
Abstract
Almond is the most important tree nut crop in terms of commercial production. Its production is limited to areas characterized by a Mediterranean climate, including regions in the Mediterranean countries, the Central Valley of California, Central Asia, the Himalayan slopes and some equivalent areas in the Southern Hemisphere, including Argentina, Australia and South Africa (Kester et al. 1975).
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

4.1 Introduction
Almond is the most important tree nut crop in terms of commercial production. Its production is limited to areas characterized by a Mediterranean climate, including regions in the Mediterranean countries, the Central Valley of California, Central Asia, the Himalayan slopes and some equivalent areas in the Southern Hemisphere, including Argentina, Australia and South Africa (Kester et al. 1975).
Cultivated almonds, often resulted from traditional seed propagation, show high levels of genetic variability due, in part, to their self-incompatibility, that makes them obligate out-crossers (Kester et al. 1991). Commercial cultivars within individual production areas, however, often show a limited genetic base due to their origin from few founder genotypes selected for their desirable regional value (Socias i Company and Felipe 1992). The range of almond species is extensive with a wide diversity of traits (Gradziel et al. 2001; Kester and Gradziel 1996). Controlled crosses of Prunus dulcis with other almond species in sections Euamygdalus and Spartiodes have been often carried out (Gradziel et al. 2001; Gradziel 2003).
One of the oldest almond germplasm collections was established at the Nikistki Botanical Garden in Yalta (Crimea), at the end of the XIX century (Rikhter 1969). Greater genetic variability and so increased breeding options for desired traits are being pursued through the incorporation of breeding material from other regions (Martìnez-Gòmez et al. 2003). Basic objectives of most almond breeding programs target increased yields, improved quality and decreased production costs. These traits have been found to be largely inherited in a quantitative manner (Spiegel-Roy and Kochba 1981). Inheritance of important breeding traits have been recently reviewed by Socias i Company et al. (2007).
Plant breeders use traditional and biotechnological techniques to create and use novel genetic variation, aimed at selecting new elite and suitable varieties, with improved traits to satisfy both farmers and consumers. Biotechnologies provide powerful tools for plant breeding , and among these ones, haploid (H) and doubled haploid (DH) technology , can effectively help to select superior plants (Seguí-Simarro 2010). Hs are sporophytes with the gametophytic chromosome number (n instead of 2n), originated from a single, male or female, immature gamete (Germanà 2011a). When spontaneous or induced chromosome duplication of a H occurs, a DH is obtained. Therefore, DHs are homozygous at all loci and can represent a new variety (in self-pollinated crops) or parental inbred line for the production of hybrid varieties (in cross-pollinated crops). Haploid plants arouse interest in the fields of genetic and developmental studies, as well as of plant breeding. Using DH technology, completely homozygous plants can be established in one step, saving thus several generations of selfing in comparison to conventional methods, by which also only partial homozygosity is obtained (Germanà 2011b). Indeed, this technique is the most rapid route to achieve homozygosity and, for self incompatible species, dioecious species and species that suffer from inbreeding depression due to self-pollination, gametic embryogenesis may be the only way to develop inbred lines (Murovec and Bohanec 2011). Particularly, microspore embryogenesis is an indispensable tool to quickly obtain homozygosis in woody plants, since these are characterized by long juvenility, high levels of heterozygosis and, often, by self-incompatibility (Germanà 2009). Unfortunately, many woody species , as well as fruit crops, are still recalcitrant to this process, generally carried out using various methods, mainly including in vitro culture of anthers (Germanà 2011a, b).
Moreover, haploid technology can be particularly effective in accelerating breeding if combined with other biotechnologies, such as ‘marker-assisted selection’ (MAS), providing a shortcut in backcross conversion to select elite lines. In fact, molecular markers are very useful for planning new crosses, predicting novel gene combinations (Tuvesson et al. 2007). DHs are also very useful for genome mapping, including the construction of genetic linkage maps and gene tagging, providing reliable information on the location of major genes for economically important traits. Nowadays, several genome sequencing programs are using haploid genome because of its simplified assembly, such as in many fruit crops like peach, pear, apple and citrus (Dunwell 2010). Obviously, the availability of an efficient and cost-effective protocol for Hs and DHs production is necessary to take advantages of combining different techniques with DH systems. There is not a universal suitable protocol and the development of new techniques is still required for many recalcitrant genotypes (Germanà 2011a, b; Cimò et al. 2017).
This chapter describes a protocol to regenerate microspore -derived embryo via microspore embryogenesis, through in vitro almond anthers culture.
4.2 Materials
-
1.
Immature flower buds of Prunus dulcis Mill., cvs. Filippo Ceo, Lauranne and Genco, at different developmental stages.
-
2.
Laminar-flow hood with ultraviolet light.
-
3.
