
Abstract
Cancer is a genetic disease, affecting many people worldwide. Chemotherapy is routinely used for cancer treatment. However, this therapeutic approach is not always effective due to the development of cell resistance and toxic effects. Plants are a reservoir of natural chemicals with chemoprotective potential against cancer and with low adverse effects. While some drugs from natural origin are currently used for cancer treatment, others are being studied. Among the compounds isolated from plants, sesquiterpene lactones are very promising anticancer agents, which are widely being studied in different models of cancer in vitro and in vivo, and some clinical trials are being performed. Sesquiterpene lactones are very attractive compounds to be used as antitumoral therapy due to the diverse mechanisms of action through which they exert their effects. Among such mechanisms are their capacity to interfere with the generation of reactive oxygen species, the epigenetic modulation of gene expression, the targeting of the sarco-/endoplasmic reticulum calcium ATPase pump, and the activation of the NF-kB and the p53 signaling pathways. The latter mechanisms could be important to reduce the development of drug resistance by tumor cells. Sesquiterpene lactones can also inhibit angiogenesis and metastasis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Cancer is a genetic disease causing high morbidity and mortality, with approximately 14 million new cases arising in 2012. The number of cases is expected to rise to almost 19 million by 2024. After cardiovascular disease, cancer represents the second leading cause of death in the world, having been responsible for 8.8 million deaths in 2015 (WHO 2017), which means that nearly 1 out of 6 deaths is due to this disease.
Cancer is caused by the deregulation of genes controlling cell growth, division, and death. The genetic changes that contribute to the development of cancer affect three main types of genes: proto-oncogenes, tumor suppressor genes, and DNA repair genes. These changes are sometimes called “drivers” of cancer. The final consequence is the generation of an imbalance between growth and death causing cells to proliferate and migrate toward distant organs, a phenomenon known as metastasis. Cancer may be either hereditary, may be the consequence of errors that occur during cell division, or may be due to DNA damage caused by certain environmental noxious agents such as the chemicals present in tobacco smoke or radiation such as ultraviolet rays (WHO 2005; Ferlay et al. 2015). Normal and tumor cells present a number of differences such as the lack of specialization of the latter, which causes them to divide continuously. Cancer cells have also lost their capacity to respond to the intracellular signals that regulate cell growth and apoptosis. Tumors are known to establish a relationship with the microenvironment, mainly through contact with normal cells, nearby molecules, and surrounding blood vessels that serve as nutrient source. Besides, tumor cells have developed mechanisms for immune evasion.
Cancers can be classified according to the organs or tissues where they originate. Thus, carcinomas are the most common type, which are constituted by epithelial cells; sarcomas are cancers generated in bones and soft tissues, including muscle, fat, blood vessels, lymph vessels, and fibrous tissue (such as tendons and ligaments); melanomas are made up of melanocyte precursors; and multiple myeloma is a cancer type of plasma cells.
Leukemia and lymphoma are a group of heterogeneous neoplastic disorders of white blood cells characterized by the uncontrolled proliferation and the blockage in the differentiation process of hematopoietic cells. While lymphomas are characterized by an abnormal lymphocyte proliferation developing as a solid tumor, most commonly in the lymph nodes of the neck, chest, armpit, or groin (Jaffe 2009), leukemias develop in the bone marrow and do not form solid tumors.
Leukemia and lymphoma stand at the fifth place of cancer-related deaths over the world (Armitage 2012). Leukemia includes acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), and chronic myeloid leukemia (CML). AML is the most common acute leukemia in adult patients, which progresses rapidly and may be lethal within weeks or months. Based on the cytogenetic/genetic features of AML, and according to the 2008 revised WHO classification system, patients can be broadly classified into three different risk groups. Treatment strategies and prognoses vary among these subtypes and are further influenced by patient-specific factors (Zeisig et al. 2012).
As stated above, lymphomas involve the uncontrolled growth of lymphocytes (T cells or B cells) and include two types: Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL), the latter is subdivided into two main types depending on the cell growth rate, that is, high-grade (or “aggressive”) and low-grade (or “indolent”) NHLs.
Taking into account patient-specific characteristics, such as age and overall health status, and each tumor type, it is obvious that the treatment strategies spectrum to be applied will be broad.
Chemotherapy is routinely used for cancer treatment; however, this strategy is not always effective due to the development of cell resistance phenomena and toxic effects (Hopfinger et al. 2012). For example, 5-fluorouracil, a common chemotherapeutic agent, is known to cause myelotoxicity and cardiotoxicity. Doxorubicin, which is another widely used drug, is known to cause cardiotoxicity, nephrotoxicity, and myelotoxicity. Similarly, bleomycin, a well-known chemotherapeutic agent, is known for its pulmonary toxicity. In addition, bleomycin causes cutaneous toxicity. Cyclophosphamide, a drug used to treat many malignant conditions, has been demonstrated to cause bladder toxicity in the form of hemorrhagic cystitis, immunosuppression, alopecia, and, at high doses, cardiotoxicity (Chabner et al. 2006).
All AML subtypes, except for the acute promyelocytic leukemia (APL), are typically treated with an induction chemotherapy using a “3 + 7” combination of daunorubicin and cytarabine (Zeisig et al. 2012). Almost all APL patients can achieve complete remission after treatment with all-trans-retinoic acid, which can effectively induce APL cell differentiation (Degos and Wang 2001). Conversely, although treatment strategies for AML have substantially progressed in recent decades, effective therapies for non-APL subtypes of AML are still urgently needed.
In this context, plants have enormous potential as a source of new drugs, constituting a reservoir of natural chemicals with low toxicity chemoprotective properties against cancer. There are four classes of commercially available plant-derived anticancer agents: the vinca alkaloids (vinblastine, vincristine, and vindesine), the epipodophyllo toxins (etoposide and teniposide), the taxanes (paclitaxel and docetaxel), and the camptothecin derivatives (camptothecin and irinotecan). The mechanisms of action of these drugs include cell cycle arrest, the induction of cell differentiation, or the induction of cell death through the activation and sensitization of the intrinsic and extrinsic pathways of apoptosis, respectively. Some drugs also interact with the cytoskeleton by either inhibiting the polymerization or by inducing depolymerization of microtubules. The latter effects also lead to a cell cycle arrest and apoptosis (Balunas and Kingborn 2005; Cragg et al. 2009).
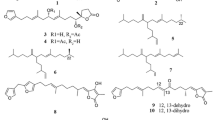
Sesquiterpene lactones are a group of plant secondary metabolites that are abundant in members of the Asteraceae family (Merfort 2011). The α, β unsaturated carbonyl function of STLs is important to exert the antiproliferative effect of these compounds. Among cytotoxic STLs, costunolide, eupatolide, parthenolide, dehydrocostuslactone, and thapsigargin can be mentioned (Fig. 13.1).

Chemical structure of cytotoxic sesquiterpene lactones
In this chapter, the cytotoxic and antitumor effects and mechanisms of action of STLs will be summarized, focusing on their effects on leukemias, lymphomas, and myelomas. Besides, some clinical trials assessing the effect of this type of compounds in other tumors will also be described. The remission obtained after the co-treatment with STLs and conventional chemotherapeutics will also be addressed.
