
Abstract
This chapter reviews our current knowledge of the role of oxidative damage mechanisms and pharmacological antioxidant neuroprotective strategies for inhibiting reactive oxygen species (ROS) and reactive nitrogen species (RNS)-mediated secondary injury following traumatic brain injury (TBI). First of all, the chemistry of the main forms of oxidative damage: lipid peroxidation, carbonylation and nitration are presented as well as the interactions of oxidative damage with other secondary injury mechanisms including glutamate-mediated excitotoxicity, intracellular calcium overload and mitochondrial dysfunction. Secondly, the general mechanistic approaches to interrupting oxidative damage are presented: decreasing ROS/RNS formation or scavenging ROS and RNS-derived radicals, inhibition of lipid peroxidation propagation, chelation of iron, which is a potent catalyst of lipid peroxidation reactions, scavenging of neurotoxic aldehydic lipid peroxidation products (‘carbonyls’), and enhancement of the expression of the pleiotopic Nrf2-antioxidant response element (ARE) pathway that controls the synthesis of several endogenous antioxidant enzymes and chemical antioxidants. Pharmacological examples of compounds that effectively inhibit oxidative damage and produce neuroprotective effects in animal TBI models by each of these various approaches are presented. Finally, the results of large phase III clinical trials with the either the radical scavenger polyethylene glycol-coupled superoxide dismutase (PEG-SOD) or the 21-aminosteroid lipid peroxidation inhibitor tirilazad are revisited in which the latter compound was found to selectively improve survival after moderate and severe TBI, particularly in male patients, suggesting that successful clinical translation of neuroprotective antioxidant compounds, or combinations of mechanistically complimentary antioxidants, should be possible.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Glutamate excitotoxicity
- Calcium overload
- Reactive oxygen species
- Lipid peroxidation
- Antioxidant
- Neuroprotection
- Traumatic brain injury
1 Introduction
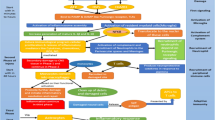
At present, there are no FDA-approved pharmacological therapies for acute treatment of traumatic brain injury (TBI) patients that are conclusively proven to mitigate the often devastating neurological effects of their injuries. However, the possibility of discovering and developing an effective ‘neuroprotective’ treatment that will limit posttraumatic brain damage and improve neurological recovery is based upon the fact that even though some of the neural injury is due to the primary mechanical injury to the parenchymal neurons, glia and vascular elements, the majority of post-traumatic neurodegeneration is due to a pathophysiological secondary injury cascade triggered initially by massive release of glutamate and its excitotoxic effects that occur during the first minutes, hours and days following the injury, which exacerbates the damaging effects of the primary injury. One of the most validated “secondary injury” mechanisms, as revealed in experimental TBI studies, that contributes to glutamate-mediated excitotoxic neurodegeneration, involves the downstream increase in reactive oxygen species (ROS) that cause oxygen radical-induced oxidative damage to brain cell lipids and proteins. This chapter outlines the key sources of reactive oxygen species (ROS), including highly reactive (i.e. rapidly oxidizing) free radicals, the pathophysiological mechanisms associated with oxidative neural damage and pharmacological antioxidants that have been shown to produce neuroprotective effects that limit excitotoxic neurodegeneration in preclinical TBI models, one of which has revealed some evidence of neuroprotective efficacy in a major pathological subset of TBI patients in a large phase III clinical trial.
2 Reactive Oxygen Species and Reactive Nitrogen Species
The term reactive oxygen species (ROS) includes oxygen-derived radicals such as the modestly reactive superoxide radical (O2 •−) and the highly reactive hydroxyl (OH•) radical as well as non-radicals such as hydrogen peroxide (H2O2) and peroxynitrite (ONOO−), the latter often referred to as a reactive nitrogen species (RNS). The cascade of posttraumatic oxygen radical reactions begins in response to the primary mechanical injury triggering neuronal depolarization, due to the voltage-dependent opening of sodium (Na+) and calcium (Ca2+) channels, which causes a massive increase in intracellular Ca2+ that stimulates rapid elevations in extracellular glutamate levels that excessively stimulates N-methyl-aspartate (NMDA) glutamate receptors, causing a further exacerbation of the injury-induced increase in intracellular Ca2+. This voltage-dependent and glutamate receptor-mediated intracellular Ca2+ overload initiates multiple downstream neurodegenerative processes, one of which is the increased generation of oxygen free radicals that initiate oxidative damage to brain cell phospholipid membranes and proteins. The primordial oxygen free radical that comes from several pathophysiological sources involves the single electron (e−) reduction of an oxygen molecule (O2) to produce superoxide (O2 •−). Superoxide can be generated from several sources; one of the main ones is O2 •− leakage from complex I of the mitochondrial electron transport chain in Ca2+-overloaded brain mitochondria. However, O2 •− is considered by many free radical chemists and biologists to be a modestly reactive radical that can potentially react with other molecules to give rise to much more reactive, and thus more potentially damaging, radical species. The reason that O2 •− is only modestly reactive is that it can act as either an oxidant by stealing an electron from another oxidizable molecule or it can act as a reductant by which it donates its unpaired electron to another radical species, thus acting as an antioxidant. However, if O2 •− reacts with a proton (H+) to form a hydroperoxyl radical (HO•2) this results in a superoxide form that is much more likely to cause oxidation (i.e. act as an electron stealer).
