy of Sciences of the United States of America, 105, 757–762.
Hillhouse, E. W., & Mosley, K. (1993). Peripheral endotoxin induces hypothalamic immunoreactive interleukin-1 beta in the rat. British Journal of Pharmacology, 109, 289–290.
Hirosumi, J., Tuncman, G., Chang, L., et al. (2002). A central role for JNK in obesity and insulin resistance. Nature, 420, 333–336.
Horwath, J. A., Hurr, C., Butler, S. D., et al. (2017). Obesity-induced hepatic steatosis is mediated by endoplasmic reticulum stress in the subfornical organ of the brain. JCI Insight. https://doi.org/10.1172/jci.insight.90170.
Hosoi, T., Okuma, Y., Kawagishi, T., et al. (2004). Bacterial endotoxin induces STAT3 activation in the mouse brain. Brain Research, 1023, 48–53.
Hosoi, T., Sasaki, M., Miyahara, T., et al. (2008). Endoplasmic reticulum stress induces leptin resistance. Molecular Pharmacology, 74, 1610–1619.
Hosoi, T., Ogawa, K., & Ozawa, K. (2010). Homocysteine induces X-box-binding protein 1 splicing in the mice brain. Neurochemistry International, 56, 216–220.
Hotamisligil, G. S. (2017). Inflammation, metaflammation and immunometabolic disorders. Nature, 542, 177–185.
Hotamisligil, G. S., Shargill, N. S., & Spiegelman, B. M. (1993). Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science, 259, 87–91.
Hotamisligil, G. S., Budavari, A., Murray, D., & Spiegelman, B. M. (1994). Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. The Journal of Clinical Investigation, 94, 1543–1549.
Hotamisligil, G. S., Peraldi, P., Budavari, A., et al. (1996). IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science, 271, 665–668.
Hu, P., Han, Z., Couvillon, A. D., et al. (2006). Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Molecular and Cellular Biology, 26, 3071–3084.
Hughes, E. G., Kang, S. H., Fukaya, M., & Bergles, D. E. (2013). Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nature Neuroscience, 16, 668–676.
Hunot, S., Vila, M., Teismann, P., et al. (2004). JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, 101, 665–670.
Iwakoshi, N. N., Lee, A.-H., Vallabhajosyula, P., et al. (2003). Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nature Immunology, 4, 321–329.
Jang, P.-G., Namkoong, C., Kang, G. M., et al. (2010). NF-kappaB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness- and leptin-induced anorexia. The Journal of Biological Chemistry, 285, 9706–9715.
Jeon, B. T., Jeong, E. A., Shin, H. J., et al. (2012). Resveratrol attenuates obesity-associated peripheral and central inflammation and improves memory deficit in mice fed a high-fat diet. Diabetes, 61, 1444–1454.
Jiang, H.-Y., Wek, S. A., McGrath, B. C., et al. (2003). Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Molecular and Cellular Biology, 23, 5651–5663.
Jin, S., Kim, J. G., Park, J. W., et al. (2016). Hypothalamic TLR2 triggers sickness behavior via a microglia-neuronal axis. Scientific Reports, 6, 29424.
Jousse, C., Oyadomari, S., Novoa, I., et al. (2003). Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. The Journal of Cell Biology, 163, 767–775.
Kanczkowski, W., Alexaki, V.-I., Tran, N., et al. (2013). Hypothalamo-pituitary and immune-dependent adrenal regulation during systemic inflammation. Proceedings of the National Academy of Sciences of the United States of America, 110, 14801–14806.
Kang, E.-B., Koo, J.-H., Jang, Y.-C., et al. (2016). Neuroprotective effects of endurance exercise against high-fat diet-induced hippocampal neuroinflammation. Journal of Neuroendocrinology. https://doi.org/10.1111/jne.12385.
Karatas, H., Erdener, S. E., Gursoy-Ozdemir, Y., et al. (2013). Spreading depression triggers headache by activating neuronal Panx1 channels. Science, 339, 1092–1095.
Kawamata, Y., Fujii, R., Hosoya, M., et al. (2003). A G protein-coupled receptor responsive to bile acids. The Journal of Biological Chemistry, 278, 9435–9440.
Keirstead, H. S., Levine, J. M., & Blakemore, W. F. (1998). Response of the oligodendrocyte progenitor cell population (defined by NG2 labelling) to demyelination of the adult spinal cord. Glia, 22, 161–170.
Kent, S., Rodriguez, F., Kelley, K. W., & Dantzer, R. (1994). Reduction in food and water intake induced by microinjection of interleukin-1 beta in the ventromedial hypothalamus of the rat. Physiology & Behavior, 56, 1031–1036.
Kent, S., Bret-Dibat, J. L., Kelley, K. W., & Dantzer, R. (1996). Mechanisms of sickness-induced decreases in food-motivated behavior. Neuroscience and Biobehavioral Reviews, 20, 171–175.
Kharitonenkov, A., Shiyanova, T. L., Koester, A., et al. (2005). FGF-21 as a novel metabolic regulator. The Journal of Clinical Investigation, 115, 1627–1635.
