
Abstract
The book “Channelopathies in Heart Disease” provides a translational overview of current state-of-the art research on ion channel (dys)function, cardiac channelopathies, and inherited arrhythmia syndromes. The latest insight on the structure and function of cardiac ion channels and the pro-arrhythmic consequences of their dysfunction is presented. Clinical and genetic characteristics of various inherited channelopathies and arrhythmia syndromes are discussed, in addition to new technologies available to this translational research field.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1.1 Channelopathies in Heart Disease: Background and Book Overview
The majority of cardiac arrhythmias occur in the setting of common (acquired) cardiovascular pathologies associated with structural cardiac abnormalities and/or metabolic dysregulation. In a subset of patients, however, cardiac arrhythmias are the consequence of an inherited arrhythmia syndrome. Mutations in genes encoding ion channels, transporters, interacting proteins, or regulatory pathways may lead to potentially life-threatening arrhythmias in relatively young and otherwise healthy individuals. During the last two decades, significant progress has been made in the identification of genetic defects underlying inherited channelopathies, which has provided some benefit through elucidation of gene-specific arrhythmia triggers and treatment. However, for many arrhythmia syndromes, clinical management is still hindered by insufficient knowledge of the functional consequences of the mutation in question, the pro-arrhythmic mechanisms involved, and hence the most optimal treatment strategy. In this book, we present the latest insight on the structure and function of cardiac ion channels and the pro-arrhythmic consequences of their dysfunction. Clinical and genetic characteristics of various inherited channelopathies and arrhythmia syndromes are discussed, in addition to new technologies available to this translational research field.
1.2 (Dys)Function of Cardiac Ion Channels
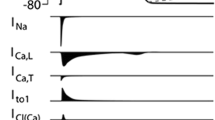
Cardiac electrical activity is a summation of sequential action potentials throughout the heart. The action potential (AP) is the consequence of an orchestrated interplay between various ion channels. Each AP is initiated by a large, rapid influx of sodium (Na+) through Na+ channels (INa), resulting in fast depolarization of the cell membrane and the AP upstroke (Fig. 1.1). Following this phase 0 of the AP, there is a brief repolarizing phase (phase 1), resulting from efflux of potassium (K+) caused by activation of the transient outward potassium current (Ito1). Next, inward flow of calcium (Ca2+) through L-type calcium channels (ICa,L) leads to the plateau phase (phase 2). Finally, the membrane repolarizes to its original state due to activation of the rapid and slow delayed rectifier K+ channels (conducting the IKr and IKs currents, respectively) in phase 3 of the AP. Adult ventricular and atrial cardiomyocytes (but not nodal cells) also exhibit phase 4 in which the resting membrane potential remains constant due to the presence of the rectifying K+ current IK1. Due to their close interrelationship, alterations in a particular ion channel (affecting one phase of the AP) will also impact on the function of other ion currents and AP phases. In Chaps. 2–4 of this book, the molecular composition, structure, and function of Na+, K+, and Ca2+ channels are described. In addition, their regulation, their role in cardiac electrophysiology, and the conditions and consequences associated with their dysfunction are discussed in these chapters. Furthermore, in Chap. 5 the funny current (If) is presented, which is the primary contributor of the diastolic pacemaker phase responsible for the automaticity of the conduction system. The chapter will detail the biophysical and modulatory properties of If and its potential as a target for pharmacological modulation of heart rate. Ion channel function is tightly interrelated with intracellular ion homeostasis, with Ca2+ influx through ICa,L eliciting the intracellular Ca2+ transient which underlies myocyte contraction. The subsequent decline of Ca2+ (required for diastolic relaxation) occurs through reuptake into the sarcoplasmic reticulum and extrusion of Ca2+ via the Na+-Ca2+ exchanger. Chapter 6 reviews the interrelation between intracellular K+, Na+, and Ca2+ and discusses the consequences of alterations in their homeostasis for cardiac electrophysiology and arrhythmogenesis.