One of the most important endogenous antioxidants is the enzyme superoxide dismutase (SOD) which rapidly catalyzes the dismutation of O2 •− into H2O2 and oxygen. At low pH, O2 •− can dismutate spontaneously. The formation of highly reactive oxygen radicals, which have unpaired electron(s) in their outer molecular orbitals, and the propagation of chain reactions are fueled by non-radical ROS, which do not have unpaired electron(s), but are chemically reactive. For example, OH• radicals are generated in the iron-catalyzed Fenton reaction, where ferrous iron (Fe2+) is oxidized to form OH• in the presence of H2O2 (Fe2+ + H2O2 → Fe3+ + OH• + OH−). Superoxide, acting as a reducing agent (i.e. an electron donor), can actually donate its unpaired electron to ferric iron (Fe3+), cycling it back to the ferrous state in the Haber-Weiss reaction (O2 •− + Fe3+ → Fe2+ + O2), thus driving subsequent Fenton reactions and increased production of OH•. Under physiological conditions, iron is tightly regulated by its transport protein, transferrin and storage protein, ferritin, both of which bind the ferric (Fe3+) form. This reversible bond of transferrin and ferritin with iron decreases with declining pH (below pH 7). Indeed, tissue acidosis is known to occur in the traumatized CNS that will cause the release of iron and initiation of iron-dependent oxygen radical production. A second source of iron comes from hemoglobin released into the blood during injury-induced hemorrhage.
Although O2 •− is much less reactive than OH• radical, its reaction with nitric oxide (NO•) radical forms the highly reactive oxidizing agent, peroxynitrite (PN: ONOO−).This reaction (O2 •− + NO• → ONOO−) occurs at a very high rate constant that out competes SOD’s ability to convert O2 •− into H2O2. Subsequently, at physiological pH, ONOO− will largely undergo protonation to form peroxynitrous acid (ONOOH) or it can react with carbon dioxide (CO2) to form nitrosoperoxycarbonate (ONOOCO2 −). The ONOOH can break down to form highly reactive nitrogen dioxide (NO•2) and OH• (ONOOH → NO• 2 + OH•). Alternatively, the ONOOCO2 − can decompose into NO• 2 and carbonate radical (CO3 •−) (ONOOCO2 − → NO•2 + CO3 •−).
3 Lipid Peroxidation
Increased production of reactive free radicals (i.e. “oxidative stress”) in the injured brain has been shown to cause “oxidative damage” to cellular lipids and proteins, leading to functional compromise and cell death in both the microvascular and brain parenchymal compartments. Extensive study shows that a major form of radical-induced oxidative damage involves oxidative attack on cell membrane polyunsaturated fatty acids, triggering the process of lipid peroxidation (LP) that is characterized by three distinct steps: initiation, propagation and termination (Gutteridge 1995), which are shown in Fig. 3.1 in the context of radical-induced LP of arachidonic acid. Initiation : LP is initiated when highly reactive oxygen radicals (e.g. OH•, NO2•, CO3 •−) react with polyunsaturated fatty acids such as arachidonic acid (AA), linoleic acid (LA), eicosapentaenoic acid (EPA) or docosahexaenoic acid (DHA), resulting in disruptions in cellular and subcellular membrane integrity. Initiation of LP begins when ROS-induced hydrogen atom (H+) and its one associated electron is abstracted from an allylic carbon. The basis for the susceptibility of the allylic carbon of the polyunsaturated fatty acid having one of its electrons stolen by a highly electrophilic free radical is that the carbon is surrounded by two relatively electronegative double bonds which tend to pull one of the electrons from the carbon. Consequently, a reactive free radical has an easy time pulling the hydrogen electron off of the carbon because the commitment of the carbon electron to staying paired with it has been weakened by the surrounding electronegative double bonds. This results in the original radical being quenched while the polyunsaturated fatty acid (L) becomes a lipid radical (L•) due to its having lost an electron. Propagation : Subsequently, in the propagation step, the unstable L• reacts with O2 to form a lipid peroxyl radical (LOO•). The LOO• in turn extracts a hydrogen atom from an adjacent polyunsaturated fatty acid, yielding a lipid hydroperoxide (LOOH) and a second L•, which sets off a series of propagation “chain” reactions.

Biochemistry involved in the initiation, propagation and termination reactions of arachidonic acid during lipid peroxidation, with the resulting formation of the aldehydic end-products 4-hydroxynonenal (4-HNE) and acrolein
Termination : These propagation reactions are terminated in the third step, when the substrate becomes depleted and a lipid radical reacts with another radical to yield potentially neurotoxic non-radical aldehydic end products. One of those endproducts that is often used to measure LP is the three carbon-containing malondialdehyde (MDA) which is mainly a stable non-toxic compound that when measured represents a LP ‘tombstone’. In contrast, two highly neurotoxic aldehydic products of LP (commonly referred to as ‘carbonyls’) are 4-hydroxynonenal (4-HNE) or 2-propenal (acrolein), both of which have been well characterized in CNS injury experimental models (Bains and Hall 2012; Hall et al. 2010; Hamann and Shi 2009). These latter two aldehydic LP end products covalently bind to proteins and amino acids (lysine, histidine, arginine) by either Schiff base or Michael addition reactions altering their structure and functional properties. Immunohistochemical and immunoblotting (western, slot, dot) techniques are commonly used to measure 4-HNE or acrolein-modified proteins (i.e. ‘protein carbonyls’) in the injured brain (Hall and Bosken 2009).
4 Free Radical-Induced Protein Carbonylation and Nitration
Free radicals can cause various forms of oxidative protein damage. Firstly, a major mechanism involves carbonylation by reaction of various free radicals with susceptible amino acids such as arginine, lysine and histidine. The protein carbonyls thus formed are measurable through immunoblotting after derivatization of the carbonyl groups with diphenylhydrazine (DNPH). Indeed, the measurement of protein carbonyls by the so-called DNPH assay has long been used to measure free radical-induced protein oxidation. However, the carbonyl assay also picks up protein carbonyls that are present due to covalent binding of LP-derived 4-HNE and acrolein to cysteine residues, in addition to those resulting from direct free radical-induced amino acid oxidation. Thus, as a result, the carbonyl assay is as much an indirect index of LP as it is of direct radical-induced protein oxidation.
Secondly, NO•2 can nitrate the three position of aromatic amino acids tyrosine or phenylalanine in proteins; 3-NT is a specific footprint of PN-induced cellular damage. Similarly, lipid peroxyl radicals (LOO•) can promote nitration of aromatic amino acids by producing initial oxidation (i.e. loss of an electron) which would enhance the ability of NO•2 to nitrate the phenyl ring. Multiple commercially available polyclonal and monoclonal antibodies are available for immunoblot or immunohistochemical measurement of proteins that have been nitrated by PN.