Kierdorf, K., Erny, D., Goldmann, T., et al. (2013). Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nature Neuroscience, 16, 273–280.
Kim, M.-S., Pak, Y. K., Jang, P.-G., et al. (2006). Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nature Neuroscience, 9, 901–906.
Kim, J. G., Suyama, S., Koch, M., et al. (2014). Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nature Neuroscience, 17, 908–910.
Kim, M. S., Yan, J., Wu, W., et al. (2015). Rapid linkage of innate immunological signals to adaptive immunity by the brain-fat axis. Nature Immunology, 16, 525–533.
Kim, G. H., Shi, G., Somlo, D. R. et al. (2018). Hypothalamic ER-associated degradation regulates POMC maturation, feeding, and age-associated obesity. Mar 1;128(3):1125–1140. https://doi.org/10.1172/JCI96420. Epub 2018 Feb 19. PMID: 29457782 Free PMC Article.
Kimata, Y., Ishiwata-Kimata, Y., Ito, T., et al. (2007). Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. The Journal of Cell Biology, 179, 75–86.
Kitamura, T., Feng, Y., Kitamura, Y. I., et al. (2006). Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nature Medicine, 12, 534–540.
Kleinridders, A., Schenten, D., Könner, A. C., et al. (2009). MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metabolism, 10, 249–259.
Kokoeva, M. V., Yin, H., & Flier, J. S. (2005). Neurogenesis in the hypothalamus of adult mice: Potential role in energy balance. Science, 310, 679–683.
Kreutzer, C., Peters, S., Schulte, D. M., et al. (2017). Hypothalamic inflammation in human obesity is mediated by environmental and genetic factors. Diabetes, 66, 2407–2415.
Kuan, C. Y., Yang, D. D., Samanta Roy, D. R., et al. (1999). The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron, 22, 667–676.
Kuan, C.-Y., Whitmarsh, A. J., Yang, D. D., et al. (2003). A critical role of neural-specific JNK3 for ischemic apoptosis. Proceedings of the National Academy of Sciences of the United States of America, 100, 15184–15189.
Kummer, J. A., Broekhuizen, R., Everett, H., et al. (2007). Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. The Journal of Histochemistry and Cytochemistry, 55, 443–452.
Ladrière, L., Igoillo-Esteve, M., Cunha, D. A., et al. (2010). Enhanced signaling downstream of ribonucleic acid-activated protein kinase-like endoplasmic reticulum kinase potentiates lipotoxic endoplasmic reticulum stress in human islets. The Journal of Clinical Endocrinology and Metabolism, 95, 1442–1449.
Lam, T. K. T., Gutierrez-Juarez, R., Pocai, A., & Rossetti, L. (2005). Regulation of blood glucose by hypothalamic pyruvate metabolism. Science, 309, 943–947.
Langlet, F., Levin, B. E., Luquet, S., et al. (2013a). Tanycytic VEGF-A boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell Metabolism, 17, 607–617.
Langlet, F., Mullier, A., Bouret, S. G., et al. (2013b). Tanycyte-like cells form a blood-cerebrospinal fluid barrier in the circumventricular organs of the mouse brain. The Journal of Comparative Neurology, 521, 3389–3405.
Lawson, L. J., Perry, V. H., Dri, P., & Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience, 39, 151–170.
Lawson, L. J., Perry, V. H., & Gordon, S. (1992). Turnover of resident microglia in the normal adult mouse brain. Neuroscience, 48, 405–415.
Layé, S., Parnet, P., Goujon, E., & Dantzer, R. (1994). Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Research. Molecular Brain Research, 27, 157–162.
Le Foll, C., & Levin, B. E. (2016). Fatty acid-induced astrocyte ketone production and the control of food intake. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 310, R1186–R1192.
Lee, S. Y., Reichlin, A., Santana, A., et al. (1997). TRAF2 is essential for JNK but not NF-kappaB activation and regulates lymphocyte proliferation and survival. Immunity, 7, 703–713.
Lee, A.-H., Iwakoshi, N. N., & Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and Cellular Biology, 23, 7448–7459.
Lee, A.-H., Scapa, E. F., Cohen, D. E., & Glimcher, L. H. (2008). Regulation of hepatic lipogenesis by the transcription factor XBP1. Science, 320, 1492–1496.
Lee, Y.-Y., Cevallos, R. C., & Jan, E. (2009). An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. The Journal of Biological Chemistry, 284, 6661–6673.
Lee, A.-H., Heidtman, K., Hotamisligil, G. S., & Glimcher, L. H. (2011). Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proceedings of the National Academy of Sciences of the United States of America, 108, 8885–8890.
Lee, D. A., Bedont, J. L., Pak, T., et al. (2012). Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nature Neuroscience, 15, 700–702.
Lehnardt, S. (2010). Innate immunity and neuroinflammation in the CNS: The role of microglia in toll-like receptor-mediated neuronal injury. Glia, 58, 253–263.
Lehnardt, S., Massillon, L., Follett, P., et al. (2003). Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proceedings of the National Academy of Sciences of the United States of America, 100, 8514–8519.