2 In Vitro Studies and Mechanism of Action
There are two mechanisms by which natural compounds can induce cytotoxicity, namely, cell death and cell differentiation.
Three types of cell death are recognized: apoptosis, necrosis, and autophagy. Apoptosis is characterized by the activation of cysteine aspartate-specific proteases (caspases), chromatin condensation, cell shrinking, and blebbing of the cell membrane. Autophagy is characterized by the formation of intracellular vacuoles, organelle degradation, and, finally, cell death. In contrast, in necrotic cells, the endoplasmic reticulum (ER), mitochondria, and cytosol continue to swell until the cell bursts and releases its contents (Krysko et al. 2003). Apoptosis is the most common mechanism of cell death by which natural compounds exert their cytotoxic effects.
Apoptosis can occur through an extrinsic pathway activated by the binding of ligands to death receptors on the cell membrane and involves caspase-8 and caspase-3 or through an intrinsic pathway activated by other stimuli involving the release of mitochondrial proteins and the activation of the caspase-9 and caspase-3. The extrinsic and intrinsic pathways are interconnected, for example, the cleavage of BH3 interacting domain death agonist (Bid) by caspase-8 results in the formation of pro-apoptotic truncated Bid (tBid), which activates the intrinsic pathway (Krysko and Vandenabeele 2009).
The mechanisms leading to the activation of apoptosis are diverse: the direct activation of caspases, the direct effect on mitochondria (cytochrome release) and consequent caspase activation, a redox imbalance and the generation of oxidative stress, a disturbance in cell cycle progression, the induction of ER stress, the activation of transcription factors that promote apoptotic proteins, the inhibition of transcription factors related to survival and proliferation such as NF-κB and STATs, and the inhibition or activation of mitogen-activated protein kinases (MAPK), which transmit extracellular signals to regulate cell growth, proliferation differentiation, and apoptosis. Three major MAPK families have been described: P-38, the extracellular signal-regulated kinase (ERK), and C-Jun N-terminal kinase (JNK). When cells are exposed to stressors, one of the major outcomes of MAPK activation is the induction of apoptosis, a process in which the transcription of the Bcl-2 family proteins, such as Bax and Bcl-2 takes place. While the inhibition of DNA methylation is related to cell differentiation and apoptosis, the inhibition of histone deacetylase 1 (HDAC1) induces apoptosis through the activation of p21 and p53 proteins.
The induction of apoptosis through the generation of oxidative stress is a mechanism by which many STLs produce either cytostatic or cytotoxic effects in tumors involving the participation of mitochondria. The mitochondrial events involve the release of apoptotic factors, including cytochrome C and second mitochondria-derived activator of caspases (Smac/Diablo). Usually, the permeabilization of the mitochondrial outer membrane leading to the release of these components is controlled by the balance between pro-apoptotic and anti-apoptotic members of the B-cell CLL/lymphoma 2 (Bcl-2) superfamily.
Over the last 5 years, many studies have been performed to assess the antiproliferative activity of STLs. It has been demonstrated that the treatment of tumor cells with STLs disrupts their redox status and causes oxidative stress, with the subsequent initiation of the intrinsic apoptosis pathway (Sun et al. 2010). For example, the treatment of tumor cells with parthenolide increases the generation of reactive oxygen species (ROS) in tumor cells through diverse mechanisms such as the reduction of the cysteine sulfhydryl group of the nonprotein antioxidant molecule glutathione (Ralstin et al. 2006), the activation of NADPH oxidase, and the downregulation of the antioxidant enzymes manganese superoxide dismutase and catalase.
Other STLs produce apoptosis by interfering with the cell cycle progression and by arresting cells in the G2/M phase (Rozenblat et al. 2008) together with an increase in the sub-G0 fraction or, conversely, by arresting the cells in the G1/S phase with the appearance of a broad sub-G1 peak (Cho et al. 2004). The activation of p53 and the subsequent inactivation of cell division control protein 2 (Cdc2) are responsible for the G2/M arrest, whereas the G1/S arrest involves the modulation of cyclin-dependent kinases and their activating partners, cyclins. Other STLs trigger apoptosis by increasing the levels of ROS, leading to the activation of either the extrinsic or the intrinsic pathway. Psilostachyin, psilostachyin C, peruvin, and cumanin exert an antiproliferative effect by inducing apoptosis on the BW5147 murine lymphoma cell line. Psilostachyin C has proved to be the most active and the less toxic pro-apoptotic compound. These effects are achieved through an impairment in the mitochondrial membrane potential and the generation of ROS. These phenomena are related to a modulation of the activity of antioxidant enzymes and by inducing cell cycle arrest in the S phase (Martino et al. 2015). Besides, psilostachyin and psilostachyin C are considered novel checkpoint inhibitors, since they cause the arrest of other cell types such as breast cancer cells (MCF-7) in the G2 phase (Sturgeon et al. 2005).
In addition, the STL EM23, isolated from Elephantopus mollis, which is a traditional herbal medicine with multiple pharmacological activities, inhibits the proliferation of human chronic myeloid leukemia (CML) K562 cells and acute myeloid leukemia (AML) HL-60 cells through the induction of apoptosis involving the translocation of the membrane-associated phospholipid phosphatidylserine, changes in cell morphology, the activation of caspases, and the cleavage of poly-(ADP-ribose) polymerase1 (PARP 1). The disruptions in the mitochondrial membrane potential observed have allowed suggesting the involvement of the mitochondrial pathway in EM23-mediated apoptosis. Mechanistic studies have indicated that EM23 caused a marked increase in the levels of ROS (Li et al. 2016).
Furthermore, five melampolide STLs (uvedalin, enhydrin, polymatin B, sonchifolin, and fluctuanin) isolated from the leaves of Smallanthus sonchifolius have been demonstrated to exert poor cytotoxic effects on peripheral blood mononuclear cells (PBMC) of healthy human subjects while being strongly cytotoxic against human T-cell acute lymphoblastic leukemia (CCRF-CEM) cells, CEM-ADR5000, and pancreatic cancer cells. These effects were achieved by triggering the generation of oxidative stress. These findings suggest that these STLs display a certain selectivity for tumor cells. It has been reported that the generation of ROS entails the induction of apoptosis in CCRF-CEM and CEM-ADR5000 cells through the activation of receptor-interacting protein kinase 1 (RIP1K). Neither apoptosis nor necroptosis was observed in human pancreatic carcinoma cell lines (MIA-PaCa-2 cells (de Ford et al. 2015)).
The dimeric STLs uvedafolin and enhydrofolin, also isolated from S. sonchifolius, have been demonstrated to have cytotoxic activity on HeLa cells with low IC50 values (2.96 and 3.17 mM, respectively) after 24 h of treatment, displaying low toxicity on normal NIH/3 T3 cells. These STLs caused cell cycle arrest in the G2/M phase and induced apoptosis through the activation of the mitochondrial pathway, with the participation of caspase-9 and caspase-3/7. In addition, the STL 2,3-dehydropsilostachyn isolated from the n-butanolic extract of Ambrosia cumanensis Kunth. has shown cytotoxic activity with different potencies in HeLa, Jurkat, and U937 cell lines. This STL was found to cause arrest in the G2/M cell cycle block (Del Socorro Jimenez-Usuga et al. 2016).
