
Abstract
Over the past 15 years, there has been intense research interest in periodontitis and its associations with several systemic conditions and how periodontitis can modify the expression of those diseases. The area that looks forward in those relationships is called periodontal medicine. Offenbacher described periodontal medicine as a discipline that focuses on the investigation of associations between periodontal diseases and systemic diseases and their biological plausibility in human populations and in animal models. It has been reported that periodontal disease may independently increase the risk of diabetes mellitus, cardiovascular disease, preterm or low-weight delivery, or rheumatoid arthritis. On the other hand, periodontitis is a chronic infection, which pathogenesis is orchestrated by multiple factors. Within those factors, genetics and epigenetics may have an important role in the pathogenesis. Epigenetics is a new area in research that is defined as genetic control by factors other than an individual’s DNA sequence via silencing certain genes while promoting others. These processes involve regulating transcription factor and access to chromatin, as well as microRNA (miRNA) and long noncoding RNA (lncRNA) regulating the expression of mRNA. In this chapter, we are going to deal with periodontitis pathogenesis, the role of epigenetics in its process, and the new connections of periodontitis and some systemic conditions by the expression of some epigenetic factors. This basic knowledge drives to know how to understand the possible connections and some targets to cope with in the future.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
3.1 Introduction
Periodontitis is a chronic inflammatory disease characterized by periodontal attachment and alveolar bone loss that eventually may lead to tooth loss. It is considered the most prevalent chronic inflammatory disease that affects 46% of adults older than 30 years in the USA [1], where the prevalence of severe periodontitis is over 11% [2]. The primary etiological factor of periodontitis is the presence of specific bacteria organized in dental biofilm that leads to the triggering of several signaling routes that prompts the initiation of immune response mechanisms. The microflora that causes periodontitis is somehow complex, in which there are over 700 different bacterial species. In the recent years, some clinical studies have defined key pathogens that present a crucial role in the initiation and progression of periodontitis [3, 4], demonstrating that their presence is a risk factor for ongoing attachment loss. Among those bacterial species, Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis are the most relevant pathogens in periodontitis. Both species exhibit a wide range of virulent factors that could activate several signaling pathways, inducing a humoral immune host response [5, 6].
While periodontopathogenic bacteria are the main cause in the activation of immune response, other elements such as genetics and environmental factors may influence or modulate the host response to bacterial burden [7,8,9].
3.1.1 Molecular Regulation of the Immune Response
In gene expression, its central dogma is based on the DNA transcription into mRNA, which serves, in turn, as template for the protein synthesis [10]. The immune response model at the transcriptional stage starts with an external stimuli, such as bacterial burden, which gives rise to a signaling cascade within the cell cytoplasm, activating the binding of promoters and enhancers that regulates some DNA sequences. These assembles activate in turn the transcription machinery as well as the expression of specific genes. At this time, certain DNA sequence alterations, mutations, and polymorphisms may come out to alter transcription factors binding to specific areas of gene expression regulation.
Regarding the protein structure, the changes might occur within the coding regions of the gene (exons), leading to changes in the position and sequence of amino acids in the protein structure. In this sense, some novel processes in this basic model have been depicted in a recent study [11] that have suggested synergistic mechanisms between transcriptional activators bound to promoters and enhancers in different layers that are superimposed, leading to different stages in gene modification that may occur.
It has been identified specific mechanisms to be rapidly turned on and off in response to that external stimuli [11]. Those have been suggested as special pathways in creating selectivity of immune response to certain bacterial stimuli, regulating the magnitude of that response and conducting to chronic inflammation [12, 13]. Therefore, it could present a potential role in how some bacterial species may activate different signaling pathways leading to a specific host immune response.
3.1.2 Epigenetic Modifications of Gene Expression
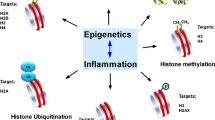
Epigenetics is defined as changes in gene expression, which are not controlled by DNA sequence but by silencing certain genes and promoting others. These processes involve alterations of the DNA and associated proteins such as DNA methylation or modifying DNA structure (e.g., chromatin alterations), as well as microRNA (miRNA) and long noncoding RNA (lncRNA) that may regulate the expression of messenger RNA (mRNA) [14]. These epigenetic alterations may be able to modulate the host response to several bacterial stimuli and regulate the magnitude and speed of those processes [7, 9].
Genetic mutations are commonly known to control the epigenome [15], inducing several changes in DNA structure and sequence alterations that affect the protein function. Nevertheless, some epigenetic modifications such as histone modulation are often strongly correlated with patterns of inherited gene expression [16], including those that lead to gene mutations. Hence, the link or boundary between genetics and epigenetics is still not clear, driving to the concept that some researchers [15] suggest that both are two sides of the same construct, being connected.
3.1.3 Immune Response in Periodontitis and Epigenetic Modifications
The immune host response to the bacterial products starts with the innate immune system activation. This is the natural host barrier defense against bacteria, which is mediated by subepithelial dendritic cells, neutrophils, macrophages, natural killer cells, and monocytes. Those cell activations are orchestrated by several families of proteins such as toll-like receptors (TLRs) that are involved in the bacterial recognition of their patterns.