Levine, J. M., & Reynolds, R. (1999). Activation and proliferation of endogenous oligodendrocyte precursor cells during ethidium bromide-induced demyelination. Experimental Neurology, 160, 333–347.
Li, J., Tang, Y., & Cai, D. (2012). IKKβ/NF-κB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nature Cell Biology, 14, 999–1012.
Li, J., Labbadia, J., & Morimoto, R. I. (2017). Rethinking HSF1 in stress, development, and organismal health. Trends in Cell Biology. https://doi.org/10.1016/j.tcb.2017.08.002.
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487.
Lindqvist, A., Mohapel, P., Bouter, B., et al. (2006). High-fat diet impairs hippocampal neurogenesis in male rats. European Journal of Neurology, 13, 1385–1388.
Lu, P. D., Harding, H. P., & Ron, D. (2004). Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. The Journal of Cell Biology, 167, 27–33.
Lu, Y., Liang, F.-X., & Wang, X. (2014). A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Molecular Cell, 55, 758–770.
Lu, W., Hagiwara, D., Morishita, Y., et al. (2016). Unfolded protein response in hypothalamic cultures of wild-type and ATF6α-knockout mice. Neuroscience Letters, 612, 199–203.
Lundgaard, I., Li, B., Xie, L., et al. (2015). Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nature Communications, 6, 6807.
Malhotra, R., Warne, J. P., Salas, E., et al. (2015). Loss of Atg12, but not Atg5, in pro-opiomelanocortin neurons exacerbates diet-induced obesity. Autophagy, 11, 145–154.
Maolood, N., & Meister, B. (2009). Protein components of the blood-brain barrier (BBB) in the brainstem area postrema-nucleus tractus solitarius region. Journal of Chemical Neuroanatomy, 37, 182–195.
Martínez, G., Vidal, R. L., Mardones, P., et al. (2016). Regulation of memory formation by the transcription factor XBP1. Cell Reports, 14, 1382–1394.
Martínez-Sánchez, N., Seoane-Collazo, P., Contreras, C., et al. (2017). Hypothalamic AMPK-ER stress-JNK1 axis mediates the central actions of thyroid hormones on energy balance. Cell Metabolism, 26, 212–229.e12.
Martinon, F., Burns, K., & Tschopp, J. (2002). The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell, 10, 417–426.
Martinon, F., Chen, X., Lee, A.-H., & Glimcher, L. H. (2010). TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nature Immunology, 11, 411–418.
Masson, G. S., Nair, A. R., Dange, R. B., et al. (2015). Toll-like receptor 4 promotes autonomic dysfunction, inflammation and microglia activation in the hypothalamic paraventricular nucleus: Role of endoplasmic reticulum stress. PLoS One, 10, e0122850.
Maurel, M., Chevet, E., Tavernier, J., & Gerlo, S. (2014). Getting RIDD of RNA: IRE1 in cell fate regulation. Trends in Biochemical Sciences, 39, 245–254.
Mauro, C., De Rosa, V., Marelli-Berg, F., & Solito, E. (2014). Metabolic syndrome and the immunological affair with the blood-brain barrier. Frontiers in Immunology, 5, 677.
Mayer, C. M., & Belsham, D. D. (2010). Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: Rescue of resistance and apoptosis through adenosine 5′ monophosphate-activated protein kinase activation. Endocrinology, 151, 576–585.
McGavigan, A. K., Henseler, Z. M., Garibay, D., et al. (2017). Vertical sleeve gastrectomy reduces blood pressure and hypothalamic endoplasmic reticulum stress in mice. Disease Models & Mechanisms, 10, 235–243.
Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature, 454, 428–435.
Meng, Q., & Cai, D. (2011). Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. The Journal of Biological Chemistry, 286, 32324–32332.
Milanski, M., Degasperi, G., Coope, A., et al. (2009). Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. The Journal of Neuroscience, 29, 359–370.
Milanski, M., Arruda, A. P., Coope, A., et al. (2012). Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes, 61, 1455–1462.
Minkiewicz, J., de Rivero Vaccari, J. P., & Keane, R. W. (2013). Human astrocytes express a novel NLRP2 inflammasome. Glia, 61, 1113–1121.
Moraes, J. C., Coope, A., Morari, J., et al. (2009). High-fat diet induces apoptosis of hypothalamic neurons. PLoS One, 4, e5045.
Morari, J., Anhe, G. F., Nascimento, L. F., et al. (2014). Fractalkine (CX3CL1) is involved in the early activation of hypothalamic inflammation in experimental obesity. Diabetes, 63, 3770–3784.
Morishita, Y., Arima, H., Hiroi, M., et al. (2011). Poly(A) tail length of neurohypophysial hormones is shortened under endoplasmic reticulum stress. Endocrinology, 152, 4846–4855.
Morita, S., Villalta, S. A., Feldman, H. C., et al. (2017). Targeting ABL-IRE1α signaling spares ER-stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metabolism, 25, 883–897.e8.
Morl, K., Ma, W., Gething, M. J., et al. (1993). A transmembrane protein with a cdc2+ cdc28-related kinase activity is required for signaling from the ER to the nucleus. Cell, 74(4), 743–756.