Petri dishes (60 × 15 mm), forceps, scalpels, glass bead sterilizer, beakers, 1–20 ml serological pipettes, and 10–1000 μl air-displacement pipettes, capped Pyrex bottles, Parafilm, magnetic stirrers.
-
4.
Ethyl alcohol 70% (v/v), sodium hypochlorite solution (0.5% active chlorine), tween-20, sterile distilled water.
-
5.
1 mg/ml of 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI).
-
6.
Microscope slides and cover glasses.
-
7.
Stereo microscope and fluorescent microscope.
-
8.
Tissue culture chamber.
-
9.
pH meter, autoclave.
-
10.
Culture media (see Tables 4.1 and 4.2).
Table 4.1 Microspore embryogenesis induction medium Table 4.2 Embryo germination medium
Basic culture induction medium composition is listed in Table 4.1. Embryo germination medium is listed in Table 4.2. pH was adjusted to 5.8 with 0.5 M KOH or 0.5 M HCl and growth regulators were added to culture medium prior to autoclave at 121 °C for 20 min. Pour 10 ml culture induction medium into 60 × 15 mm Petri dishes.
4.3 Methods
The microspore embryogenesis procedure includes three steps: (1) flower bud size and microspore stage correlation, (2) flower bud sterilization and anther culture, (3) embryo production and maturation.
4.3.1 Flower Bud Size and Microspore Stage Correlation
The microspore development stage is a key factor influencing their ability to turn totipotent. Therefore, selection of buds with the maximum proportion of competent microspores (late uninucleated-vacuolated stage) is essential for efficient microspore-derived embryo yield. For this reason, collect one year almond shoots bearing flower buds at different developmental stages to determine the microspore developmental stage.
-
1.
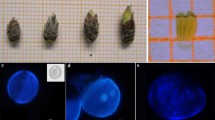
Select flower buds of different sizes (Fig. 4.1a1–d1) and divide them into groups.
Fig. 4.1 
Bud size and microspore development stage correlation. a1 Inflorescence bud swelling: bud closed, light brown scales visible (BBCH 5.1); b1 Bud burst: scales separated, light green bud sections visible (BBCH 5.3) *(best flower stage); c1 Sepals open: white or pink petal tips visible (BBCH 5.7); d1 Flowers with petals forming a hollow ball (BBCH 5.9); a2 Middle microspore ; b2 Polarized uninucleated-vacuolated microspore; c2 Early bicellular pollen ; d2 Mature pollen grain. Growth Stage numbering system is in accordance to the extended BBCH scale (www.agvita.com.au/pdf/sampling/Almond_GS_V2.pdf)
-
2.
Excise anthers separately from each bud size group and squash them in a few drops of DAPI staining solution to determine the microspore developmental stage.
-
3.
Observe the different microspore developmental stages under a fluorescent microscope (Fig. 4.1a2–d2).
-
4.
For the in vitro anthers culture, use only flower buds of the correct size, generally 7–9 mm in length depending on the cultivar (Fig. 4.1b1), containing anthers mostly with microspores at the polarized uninucleated-vacuolated stage (Fig. 4.1b2).

4.3.2 Flower Bud Sterilization and Anther Culture
Store selected flower buds at 4 °C for one week before use (cold-pretreatment).
-
1.
Perform sterilization under a laminar flow hood by immersion of the flower buds in 70% (v/v) ethyl alcohol for 5 min, followed by immersion in a sodium hypochlorite solution (0.5% active chlorine), with a few drops of Tween-20 for 20 min and finally rinse three times in sterile distilled water.
-
2.
Aseptically remove sepals, petals and carefully excise anthers using small forceps.
-
3.
Place immediately the anthers in sterilized Petri dishes containing about 10 mL of solidified culture induction medium (Table 4.1).
-
4.
Incubate the Petri dishes in a grow chamber at 25 ± 1 °C in the dark for 30 days.
-
5.
After one month in the dark, transfer the Petri dishes under cool white fluorescent lamps, with a photosynthetic photon flux density of 35 µmol m−2 s−1 and a photoperiod of 16 h light.
4.3.3 Embryo Production and Maturation
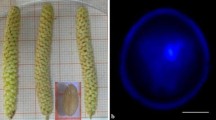
Different features have been described in almond anther culture as first signs of a change in the developmental pathway (from gametophytic to sporophytic) and of a morphogenic response. After two weeks of in vitro culture , anthers start to increase their dimension and produce calli (Fig. 4.2a) and, after one month, it will be already possible to observe the presence of multinucleated structures (Fig. 4.2b). In addition, when transferred under cool white light, embryo production (Fig. 4.3) will be also achieved.