5 Interaction of Oxidative Damage with Other Secondary Injury Mechanisms
The impact of ROS/RNS production is heightened when oxygen radicals feed back and amplify other secondary injury pathways creating a continuous cycle of ion imbalance, Ca2+ buffering impairment, mitochondrial dysfunction, glutamate-induced excitotoxicity and microvascular disruption. One example of ROS-induced ionic disruption arises from LP-induced damage to the plasma membrane ATP-driven Ca2+ pump (Ca2+-ATPase) and Na+ pump (Na+/K+-ATPase), which contributes to increases in intracellular Ca2+ concentrations, mitochondrial dysfunction and additional ROS production. Both Ca2+-ATPase and Na+/K+-ATPase disruptions result in further increases in intracellular Ca2+ and Na+ accumulation respectively (Bains and Hall 2012), the latter causing reversal of the Na+/Ca2+ exchanger which further exacerbates intracellular Ca2+ (Rohn et al. 1993, 1996). As already noted above, PN formed from mitochondrial Ca2+ overload also contributes to mitochondrial dysfunction. Specifically, nitric oxide (NO•), formed from mitochondrial NOS, which in turn reacts with O2 •− to produce the highly toxic PN, which impairs respiratory and Ca2+ buffering capacity via its derived free radicals (Bringold et al. 2000). Indeed increased PN-derived 3NT and 4HNE has been detected during the time of mitochondrial dysfunction and correlates with respiratory and Ca2+ buffering impairment (Sullivan et al. 2007). Increased synaptosomal 4-HNE content is associated with impaired synaptosomal glutamate and amino acid uptake (Carrico et al. 2009; Zhang et al. 1996). Glutamate and NMDA- induced damage in neuronal cultures is attenuated with LP inhibition, confirming LP and oxidative damage as promoters of glutamate excitotoxicity (Monyer et al. 1990; Pellegrini-Giampietro et al. 1990).
6 Mechanisms for Pharmacological Inhibition of Oxidative Damage
Based upon the discussion above concerning oxidative stress (increased ROS/RNS) and oxidative damage (LP, protein oxidation and nitration), a number of potential mechanisms for its inhibition are apparent which fall into five categories. The first category includes compounds that inhibit the initiation of LP and other forms of oxidative damage by attenuating the formation of ROS or RNS species. For instance, nitric oxide synthase (NOS) inhibitors exert an indirect antioxidant effect by limiting NO• production and thus PN formation. However, they also have the potential to interfere with the physiological roles that NO• is responsible for, including antioxidant effects which are due to its important role as a scavenger of lipid peroxyl radicals (e.g. LOO• + NO• → LOONO) (Hummel et al. 2006). Another approach to blocking posttraumatic radical formation is the inhibition of the enzymatic (e.g. cyclooxygenase, 5-lipoxygenases) arachidonic acid (AA) cascade during which O2 •− is produced as a by-product of prostanoid and leukotriene synthesis. Kontos and colleagues (Kontos 1989; Kontos and Wei 1986) and Hall (1986) have shown that cyclooxygenase-inhibiting non-steroidal anti-inflammatory agents (e.g. indomethacin, ibuprofen) are vaso- and neuro-protective in TBI models.
Another example of an indirect approach for reducing the formation of ROS/RNS in the injured brain is via the inhibition of brain mitochondrial functional failure with the drug cyclosporine A which has been shown to reduce mitochondrial permeability transition pore (mPTP) formation by blocking cyclophilin D interaction with other components of the pathological pore which has been shown to lessen mitochondrial free radical formation and consequently attenuate LP and nitrative mitochondrial protein oxidative damage (Mbye et al. 2008; Sullivan et al. 1999).
A second indirect LP inhibitory approach involves chemically scavenging the radical species (e.g. O2 •−, OH•, NO•2, CO3 •−) before they have a chance to steal an electron from a polyunsaturated fatty acid and thus initiate LP. The use of pharmacologically-administered SOD represents an example of this strategy. Another example concerns the use of the nitroxide antioxidant tempol which has been shown to catalytically scavenge the PN-derived free radicals NO•2 and CO3•− (Carroll et al. 2000). In either case, a general limitation to these first two approaches and antioxidant agents that work by this mechanism is that they would be expected to have a short therapeutic window and would have to be administered rapidly in order to have a chance to interfere with the initial posttraumatic “burst” of free radical production that has been documented in TBI models (Kontos and Wei 1986; Hall et al. 1993). While it is believed that ROS, including PN production, persists several hours after injury, the major portion is an early event that peaks in the first 60 min after injury, making it clinically impractical to pharmacologically inhibit, unless the antioxidant compound is already “on board” when the TBI occurs (Fig. 3.2).