Another mechanism through which STLs activate the apoptotic intrinsic pathway is the interaction with ER through the ER stress-dependent and ER stress-independent pathways related to proteasome inhibition. The ER is responsible for the folding and the posttranslational modification of secreted proteins. When the ER becomes stressed due to the accumulation of newly synthesized unfolded or misfolded proteins, a complex intracellular signal transduction pathway, named the unfolded protein response (UPR), is activated, leading to the activation of the intrinsic pathway of apoptosis by phosphorylation and inactivation of Bcl-2, repression of Bcl-2 expression, or translocation of Bax/Bak to the mitochondria (Krysko and Vandenabeele 2009). The dehydrocostus lactone activates the ER-resident transmembrane protein sensors, which lead to the activation of a transcription factor that regulates UPR target gene expression and upregulates the pro-apoptotic transcription factor growth arrest and GADD153/C/EBP protein (Hung et al. 2010).The guaianolide thapsigargin induces ER stress by decreasing the Ca2+concentration in the ER by blocking the capacity of the sarco-/endoplasmic reticulum calcium ATPase (SERCA) pump to transfer Ca2+ from the cytosol to the ER, thus increasing the cytosolic Ca2+concentration. The latter phenomenon leads to the generation of ROS and subsequent cell death (Denmeade and Isaacs 2005).
Costunolide has antiproliferative effects on human bone osteosarcoma epithelial cells (U2OS) by inducing the loss of the mitochondrial transmembrane potential. This STL decreases the Bcl-2/Bax ratio, induces cytochrome C release, and activates caspases. All these effects are related to the generation of ROS and the ER stress-induced mitochondrial dysfunction, which activate kinases such as c-Jun N-terminal kinase (JNK) and induce apoptosis (Zhang et al. 2016).
It has also been demonstrated that the balance between apoptosis and survival can be affected by the reactivation of tumor suppressor genes. Tumor suppressor genes are silenced in tumor cells by hypermethylation. Therefore, inhibition of DNA methylation results either in the differentiation of cancer cells or in apoptosis. In this sense, the inhibition of DNA methyl transferase 1 by parthenolide results in significant hypomethylation and reactivation of histidine nucleotide-binding protein 1 (HIN-1) suppressor gene (Liu et al. 2009). Moreover, the activation of HIN-1 results in the inhibition of AKT signaling, which leads to the dephosphorylation of Bad, allowing it to bind anti-apoptotic Bcl-2 members to neutralize them. Parthenolide also affects histone acetylation by inhibiting histone deacetylase 1 (HDAC1), leading to p21 and p53 activation and subsequent apoptosis (Gopal et al. 2009).
It has also been demonstrated that rapidly proliferating cancer cells express elevated levels of transferring receptors on their cell surface and have higher amounts of intracellular iron than normal or slowly proliferating cells. In this regard, artemisinin has an endoperoxide bridge that is cleaved upon binding to Fe (II), producing toxic C-4 and seco-C-4 free radicals that destroy tumor cells. The addition of holotransferrin and iron sources to artemisinin increases its antitumor properties and targeted delivery (Nakase et al. 2009).
It is noteworthy that some STLs produce sensitization of the extrinsic pathway of apoptosis. This pathway is activated upon binding of membrane death receptors to their extracellular ligands, such as TNF and TNF-related apoptosis-inducing ligand (TRAIL). For example, the germacranolide eupatolide sensitizes cancer cells to TRAIL-induced apoptosis by downregulating the cellular FLICE-like inhibitory protein (c-FLIP). The treatment of human breast cancer cells with TRAIL in combination with subtoxic concentrations of eupatolide enhanced the TRAIL-induced cytotoxicity, as compared with the effect exerted by each STL alone (Lee et al. 2010). Dehydrocostus lactone renders human leukemia HL-60 cells susceptible to TNF-induced apoptosis by enhancing caspase-3 and caspase-8 activities (Oh et al. 2004). Parthenolide sensitizes cancer cells for TNF-induced apoptosis by downregulating anti-apoptotic genes and sensitizes hepatocellular carcinoma cells to TRAIL by inducing the expression of death receptors through the inhibition of STAT3 (Carlisi et al. 2011).
However, the most important mechanism by which STLs exert their antiproliferative effect and apoptosis involves the inhibition of NF-κB or STATs transcription factors. NF-κB and STAT3 are two important pathways involved in cell survival and proliferation that are constitutively activated in neoplastic cells in response to autocrine and paracrine factors that are produced within the tumor microenvironment (Takeuchi and Akira 2010). The NF-kB activation favors angiogenesis and tumorigenesis and plays a key role in the expression of several inflammatory genes and anti-apoptotic genes. Thus, NF-κB has been considered an important link between inflammation and cancer.
Parthenolide is one STL that is capable of inhibiting NF-κB transcription, presenting antiproliferative activity on leukemia cells. This STL can be found in different medicinal plants, particularly in feverfew (Tanacetum parthenium). It has been demonstrated that parthenolide induces apoptosis in acute myeloid leukemia (AML) stem and progenitor cells, without causing significant toxicity to normal hematopoietic stem cells (HSCs). AML growth is hierarchical and originates from leukemic stem cells (LSCs). All these findings have anointed parthenolide as the prototypical member of next-generation therapies for the elimination of LSCs (Guzman et al. 2005).
Parthenolide also suppresses the growth of the Raji cell line and activates the transcription of BZLF1 by inhibiting the activity of NF-κB. BL has been reported to be strongly associated with EBV infection. When parthenolide was used in combination with ganciclovir, the cytotoxic effect of first drug was enhanced. These data suggest that the induction of EBV-mediated lysis achieved with this drugs combination may constitute an efficient viral-targeted therapy for EBV-associated BL (Li et al. 2012). In addition, the STLs honokiol, magnolol, and parthenolide, isolated from Magnolia grandiflora, have proved to induce apoptosis and to exert cytotoxic effects in vitro on the atypical lymphocytes follicular Non-Hodgkin lymphoma cells (NHL) obtained from a patient (Marin et al. 2013). The increase in Iĸ-B and the consequent decrease in NF-κB DNA binding, the inhibition of the EGFR/PI3K/Akt signaling pathway, and the inhibition of telomerase are possible mechanisms proposed by the authors to explain this effect. The same authors had previously demonstrated that parthenolide has an antitumor activity in a lymphocyte malignancy animal model.
Another antitumor mechanism exerted by STLs involves NF-kB-driven transcription of matrix metalloproteinase 9, a crucial angiogenesis-related transcript that is suppressed by treatment with parthenolide (Oka et al. 2007) and by the germacranolide deoxyelephantopin (Huang et al. 2010). These antiangiogenic treatments reduce the occurrence of metastasis.