Nadeau, S., & Rivest, S. (1999). Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: A view from the blood-brain barrier. Neuroscience, 93, 1449–1464.
Nakamura, D., Tsuru, A., Ikegami, K., et al. (2011). Mammalian ER stress sensor IRE1β specifically down-regulates the synthesis of secretory pathway proteins. FEBS Letters, 585, 133–138.
Naranjo, J. R., Zhang, H., Villar, D., et al. (2016). Activating transcription factor 6 derepression mediates neuroprotection in Huntington disease. The Journal of Clinical Investigation, 126, 627–638.
Naznin, F., Toshinai, K., Waise, T. M. Z., et al. (2015). Diet-induced obesity causes peripheral and central ghrelin resistance by promoting inflammation. The Journal of Endocrinology, 226, 81–92.
Nelson, G., Wilde, G. J. C., Spiller, D. G., et al. (2003). NF-kappaB signalling is inhibited by glucocorticoid receptor and STAT6 via distinct mechanisms. Journal of Cell Science, 116, 2495–2503.
Neumann, H., Cavalié, A., Jenne, D. E., & Wekerle, H. (1995). Induction of MHC class I genes in neurons. Science, 269, 549–552.
Nguyen, J. C. D., Killcross, A. S., & Jenkins, T. A. (2014). Obesity and cognitive decline: Role of inflammation and vascular changes. Frontiers in Neuroscience, 8, 375.
Nishiyama, A., Komitova, M., Suzuki, R., & Zhu, X. (2009). Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nature Reviews. Neuroscience, 10, 9–22.
Novoa, I., Zeng, H., Harding, H. P., & Ron, D. (2001). Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. The Journal of Cell Biology, 153, 1011–1022.
Ogimoto, K., Harris, M. K., Jr., & Wisse, B. E. (2006). MyD88 is a key mediator of anorexia, but not weight loss, induced by lipopolysaccharide and interleukin-1 beta. Endocrinology, 147, 4445–4453.
Okin, D., & Medzhitov, R. (2016). The effect of sustained inflammation on hepatic mevalonate pathway results in hyperglycemia. Cell, 165, 343–356.
Olivares, S., & Henkel, A. S. (2015). Hepatic Xbp1 gene deletion promotes endoplasmic reticulum stress-induced liver injury and apoptosis. The Journal of Biological Chemistry, 290, 30142–30151.
Ono, H., Pocai, A., Wang, Y., et al. (2008). Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. The Journal of Clinical Investigation, 118, 2959–2968.
Oral, E. A., Reilly, S. M., Gomez, A. V., et al. (2017). Inhibition of IKKɛ and TBK1 improves glucose control in a subset of patients with type 2 diabetes. Cell Metabolism, 26, 157–170.e7.
Outinen, P. A., Sood, S. K., Liaw, P. C., et al. (1998). Characterization of the stress-inducing effects of homocysteine. The Biochemical Journal, 332(Pt 1), 213–221.
Ozcan, U., Cao, Q., Yilmaz, E., et al. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science, 306, 457–461.
Padilla, S. L., Reef, D., & Zeltser, L. M. (2012). Defining POMC neurons using transgenic reagents: Impact of transient Pomc expression in diverse immature neuronal populations. Endocrinology, 153, 1219–1231.
Pakos-Zebrucka, K., Koryga, I., Mnich, K., et al. (2016). The integrated stress response. EMBO Reports, 17, 1374–1395.
Paolicelli, R. C., Bolasco, G., Pagani, F., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science, 333, 1456–1458.
Park, H. R., Park, M., Choi, J., et al. (2010). A high-fat diet impairs neurogenesis: Involvement of lipid peroxidation and brain-derived neurotrophic factor. Neuroscience Letters, 482, 235–239.
Pascual, O., Ben Achour, S., Rostaing, P., et al. (2012). Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proceedings of the National Academy of Sciences of the United States of America, 109, E197–E205.
Perello, M., Cakir, I., Cyr, N. E., et al. (2010). Maintenance of the thyroid axis during diet-induced obesity in rodents is controlled at the central level. American Journal of Physiology. Endocrinology and Metabolism, 299, E976–E989.
Pérez-Domínguez, M., Tovar-Y-Romo, L. B., & Zepeda, A. (2017). Neuroinflammation and physical exercise as modulators of adult hippocampal neural precursor cell behavior. Reviews in the Neurosciences. https://doi.org/10.1515/revneuro-2017-0024.
Pistell, P. J., Morrison, C. D., Gupta, S., et al. (2010). Cognitive impairment following high fat diet consumption is associated with brain inflammation. Journal of Neuroimmunology, 219, 25–32.
Posey, K. A., Clegg, D. J., Printz, R. L., et al. (2009). Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. American Journal of Physiology. Endocrinology and Metabolism, 296, E1003–E1012.
Pozzan, T., Rizzuto, R., Volpe, P., & Meldolesi, J. (1994). Molecular and cellular physiology of intracellular calcium stores. Physiological Reviews, 74, 595–636.