(a) Chemical scavenging mechanism involved in the reactivity of the hydrazine-containing compound phenelzine with 4-HNE. (b) (Top): Effects of repeated phenelzine (PZ: 10 mg/kg s.c. 15 min after injury followed by maintenance dosing (5 mg/kg s.c.) every 12 h) on cortical mitochondrial bioenergetics 72 h following severe controlled cortical impact TBI. Mitochondrial respiration was measured with a Clark-type electrode expressed as respiratory control ratio (RCR). The RCR is rate of oxygen consumption during State III divided by State IV respiration. Animals received PZ rats were euthanized at 72 h. Sham and Sham + PZ groups were significantly different compared to Vehicle groups. RCR of PZ treatment was significantly increased compared to vehicle and not significantly different from either sham control variant (Sham or Sham + PZ). One-way ANOVA (F = 7.7, df = 3, 24, P < 0.009) followed by Student Newman-Keuls post-hoc test. *p < 0.05. Error bars represent ±SD; n = 8–9 rats per group except sham where n = 5 rats per group. (b) (Bottom): Repeated PZ reduces 4-hydroxynonenal (4-HNE) accumulation in mitochondria 72 h after TBI. As revealed by quantitative western blot (see sample blot and bar graph), 4-HNE-modified proteins were significantly elevated in the Vehicle group compared to both Sham groups. PZ treatment group exhibited significantly reduced oxidative damage compared to Vehicle group, but did not return to Sham levels. ANOVA (F = 9.9, df = 3, 24, p < 0.0002) followed by Student Newman-Keuls post-hoc test. *p < 0.05. (c) Repeated PZ reduces cortical neurodegeneration 72 h after TBI: Coronal sections of ipsilateral rat brains rat taken at 1.2× magnification. Left: Vehicle (0.9% saline) treated rat brain injected 15 min after TBI; Center: Phenelzine (PZs) single 10 mg/kg s.c. dose treated animal; Right: Rat brain of PZ-treated with a multiple dosing paradigm (PZm): single subcutaneous injection of PZ 15 min after injury, followed by maintenance dosing of 5 mg/kg every 12 h thereafter. All groups (Vehicle, PZ(S), PZ(M)) were euthanized 72 h after first injection. Black bar under the photomicrographs represents 1 mm. The graph below the photos shows percent of cortical tissue sparing followed by either Vehicle (saline), PZ(S), or PZ(M) treatment. Rats were euthanized in all treatment paradigms at 72 h after first injection. PZs did not exhibit a statistically significant amount of cortical tissue sparing when compared to Vehicle. However, PZm significantly increased the total volume of spared cortical tissue. One-way ANOVA followed by Dunnett’s post-hoc test. *p < 0.05 compared to Vehicle. Error bars represent mean ± SD; n = 6 rats for vehicle group; n = 8 rats per group for drug-treated rats. These data are reproduced with permission from Cebak et al. (2017)
In contrast to the above indirect-acting antioxidant mechanisms, the third category involves stopping the “chain reaction” propagation of LP once it has begun. The most demonstrated way to accomplish this is by scavenging of lipid peroxyl (LOO•) radicals. The prototype scavenger of these lipid radicals is alpha tocopherol or vitamin E (Vit E) which can donate an electron from its phenolic hydroxyl moiety to quench a LOO•. However, the scavenging process is stoichiometric (1 Vit E can only quench 1 LOO•) and in the process vitamin E loses its antioxidant efficacy and becomes Vitamin E radical (LOO• + Vit E → LOOH + Vit E•). Although Vit E• is relatively unreactive (i.e. harmless), it also cannot scavenge another LOO• until it is reduced back to its active form by receiving an electron from other endogenous antioxidant reducing agents such as ascorbic acid (Vitamin C) or glutathione (GSH). While this tripartite LOO• antioxidant defense system (Vit E, Vit C, GSH) works fairly effectively in the absence of a major oxidative stress, numerous studies have shown that each of these antioxidants are rapidly consumed during the early minutes and hours after CNS injury (Hall et al. 1989, 1992). Thus, it has long been recognized that more effective brain penetrable pharmacological LOO• scavengers are needed. Furthermore, compared to antioxidants that are scavengers of the initial post-TBI oxygen radical burst, it is reasonable to theorize that antioxidants that interrupt the LP process after it has begun would be able to exert a more clinically practical neuroprotective effect (i.e. possess a longer antioxidant therapeutic window).
An additional approach to inhibiting the propagation of LP reactions is to chelate free iron, either ferrous (Fe2+) or ferric (Fe3+), which potently catalyzes the breakdown of lipid hydroperoxides (LOOH), an essential event in the continuation of LP chain reactions in cellular membranes. The prototypical iron-chelating drug which chelates Fe3+, is the tri-hydroxamic acid compound deferoxamine.
The fourth antioxidant category that has begun to be explored for neuroprotection following TBI concerns pharmacological scavenging of LP-derived aldehydic (carbonyl-containing) breakdown products 4-HNE and acrolein. As introduced earlier, these highly neurotoxic compounds have high affinity for covalently binding to basic amino acid residues including histidine, lysine, arginine and cysteine. These modifications have been shown to inhibit the activities of a variety of enzymatic proteins (Halliwell and Gutteridge 2008). Also, 4-HNE and acrolein, formed by LP oxidative damage, are also associated with stimulating additional free radical generation (i.e. oxidative stress) in injured CNS tissue (Hamann and Shi 2009). Several compounds have been identified that are able to antagonize this “carbonyl stress” by covalently binding to reactive LP-derived aldehydes. Two commercially available FDA-approved drugs that have been tested in TBI models are d-penicillamine and phenelzine, whose neuroprotective effects will be briefly discussed in the next section of this chapter.
A fifth antioxidant category that is theoretically an attractive broad spectrum mechanistic approach for achieving neuroprotection in TBI involves pharmacologically activating the body’s endogenous pleiotropic antioxidant defense system that is largely regulated by nuclear factor E2-related factor 2/antioxidant response element (Nrf2/ARE) signaling at the transcriptional level (Kensler et al. 2007). As will be discussed below, Nrf2 activation and the up-regulation of antioxidant and anti-inflammatory genes, which has been previously described in experimental models of stroke and neurodegenerative disease (Shih et al. 2003), appears to be particularly promising in TBI models. Indeed, it has been documented that in the mouse controlled cortical impact TBI paradigm the injury itself upregulates Nrf2 and antioxidant gene expression. However, the time course of that antioxidant response occurs simultaneously with the time course of posttraumatic LP in brain tissue (Miller et al. 2014). Thus, what is needed is a compound that speeds up and increases the magnitude of the post-TBI Nrf2/ARE activation in the injured brain so that is has a chance to attenuate the peak of posttraumatic oxidative neural damage. Two such naturally occurring compounds that have been shown to be protective in TBI models are sulforaphane, found in high concentrations in broccoli, and carnosic acid, found in the herb rosemary.