More recently, the chemical optimization of parthenolide on the α-methylene-γ-butyrolactone has been done to improve the potency and pharmacokinetic parameters. The modification in C1-C10 olefinic groups of parthenolide leads to the generation of the parthenolide analogues cyclopropane 4 and micheliolide, which were found to have inhibitory activity on drug resistant AML cells and low toxicity to healthy bone marrow cells. These analogues are also known to induce the generation of ROS (Kempema et al. 2015).
Not only do STLs inhibit NF-κB, but also they exert inhibition of STATs. Using the luciferase reporter system, some authors have observed that two STLs, damsin and coronopilin, isolated from Ambrosia arborescens, are capable of inhibiting NF-κB and STAT3 transcriptional activities in Jurkat and HeLa cells. The expression of NF-κB and STAT3 were induced by TNFα and IFNγ, respectively. In both cases, the IC50 values for damsin were lower than those of coronopilin, indicating that damsin is more effective than coronopilin. The authors hypothesized that the NF-κB signaling pathway can be inhibited by multiple mechanisms, including the activation of the inhibitor of NF-κB kinase (IKK) activity, the activation of the inhibitor of NF-κB (IκB) phosphorylation through IκB degradation, and DNA binding. Some STLs, such as ergolide, artemisinin, costunolide, and zerumbone, have also been reported to have inhibitory effects on the NF-κB pathway (Villagomez et al. 2013).
Leptocarpin (LTC) is a STL isolated from a native Chilean plant, Leptocarpha rivularis, which has been widely used in traditional medicine by Mapuche peoples. It decreases cell viability of cancer cell lines (such as HT-29, PC-3, DU-145, MCF7, and MDA-MB-231) by inducing caspase-dependent apoptosis and through the inhibition of the NF-kB factor, the latter effect being associated with a decreased DNA-binding capacity of this transcription factor. However, the specific mechanism underlying this effect remains unknown (Bosio et al. 2015). Four new STLs, 8α-(2′Z-tigloyloxy)-hirsutinolide, 8α-(2′Z-tigloyloxy)- hirsutinolide-13-O-acetate, 8α-(4-hydroxytigloyloxy)-hirsutinolide, and 8α-hydroxy- 13-O-tigloyl-hirsutinolide, isolated from Vernonia cinerea leaves and steams, are capable of inhibiting the proliferation of U251MG glioblastoma and MDA-MB-231 breast cancer cells by inhibiting aberrant active STAT3 (Joung Youn et al. 2014).
It has been observed that the inhibition of NF-κB and STATs by STLs sensitize tumoral cells to the action of chemotherapeutic agents. When STLs are combined with other treatments, they also sensitize the extrinsic apoptosis pathway by interfering with anti-apoptotic gene expression or by increasing the expression of death receptors. Parthenolide has potent antitumor activity against cancer stem cells (Guzman et al. 2005) and acts synergistically with the treatment of pancreatic adenocarcinoma and on a xenograft model of breast cancer when used in combination with docetaxel or sulindac, respectively (Yip-Schneider et al. 2005; Sweeney et al. 2005).
It is noteworthy that STLs are active on cells that have become resistant to many chemotherapeutic agents. The multidrug resistance is a prevailing phenomenon leading to chemotherapy treatment failure in cancer patients. In this regard, two STLs, neoambrosin and damsin, isolated from Ambrosia maritima (Asteraceae), have been studied on MDR P-gp CEM/ADR 5000 cells P-gp-overexpressing CEM/ADR5000 leukemic cells. The compounds inhibited cell proliferation by silencing the proto-oncogene tyrosine kinase (c-Src) which is a non-receptor tyrosine kinase protein that in humans is encoded by the SRC gene. This protein phosphorylates specific tyrosine residues in other proteins, for example, in tumor cells, the overexpression of c-Src leads to the constitutive activation of PI3K (phosphatidylinositol-3-kinase)/Akt-NF-kB and STATs (STAT3 and STAT5) pathways. As described above, this activation governs cancer development and progression (Saeed et al. 2015).
Sesquiterpene lactones can also exert antiproliferative effects through the inhibition of oncogenes or the inhibition of tumor suppressor gene p53 (related to apoptosis). It has been demonstrated that STLs inhibit the oncogene nucleophosmin-anaplastic lymphoma kinase (NPM/ALK) chimera, which is involved in the majority of anaplastic large cell lymphomas (ALCLs). The furanoheliangolide STL lobatin B, isolated from Neurolaena lobata, downregulates NPM-ALK, and as a consequence, an inhibition of expression of Jun B (which is a transcription factor of the tyrosine receptor kinase PDGF-Rβ) is observed (Kiss et al. 2015).
The other mechanism by which STLs induce death is necrosis. Through this mechanism, some STLs, such as the pseudoguainolide helenalin, kill apoptosis-resistant cells. Helenalin induces necrosis in three cell lines overexpressing Bcl-2 by mechanisms involving free intracellular iron, ROS, and inhibition of NF-kB. This form of necrosis has proved to be independent of receptor-interacting protein kinase 1 (RIPK1) and poly-(ADP-ribose) polymerase1 (PARP1) (Hoffmann et al. 2011).
Differentiation is another mechanism exerted by antitumoral drugs. The STL arsantin, present in Artemisia santolina, induces cell differentiation of the human promyelocytic leukemia HL-60 cell line in a concentration-dependent manner. A cytofluorometric analysis indicated that arsantin induces HL-60 cell differentiation predominantly into granulocytes by increasing the levels of protein kinase C alpha (PKCα) and protein kinase β (PKCβII) isoforms and by increasing the phosphorylation rate MAPKs in HL-60 cells. In addition, arsantin synergistically enhanced the differentiation of HL-60 cells in a dose-dependent manner when combined with low doses of 1–25-(OH)2D3 (Kweon et al. 2015).
In many cases, the antiproliferative activity of STLs in leukemic cells is shown without describing the mechanism of action. This is a case of an amino conjugate of a sulfated guaiane STL isolated from the roots of Scorzonera divaricata. This STL exhibits significant in vitro cytotoxic activity against human cancer cell lines such HL60, HeLa, HepG2, and SMMC-7721, among others (Yang et al. 2016).
In addition, the STL dehydroleucodine, isolated from Gynoxys verrucosa, a species used in traditional medicine in Southern Ecuador, has proved to have an antiproliferative effect against eight acute myeloid leukemia (AML) cell lines, with LD50 values ranging from 5.0 to 18.9 μM. Besides, this STL was active against human AML cell samples from five patients, with an average LD50 of 9.4 μM. It is noteworthy that the compound does not affect the proliferation of normal peripheral blood mononuclear cells. The authors concluded that an exocyclic methylene in the lactone ring is required for the cytotoxic activity (Ordóñez et al. 2016).
The mechanism of action of the three more studied STLs, parthenolide, artemisinin, and thapsigargin, is shown in Fig. 13.2.