Prada, P. O., Zecchin, H. G., Gasparetti, A. L., et al. (2005). Western diet modulates insulin signaling, c-Jun N-terminal kinase activity, and insulin receptor substrate-1ser307 phosphorylation in a tissue-specific fashion. Endocrinology, 146, 1576–1587.
Puig, J., Blasco, G., Daunis-I-Estadella, J., et al. (2015). Hypothalamic damage is associated with inflammatory markers and worse cognitive performance in obese subjects. The Journal of Clinical Endocrinology and Metabolism, 100, E276–E281.
Purkayastha, S., Zhang, G., & Cai, D. (2011a). Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-β and NF-κB. Nature Medicine, 17, 883–887.
Purkayastha, S., Zhang, H., Zhang, G., et al. (2011b). Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proceedings of the National Academy of Sciences of the United States of America, 108, 2939–2944.
Quagliarello, V. J., Wispelwey, B., Long, W. J., Jr., & Scheld, W. M. (1991). Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in the rat. Characterization and comparison with tumor necrosis factor. The Journal of Clinical Investigation, 87, 1360–1366.
Quarta, C., Clemmensen, C., Zhu, Z., et al. (2017). Molecular integration of incretin and glucocorticoid action reverses immunometabolic dysfunction and obesity. Cell Metabolism, 26, 620–632.e6.
Ramírez, S., Gómez-Valadés, A. G., Schneeberger, M., et al. (2017). Mitochondrial dynamics mediated by Mitofusin 1 is required for POMC neuron glucose-sensing and insulin release control. Cell Metabolism, 25, 1390–1399.e6.
Ramos, H. J., Lanteri, M. C., Blahnik, G., et al. (2012). IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathogens, 8, e1003039.
Ray, A., & Prefontaine, K. E. (1994). Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proceedings of the National Academy of Sciences of the United States of America, 91, 752–756.
Reilly, S. M., Chiang, S.-H., Decker, S. J., et al. (2013). An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nature Medicine, 19, 313–321.
Reimold, A. M., Etkin, A., Clauss, I., et al. (2000). An essential role in liver development for transcription factor XBP-1. Genes & Development, 14, 152–157.
Reimold, A. M., Iwakoshi, N. N., Manis, J., et al. (2001). Plasma cell differentiation requires the transcription factor XBP-1. Nature, 412, 300–307.
Ren, H., Orozco, I. J., Su, Y., et al. (2012). FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell, 149, 1314–1326.
Riediger, T. (2012). The receptive function of hypothalamic and brainstem centres to hormonal and nutrient signals affecting energy balance. The Proceedings of the Nutrition Society, 71, 463–477.
Rojas, M., Vasconcelos, G., & Dever, T. E. (2015). An eIF2α-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2α. Proceedings of the National Academy of Sciences of the United States of America, 112, E3466–E3475.
Rolls, A., Shechter, R., London, A., et al. (2007). Toll-like receptors modulate adult hippocampal neurogenesis. Nature Cell Biology, 9, 1081–1088.
Ropelle, E. R., Flores, M. B., Cintra, D. E., et al. (2010). IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biology. https://doi.org/10.1371/journal.pbio.1000465.
Rottkamp, D. M., Rudenko, I. A., Maier, M. T., et al. (2015). Leptin potentiates astrogenesis in the developing hypothalamus. Mol Metab, 4, 881–889.
Russo, I., Barlati, S., & Bosetti, F. (2011). Effects of neuroinflammation on the regenerative capacity of brain stem cells. Journal of Neurochemistry, 116, 947–956.
Sabio, G., Das, M., Mora, A., et al. (2008). A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science, 322, 1539–1543.
Sabio, G., Cavanagh-Kyros, J., Ko, H. J., et al. (2009). Prevention of steatosis by hepatic JNK1. Cell Metabolism, 10, 491–498.
Sabio, G., Cavanagh-Kyros, J., Barrett, T., et al. (2010a). Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes & Development, 24, 256–264.
Sabio, G., Kennedy, N. J., Cavanagh-Kyros, J., et al. (2010b). Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Molecular and Cellular Biology, 30, 106–115.
Sachot, C., Poole, S., & Luheshi, G. N. (2004). Circulating leptin mediates lipopolysaccharide-induced anorexia and fever in rats. The Journal of Physiology, 561, 263–272.
Sacoccio, C., Dornand, J., & Barbanel, G. (1998). Differential regulation of brain and plasma TNFalpha produced after endotoxin shock. Neuroreport, 9, 309–313.
Sagar, S. M., & Sharp, F. R. (1993). Early response genes as markers of neuronal activity and growth factor action. Advances in Neurology, 59, 273–284.
Schaap, F. G., Kremer, A. E., Lamers, W. H., et al. (2013). Fibroblast growth factor 21 is induced by endoplasmic reticulum stress. Biochimie, 95, 692–699.
Schenk, S., Saberi, M., & Olefsky, J. M. (2008). Insulin sensitivity: Modulation by nutrients and inflammation. The Journal of Clinical Investigation, 118, 2992–3002.
Schneeberger, M., Dietrich, M. O., Sebastián, D., et al. (2013). Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell, 155, 172–187.