Finally, a sixth strategy for achieving antioxidant neuroprotection in injured brain tissue involves protecting the neural mitochondrion which is essential for maintaining ATP production via the multi-complex electron transport chain as well as for its role in excessive post-TBI cytoplasmic Ca2+ accumulation. While mitochondrial Ca2+ buffering is one of the major functions of cellular mitochondria, as the intra-mitochondrial Ca2+ concentration increases, this leads to increases in mitochondrial O2 •− as well as activation of a mitochondrial NOS that produces NO•. These two radicals rapidly combine to generate peroxynitrite and its derived highly reactive radicals (OH•, NO•2, CO3 •−) which cause damage NO• leakage, causing oxidative damage to the electron transport chain (Bains and Hall 2012). Ultimately, mitochondrial dysfunction triggers the formation of the multi-component mitochondrial permeability transition pore (mPTP), which when it opens triggers mitochondrial permeability transition (mPT) loss of ionic gradients and leakage of important mitochondrial proteins (e.g. cytochrome C). Cyclosporine A (CsA), in addition to its immunosuppressive properties caused by inhibition of calcineurin, also has the ability to prevent mPTP formation by binding to one of the mPTP components, cyclophilin D, preventing the latter from joining mPTP complex, which is required in order for mPT to take place. Consequently, CsA acts to rescue the mitochondrion, preserves membrane potential and lessens additional ROS generation and oxidative damage. This has repeatedly been demonstrated in TBI models (Mbye et al. 2008; Sullivan et al. 1999). Because CsA is not an electron-donor or radical scavenger, its antioxidant action is consequently indirect. In other words, by preventing mPT from occurring it decreases mitochondrial ROS generation and thus indirectly limits oxidative damage. That this protective effect of CsA has little or nothing to do with its inhibition of calcineurin is due to the demonstration that the non-immunosuppressive CsA analog NIM811 does not inhibit calcineurin, but does bind to cyclophilin D, is just as protective as CsA in terms of mitochondrial function in the injured brain and able to reduce lesion volume (Mbye et al. 2008, 2009; Readnower et al. 2011)
7 Neuroprotective Effects of Pharmacological Antioxidants in TBI Patients and Models
TBI Clinical Trial Results with PEG-SOD and Tirilazad: During the past 30 years, there has been an intense effort to discover and develop pharmacological agents for acute treatment of TBI. This has included two compounds that possess free radical scavenging/antioxidant properties, including polyethylene glycol-conjugated superoxide dismutase (PEG-SOD) and the LP inhibitor tirilazad, that were tested in phase III clinical trials in a pathologically heterogeneous population of TBI patients (Langham et al. 2000; Marshall et al. 1998; Narayan et al. 2002). However, each of these trials was a therapeutic failure in that no overall benefit was documented. These failures have been hypothesized to be due to several factors (Narayan et al. 2002).
PEG-SOD: As mentioned earlier, the initial studies of free radical scavenging compounds in TBI models were carried out with Cu/Zn SOD based upon the work of Kontos and colleagues, who showed that post-traumatic microvascular dysfunction was initiated by O2 •− generated as a by-product of the arachidonic acid cascade, which is massively activated during the first minutes and hours after TBI (Kontos 1989; Kontos and Wei 1986; Kontos and Povlishock 1986). Their work showed that administration of SOD prevented the post-traumatic microvascular dysfunction. This led to clinical trials in which the more metabolically stable polyethylene glycol (PEG)-conjugated SOD was examined in moderate and severe TBI patients when administered within the first 8 h after injury. Although an initial small phase II study showed a positive trend, subsequent multi-center phase III studies failed to show a significant benefit in terms of increased survival or improved neurological outcomes (Muizelaar et al. 1995). One theoretical reason may be that a large protein like SOD is unlikely to have much brain penetrability and therefore its radical scavenging effects may be limited to the microvasculature. A second reason may be that attempting to scavenge the short-lived inorganic radical O2 •− may be associated with a very short therapeutic window, as suggested above. As pointed out earlier, the time course of measurable post-traumatic OH• formation in the injured rodent brain has been shown to largely run its course by the end of the first hour after TBI (Hall et al. 1993; Smith et al. 1994). A more rational strategy would be to inhibit the LP that is triggered by the initial burst of inorganic radicals. A comparison of the time course of LP with that of post-traumatic OH• shows that LP reactions continue to build beyond the first post-traumatic hours (Smith et al. 1994) and may continue for 3–4 days (Du et al. 2004; Miller et al. 2014; Hall et al. 2012). Despite the failure of PEG-SOD in human TBI, experimental studies have shown that transgenic mice that over-express Cu/Zn SOD are significantly protected against post-TBI pathophysiology and neurodegeneration (Chan et al. 1995; Gladstone et al. 2002; Lewen et al. 2000; Mikawa et al. 1996; Xiong et al. 2005). This fully supports the importance of post-traumatic O2 •− in post-traumatic secondary injury, despite the fact that targeting this primordial radical is probably not the best antioxidant strategy for acute TBI compared to trying to stop the downstream LP process that is initiated by the early increases in OH•, NO•2 and CO3 •−.
Tirilazad: Consistent with that rationale, the 21-aminosteroid LP inhibitor tirilazad was discovered, which inhibits free radical-induced LP by a combination of LOO• scavenging and a membrane-stabilizing action that limits the propagation of LP reactions between an LOO• and an adjacent polyunsaturated fatty acid (Hall et al. 1994). The protective efficacy of tirilazad has been demonstrated in multiple animal models of acute TBI in mice (Hall et al. 1988), rats (McIntosh et al. 1992) and cats (Dimlich et al. 1990). While the compound is largely localized in the microvascular endothelium, the post-traumatic disruption of the BBB is known to allow the successful penetration of tirilazad into the brain parenchyma as noted earlier (Hall et al. 1992). Other mechanistic data derived from the rat controlled cortical impact and the mouse diffuse concussive head injury models have definitively shown that a major effect of tirilazad is to lessen post-traumatic microvascular damage including BBB opening (Hall et al. 1992; Smith et al. 1994).