Mechanism of action of sesquiterpene lactones on tumoral cells. The mechanism of action of the three more studied STLs is shown. STLs diffuse through the plasma membrane and selectively target the SERCA pump, high iron content and cell surface transferrin receptors, NF-kB signaling, and epigenetic mechanisms. Thapsigargin can diffuse into the SERCA pump blocking its ability to transfer Ca2+ from the cytosol into the ER. High cytosolic Ca2+ concentrations lead to the generation of ROS and subsequent cell death. Many tumoral cells have high intracellular Fe2+ contents and high levels of transferrin receptors. The cytosolic Fe2+ binds to the endoperoxide bridge of artemisinin, leading to its activation and the consequent generation of toxic ROS. Parthenolide directly inhibits DNMT1 and causes ubiquitin-mediated proteasomal degradation of HDAC1, leading to expression of HIN-1 and p21, respectively. Parthenolide directly inhibits the NF-kB p65 subunit and the IKK complex preventing IKK-mediated phosphorylation and proteasomal degradation of IkB. Artemisinin also inhibits NF-kB activity. Abbreviations: ER sarcoplasmic/endoplasmic reticulum, HDAC1 histone deacetylase 1, IKK, IkB kinase, IkB inhibitor of NF-kB, NF-kB nuclear factor-kB, ROS reactive oxygen species, SERCA, ER calcium ATPase, Ub ubiquitin. Dashed and solid lines indicate translocation and activation, respectively
Apart from blood cell malignancies, STLs also have activity on other tumors. For example, parthenolide presents activity against breast cancer and pancreatic carcinoma (Carlisi et al. 2016; Liu et al. 2017); the prodrug of thapsigargin, mipsagargin, has proved to induce apoptosis in prostatic tumors (Mahalingam et al. 2016).
Moreover, eupatolide induces apoptosis in breast cancer cells, and artemisinin is being evaluated in clinical trials for breast and colorectal cancer (see below).
Furthermore, dehydrocostus lactone and costunolide are active on hepatocellular carcinoma and on sarcomas (SW-872, SW-982, and TE-671) (Lohberger et al. 2013). Furthermore, deoxyelephantopin exerts antitumoral activity on breast adenocarcinoma in mice (Kreuger et al. 2012; Merfort 2011). Psilostachyin, psilostachyin C, helenalin, and mexicanin I are active on different tumor cells apart from white blood cells (Martino et al. 2015; Kreuger et al. 2012; Merfort 2011; Sturgeon et al. 2005; Bujnicki et al. 2012).
3 In Vivo Studies
The “in vivo” antitumoral activity of STLs had been demonstrated in animal models. For example, goyazensolide exerts antitumoral activity in an in vivo hollow fiber assay employing HT 29 and MDA-MB 435 human tumoral cells. The fibers were implanted into immunodeficient female NCr nu/nu mice. On the contrary, the STLs 15-deoxygoyazensolide and ereglomerulide did not present antitumoral activity in the same system (Muñoz Acuña et al. 2013). Japonicone A, which is found in the traditional herb Inula japonica, is considered a promising compound. This STL has proved to have potent in vitro and in vivo antitumor activity against Burkitt’s lymphoma cells (Wang et al. 2014). Besides, it has been demonstrated that the treatment of BALB/c mice bearing Ehrlich tumors with a daily dose of the STL diacetyl piptocarphol, isolated from Vernonia scorpioides, produced a decrease on the tumor size and prevented the development of the ascitic tumor. Furthermore, the dichloromethane fraction of Moquiniastrum polymorphum subsp. floccosum, which is rich in STLs, has shown activity against Walker-256 carcinosarcoma in rats. The treatment with the fraction significantly reduced the tumor volume and weight. The fraction also modified hepatic glutathione and superoxide dismutase levels and normalized plasma glucose, alkaline phosphatase, and amylase and the nitric oxide levels in the tumoral tissue. Moreover, the fraction induced apoptosis through the upregulation of the p53 and Bax gene expression. It is noteworthy that no clinical signs of toxicity or death were observed in the rats treated with the fraction (Gomes Martins et al. 2015)
Angiogenesis and metastasis are also two important targets for antitumoral drugs in vivo. Artemisinin has proved to prevent angiogenesis by inhibiting human vein endothelial cell proliferation and vascular endothelial growth factor and receptor expression. DNA microarray analysis correlates tumor inhibition exerted by artemisinin with the reduced expression of crucial angiogenesis-related transcripts, namely, vascular endothelial growth factor, fibroblast growth factor, several matrix metalloproteinases, and hypoxia-inducing factor (Anfosso et al. 2006). Thapsigargin also inhibits microvessel formation and proliferation of human artery endothelial cells in vitro (Shukla et al. 2001). Furthermore, parthenolide has been found to inhibit the expression of matrix metalloproteinase-9 and urinary plasminogen activator and the migration of carcinoma cells in vitro, as well as osteolytic bone metastasis in vivo (Idris et al. 2009).
4 Clinical Trials
Nowadays, some STLs such as artemisinin from Artemisia annua, thapsigargin from Thapsia genus (Apiaceae), and parthenolide from Tanacetum parthenium (feverfew) and/or many of their synthetic derivatives are under evaluation in clinical trials.
These compounds have been selected for their selectivity, which is attributed to their ability to target the sarco-/endoplasmic reticulum (ER) calcium ATPase (SERCA) pump, particular proteases secreted by cancer cells, high iron content and cell surface transferrin receptors, nuclear factor-kB (NF-kB) signaling, MDM2 degradation and p53 activation, angiogenesis, metastasis, and epigenetic mechanisms.
Clinical evidence indicates that artemisinin-derived drugs are promising for laryngeal carcinoma, uveal melanoma, and pituitary macroadenoma. These drugs are in phase I–II trials against lupus nephritis and breast, colorectal, and non-small cell lung cancers (Singh and Panwar 2006). In addition, the artemisinin derivatives, artemether and artesunate, have been subjected for evaluation in cancer clinical trials (Chen et al. 2004). Besides, artesunate was then also incorporated in several clinical trials: in 2008, it was evaluated for the treatment of metastatic breast cancer (Table 13.1); in 2015, a study was undertaken to assess the effect of artesunate administered intravaginally for the HPV-positive high-grade cervical intraepithelial neoplasia (CIN2/3) (Table 13.1); and in 2016, this STL was incorporated in a phase I study involving patients with solid tumors to evaluate its effect after intravenous administration (Table 13.1). In the same year, the safety and pharmacokinetics parameters of artesunate were evaluated in a phase I dose-escalation study in patients with hepatocellular carcinoma. The study was completed in December 2016; however, the results have not been published (Table 13.1). Artesunate will also be evaluated in two clinical trials: one of the trials is not in the recruiting phase yet (March 2017), and the other is already recruiting participants (October 2016) to study the safety and effectiveness of this STL on colorectal cancer and stage II/III bowel cancer (Table 13.1). Furthermore, artemether will also be assessed in a clinical trial (not yet recruiting participants, March 2017) to study its effect on solid tumors (Table 13.1). The orally bioavailable parthenolide analogue dimethyl amino-parthenolide or LC-1 has been subjected to phase I clinical trials against acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), and other blood and lymph node cancers (Guzman et al. 2007).