Schneeberger, M., Gómez-Valadés, A. G., Altirriba, J., et al. (2015). Reduced α-MSH underlies hypothalamic ER-stress-induced hepatic gluconeogenesis. Cell Reports, 12, 361–370.
Sebastián, D., Hernández-Alvarez, M. I., Segalés, J., et al. (2012). Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proceedings of the National Academy of Sciences of the United States of America, 109, 5523–5528.
Serhan, C. N., Brain, S. D., Buckley, C. D., et al. (2007). Resolution of inflammation: State of the art, definitions and terms. The FASEB Journal, 21, 325–332.
Shan, B., Wang, X., Wu, Y., et al. (2017). The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nature Immunology, 18, 519–529.
Shen, J., Chen, X., Hendershot, L., & Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Developmental Cell, 3, 99–111.
Shi, H., Kokoeva, M. V., Inouye, K., et al. (2006). TLR4 links innate immunity and fatty acid-induced insulin resistance. The Journal of Clinical Investigation, 116, 3015–3025.
Shi, G., Somlo, D., Kim, G. H., et al. (2017). ER-associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. The Journal of Clinical Investigation, 127, 3897–3912.
Shimazaki, T., Shingo, T., & Weiss, S. (2001). The ciliary neurotrophic factor/leukemia inhibitory factor/gp130 receptor complex operates in the maintenance of mammalian forebrain neural stem cells. The Journal of Neuroscience, 21, 7642–7653.
Sidrauski, C., Acosta-Alvear, D., Khoutorsky, A., et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife, 2, e00498.
Silverman, W. R., de Rivero Vaccari, J. P., Locovei, S., et al. (2009). The pannexin 1 channel activates the inflammasome in neurons and astrocytes. The Journal of Biological Chemistry, 284, 18143–18151.
Smith, C. A., Farrah, T., & Goodwin, R. G. (1994). The TNF receptor superfamily of cellular and viral proteins: Activation, costimulation, and death. Cell, 76, 959–962.
Song, L., Pei, L., Yao, S., et al. (2017). NLRP3 inflammasome in neurological diseases, from functions to therapies. Frontiers in Cellular Neuroscience, 11, 63.
Spencer, S. J., D’Angelo, H., Soch, A., et al. (2017). High-fat diet and aging interact to produce neuroinflammation and impair hippocampal- and amygdalar-dependent memory. Neurobiology of Aging, 58, 88–101.
Speretta, G. F., Silva, A. A., Vendramini, R. C., et al. (2016). Resistance training prevents the cardiovascular changes caused by high-fat diet. Life Sciences, 146, 154–162.
Sriburi, R., Jackowski, S., Mori, K., & Brewer, J. W. (2004). XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. The Journal of Cell Biology, 167, 35–41.
Steiner, A. A., Dogan, M. D., Ivanov, A. I., et al. (2004). A new function of the leptin receptor: Mediation of the recovery from lipopolysaccharide-induced hypothermia. The FASEB Journal, 18, 1949–1951.
Stutz, A., Golenbock, D. T., & Latz, E. (2009). Inflammasomes: Too big to miss. The Journal of Clinical Investigation, 119, 3502–3511.
Takatsuki, A., & Tamura, G. (1971). Effect of tunicamycin on the synthesis of macromolecules in cultures of chick embryo fibroblasts infected with Newcastle disease virus. The Journal of Antibiotics, 24, 785–794.
Taniuchi, S., Miyake, M., Tsugawa, K., et al. (2016). Integrated stress response of vertebrates is regulated by four eIF2α kinases. Scientific Reports, 6, 32886.
Taylor, R. C., & Dillin, A. (2013). XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell, 153, 1435–1447.
Thaler, J. P., Yi, C.-X., Schur, E. A., et al. (2012). Obesity is associated with hypothalamic injury in rodents and humans. The Journal of Clinical Investigation, 122, 153–162.
Tsaousidou, E., Paeger, L., Belgardt, B. F., et al. (2014). Distinct roles for JNK and IKK activation in agouti-related peptide neurons in the development of obesity and insulin resistance. Cell Reports, 9, 1495–1506.
Unger, E. K., Piper, M. L., Olofsson, L. E., & Xu, A. W. (2010). Functional role of c-Jun-N-terminal kinase in feeding regulation. Endocrinology, 151, 671–682.
Upton, J.-P., Wang, L., Han, D., et al. (2012). IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science, 338, 818–822.
Urano, F., Wang, X., Bertolotti, A., et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science, 287, 664–666.
Uysal, K. T., Wiesbrock, S. M., Marino, M. W., & Hotamisligil, G. S. (1997). Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature, 389, 610–614.
Valdearcos, M., Robblee, M. M., Benjamin, D. I., et al. (2014). Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Reports, 9, 2124–2138.
Valdearcos, M., Douglass, J. D., Robblee, M. M., et al. (2017). Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metabolism, 26, 185–197.e3.