Innate system does not require any previous exposure to a microbial product, so thus, it is so important in initiating the host response to new microbial challenges. However, some bacteria have the capacity to evade some host response mechanisms as a result from adaptation to the hostile environment [17]. Shifts within the periodontal tissues have been ascribed to the interaction between biofilms and their products and some specific receptors such as TLRs.
Afterward, when innate response is not able to control the bacterial aggression, adaptive immune response is activated. This host response is normally mediated by T- and B-cells that will provide to the host the so-called immunological memory. This adaptive response may play a crucial role in established periodontitis lesions that are characterized by high proportions of plasma cells [18], which in turn are going to generate antibodies against microbial products.
Nevertheless, the initiation and the magnitude of this interaction between biofilms, its products, and the host response may be altered by genetic traits but also by some systemic conditions and environmental factors such as smoking or stress. Within this interaction, epigenetic modifications may modify the interplay between environmental factors, genetic traits, and host immune response.
3.1.4 DNA Methylation, Histone Modulation, and Other Gene Alterations
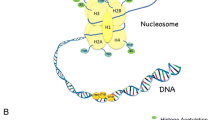
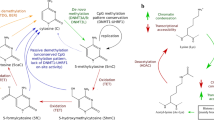
The genome modification in host cells can often occur via DNA methylation . It normally takes place at the 5′ position of cytosine in CpG dinucleotides, called CpG islands. CpG islands are dinucleotides that involve the connection between a cytosine nucleotide and a guanine nucleotide by a phosphodiester bond. Clusters of CpG sequences appear in the promoter regions, prevent transcriptional initiation, and silence the genes [31]. There are three different isoforms of DNA methyltransferases by which DNA methylation can occur. DNA methyltransferase 3a and DNA methyltransferase 3b normally present a crucial role in “de novo” methylation of CpG residues, while DNA methyltransferase 1 (DNMT1) regulates the methylation pattern copying to new synthetized DNA strand during replication. DNA methylation is necessary for normal cell development, and it is essential in tissue-specific gene transcription [19]. Differential methylation patterns associated with lipopolysaccharide (LPS) signaling , cell adhesion, and other processes such as apoptosis or oncogenesis have been observed in untreated periodontitis tissues [20]. Recent studies have suggested that bacteria have the potential to cause alterations in the DNA methylation status, but also the environment, aging, and stress may also play a role on modifying the expression of periodontitis, involving epigenetic changes that affect disease expression or cause oral dysbiosis.
Depending on the health status of periodontal tissues, CpG islands are differentially methylated. Recent data that investigated the differences in DNA methylations between healthy gingival tissues from healthy subjects and inflamed periodontal tissues from chronic periodontitis tissues identified some CpG sites methylated in inflamed tissues different from healthy tissues [20]. Some genes such as SOCS3, VDR, MMP25, BMP4, RUNX3, interleukin-17, TNFRSF18, ZNF277, ZNF501, CADM3, and BDNF were observed hypermethylated in inflamed gingival tissues and others hypomethylated compared to healthy gingival tissues [20]. The authors suggested that these methylated CpG regions might confirm a linkage between epigenetic modifications and the immune host response. One example of DNA methylation in periodontitis is the DNA hypermethylation of prostaglandin-endoperoxidase synthase 2 promoter in chronic periodontitis lesions. This pattern is associated with a high expression of cyclooxygenase-2 in chronic periodontitis tissues, increasing the inflammation status of the tissues. Those epigenetic modifications of gingival tissues are suggested to mainly occur within the biofilm-sulcular epithelium interface. Within this interaction bacteria may have the capacity to induce some DNA methylations (meter ref). It has been described that Porphyromonas gingivalis could cause the hypermethylation of a protein that regulates the chromatin remodeling, called as GATA binding protein [21]. This protein could also be hypomethylated by a Fusobacterium nucleatum infection, driving to the concept that one epigenetic modification may occur depending on the presence of specific bacteria.
Other epigenetic change related to oral dysbiosis is histone acetylation. A recent study conducted by Martins and co-workers [22] reported that both regulation of DNMT1 and histone acetylation were shown in oral dysbiosis. This study has proven that epigenetic changes may indeed be associated with oral dysbiosis.
In general, histone acetylation is associated with enhanced transcription of genes [31], nucleosome assembly, chromatin folding, DNA damage repair, and replication [23]. It is usually related to the chromatin structure relaxing, leading to an enhanced transcription of inflammatory genes, such as nuclear factor kappa B (NF-κB) target gene or several genes of pro-inflammatory cytokines, which are commonly upregulated in periodontitis. Normally, NF-κB is able to activate innate immune system mediated by TLRs [24], and its chronic activation may induce osteoclastogenesis that would lead to bone resorption [25]. In periodontitis lesions, it has been demonstrated that Porphyromonas gingivalis and Fusobacterium nucleatum infection may be able to induce epigenetic modifications such as histone 3 acetylation and the downregulation of DNMT1.