Thapsigargin-derived drugs are currently being studied in phase I clinical trials for breast, kidney, and prostate cancer treatments. For example, a thapsigargin prodrug (G-202 mipsagargin, which consists of a thapsigargin molecule coupled with a masking peptide that is cleaved at the tumor site) is in phase I clinical trials for advanced solid tumors (Christensen et al. 2009). Mipsagargin has also been evaluated in several clinical trials for the treatment of multiform glioblastoma, advanced adult hepatocellular carcinoma, clear renal cell carcinoma, and prostatic neoplasm (Table 13.1) (Quynh Doan and Christensen 2015). For example, in a study carried out with patients affected with solid tumors [14 patients (32%) with colorectal, 9 (20%) with prostate, and 6 (14%) with hepatocellular carcinoma], the drug presented a good pharmacokinetic profile and was well tolerated. Only 12 (28.6%) of the patients presented a disease stabilization (Mahalingam et al. 2016). The clinical trials with STLs are shown in Table 13.1.
Some other STLs are currently being used for the treatment of different cancer types, for example, arglabin, isolated from Artemisia glabella (Asteraceae), is used for liver, breast, ovarian, and uterus cancer (Adekenov 2016).
5 Conclusion
Sesquiterpene lactones are very attractive compounds to be used in antitumor therapy due to the diverse mechanisms of action they exert, such as ROS generation, epigenetic modulation of gene expression, and targeting of the sarco-/endoplasmic reticulum calcium ATPase (SERCA) pump, the NF-kB signaling pathway, and the p53 pathway. Because of this, sesquiterpene lactones could be useful to treat resistant tumors. In addition, sesquiterpene lactones have proved to inhibit angiogenesis and metastasis. Promising results have also been achieved in animal models, and some sesquiterpene lactones are under study in clinical trials.
It can be concluded that sesquiterpene lactones can exert the antitumor activity mainly by inducing apoptosis through different pathways. All these considerations make these compounds very attractive to be used as antitumor drugs to be used either alone or in combination with other traditional chemotherapeutic drugs.
Abbreviations
- ALCLs:
-
Anaplastic large cell lymphomas
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- APL:
-
Acute promyelocytic leukemia
- Bax:
-
Apoptosis regulator BAX
- Bcl- 2:
-
B-cell CLL/lymphoma 2
- Bid:
-
BH3 interacting domain death agonist
- BL:
-
Burkitt lymphoma
- BZLF1:
-
Human herpes virus (E′stein-Barr virus) gene
- Caspases:
-
Cysteine aspartate-specific proteases
- CCRF-CEM cells:
-
Cellosaurus acute lymphoblastic leukemia cell line
- CCRF/ADR 5000:
-
Cellosaurus cell line, a doxorubicin-resistant sub-line derived from drug-sensitive, parental CCRF-CEM cells
- Cdc2:
-
Cell division control protein 2
- c-FLIP:
-
Cellular FLICE-like inhibitory protein
- CLL:
-
Chronic lymphocytic leukemia
- CML:
-
Chronic myeloid leukemia
- c-Src:
-
Proto-oncogene tyrosine-protein kinase
- C/EBP:
-
CHOP homologus protein
- GADD 153:
-
DNA damage inducible gene 153
- DU-145:
-
Human prostate carcinoma cell line
- EGFR:
-
EGFR/PI3K/Akt signaling pathway
- EBV:
-
Raji cell line: Epstein Barr virus (EBV)-positive Burkitt lymphoma (BL) cell line
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- HDAC1:
-
Histone deacetylase 1
- HeLa:
-
Immortal cell line
- HepG2:
-
Human liver cancer cell line
- HIN-1:
-
Histidine nucleotide-binding protein 1
- HL:
-
Hodgkin lymphoma
- HL-60 :
-
Acute myeloid leukemia
- HSCs:
-
Healthy hematopoietic stem cells
- HT 29:
-
Human colorectal adenocarcinoma
- CML:
-
Human chronic myeloid leukemia (CML) K562 cells
- HL:
-
Human leukemia HL-60 cells
- IkB:
-
Inhibitor of NF-kB
- IKK:
-
NF-κB kinase
- JNK:
-
C-Jun N-terminal kinase
- Jun B:
-
Transcription factor of the tyrosine receptor kinase PDGF-Rβ
- LSCs:
-
Leukemic stem cells
- MAPK:
-
Mitogen activated protein kinases
- MCF-7:
-
Breast cancer cells
- MCF7:
-
Human breast adenocarcinoma cell line
- MDA-MB 435:
-
Melanoma cell line
- MDA-MB-231:
-
Human breast cancer cell line
- MDM2:
-
Mouse double minute 2 homolog protein
- MIA-PaCa-2 cells:
-
Human pancreatic carcinoma cell lines
- MDR P-gp CEM/ADR5000 cells:
-
Multidrug-resistant P-gp over expressing CEM/ADR5000 leukemic cells
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NHL:
-
Non-Hodgkin lymphoma
- NIH/3T3:
-
Normal fibroblast cell line
- NPM-ALK:
-
Nucleophosmin-anaplastic lymphoma kinase
- p21:
-
Protein P21
- P-38:
-
Protein kinase P-38
- p53:
-
Protein P53
- PARP 1:
-
Poly-(ADP-ribose) polymerase1
- PBMC:
-
Peripheral blood mononuclear cells
- PC-3:
-
Human prostate grade IV adenocarcinoma cell line
- PDGF-Rβ:
-
Beta-type platelet-derived growth factor receptor
- PI3K:
-
Phosphatidylinositol-3-kinase
- PKCα:
-
Protein kinase C alpha
- PKCβII:
-
Protein kinase β
- RIPK1:
-
Receptor interacting protein kinase 1
- ROS:
-
Reactive oxygen species
- SW872:
-
Cellosaurus cell line SW 872, liposarcoma
- SW982:
-
Cellosaurus cell line SW 982, biphasic synovial sarcoma
- TE-671:
-
Cellosaurus cell line T-671, a human rhabdomyosarcoma
- SER:
-
Sarcoplasmic/endoplasmic reticulum
- SERCA:
-
Sarco/endoplasmic reticulum calcium ATPase
- Smac/Diablo:
-
Mitochondria-derived activator of caspases
- SMMC-7721:
-
Human hepatocarcinoma cell line
- STAT:
-
Signal transducer and activator of transcription
- TNFα:
-
Tumor necrosis factor-α
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- U251MG:
-
Glioblastoma cell line
- U937:
-
Histiocytic lymphoma cell line
- Ub:
-
Ubiquitin
- UPR:
-
Unfolded protein response
- Walker-256:
-
Rat breast carcinoma cell line
References
Adekenov SM (2016) Chemical modification of arglabin and biological activity of its new derivatives. Fitoterapia 110:196–205
Anfosso L, Efferth T, Albini A et al (2006) Microarray expression profiles of angiogenesis-related genes predict tumor cell response to artemisinins. Pharmacogenomics J 6:269–278
Armitage JO (2012) The aggressive peripheral T-cell lymphomas: update on diagnosis, risk stratification and management. Am J Hematol 87:511–519