Indications of the morphogenic response in anther culture and of the change of developmental pathway in microspore : a Anther producing callus after 2 weeks of culture; b Multinucleated structure after one month of culture

Embryo maturation and germination a Direct embryogenesis from almond anther culture ; b, c Early cotyledonary embryos coming out from almond anthers; d Embryo developing cotyledons
To obtain the germination of the microspore -derived embryos , they have to be transferred to a fresh medium containing Murashige and Skoog basal salt and vitamins, sucrose, zeatin (Z), naphthaleneacetic acid (NAA ) and indolbutyric acid (IBA) (Table 4.2).
Transfer globular (Fig. 4.3a) or early cotyledonary embryos (Fig. 4.3b, c) into the germination medium reported in Table 4.2, to induce their maturation and development (Fig. 4.2d).
4.3.4 Ploidy Analysis and Detection of Homozygosity
Through anther culture , also somatic and heterozygous embryos can be obtained. For this reason, after successful embryo regeneration , their evaluation is needed to determine their origins (assessing homozygosity), and to distinguish between spontaneously doubled haploids (obtained through microspore embryogenesis) and heterozygous diploids (resulted from somatic embryogenesis ). Many direct and indirect approaches are available for determining the ploidy level of regenerated plants. Direct methods are more reliable and include conventional cytological techniques, such as counting the chromosome number (Maluszynska 2003) or measurement of DNA content using flow cytometry (Fig. 4.4), that allows the analysis of a large number of target plants in a shorter period of time (Ochatt 2008).

Flow cytometry histogram. The relative fluorescence of the nuclear mixture is 2-fold lower (≈34) in haploids when compared to diploid (≈65) controls
Moreover, because of the possible spontaneous chromosome doubling occurring during the microspore embryogenesis, ploidy level analysis cannot always identify pollen -derived plants. To detect homozygosity and distinguish between gametic and somatic diploids, DNA molecular markers and microsatellites can be employed (De Vienne 2003). Simple Sequence Repeat (SSR) markers can be adopted to assess the homozygosity, to determine embryo origin (gametic or somatic) and to discriminate homozygous from heterozygous individuals (Fig. 4.5).

Characterization of anther culture regenerants. Microsatellite analysis: Pherograms of the microsatellite markers BPPCT-007 profiles of the mother plant (top) and of one ‘Filippo Ceo’ microspore -derived embryo. The mother plant is heterozygous and carries two alleles, the embryo shows only one of the mother plant alleles, considered as support for the gametic origin of the embryo
SSR polymorphic microsatellites, selected for being heterozygous in the donor plant and because they amplified in almond (i.e. PaCITA-21, MA031a, BPPCT-007, UDAP-468, EPPCU-5990 and PaCITA-23) can be employed for the analysis (Cimò et al. 2017).
The polymerase chain reaction (PCR) is carried out with three primers: the specific forward primer of each microsatellite with M13(-21) tail at its 5’ end, the sequence specific reverse primer and the universal fluorescent-labeled M13(-21) primer (Schuelke 2000). PCR reactions performed in a GeneAmp® PCR System 9700 thermal cycler (Perkin-Elmer Corp, Freemont, CA) in a final volume of 25 µL, containing 2.5 µL of 10X PCR buffer, 0.2 µL MgCl2, 2 µL dNTPs, 0.5 µL of the forward primer (5 µM), 0.5 µL of the M13(-21) primer, 1 µL of the reverse primer, 0.2 µL Taq polymerase (Invitrogen) and 20 ng of genomic DNA. PCR thermal profile as follows: an initial denaturation step at 95 °C for 60 s, followed by the annealing for 20 cycles at 60 °C (−0.5 °C/cycle) for 60 s, and extension at 72 °C for 90 s; followed by a second thermal profile of one cycle at 95 °C for 60 s; 40 cycles at 50 °C for 60 s, 72 °C for 90 s, finishing with 72 °C for 30 min. Three µL of desalted PCR product mixed with 12 µL of loading solution (70% formamide and 1 mM EDTA), 0.3 µL of LIZ dye, denatured at 95 °C for 5 min, and cooled on ice. Electrophoresis performed on a ABI PRISM 3130 Genetic Analyzer capillary system (Applied Biosystems, Warrington, UK). Allele lengths determined using an ABI Prism 3130 Genetic Analyzer with the GeneMapper software, version 4.0 (Applied Biosystems).
4.4 Identify Steps Required Further Protocol Modifications
In this chapter, microspore -derived embryo regeneration in almond , through in vitro anther culture , is described. However, further protocol modifications are needed. These include: (1) increase of the frequency of embryo recovery; (2) achievement of embryo conversion into plantlets ; (3) optimization of embryo maturation and germination and (4) enlargement of the number of respondent almond cultivars.