Tirilazad was taken into clinical development in the early 1990s, and following a small phase II dose-escalation study that demonstrated the drug’s safety in TBI patients, it was evaluated in two phase III multi-center clinical trials for its ability to improve neurological recovery in moderately and severely injured closed TBI patients. One trial was conducted in North America and the other in Europe. In both trials, TBI patients were treated within 4 h after injury with either vehicle or tirilazad (10 mg/kg i.v. q6h for 5 days). The North American trial was never published, due to a major confounding imbalance in the randomization of the patients to placebo or tirilazad in regards to injury severity and pre-treatment neurological status. In contrast, the European trial that 1120 enrolled had much better randomization balance and was published (Marshall et al. 1998). As observed for PEG-SOD, tirilazad failed to show a significant beneficial effect of tirilazad in either moderate (GCS = 9–12) or severe (GCS = 4–8) patient categories. However, a post hoc analysis showed that moderately-injured male TBI patients with traumatic subarachnoid hemorrhage (tSAH) had a significantly lower incidence of 6 month mortality after treatment with tirilazad (6%) compared to placebo (24%, p < 0.042). In severely injured males with tSAH, tirilazad also lessened mortality from 43% in placebo-treated to 34% (p < 0.026). This result is consistent with the fact that tirilazad is also highly effective in reducing SAH-induced brain edema and vasospasm in animal models of SAH (Hall et al. 1994). Nevertheless, additional trials would have been required in order to establish the neuroprotective utility of tirilazad in tSAH patients in order to gain FDA approval. However, the sponsoring company Pharmacia & Upjohn opted not to continue the compound’s development for TBI although tirilazad was successfully approved and marketed for use in aneurysmal SAH (aSAH) in several western European countries, Australia, New Zealand and South Africa, based upon its demonstrated efficacy in phase III aSAH trials conducted in those countries (Kassell et al. 1996; Lanzino and Kassell 1999). Therefore, the apparent post hoc-identified benefit in tSAH patients is consistent with tirilazad’s prospectively demonstrated efficacy in aSAH patients, also mainly observed in males.
Effects of Other Direct and Indirect-Acting Lipid Peroxidation Inhibitors: In addition to tirilazad, several other LP inhibitors have been reported to be effective neuroprotectants in TBI models. These include the lipid peroxyl radical (LOO•) scavenging 2-methylaminochromans U-78517F and U-83836E (Hall et al. 1991; Mustafa et al. 2010, 2011), the pyrrolopyrimidine U-101033E (Hall et al. 1997; Xiong et al. 1997, 1998), OPC-14117 (Mori et al. 1998) and the naturally-occurring LOO• scavengers curcumin (Sharma et al. 2009; Wu et al. 2006) and resveratrol (Ates et al. 2007; Sonmez et al. 2007), the indoleamine melatonin (Beni et al. 2004; Cirak et al. 1999; Mesenge et al. 1998; Ozdemir et al. 2005a, b) and lastly, the endogenous antioxidant lipoic acid (Toklu et al. 2009). In the case of curcumin and resveratrol, these are potent LOO• scavengers due to their possession of multiple phenolic hydroxyl groups that can donate electrons to LOO• radicals. Melatonin also has LOO• scavenging capability (Longoni et al. 1998), but in addition appears to react with PN (Zhang et al. 1999). Lipoic acid may also have LOO• scavenging effects, but these are more likely to be indirect via the regeneration (i.e. re-reduction) of other endogenous electron-donating antioxidants, including vitamin E, glutathione and vitamin C.
Among these LP inhibitors, arguably the most potent and effective LOO• scavenging LP inhibitor yet discovered is the 2-methylaminochroman compound U-83836E which combines the LOO• scavenging antioxidant chroman ring structure of vitamin E with the bis-pyrrolopyrimidine moiety of tirilazad. The phenolic chroman antioxidant moiety, after it sacrifices it phenolic electron to scavenge an LOO•, can be re-reduced by endogenous ascorbic acid (vitamin C) or glutathione (GSH) making it able to quench a second and then a third LOO•, etc. The bis-pyrrolopyrimidine moiety, on the other hand, can also scavenge multiple moles of LOO• by a true catalytic mechanism (Hall et al. 1991; Hall et al. 1995). Thus, U-83836E, is a dual functionality LOO• scavenger that is understandably more effective than either vitamin E, tirilazad (Hall et al. 1991) and possibly the other naturally-occurring LOO• scavengers such as curcumin, resveratrol, melatonin and lipoic acid. Furthermore, U-83836E possesses a high degree of lipophilicity endowing it with a high affinity for membrane phospholipids where LP takes place. Studies from the authors’ laboratory in the mouse CCI-TBI model have shown that U-83836E is able to reduce post-traumatic LP and protein nitration and preserve mitochondrial respiratory function, and lessen calpain-mediated neuronal cytoskeletal degradation and decrease injured tissue (Mustafa et al. 2010, 2011).
Nitroxide Antioxidants and Peroxynitrite Scavengers: In addition to the lipid peroxyl (LOO•) radical scavengers, the neuroprotective effects of a family of nitroxide-containing antioxidants have also been examined in experimental TBI models. These are sometimes referred to as “spin-trapping agents” and include α-phenyl-tert-butyl nitrone (PBN) and its thiol analog NXY-059 and tempol. Both PBN and tempol have been shown to be protective in rodent TBI paradigms (Awasthi et al. 1997; Marklund et al. 2001). As mentioned earlier, tempol has been shown by the author and colleagues to catalytically scavenge PN-derived NO•2 and CO3 •− (Carroll et al. 2000; Bonini et al. 2002), and to reduce post-traumatic oxidative damage (both LP and protein nitration), preserve mitochondrial function, decrease calcium-activated, calpain-mediated cytoskeletal damage and reduce neurodegeneration in mice subjected to a severe controlled cortical impact-induced focal TBI (Deng-Bryant et al. 2008). Earlier, another laboratory reported that tempol can reduce post-traumatic brain edema and improve neurological recovery in a rat contusion injury model (Beit-Yannai et al. 1996; Zhang et al. 1998). However, the neuroprotective effect of tempol, administered alone, is associated with a therapeutic window of an hour or less in the mouse controlled cortical impact TBI (CCI-TBI) model. Moreover, tempol is not effective at directly inhibiting LP in the latter model (Deng-Bryant et al. 2008).