Balunas MJ, Kingborn AD (2005) Drug discovery from medicinal plants. Life Sci 78:431–441
Bosio C, Tomasoni G, Martínez R et al (2015) Cytotoxic and apoptotic effects of leptocarpin, a plant-derived sesquiterpene lactone, on human cancer cell lines. Chem Biol Interact 242:415–421
Bujnicki T, Wilczek C, Schomburg C et al (2012) Inhibition of Myb-dependent gene expression by the sesquiterpene lactone mexicanin-I. Leukemia 26(4):615–622
Carlisi D, D’Anneo A, Angileri L et al (2011) Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by inducing the expression of death receptors through inhibition of STAT3activation. J Cell Physiol 226:1632–1641
Carlisi D, Buttitta G, Di Fiore R et al (2016) Parthenolide and DMAPT exert cytotoxic effects on breast cancer stem-like cells by inducing oxidative stress, mitochondrial dysfunction and necrosis. Cell Death Dis 7(4):e2194
Chabner BA, Amrein PC, Druker BJ et al (2006) Antineoplastic agents. In: Goodman, Gilman’s (eds) The pharmacological basis of the therapeutics, 11th edn. The McGraw-Hill Companies Inc, New York, pp 1731–1755
Chen HH, Zhou HJ, Wu GD et al (2004) Inhibitory effects of artesunate on angiogenesis and on expressions of vascular endothelial growth factor and VEGF receptor KDR/flk-1. Pharmacology 71:1–9
Cho JY, Kim AR, Jung JH et al (2004) Cytotoxic and pro-apoptotic activities of cynaropicrin, a sesquiterpene lactone on the viability of leukocyte cancer cell lines. Eur J Pharmacol 492:85–94
Christensen SB, Skytte DM, Denmeade SR et al (2009) A trojan horse in drug development: targeting of thapsigargins towards prostate cancer cells. Anti Cancer Agents Med Chem 9:276–294
Cragg GM, Grothaus PG, Newman DJ (2009) Impact of natural products on developing new anti-cancer agents. Chem Rev 109:3012–3043
De Ford C, Ulloa JL, Catalán CA et al (2015) The sesquiterpene lactone polymatin B from Smallanthus sonchifolius induces different cell death mechanisms in three cancer cell lines. Phytochemistry 117:332–339. https://doi.org/10.1016/j.phytochem.2015.06.020
Degos L, Wang ZY (2001) All trans retinoic acid in acute promyelocytic leukemia. Oncogene 20:7140–7145. https://doi.org/10.1038/sj.onc.1204763
Del Socorro Jimenez Usuga N, Malafronte N, Osorio Durango EJ et al (2016) Phytochemical investigation of Pseudelephantopus spiralis (Less.) Cronquist. Phytochem Lett 15:256–259
Denmeade SR, Isaacs JT (2005) The SERCA pump as a therapeutic target: making a ‘smart bomb’ for prostate cancer. Cancer Biol Ther 4:14–22
Ferlay J, Soerjomataram I, Dikshit R et al (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136(5):E359–E386
Ghantous A, Gali-Muhtasib H, Vuorela H et al (2010) What made sesquiterpene lactones reach cancer clinical trials? Drug Discov Today 15:668–678
Gomes Martins G, dos Reis Lívero FA, Stolf AM et al (2015) Sesquiterpene lactones of Moquiniastrum polymorpha subsp. floccosum have antineoplastic effects in Walker-256 tumor-bearing rats. Chem Biol Interact 228:46–56
Gopal YN, Chanchorn E, Van Dyke MW (2009) Parthenolide promotes the ubiquitination of MDM2 and activates p53 cellular functions. Mol Cancer Ther 8:552–562
Guzman ML, Rossi RM, Karnischky L et al (2005) The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 105:4163–4169
Guzman ML, Rossi RM, Neelakantan S et al (2007) An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 110:4427–4435
Hoffmann R, von Schwarzenberg K, Lopez-Anton N et al (2011) Helenalin by passes Bcl-2-mediated cell death resistance by inhibiting NF-kappaB and promoting reactive oxygen species generation. Biochem Pharmacol 82:453–463
Hopfinger G, Griessl R, Sifft E et al (2012) Novel treatment avenues for peripheral T-cell lymphomas. Expert Opin Drug Discovery 7:1149–1163
Huang CC, Lo CP, Chiu CY et al (2010) Deoxyelephantopin, a novel multifunctional agent, suppresses mammary tumour growth and lungmetastasis and doubles survival time in mice. Br J Pharmacol 159:856–871
Hung JY, Hsu YL, Ni WC et al (2010) Oxidative and endoplasmic reticulum stress signaling are involved in dehydrocostuslactone-mediated apoptosis in human non-small cell lungcancer cells. Lung Cancer 68:355–365
Idris AI, Libouban H, Nyangoga H et al (2009) Pharmacologic inhibitors of IkappaB kinase suppress growth and migration of mammary carcinosarcoma cells in vitro and prevent osteolyticbone metastasis in vivo. Mol Cancer Ther 8:2339–2347
Jaffe ES (2009) The 2008 WHO classification of lymphomas: implications for clinical practice and translational research. Hematology Am Soc Hematol Educ Program:523–531. https://doi.org/10.1182/asheducation-2009.1.523
JoungYoun U, Miklossy G, Chai X et al (2014) Bioactive sesquiterpene lactones and other compounds isolated from Vernonia cinerea. Fitoterapia 93:194–200
Kempema AM, Widen JC, Hexum JK et al (2015) Synthesis and antileukemic activities of C1–C10-modifiedparthenolide analogues. Bioorg Med Chem 23:4737–4745
Kiss I, Unger C, Huu CN et al (2015) Lobatin B inhibits NPM/ALK and NF-κB attenuating anaplastic-large cell-lymphomagenesis and lymph endothelial tumour in travasation. Cancer Lett 356:994–1006
Kreuger M, Grootjans S, Biavatti MW et al (2012) Sesquiterpene lactones as drugs with multiple targets in cancer treatment: focus on parthenolide. Anti-Cancer Drugs 23(9):883–896
Krysko DV, Vandenabeele P (2009) Part I-molecular mechanisms of phagocytosis of dying cells. In: Krysko DV, Vandenabeele P (eds) Phagocytosis of dying cells from molecular mechanisms to human diseases. Springer, Dordrecht, pp 3–31
Krysko DV, Brouckaert G, Kalai M et al (2003) Mechanisms of internalization of apoptotic and necrotic L929 cells by amacrophage cell line studied by electron microscopy. J Morphol 258:336–345
Kweon SH, Song JH, Kim HJ et al (2015) Induction of human leukemia cell differentiation via PKC/MAPK pathways by arsantin, a sesquiterpene lactone from Artemisia santolina. Arch Pharm Res 38(11):2020–2028. https://doi.org/10.1007/s12272-015-0609-4
Lee J, Hwangbo C, Lee JJ et al (2010) The sesquiterpene lactone eupatolide sensitizes breast cancer cells to TRAIL through down-regulationof c-FLIP expression. Oncol Rep 23:229–237
Li Y, Zhang Y, Fu M et al (2012) Parthenolide induces apoptosis and lytic cytotoxicity in Epstein-Barr virus-positive Burkitt lymphoma. Mol Med Rep 6:477–482