Moreover, histone acetylation by lipopolysaccharides (LPS) could influence p300/CBP activation. Its activation is related to the transcriptional stimulation of some pro-inflammatory cytokines such as IL-1, IL-2, IL-8, and IL-12, commonly found in periodontally affected tissues [22].
3.2 Role of MicroRNAs
MicroRNAs (miRNAs) are a group of small, noncoding RNAs that play key roles in epigenetic regulation by controlling the translation and stability of mRNAs [26]. They are crucial in developmental processes, apoptosis, and cell proliferation [27]. However, this regulation depends on the activities of other cofactors, DNA methylation and/or histone acetylation. The other cofactors include RNA-binding protein, CREB-binding protein or E1A binding protein p300, and cyclic AMP response element-binding protein (CBP) . This regulation indirectly inhibits or promotes mRNA expression. Hence, these molecules may play a significant role in inflammatory processes, affecting both innate and humoral immune response to the microbial challenge [7, 26]. In addition, autoimmune diseases may be also affected by these molecules [26]. However, periodontitis is the gold standard in oral chronic inflammatory diseases, where miRNA might have a specific and detrimental role in its pathogenesis [7]. Recently, Kebschull and Papapanou have performed an extensive review about the role of miRNA in periodontal disease [7]. They have observed that miRNAs could affect different immune processes at different stages of the inflammatory response against the bacterial insult in periodontal disease.
As it has been explained, the inflammatory process starts with a bacterial challenge. The first line in host response involves the pathogen pattern recognition mediated by TLRs. The most common TLRs in periodontitis are TLR-2 and TLR-4, which have the capacity to interact with specific bacterial species and their virulent factors. Aggregatibacter actinomycetemcomitans and the lipopolysaccharide (LPS) from Porphyromonas gingivalis are the main bacterial species and products that are able to activate those receptors. After their binding, NF-κB pathway is activated. NF-κB is a family of rapid responder transcription factors that are able to induce changes in target gene expression. As a consequence of TLR activation, different cell types are triggered, such as macrophages, neutrophils, natural killer cells, or dendritic cells, developing the comprehensive innate immune response.
Several miRNAs are able to induce changes in the NF-κB signaling pathways. One of them is the miRNA-146a, which is itself regulated after NF-κB activation via TLR-2, TLR-4, TLR-5, and TLR-9 in response to the bacterial challenge, driving to a downregulation of two important cytokine receptors, receptors of IL-1 and TNFα. This miRNA was found to be upregulated in periodontally affected gingival tissues [28, 29]. miR-146a is also able to suppress TLR-2 expression in keratinocytes [30] and macrophages [31], leading to a weak inflammatory response. However, in macrophages, the induction of miRNA-146a by P. gingivalis LPS does not result in a lower cytokine production [32], driving to the concept of a counteracting effect by other miRNAs [7].
One of the most important miRNAs in periodontitis is miR-155, which acts at different stages in the host response. This miRNA is able to downregulate NF-κB signaling pathway, as well as to promote cell differentiation [33]. It also mediates the response to infection by type I interferon production [34], a mechanism possibly connected to aggressive periodontitis [35], increasing inflammation and periodontal tissue destruction. Furthermore, it is able to induce the expansion and activation of natural killer cells [36] and high production of interferon-γ [37]. In macrophages, it was shown that miR-155 increases the levels of leukotriene B4, resulting in high responsiveness of TLRs [38]. However, the dysregulation of its expression and also the miR-146 expression in epithelial cells infected by P. gingivalis was observed to turn into an increased sensitivity of TLR signaling pathway, exponentially expanding the immune response [39, 40].
In natural killer cells , miR-30e and miR-200a are downregulated in periodontally affected tissues that lead to an increased activation of natural killer cells and a higher production of interferon-γ, driving to a higher tissue destruction [41, 42]. Other miRNAs such as miR-451, which has the ability to suppress neutrophil chemotaxis [43], or miR-486, which leads to an overexpression of NF-κB signaling [44], are upregulated in periodontitis lesions.
Dendritic cells play an essential role in the innate system, bridging the innate system and adaptive immune response. They are able to detect some bacterial species and their products, producing specific inflammatory cytokines, critical in the immune response. Recent investigations have pointed out that their functions are tightly controlled by miRNAs [45].
One example is miR-155, a miRNA that was found to control other innate system processes (see above). It was shown to have the ability to affect the function and maturation of dendritic cells, influencing the cytokine signaling and thus the inflammatory host response [46]. On the other hand, miR-451 has exhibited to reduce cytokine production by dendritic cells, which has responded to bacterial infection [47], and miR-148a was shown to damage the innate response and the antigen presentation mechanism by dendritic cells [48], both overexpressed in periodontally affected gingival tissues.