Effects of the Iron Chelator Deferoxamine: The prototype iron chelator deferoxamine, which binds ferric (Fe3+) iron and thereby would lessen the catalytic effects of iron on LP, has also been reported to have beneficial actions in preclinical TBI or TBI-related models (Gu et al. 2009; Long et al. 1996). However, deferoxamine is hindered by its limited brain penetration and rapid plasma elimination rate. To counter the latter limitation, a dextran-coupled deferoxamine has been synthesized that has been reported to significantly improve early neurological recovery in a mouse diffuse TBI model (Panter et al. 1992). Much of this activity, however, is probably due to microvascular antioxidant protection because of limited brain penetrability. Another caveat to the iron-chelation antioxidant neuroprotective approach that is at least relevant to the ferric iron chelators such as deferoxamine is that at they can cause a pro-oxidant effect in that their binding of Fe3+ can actually drive the oxidation of ferrous to ferric iron which can increase superoxide radical formation in the process (Fe2+ + O2 → Fe3+ + O2 •−).
Effects of Carbonyl Scavengers: We have previously demonstrated that d-penicillamine is able to scavenge PN (Althaus et al. 1994) and to protect brain mitochondria from PN-induced respiratory dysfunction in isolated rat brain mitochondria (Singh et al. 2007). d-Penicillamine has also been documented to form an irreversible bond to primary aldehydes, enabling it to scavenge neurotoxic LP-derived carbonyl compounds such as 4-HNE and acrolein (Wood et al. 2008). Consistent with that mechanism of action, d-penicillamine was shown to attenuate the levels 4-HNE-modified brain mitochondrial proteins after exposure of isolated mitochondria to 4-HNE (Singh et al. 2007). The PN scavenging action of d-penicillamine along with its carbonyl scavenging capability may jointly explain our previous findings that acutely administered penicillamine can improve early neurological recovery of mice subjected to moderately severe concussive TBI (Hall et al. 1999).
More recently, it has been demonstrated that a variety of FDA-approved hydrazine (–NH–NH2)-containing compounds including the anti-hypertensive agent hydralazine and the anti-depressant phenelzine can react with the carbonyl moieties of 4-HNE or acrolein, which prevents the latter from binding to susceptible amino acids in proteins (Galvani et al. 2008). Most impressive is the fact that the application of hydrazines can rescue cultured cells from 4-HNE toxicity even when administered after the 4-HNE has already covalently bound to cellular proteins (Galvani et al. 2008). Consistent with this effect being neuroprotective, others have shown that hydralazine inhibits either compression or acrolein-mediated injuries to ex vivo spinal cord (Hamann et al. 2008). However, hydralazine, which is a potent vasodilator, would be difficult to administer in vivo after either spinal cord injury or TBI in which hypotension is already a common pathophysiological problem. In contrast, another FDA-approved hydrazine-containing drug phenelzine, used for certain depressive patients, should not compromise blood pressure as readily as hydralazine. Accordingly, a recently published paper has shown that phenelzine administration to rats subjected to acute contusion SCI mitigated post-SCI neuropathic pain, reduces motor deficits and improves spinal cord tissue sparing (Chen et al. 2016). Earlier studies have demonstrated neuroprotective efficacy in a rodent ischemia-reperfusion stroke model that were attributed to reducing ‘aldehyde load’ in the stroke-injured brain (Wood et al. 2006).
In vitro studies in our laboratory have documented the ability of phenelzine to protect isolated rat brain mitochondria from the respiratory depressant effects of 4-HNE, together with a concentration-related attenuation of the accumulation of 4-HNE modified mitochondrial proteins. More recently, we have observed that phenelzine is able to protect isolated mitochondria from respiratory functional depression and modification of mitochondrial proteins following application of the more highly reactive aldehyde acrolein (Cebak et al. 2017). Subsequent in vivo studies in the rat controlled cortical impact TBI model have found that a single 10 mg/kg s.c. dose of phenelzine can also reduce early (3 h) posttraumatic mitochondrial respiratory failure as well as reducing cortical lesion volume at 14 days post-injury (Singh et al. 2013). To better define the optimal neuroprotective use of phenelzine, additional in vivo TBI studies have demonstrated that repeated dosing with phenelzine over a 60 h post-TBI period is capable of protecting delayed mitochondrial failure at its peak at 72 h in the same TBI model along with a reduction in cortical lesion volume that is greater than that seen with a single early dose. This makes sense in that the adequate carbonyl-scavenging drug levels logically need to be maintained during the 72 h long time course of posttraumatic generation of LP-derived neurotoxic aldehydes (Cebak et al. 2017).
Effects of Nrf2/ARE Signaling Activators: The body’s endogenous antioxidant defense system is largely regulated by nuclear factor E2-related factor 2/antioxidant response element (Nrf2/ARE) signaling at the transcriptional level (Zhang 2006; Kensler et al. 2007). Nrf2 activation and the up-regulation of antioxidant and anti-inflammatory genes represents a valid neurotherapeutic intervention in CNS injury and has been previously described in various experimental models of stroke and neurodegenerative diseases (Shih et al. 2003). More recently, the role of Nrf2/ARE activation in SCI has been explored as a targeted neuroprotective strategy for both TBI and SCI. Indeed, studies in Nrf2 (−/−) mice demonstrated increased spinal cord edema and expression of inflammatory cytokines compared to wild-type Nrf2 mice following SCI (Mao et al. 2010; Mao et al. 2011). In mild rat thoracic SCI, it has been reported that Nrf2 levels increase as early as 30 min post-injury and remain elevated through 3 days. In the same study, application of the natural product sulforaphrane, a Nrf2/ARE signaling activator, significantly reduced contusion volume and increased post-SCI coordination. These positive outcomes were a result of sulforaphrane-induced increases in Nrf2, glutamine and decreases in inflammatory cytokines, IL-1β and TBFα (Wang et al. 2012).