Valdés, P., Mercado, G., Vidal, R. L., et al. (2014). Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proceedings of the National Academy of Sciences of the United States of America, 111, 6804–6809.
van Oosten-Hawle, P., & Morimoto, R. I. (2014). Organismal proteostasis: Role of cell-nonautonomous regulation and transcellular chaperone signaling. Genes & Development, 28, 1533–1543.
van Oosten-Hawle, P., Porter, R. S., & Morimoto, R. I. (2013). Regulation of organismal proteostasis by transcellular chaperone signaling. Cell, 153, 1366–1378.
Vandanmagsar, B., Youm, Y.-H., Ravussin, A., et al. (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature Medicine, 17, 179–188.
Varatharaj, A., & Galea, I. (2017). The blood-brain barrier in systemic inflammation. Brain, Behavior, and Immunity, 60, 1–12.
Vattem, K. M., & Wek, R. C. (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America, 101, 11269–11274.
Vernia, S., Cavanagh-Kyros, J., Garcia-Haro, L., et al. (2014). The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metabolism, 20, 512–525.
Vernia, S., Morel, C., Madara, J. C., et al. (2016). Excitatory transmission onto AgRP neurons is regulated by cJun NH2-terminal kinase 3 in response to metabolic stress. eLife, 5, e10031.
Veronese, N., Facchini, S., Stubbs, B., et al. (2017). Weight loss is associated with improvements in cognitive function among overweight and obese people: A systematic review and meta-analysis. Neuroscience and Biobehavioral Reviews, 72, 87–94.
Wake, H., Moorhouse, A. J., Miyamoto, A., & Nabekura, J. (2013). Microglia: Actively surveying and shaping neuronal circuit structure and function. Trends in Neurosciences, 36, 209–217.
Wallingford, N., Perroud, B., Gao, Q., et al. (2009). Prolylcarboxypeptidase regulates food intake by inactivating alpha-MSH in rodents. The Journal of Clinical Investigation, 119, 2291–2303.
Walton, N. M., Sutter, B. M., Laywell, E. D., et al. (2006). Microglia instruct subventricular zone neurogenesis. Glia, 54, 815–825.
Wang, Y., Vera, L., Fischer, W. H., & Montminy, M. (2009). The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature, 460, 534–537.
Wang, S., Chen, Z., Lam, V., et al. (2012). IRE1α-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metabolism, 16, 473–486.
Wang, S., Huang, X.-F., Zhang, P., et al. (2017). Dietary teasaponin ameliorates alteration of gut microbiota and cognitive decline in diet-induced obese mice. Scientific Reports, 7, 12203.
Weissmann, L., Quaresma, P. G. F., Santos, A. C., et al. (2014). IKKε is key to induction of insulin resistance in the hypothalamus, and its inhibition reverses obesity. Diabetes, 63, 3334–3345.
Welch, W. J., & Brown, C. R. (1996). Influence of molecular and chemical chaperones on protein folding. Cell Stress & Chaperones, 1, 109–115.
Wilkins, A., Chandran, S., & Compston, A. (2001). A role for oligodendrocyte-derived IGF-1 in trophic support of cortical neurons. Glia, 36, 48–57.
Wilkins, A., Majed, H., Layfield, R., et al. (2003). Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechanisms: A novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. The Journal of Neuroscience, 23, 4967–4974.
Williams, K. W., Liu, T., Kong, X., et al. (2014). Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metabolism, 20, 471–482.
Won, J. C., Jang, P.-G., Namkoong, C., et al. (2009). Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity, 17, 1861–1865.
Wong, M. L., Bongiorno, P. B., Rettori, V., et al. (1997). Interleukin (IL) 1beta, IL-1 receptor antagonist, IL-10, and IL-13 gene expression in the central nervous system and anterior pituitary during systemic inflammation: Pathophysiological implications. Proceedings of the National Academy of Sciences of the United States of America, 94, 227–232.
Woo, C. W., Cui, D., Arellano, J., et al. (2009). Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nature Cell Biology, 11, 1473–1480.
Wu, K. L. H., Chan, S. H. H., & Chan, J. Y. H. (2012). Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. Journal of Neuroinflammation, 9, 212.
Wunderlich, F. T., Luedde, T., Singer, S., et al. (2008). Hepatic NF-kappa B essential modulator deficiency prevents obesity-induced insulin resistance but synergizes with high-fat feeding in tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America, 105, 1297–1302.
Xiao, Y., Deng, Y., Yuan, F., et al. (2017a). ATF4/ATG5 signaling in hypothalamic proopiomelanocortin neurons regulates fat mass via affecting energy expenditure. Diabetes, 66, 1146–1158.
Xiao, Y., Deng, Y., Yuan, F., et al. (2017b). An ATF4-ATG5 signaling in hypothalamic POMC neurons regulates obesity. Autophagy, 13, 1088–1089.
Xu, H., Barnes, G. T., Yang, Q., et al. (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of Clinical Investigation, 112, 1821–1830.
Xu, T., Yang, L., Yan, C., et al. (2014). The IRE1α-XBP1 pathway regulates metabolic stress-induced compensatory proliferation of pancreatic β-cells. Cell Research, 24, 1137–1140.