As the innate response can be regulated by several miRNAs, some of those might also influence certain adaptive response processes. B- and T-cells are the main cell strains in the adaptive immune system. This system involves their expansion and provides immunological memory. Some miRNAs, such as miR-146a, miR-650, miR-155, miR-210, or miR-455, may play a role in the control of some adaptive immune processes. Other miRNAs are upregulated in periodontitis lesions such as miR-650 that was found to influence the proliferative capacity of B-cells [49]. As seen above, miR-155, an upregulated miRNA in periodontally affected tissues, has different functions in host immune regulation. In adaptive immune, this miRNA could also have the capacity to control CD8 T-cell response [50], as well as to influence in the indirect activation of T-helper cell 17 response by dendritic cell signaling [51]. Nevertheless, others are downregulated in periodontitis lesions such as miR-210, which are related to the increase of T-cell signaling, being associated with the etiopathogenesis of periodontitis [52, 53].
In summary, miRNAs are epigenetic modifications that are crucial in immune response regulation, being bridges between different exogenous challenges and host response. While further research in miRNA functions and role in chronic inflammatory disease such as periodontal disease is needed, they are a very promising source of connection between different immune processes in different oral and systemic conditions.
3.3 Role of Long Noncoding RNAs
Long noncoding RNAs (lncRNAs) modulate cell proliferation, senescence, migration, and apoptosis. They also interact with DNA, RNA, and other proteins and regulate gene expression and other miRNA activities. A recent publication performed by Zou et al. [54] has demonstrated the presence of some lncRNAs in chronic periodontitis lesions. As miRNAs, lncRNAs could be up- or downregulated, affecting different host immune pathways and miRNA functions. Some lncRNAs such as HOTAIR, PRDX6, IFNG, or TIRAP are associated with periodontitis lesions [54]. Periodontitis was shown to express the upregulation of HOTAIR, PI3 (lncRNAs RP3-461P17 and RP1-300I2.2), PRDX6, TIRAP, lncRNA CDKN2A, and CDKN2B. Conversely, lncRNAs NR_003716, RP11–29014.3, IFNG, lincRNA-CDON-1, and CDKN2BAS were shown to be downregulated in periodontally affected tissues. Certain lncRNAs such as lincRNA-CDON-1 appear to be involved in signaling pathways in TLR expression, crucial in the etiopathogenesis of periodontitis. Likewise, further studies to understand the role and functions of those lncRNAs in periodontitis pathogenesis are needed.
It has been found that lncRNAs possess transcribed ultraconserved regions (T-UCRs), which are a segment of DNA and considered as a novel class of noncoding RNAs. UCRs are conserved, i.e., unchanged between the species. Therefore, alteration in this area is unlikely to occur due to chance, and differential expressions have been observed in several systemic conditions such as cancers or cardiovascular diseases [55].
3.4 Periodontitis, Epigenetic Alterations, and Systemic Diseases
Nowadays, periodontitis is considered a noncommunicable chronic inflammatory infectious disease which is connected to some systemic conditions. Recent epidemiological studies have remarked important associations between periodontitis and some systemic diseases such as diabetes, cardiovascular disease, rheumatoid arthritis, or cancer [56, 57], where systemic inflammation and bacteremia are the main mechanisms. However, some epigenetic modifications commonly found in periodontitis are also related to some systemic conditions, leading to another biological mechanism of connection between periodontitis and systemic diseases.
As it has been discussed in previous items, oral dysbiosis, commonly related to periodontitis, is able to produce epigenetic modifications that, in turn, have the capacity to induce some changes in different levels of immune response, both in the innate and humoral system, leading to a chronic response that could be associated with other systemic immune responses or even changes in other sites of the human body.
3.4.1 Obesity
The association between periodontitis and obesity has been studied along years. While a systematic review published by Suvan et al. [58] has established that there are clinical studies that support this association, the magnitude of that association is still unclear, and there is a need for more prospective and intervention studies to understand the connection between both diseases. Despite the fact that recent data has demonstrated that obesity could be a risk factor of clinical attachment loss [59], the biological plausibility of that connection is still not clarified. It has been hypothesized that the high secretion of adipokines in obesity creates a pro-inflammatory state that has been positively associated with periodontitis [60]. A recent publication [61] has showed that the stimulation of macrophages with adiponectin causes the expression of miR-155, an important miRNA in the pathogenesis of periodontitis [62]. Moreover, it has been demonstrated that some miRNAs such as miR-185 were found to be strongly expressed in obese patients with periodontitis compared to nonobese periodontitis patients [62], suggesting that an obese status may aggravate periodontal tissue destruction.
3.4.2 Cardiovascular Disease
The association between periodontitis and cardiovascular disease (CVD) has been a focus of research along the last decades. Last data have established that periodontitis may be a risk factor to control in CVD patients [57, 63,64,65,66]. Bacteremia has been hypothesized as the main biological mechanism that connects both diseases [67, 68]. Nevertheless, recent publications have pointed out a genetic susceptibility contributing to periodontitis and CVD [69]. This publication has remarked some genes that are presented in both conditions. ANRIL, CDKN2A, CDKN2B, and PLG are the most relevant genes [69,70,71,72]. In that sense, mRNA transcription of ANRIL, lncRNA, and ANRIL has been associated with atherosclerosis, periodontitis, and several types of cancers [70]. Therefore, it is important to keep investigating the influence of those lncRNAs and genes to fully understand the connection and possible target to treat consequences in both conditions.