References
Chu C (1978) The N6 medium and its applications to anther culture of cereal crops. In: Proceedings of symposium on plant tissue culture. Science Press, Peking, pp 43–50
Cimò G, Marchese A, Germanà MA (2017) Microspore embryogenesis induced through in vitro anther culture of almond (Prunus dulcis Mill.). Plant Cell Tiss Org 128(1):85–95
De Vienne D (2003) Molecular markers in plant genetics and biotechnology. Science Publishers, Inc., Enfield, NH
Dunwell JM (2010) Haploids in flowering plants: origins and exploitation. Plant Biotechnol J 8:377–424
Germanà MA (2009) Haploids and doubled haploids in fruit trees. In: Advances in haploid production in higher plants. Springer, Netherlands, pp 241–263
Germanà MA (2011a) Anther culture for haploid and doubled haploid production. Plant Cell Tiss Org 104:283–300
Germanà MA (2011b) Gametic embryogenesis and haploid technology as valuable support to plant breeding. Plant Cell Rep 30:839–857
Germanà MA, Scarano MT, Crescimanno FG (1996) First results on isolated microspore culture of Citrus. Proc Int Soc Citricul 2:882–885
Gradziel TM (2003) Interspecific hybridizations and subsequent gene introgression within Prunus subgenus Amygdalus. Acta Hort 622:249–255
Gradziel TM, Dicenta F, Kester DE (2001) The utilization of related almond species for almond variety improvement. J Amer Pomol Soc 55:100–108
Growth Stage numbering system is in accordance to the extended BBCH, a uniform coding of phenologically similar growth stages for all plant species. Almond: Sampling & Growth Stages. www.agvita.com.au/pdf/sampling/Almond_GS_V2.pdf
Kester DE, Asay R, Almonds. in: Janick J, Moore, JN (eds) (1975) Advances in fruit breeding. Purdue Univ. Press, West Lafayette, IN, USA. pp 387–419
Kester DE, Gradziel TM, Grasselly C (1991) Almonds (Prunus). In: Moore JN, Ballington HJ (eds) Genetic resources of temperate fruit and nut crops. International Society for Horticultural Science, The Netherlands, pp 701–758
Kester DE, Gradziel TM (1996) Almonds. In: Janick J, Moore JN (eds), Fruit breeding, vol 3. John Wiley & Sons, New York, USA. pp 1–97
Li J, Wang Y, Lin L, Zhou L, Luo N, Deng Q, Xian J, Hou C, Qiu Y (2008) Embryogenesis and plant regeneration from anther culture in loquat (Eriobotrya japonica L.). Sci Hortic-Amsterdam 115:329–336
Maluszynska J (2003) Cytogenetic tests for ploidy level analyses - chromosome counting. In: Doubled haploid production in crop plants: a manual. Kluwer Academic Publishers, Dordrecht, pp 391–395. ISBN 1-4020-1544-5
Martìnez-Gòmez P, Sozzi GO, Sánchez-Pérez R, Rubio M, Gradziel TM (2003) New approaches to Prunus tree crop breeding. J Food Agr Env 1:52–63
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plantarum 15:473–497
Murovec J, Bohanec B (2011) Haploids and doubled haploids in plant breeding. Plant breeding. InTech, Rijeka, Croatia. pp 87–106
Nitsch JP, Nitsch C (1969) Haploid plants from pollen grains. Science 163:85
Ochatt SJ (2008) Flow cytometry in plant breeding. Cytometry 73A:581–598
Rikhter AA (1969) Ways and methods of almond breeding. Tr Gos Nikit Bot Sad 43:81–94
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Seguí-Simarro JM (2010) Androgenesis revisited. Bot Rev 76:377–404
Socias i Company R, Felipe AJ (1992) Self-compatibility and autogamy in ‘Guara’almond. J Hortic Sci 67(3):313–317
Socias i Company R, Kodad O, Alonso JM, Gradziel JTM (2007) Almond quality: a breeding perspective. In: Janick J (ed), Horticultural reviews, vol 33, pp 1–33
Spiegel-Roy P, Kochba J (1981) Inheritance of nut and kernel traits in almond (Prunus amygdalus Batsch). Euphytica 30:167–174
Tuvesson S, Dayteg C, Hagberg P, Manninen O, Tanhuanpaa P, Tenhola-Roininen T, Kiviharju E, Weyen J, Forster J, Schondelmaier J, Lafferty J, Marn M, Fleck A (2007) Molecular markers and doubled haploids in European plant breeding. Euphytica 158:305–312
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Cimò, G., Germanà, M.A. (2018). Microspore Embryogenesis in Almond (Prunus dulcis Mill.). In: Jain, S., Gupta, P. (eds) Step Wise Protocols for Somatic Embryogenesis of Important Woody Plants. Forestry Sciences, vol 85. Springer, Cham. https://doi.org/10.1007/978-3-319-79087-9_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-79087-9_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-79086-2
Online ISBN: 978-3-319-79087-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)