Yagishita, N., Ohneda, K., Amano, T., et al. (2005). Essential role of synoviolin in embryogenesis. The Journal of Biological Chemistry, 280, 7909–7916.
Yamamoto, K., Sato, T., Matsui, T., et al. (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Developmental Cell, 13, 365–376.
Yamawaki, Y., Kimura, H., Hosoi, T., & Ozawa, K. (2010). MyD88 plays a key role in LPS-induced Stat3 activation in the hypothalamus. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 298, R403–R410.
Yamazaki, H., Hiramatsu, N., Hayakawa, K., et al. (2009). Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. Journal of Immunology, 183, 1480–1487.
Yan, J., Zhang, H., Yin, Y., et al. (2014). Obesity- and aging-induced excess of central transforming growth factor-β potentiates diabetic development via an RNA stress response. Nature Medicine, 20, 1001–1008.
Yang, L., Qi, Y., & Yang, Y. (2015). Astrocytes control food intake by inhibiting AGRP neuron activity via adenosine A1 receptors. Cell Reports, 11, 798–807.
Yanguas-Casás, N., Barreda-Manso, M. A., Nieto-Sampedro, M., & Romero-Ramírez, L. (2017). TUDCA: An agonist of the bile acid receptor GPBAR1/TGR5 with anti-inflammatory effects in microglial cells. Journal of Cellular Physiology, 232, 2231–2245.
Yao, J. H., Ye, S. M., Burgess, W., et al. (1999). Mice deficient in interleukin-1beta converting enzyme resist anorexia induced by central lipopolysaccharide. The American Journal of Physiology, 277, R1435–R1443.
Yao, T., Deng, Z., Gao, Y., et al. (2017). Ire1α in Pomc neurons is required for thermogenesis and Glycemia. Diabetes, 66, 663–673.
Ye, J., Rawson, R. B., Komuro, R., et al. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Molecular Cell, 6, 1355–1364.
Yehuda, S., Rabinovitz, S., & Mostofsky, D. I. (2005). Mediation of cognitive function by high fat diet following stress and inflammation. Nutritional Neuroscience, 8, 309–315.
Yi, C.-X., Al-Massadi, O., Donelan, E., et al. (2012a). Exercise protects against high-fat diet-induced hypothalamic inflammation. Physiology & Behavior, 106, 485–490.
Yi, C.-X., Gericke, M., Krüger, M., et al. (2012b). High calorie diet triggers hypothalamic angiopathy. Mol Metab, 1, 95–100.
Yi, C.-X., Tschöp, M. H., Woods, S. C., & Hofmann, S. M. (2012c). High-fat-diet exposure induces IgG accumulation in hypothalamic microglia. Disease Models & Mechanisms, 5, 686–690.
Yi, C.-X., Walter, M., Gao, Y., et al. (2017). TNFα drives mitochondrial stress in POMC neurons in obesity. Nature Communications, 8, 15143.
Yin, Y., Yan, Y., Jiang, X., et al. (2009). Inflammasomes are differentially expressed in cardiovascular and other tissues. International Journal of Immunopathology and Pharmacology, 22, 311–322.
Yoshida, H., Matsui, T., Yamamoto, A., et al. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell, 107, 881–891.
Yoshida, H., Matsui, T., Hosokawa, N., et al. (2003). A time-dependent phase shift in the mammalian unfolded protein response. Developmental Cell, 4, 265–271.
Yoshida, H., Oku, M., Suzuki, M., & Mori, K. (2006). pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. The Journal of Cell Biology, 172, 565–575.
Yoshikawa, A., Kamide, T., Hashida, K., et al. (2015). Deletion of Atf6α impairs astroglial activation and enhances neuronal death following brain ischemia in mice. Journal of Neurochemistry, 132, 342–353.
Youm, Y.-H., Grant, R. W., McCabe, L. R., et al. (2013). Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metabolism, 18, 519–532.
Zabolotny, J. M., Kim, Y.-B., Welsh, L. A., et al. (2008). Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. The Journal of Biological Chemistry, 283, 14230–14241.
Zhang, K., Shen, X., Wu, J., et al. (2006). Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell, 124, 587–599.
Zhang, X., Zhang, G., Zhang, H., et al. (2008). Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell, 135, 61–73.
Zhang, G., Li, J., Purkayastha, S., et al. (2013a). Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature, 497, 211–216.
Zhang, Q., Yu, J., Liu, B., et al. (2013b). Central activating transcription factor 4 (ATF4) regulates hepatic insulin resistance in mice via S6K1 signaling and the vagus nerve. Diabetes, 62, 2230–2239.
Zhang, Y., Chen, K., Sloan, S. A., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of Neuroscience, 34, 11929–11947.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Cakir, I., Nillni, E.A. (2018). Brain Inflammation and Endoplasmic Reticulum Stress. In: Nillni, E. (eds) Textbook of Energy Balance, Neuropeptide Hormones, and Neuroendocrine Function. Springer, Cham. https://doi.org/10.1007/978-3-319-89506-2_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-89506-2_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-89505-5
Online ISBN: 978-3-319-89506-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)