3.4.3 Rheumatoid Arthritis
Rheumatoid arthritis(RA) is a chronic inflammatory autoimmune disease that has been extensively related to periodontitis [73, 74]. It has been hypothesized that oral bacteria, such as Porphyromonas gingivalis, among others [75], may play a key role in protein citrullination and ACPA formation in RA patients [76, 77], initiating and perpetuating the immune response in RA. Recent reports have shown that also Aggregatibacter actinomycetemcomitans, another well-established periodontal pathogen [78], is able to increase chronic exposure to citrullinated proteins and the development of autoantibodies. A number of evidence have shown that periodontitis and RA share a number of genetic and environmental risk factors [79]. The main genetic risk factor is the HLA-DRB1 allele of the class II major histocompatibility complex (MHC-II), and the smoking habit is a common risk factor between periodontitis and RA [75].
Nonetheless, recent data have shown that periodontitis patients are exposed to citrullinated histone H3 in inflamed gingival tissues, which drives to other exposure target for the autoantibodies presented in RA [80]. It means that not only proteins citrullinated by bacteria are subjected to be a target for autoantibodies, but some epigenetic modifications of that oral dysbiosis increase targets and sources of perpetuating inflammation and citrullination.
3.4.4 Cancer
The role of oral infections in oncogenesis remains changing over time. As discussed above, chronic inflammation such as in periodontitis may have the potential to provoke epigenetic modifications [20] leading to DNA and histone methylations that contribute to oncogenesis. However, any bone modulation will involve these histone modifications [81], and periodontitis, which involves bone loss, may also cause histone modulation [14].
A recent publication by our group [14] has extensively reviewed the plausible role of oral infections such as periodontitis in oncogenesis. We have found different population-based studies, by which a plausible association between periodontitis and different types of cancer could be explained. A longitudinal study reported that serum P. gingivalis antibody increased the risk of orodigestive cancer mortality [82]. Likewise, some data showed antibodies to several oral pathogens to pancreatic cancer [83]. It was found that the antibodies to the commensals were associated with lower risk of pancreatic cancer suggesting that dysbiosis may be a more appropriate risk marker than the role of a few pathogens. Dysbiosis on the other hand can be a marker for abnormal immunity which predisposes to cancer development [84]. Moreover, a prospective cohort study that assessed periodontal treatment was associated with lower risk of subsequent cancers, but this study did not adjust for confounding factors such as smoking, alcohol consumption, or genetics [85]. Despite some methodological flaws of those studies, there is a plausible role of periodontal disease in the oncogenesis process.
As it has been discussed above, epigenetics might play a significant role in different host immune processes as well as other cell functions. During the process of tumor initiation and progression, the cancer epigenome is remodeled via global hypomethylation, increased promoter methylation at CpG islands, global downregulation of miRNAs and lncRNAs, or interactions between them and alterations in the nucleosome. The imbalance between transcriptionally permissive and repressive chromatin modifications may alter gene expression and lead to cancer [14]. While DNMT1 is overexpressed in many cancers [86], oral dysbiosis drives to a reduced expression of DNMT1. Thus, the assumption that oral dysbiosis may foster oncogenesis might not be supported. On the other hand, histone acetylation has been shown to regulate tumor suppressor gene p53 or proto-oncogene c-Myb. These indicate that histone acetylation can up- or downregulate oncogenesis [87].
Notably, the role of miRNAs in oncogenesis varies depending on the mRNA they regulate and thus can be promoters or suppressors of oncogenesis [88]. Some miRNAs commonly found in periodontitis have been related to different types of cancers. MiR-31 may be found in pancreatic cancer or oral potentially malignant disorder [89, 90]. Moreover, miRNAs, miR-146a and miR-155, were related to head and neck squamous cell carcinoma [91]. Nonetheless, certain lncRNAs have been also correlated with different types of cancers. The lncRNA called HOTAIR, upregulated in periodontitis lesions, has been associated with tumor metastasis, recurrence, and prognosis in breast, colon, and liver cancers and oral squamous cell carcinoma [54, 92, 93].
However, further studies are needed to understand how they may interact between both conditions and to examine the role of oral infections in carcinogenesis via the holistic approach considering the multisystem in the whole human body (Tables 3.1, 3.2, 3.3, 3.4, and 3.5).
3.5 Concluding Remarks
Epigenetics is a new field that bridges genetics with environment, adapting the host response to the bacterial infection. The interaction between genetics, immunity epigenetics, and inflammation might play major roles in periodontitis, connecting it with systemic conditions. However, further studies are needed to explore these bridges, their impact and functions, not only in periodontitis and systemic conditions that have been drawn in this chapter but also with other autoimmune diseases.
References
Eke PI, et al. Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol. 2015;86(5):611–22.
Kassebaum NJ, et al. Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J Dent Res. 2014;93(11):1045–53.
Roberts FA, Darveau RP. Microbial protection and virulence in periodontal tissue as a function of polymicrobial communities: symbiosis and dysbiosis. Periodontology 2000. 2015;69(1):18–27.
Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontology 2000. 2005;38:135–87.
Henderson B, Ward JM, Ready D. Aggregatibacter (Actinobacillus) actinomycetemcomitans: a triple A* periodontopathogen? Periodontology 2000. 2010;54(1):78–105.
Pathirana RD, O’Brien-Simpson NM, Reynolds EC. Host immune responses to Porphyromonas gingivalis antigens. Periodontology 2000. 2010;52(1):218–37.
Kebschull M, Papapanou PN. Mini but mighty: microRNAs in the pathobiology of periodontal disease. Periodontol. 2015;69(1):201–20.
Kornman KS. Mapping the pathogenesis of periodontitis: a new look. J Periodontol. 2008;79(8 Suppl):1560–8.
Larsson L, Castilho RM, Giannobile WV. Epigenetics and its role in periodontal diseases: a state-of-the-art review. J Periodontol. 2015;86(4):556–68.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81(1):145–66.
Smale ST. Transcriptional regulation in the innate immune system. Curr Opin Immunol. 2012;24(1):51–7.
O’Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30(1):295–312.
Rebane A, Akdis CA. MicroRNAs: essential players in the regulation of inflammation. J Allergy Clin Immunol. 2013;132(1):15–26.
Janket S-J, Qureshi M, Bascones-Martinez A, González-Febles J, Meurman JH. Holistic paradigm in carcinogenesis: genetics, epigenetics, immunity, inflammation and oral infections. World J Immunol. 2017;7(2):11.
You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22(1):9–20.
Ptashne M. On the use of the word ‘epigenetic’. Curr Biol. 2007;17(7):R233–6.
Marsh PD, Zaura E. Dental biofilm: ecological interactions in health and disease. J Clin Periodontol. 2017;44:S12–22.
Berglundh T, Donati M. Aspects of adaptive host response in periodontitis. J Clin Periodontol. 2005;32(Suppl 6):87–107.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–92.
Barros SP, Offenbacher S. Modifiable risk factors in periodontal disease: epigenetic regulation of gene expression in the inflammatory response. Periodontology 2000. 2014;64(1):95–110.
Yin L, Chung WO. Epigenetic regulation of human β-defensin 2 and CC chemokine ligand 20 expression in gingival epithelial cells in response to oral bacteria. Mucosal Immunol. 2011;4(4):409–19.
Martins MD, et al. Epigenetic modifications of histones in periodontal disease. J Dent Res. 2015;95(2):215–22. Available at: http://journals.sagepub.com/doi/10.1177/0022034515611876
Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76(1):75–100.
Dev A, et al. NF-κB and innate immunity. Curr Top Microbiol Immunol. 2011;349(Chapter 102):115–43.
Abu-Amer Y. NF-κB signaling and bone resorption. Osteoporos Int. 2013;24(9):2377–86.
Meurman JH. Genetic regulation of inflammation – micro-RNA revolution? Oral Dis. 2017;23(1):1–2.
Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12(11):847–65.
Nahid MA, et al. Polymicrobial infection with periodontal pathogens specifically enhances microRNA miR-146a in ApoE−/− mice during experimental periodontal disease. Infect Immun. 2011;79(4):1597–605.
Taganov KD, et al. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481–6.
Meisgen F, et al. MiR-146a negatively regulates TLR2-induced inflammatory responses in keratinocytes. J Invest Dermatol. 2014;134(7):1931–40.
Quinn EM, et al. MicroRNA-146a is upregulated by and negatively regulates TLR2 signaling. PLoS One. 2013;8(4):e62232.
Honda T, et al. Porphyromonas gingivalis lipopolysaccharide induces miR-146a without altering the production of inflammatory cytokines. Biochem Biophys Res Commun. 2012;420(4):918–25.
Hung P-S, et al. miR-146a induces differentiation of periodontal ligament cells. J Dent Res. 2010;89(3):252–7.
Wang P, et al. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. 2010;185(10):6226–33.
Nowak M, et al. Activation of invariant NK T cells in periodontitis lesions. J Immunol. 2013;190(5):2282–91.
Trotta R, et al. Overexpression of miR-155 causes expansion, arrest in terminal differentiation and functional activation of mouse natural killer cells. Blood. 2013;121(16):3126–34.
Trotta R, et al. miR-155 regulates IFN-γ production in natural killer cells. Blood. 2012;119(15):3478–85.
Wang Z, et al. Leukotriene B4 enhances the generation of proinflammatory microRNAs to promote MyD88-dependent macrophage activation. J Immunol. 2014;192(5):2349–56.
Hou J, et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. 2009;183(3):2150–8.
Ruggiero T, et al. LPS induces KH-type splicing regulatory protein-dependent processing of microRNA-155 precursors in macrophages. FASEB J. 2009;23(9):2898–908.
Chaushu S, et al. Direct recognition of Fusobacterium nucleatum by the NK cell natural cytotoxicity receptor NKp46 aggravates periodontal disease. PLoS Pathog. 2012;8(3):e1002601.