Li H, Li M, Wang G et al (2016) EM23, a natural sesquiterpene lactone from Elephantopus mollis, induces apoptosis in human myeloid leukemia cells through thioredoxin- and reactive oxygen species-mediated signaling pathways. Front Pharmacol. https://doi.org/10.3389/fphar.2016.00077
Liu Z, Liu S, Xie Z et al (2009) Modulation of DNA methylation by a sesquiterpene lactone parthenolide. J Pharmacol Exp Ther 329:505–514
Liu W, Wang X, Sun J et al (2017) Parthenolide suppresses pancreatic cell growth by autophagy-mediated apoptosis. Onco Targets Ther 10:453–461
Lohberger B, Rinner B, Stuendl N et al (2013) Sesquiterpene lactones downregulate G2/M cell cycle regulator proteins and affect the invasive potential of human soft tissue sarcoma cells. PLoS One 8:1–9
Mahalingam D, Wilding G, Denmeade S et al (2016) Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br J Cancer 114:986–994. https://doi.org/10.1038/bjc.2016.72
Marin GH, Mansilla E, Ciocchini S et al (2013) Sesquiterpene lactone extract from native American herbs demonstrated antineoplastic activity against non Hodgkin lymphoma cells. Annalen der Chemi Forschung 1(2):50–55
Martino R, Beer MF, Anesini C et al (2015) Sesquiterpene lactones from Ambrosia spp.are active against a murine lymphoma cell line by inducing apoptosis and cell cycle arrest. Toxicol In Vitro 29:1529–1536
Merfort I (2011) Perspectives on sesquiterpene lactones in inflammation and cancer. Curr Drug Targets 12:1560–1573
Muñoz Acuña U, Shen Q, Ren Y et al (2013) Goyazensolide induces apoptosis in cancer cells in vitro and in vivo. Int J Cancer Res 9(2):36–53. https://doi.org/10.3923/ijcr.2013.36.53
Nakase I, Gallis B, Takatani-Nakase T et al (2009) Transferrin receptor-dependent cytotoxicity of artemisinin–transferrin conjugates on prostate cancer cells and induction of apoptosis. Cancer Lett 274:290–298
Oh GS, Pae HO, Chung HT et al (2004) Dehydrocostuslactone enhances tumor necrosis factor-alpha-inducedapoptosis of human leukemia HL-60 cells. Immunopharmacol Immunotoxicol 26:163–175
Oka D, Nishimura K, Shiba M et al (2007) Sesquiterpene lactone parthenolide suppresses tumor growth in axenograft model of renal cell carcinoma by inhibiting the activation of NF-kappaB. Int J Cancer 120:2576–2581
Ordóñez PE, Sharma KK, Bystrom LM et al (2016) Dehydroleucodine, a sesquiterpene lactone from Gynoxys verrucosa, demonstrates cytotoxic activity against Human Leukemia Cells. J Nat Prod 79(4):691–696
Quynh D NT, Christensen SB (2015). Thapsigargin, Origin, Chemistry, Structure-Activity Relationships and Prodrug Development. Curr Pharm Des. 21(38):5501–5517
Ralstin MC, Gage EA, Yip-Schneider MT et al (2006) Parthenolide cooperates with NS398 to inhibit growth of humanhepatocellular carcinoma cells through effects on apoptosis and G0-G1 cellcycle arrest. Mol Cancer Res 4:387–399
Rozenblat S, Grossman S, Bergman M et al (2008) Induction of G2/M arrest and apoptosis by sesquiterpene lactones inhuman melanoma cell lines. Biochem Pharmacol 75:369–382
Saeed M, Jacob S, Sandjo LP et al (2015) Cytotoxicity of the sesquiterpene lactones neoambrosin and damsin from Ambrosia maritime against multidrug-resistant cancer cells. Front Pharmacol 6:267. https://doi.org/10.3389/fphar.2015.00267
Shukla N, Freeman N, Gadsdon P et al (2001) Thapsigargin inhibits angiogenesis in the rat isolated aorta:studies on the role of intracellular calcium pools. Cardiovasc Res 49:681–689
Singh NP, Panwar VK (2006) Case report of a pituitary macro adenoma treated with artemether. Integr Cancer Ther 5:391–394
Sturgeon CM, Craig K, Brown C et al (2005) Modulation of the G2 cell cycle checkpoint by sesquiterpene lactones psilostachyins A and C isolated from the common ragweed Ambrosia artemisiifolia. Planta Med 71(10):938–943
Sun Y, St Clair DK, Xu Y et al (2010) A NADPH oxidase dependent redox signaling pathway mediates the selective radio sensitization effect of parthenolide in prostate cancer cells. Cancer Res 70:2880–2890
Sweeney CJ, Mehrotra S, Sadaria MR et al (2005) The sesquiterpene lactone parthenolide in combination with docetaxel reduces metastasis and improves survival in a xenograft model of breast cancer. Mol Cancer Ther 4:1004–1012
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
Villagomez R, Rodrigo GC, Collado IG et al (2013) Multiple anticancer effects of damsin and coronopilin isolated from Ambrosia arborescens on cell cultures. Int J Anticancer Res 33:3799–3806
Wang GW, Qin JJ, Cheng XR et al (2014) Inula sesquiterpenoids: structural diversity, cytotoxicity and anti-tumor activity. Expert Opin Investig Drugs 23(3):317–345. https://doi.org/10.1517/13543784.2014.868882
World Health Organization (WHO) (2005) Preventing chronic diseases: a vital investment. Geneva: WHO Global report. http://www.who.int/chp/chronic_disease_report/en/. Accessed 14 Aug 2017
World Health Organization (WHO) (2017) http://www.who.int/cancer/en/. Accessed 14 Aug 2017
Yang YJ, Yao J, Jin X et al (2016) Sesquiterpenoids and tirucallane triterpenoids from the roots of Scorzonera divaricata. Phytochemistry 124:86–98
Yip-Schneider MT, Nakshatri H, Sweeney CJ et al (2005) Parthenolide and sulindac cooperate to mediate growth suppression and inhibit the nuclear factor-kB pathway in pancreatic carcinoma cells. Mol Cancer Ther 4:587–594
Zeisig BB, Kulasekararaj AG, Mufti GJ et al (2012) Snap shot: acute myeloid leukemia. Cancer Cell 22:691–698. https://doi.org/10.1016/j.ccr.2012.10.017
Zhang C, Lu T, Wang GD et al (2016) Costunolide, an active sesquiterpene lactone, induced apoptosis via ROS-mediated ER stress and JNK pathway in human U2OS cells. Biomed Pharmacother 80:253–259
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Anesini, C.A., Alonso, M.R., Martino, R.F. (2018). Antiproliferative and Cytotoxic Activities. In: Sülsen, V., Martino, V. (eds) Sesquiterpene Lactones. Springer, Cham. https://doi.org/10.1007/978-3-319-78274-4_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-78274-4_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-78273-7
Online ISBN: 978-3-319-78274-4
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)