Overview: Primary ionic currents underlying ventricular and atrial action potentials (AP) in the heart (reproduced with permission from Shah et al. 2005)
1.3 Cardiac Channelopathies: Clinical and Genetic Findings
Genetic defects in ion channels and their interacting proteins are associated with various types of clinical arrhythmia syndromes, with mutations in specific ion channels related to different clinical symptoms. Moreover, different types or locations of mutations may be associated with different phenotypes, severity of disease, and treatment strategy and efficacy. During the last two decades, significant progress has been made in genetic studies, which has facilitated the identification of family members and patients at risk. Research on ion channel mutations have also provided essential molecular and biophysical insight into (normal) ion channel function. Furthermore, in some cases elucidation of gene-specific arrhythmia triggers and pharmacology has guided patient treatment and management. However, reduced penetrance and variable disease severity and expressivity among mutation carriers remain problematic. In Chaps. 7–12, the most prevalent inherited cardiac channelopathies and arrhythmia syndromes are reviewed, i.e., long QT syndrome, Brugada syndrome, sinus node disease, Lev-Lenègre syndrome, progressive cardiac conduction disease, catecholaminergic polymorphic ventricular tachycardia, idiopathic ventricular fibrillation, early repolarization syndrome, and atrial fibrillation. For each syndrome, current knowledge on their genetic basis, clinical presentation, diagnosis, risk stratification, and therapy is discussed. Chapter 13 presents an overview of genetic testing and familial clinical screening in patients with (suspected) inherited arrhythmia syndrome in addition to a proposed screening hierarchy according to phenotype. Furthermore, the impact of new sequencing technologies is discussed.
1.4 Research into Cardiac Channelopathies: New Avenues
In recent years, a number of new technologies and innovations have provided essential novel insight into ion channel function and the mechanisms involved in cardiac channelopathies. On the molecular level, novel imaging techniques are significantly improving our knowledge on cardiac nanoscale architecture, ion channel distribution, and function. The methodologies and advantages of one of these techniques, super-resolution fluorescence microscopy, are discussed in Chap. 14, in addition to its application and future potential in cardiac research in general and ion channel (dys)function in particular. Functionally, studies employing transgenic mouse models have demonstrated that heterologous expression systems, traditionally employed for investigating the consequences of ion channel mutations, are not necessarily representative of the situation within the cardiomyocyte environment. As discussed in Chap. 15, transgenic mice allow the investigation of mutation effects in different regions and cell types of the heart, as well as investigation of disease progression. More recently, cardiomyocytes derived from human-induced pluripotent stem cells (hiPSC) have been employed to study mutations in a more physiological environment. These hiPSC, which are reviewed in Chap. 16, appear suitable for the investigation of patient- and disease-specific pharmacology and may provide a tool for studying the role of genetic background.
1.5 Concluding Remark
The current book provides a translational overview of current state-of-the art research on ion channel (dys)function, cardiac channelopathies, and inherited arrhythmia syndromes, bringing together clinical, genetic, and basic science experts.
Reference
Shah M, Akar FG, Tomaselli GF. Molecular basis of arrhythmias. Circulation. 2005;112:2517–29.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
Sources of Funding
This work was funded by a Priority Medicines Rare Diseases and Orphan Drugs grant (PM-Rare, 113303006 to C.A.R.) from The Netherlands Organization for Health Research and Development (ZonMw) and an Innovational Research Incentives Scheme Vidi grant from ZonMw (grant no. 91714371 to C.A.R.), by the German Cardiac Society and the Hengstberger Foundation (Klaus-Georg and Sigrid Hengstberger Scholarship to D.T.), the German Heart Foundation/German Foundation of Heart Research (F/08/14 to D.T.), the Joachim Siebeneicher Foundation (to D.T.), and the Baden-Wuerttemberg Ministry of Science, Research and Art (Sonderlinie Medizin to D.T.).
Conflict of Interest
C.A.R. has previously received research grants from Gilead Sciences. D.T. reports receiving lecture fees/honoraria from Bayer Vital, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, Medtronic, Pfizer Pharma, Sanofi-Aventis, St. Jude Medical, and ZOLL CMS and research grant support from Daiichi Sankyo. D.T. filed a patent application for the use of K2P potassium channels for altering cardiac electrophysiology.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Remme, C.A., Thomas, D. (2018). Channelopathies in Heart Disease. In: Thomas, D., Remme, C. (eds) Channelopathies in Heart Disease . Cardiac and Vascular Biology, vol 6. Springer, Cham. https://doi.org/10.1007/978-3-319-77812-9_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-77812-9_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-77811-2
Online ISBN: 978-3-319-77812-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)