Krämer B, et al. Role of the NK cell-activating receptor CRACC in periodontitis. Infect Immun. 2013;81(3):690–6.
Yang J-S, et al. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci U S A. 2010;107(34):15163–8.
Song L, et al. miR-486 sustains NF-κB activity by disrupting multiple NF-κB-negative feedback loops. Cell Res. 2013;23(2):274–89.
Smyth LA, et al. MicroRNAs affect dendritic cell function and phenotype. Immunology. 2015;144(2):197–205.
Dunand-Sauthier I, et al. Silencing of c-Fos expression by microRNA-155 is critical for dendritic cell maturation and function. Blood. 2011;117(17):4490–500.
Rosenberger CM, et al. miR-451 regulates dendritic cell cytokine responses to influenza infection. J Immunol. 2012;189(12):5965–75.
Liu X, et al. MicroRNA-148/152 impair innate response and antigen presentation of TLR-triggered dendritic cells by targeting CaMKIIα. J Immunol. 2010;185(12):7244–51.
Mraz M, et al. MicroRNA-650 expression is influenced by immunoglobulin gene rearrangement and affects the biology of chronic lymphocytic leukemia. Blood. 2012;119(9):2110–3.
Gracias DT, et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat Immunol. 2013;14(6):593–602.
O’Connell RM, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33(4):607–19.
Parachuru VPB, et al. Forkhead box P3-positive regulatory T-cells and interleukin 17-positive T-helper 17 cells in chronic inflammatory periodontal disease. J Periodontal Res. 2014;49(6):817–26.
Zhao M, et al. Up-regulation of microRNA-210 induces immune dysfunction via targeting FOXP3 in CD4(+) T cells of psoriasis vulgaris. Clin Immunol. 2014;150(1):22–30.
Zou Y, et al. lncRNA expression signatures in periodontitis revealed by microarray: the potential role of lncRNAs in periodontitis pathogenesis. J Cell Biochem. 2015;116(4):640–7.
Goto K, et al. The transcribed-ultraconserved regions in prostate and gastric cancer: DNA hypermethylation and microRNA-associated regulation. Oncogene. 2016;35(27):3598–606.
Taylor JJ, Preshaw PM, Lalla E. A review of the evidence for pathogenic mechanisms that may link periodontitis and diabetes. J Periodontol. 2013;84(4 Suppl):S113–34.
Tonetti MS, Van Dyke TE, Working Group 1 of the Joint EFP/AAP Workshop. Periodontitis and atherosclerotic cardiovascular disease: consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Clin Periodontol. 2013;40(Suppl 14):S24–9.
Suvan J, et al. Association between overweight/obesity and periodontitis in adults. A systematic review. Obes Rev. 2011;12(5):e381–404.
Suvan J, et al. Association between overweight/obesity and increased risk of periodontitis. J Clin Periodontol. 2015. https://doi.org/10.1111/jcpe.12421.
Fantuzzi G. Adiponectin in inflammatory and immune-mediated diseases. Cytokine. 2013;64(1):1–10.
Subedi A, Park P-H. Autocrine and paracrine modulation of microRNA-155 expression by globular adiponectin in RAW 264.7 macrophages: involvement of MAPK/NF-κB pathway. Cytokine. 2013;64(3):638–41.
Perri R, et al. MicroRNA modulation in obesity and periodontitis. J Dent Res. 2012;91(1):33–8.
Dietrich T, et al. The epidemiological evidence behind the association between periodontitis and incident atherosclerotic cardiovascular disease. J Clin Periodontol. 2013;40(Suppl 14):S70–84.
Liljestrand JM, et al. Missing teeth predict incident cardiovascular events, diabetes, and death. J Dent Res. 2015;94(8):1055–62.
Mäntylä P, et al. Acute myocardial infarction elevates serine protease activity in saliva of patients with periodontitis. J Periodontal Res. 2012;47(3):345–53.
Widén C, et al. Systemic inflammatory impact of periodontitis on acute coronary syndrome. J Clin Periodontol. 2016;43(9):713–9.
Kebschull M, et al. “Gum bug, leave my heart alone!”—epidemiologic and mechanistic evidence linking periodontal infections and atherosclerosis. J Dent Res. 2010;89(9):879–902.
Reyes L, et al. Periodontal bacterial invasion and infection: contribution to atherosclerotic pathology. J Clin Periodontol. 2013;40(Suppl 14):S30–50.
Aarabi G, et al. Genetic susceptibility contributing to periodontal and cardiovascular disease. J Dent Res. 2017;96(6):610–7.
Bochenek G, et al. The large non-coding RNA ANRIL, which is associated with atherosclerosis, periodontitis and several forms of cancer, regulates ADIPOR1, VAMP3 and C11ORF10. Hum Mol Genet. 2013;22(22):4516–27.
Schaefer AS, et al. Genetic evidence for PLASMINOGEN as a shared genetic risk factor of coronary artery disease and periodontitis. Circ Cardiovasc Genet. 2015;8(1):159–67.
Teeuw WJ, et al. A lead ANRIL polymorphism is associated with elevated CRP levels in periodontitis: a pilot case-control study. PLoS One. 2015;10(9):e0137335.
Kaur S, White S, Bartold PM. Periodontal disease and rheumatoid arthritis: a systematic review. J Dent Res. 2013;92(5):399–408. Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=23525531&retmode=ref&cmd=prlinks
Koziel J, Mydel P, Potempa J. The link between periodontal disease and rheumatoid arthritis: an updated review. Curr Rheumatol Rep. 2014;16(3):408.
de Pablo P, et al. Periodontitis in systemic rheumatic diseases. Nat Rev Rheumatol. 2009;5(4):218–24. Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19337286&retmode=ref&cmd=prlinks
Mikuls TR, et al. Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014;66(5):1090–100.
Ziebolz D, et al. Clinical periodontal and microbiologic parameters in patients with rheumatoid arthritis. J Periodontol. 2011;82(10):1424–32.
Konig MF, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. 2016;8(369):369ra176.
Marotte H. The association between periodontal disease and joint destruction in rheumatoid arthritis extends the link between the HLA-DR shared epitope and severity of bone destruction. Ann Rheum Dis. 2005;65(7):905–9.
Janssen KMJ, et al. Autoantibodies against citrullinated histone H3 in rheumatoid arthritis and periodontitis patients. J Clin Periodontol. 2017;44(6):577–84.
Gordon JAR, et al. Chromatin modifiers and histone modifications in bone formation, regeneration, and therapeutic intervention for bone-related disease. Bone. 2015;81:739–45.
Ahn J, et al. Periodontal disease, Porphyromonas gingivalis serum antibody levels and orodigestive cancer mortality. Carcinogenesis. 2012;33(5):1055–8.
Michaud DS, et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut. 2013;62(12):1764–70.
Chang JS, et al. Investigating the association between periodontal disease and risk of pancreatic cancer. Pancreas. 2016;45(1):134–41.
Hwang IM, et al. Periodontal disease with treatment reduces subsequent cancer risks. QJM. 2014;107(10):805–12.
Li A, et al. Pancreatic cancer DNMT1 expression and sensitivity to DNMT1 inhibitors. Cancer Biol Ther. 2010;9(4):321–9.
Archer SY, Hodin RA. Histone acetylation and cancer. Curr Opin Genet Dev. 1999;9(2):171–4.
Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. 2009;4:199–227.
Hung K-F, et al. MicroRNA-31 upregulation predicts increased risk of progression of oral potentially malignant disorder. Oral Oncol. 2016;53:42–7.
Kent OA, et al. Transcriptional regulation of miR-31 by oncogenic KRAS mediates metastatic phenotypes by repressing RASA1. Mol Cancer Res. 2016;14(3):267–77.
Lerner C, et al. Characterization of miR-146a and miR-155 in blood, tissue and cell lines of head and neck squamous cell carcinoma patients and their impact on cell proliferation and migration. J Cancer Res Clin Oncol. 2016;142(4):757–66.
Wu J, Xie H. Expression of long noncoding RNA-HOX transcript antisense intergenic RNA in oral squamous cell carcinoma and effect on cell growth. Tumour Biol. 2015;36(11):8573–8.
Wu Y, et al. Long non-coding RNA HOTAIR promotes tumor cell invasion and metastasis by recruiting EZH2 and repressing E-cadherin in oral squamous cell carcinoma. Int J Oncol. 2015;46(6):2586–94.
Cannuyer J, et al. A gene expression signature identifying transient DNMT1 depletion as a causal factor of cancer-germline gene activation in melanoma. Clin Epigenetics. 2015;7:114.
Merry CR, et al. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum Mol Genet. 2015;24(21):6240–53.
Mariotti A, et al. The DNA methyltransferase DNMT1 and tyrosine-protein kinase KIT cooperatively promote resistance to 5-Aza-2′-deoxycytidine (decitabine) and midostaurin (PKC412) in lung cancer cells. J Biol Chem. 2015;290(30):18480–94.
Tian H-P, et al. DNA methylation affects the SP1-regulated transcription of FOXF2 in breast cancer cells. J Biol Chem. 2015;290(31):19173–83.
Chan H-L, et al. MiR-376a and histone deacetylation 9 form a regulatory circuitry in hepatocellular carcinoma. Cell Physiol Biochem. 2015;35(2):729–39.
Karczmarski J, et al. Histone H3 lysine 27 acetylation is altered in colon cancer. Clin Proteomics. 2014;11(1):24.
Phi van DK, et al. Histone deacetylase HDAC1 downregulates transcription of the serotonin transporter (5-HTT) gene in tumor cells. Biochim Biophys Acta. 2015;1849(8):909–18.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Bascones-Martinez, A., González-Febles, J. (2018). Epigenetics and Periodontitis: A Source of Connection to Systemic Diseases. In: Meurman, J. (eds) Translational Oral Health Research. Springer, Cham. https://doi.org/10.1007/978-3-319-78205-8_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-78205-8_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-78204-1
Online ISBN: 978-3-319-78205-8
eBook Packages: MedicineMedicine (R0)