The mRNA levels of Nrf2-regulated antioxidant enzymes, heme oxygenase (HO-1) and NADPH:quinone oxidoreductase-1 (NQO1), are up-regulated 24 h post TBI (Yan et al. 2008). In addition, Nrf2-knockout mice are susceptible to increased oxidative stress and neurologic deficits following TBI compared to their wild-type counterparts (Hong et al. 1994). Administration of sulforaphane is also neuroprotective in various animal models of TBI, specifically reducing cerebral edema and oxidative stress and improving BBB function and cognitive deficits (Dash et al. 2009). Studies by Chen et al. (2011) demonstrated increased expression of Nrf2 and HO-1 in the cortex of the rat subarachnoid hemorrhage model. Treatment with sulforaphane further increased the expression of Nrf2, HO-1, NQ01 and glutathione S-transferase-α1 (GST-α1), resulting in the reduction of brain edema, cortical neuronal death and motor deficits (Chen et al. 2011). Tert-butylhydroquinone, another activator of Nrf2, protects against TBI-induced inflammation and damage via reduction in NF-KB activation and TNFα and IL-1β production following injury in the mouse closed head injury model (Jin et al. 2011). Collectively these studies demonstrate a significant neuroprotective role of Nrf2 signaling through the activation of antioxidant enzymes and reduction of oxidative secondary injury responses following CNS injury. Thus, Nrf2 activation may be a prime candidate for the attenuation of oxidative stress and subsequent neurotoxicity in TBI via the development of small-molecule activators of the Nrf2/ARE pathway.
Recent work in our laboratory has revealed that following controlled cortical impact TBI in mice, there is indeed a progressive activation of the Nrf2-ARE system in the traumatically-injured brain as evidenced by an increase in HO-1 mRNA and protein that peaks at 72 h after TBI. However, this effect does not precede, but rather it is coincident with the post-injury increase in LP-related 4-HNE (Miller et al. 2014). Therefore, it is apparent that this endogenous neuroprotective antioxidant response needs to be pharmacologically enhanced and/or sped up if it is to be capable of exerting acute post-TBI neuroprotection. Our laboratory is currently studying another Nrf2-ARE activator, natural product carnosic acid, that has been shown by others to more effectively induce this antioxidant defense system than the prototype sulforaphane (Satoh et al. 2008). We have shown that administration of carnosic acid to non-TBI mice is able to significantly increase the resistance of cortical mitochondria harvested 48 h later to the respiratory depressant effects of the in vitro applied 4-HNE together with in decrease in 4-HNE modification of mitochondrial proteins (Miller et al. 2013). Subsequently, we have administered a single 1 mg/kg i.p. dose of carnosic acid to mice at 15 min after controlled cortical impact TBI and observed that the compound is able to significantly preserve respiratory function along with a reduction in the level of LP-mediated damage in mitochondria harvested from the injured cortex at 24 h after TBI (Miller et al. 2015). Furthermore, carnosic acid’s antioxidant effects were still apparent at 48 h post-injury in terms of an attenuation of 4-HNE and 3-NT in the injured cortical tissue together with a decrease in Ca2+-activated, calpain-mediated neuronal cytoskeletal degradation. In regards to the latter neuronal protective effect, a decrease in 48 h cytoskeletal degradation was also shown to occur even with a post-TBI treatment delay of 8 h. Ongoing studies are evaluating the behavioral recovery and tissue protective effects of carnosic acid whether these are achievable with a clinically practical therapeutic window.
8 Combination Antioxidant Treatment of Traumatic Brain Injury
Antioxidant neuroprotective therapeutic discovery directed at acute TBI has consistently been focused upon attempting to inhibit the secondary injury cascade by pharmacological targeting of a single oxidative damage mechanism. As presented above, these efforts have included either enzymatic scavenging of superoxide radicals with SOD (Muizelaar et al. 1995) or inhibition of LP with tirilazad (Marshall et al. 1998). While each of these strategies alone has shown protective efficacy in animal models of TBI, phase III clinical trials with either compound failed to demonstrate a statistically significant positive effect although post hoc subgroup analysis suggests that the microvascularly localized tirilazad may have efficacy in moderate and severe TBI patients with tSAH (Marshall et al. 1998). While many reasons have been identified as possible contributors to the failure, one logical explanation has to do with the possible need to interfere at multiple points in the oxidative damage portion of the secondary injury cascade either simultaneously or in a phased manner in order to achieve a clinically demonstrable level of neuroprotection. To begin to address this hypothesis, we are currently exploring the possibility that reducing posttraumatic oxidative damage more completely and less variably might be achievable by combined treatment with two or more mechanistically complimentary antioxidant compounds. Figure 3.3 summarizes the overall rationale for a multi-mechanistic antioxidant therapy for TBI. It is anticipated that the combination of two or three antioxidant mechanistic strategies may improve the extent of neuroprotective efficacy, lessen the variability of the effect and possibly provide a longer therapeutic window of opportunity compared to the window for the individual strategies. Figure 3.4 shows preliminary, not yet published data, suggesting that combination treatment of the PN radical scavenger tempol with the LP inhibitor U-83836E in mice subjected to controlled cortical impact TBI was more effective in reducing 7 day post-TBI cortical tissue damage as well as resulting in a reduction in the variability of the data to half of that seen in the parallel groups treated with the either of the two drugs alone.

Rationale for the combination of two or more antioxidant strategies to achieve a more effective and consistent (i.e. less variable) neuroprotective effect in the injured brain

Preliminary data on the neuroprotective effects of 15 min post-injury administration of tempol, U83836E or the combination on cortical tissue sparing. U83836E and the combination both significantly improved tissue sparing whereas tempol in this experiment did not. However, only the combination significantly out-performed tempol. As in scatter plot on the right, the variability in the combination group was considerably lower than in the single treatment groups. All values = mean ± SEM for N = 8/group; #p < 0.05 vs sham; *p < 0.05 vs. vehicle, @p < 0.05 tempol alone vs. combination
In other published studies, we have observed that combined treatment with an LP inhibitor with an inhibitor of excitotoxic glutamate release increases the neuroprotective therapeutic window. Using an infant rat model of shaken baby-induced brain damage model, we have documented that treatment with riluzole, an inhibitor of excitotoxic glutamate release, attenuated cortical neurodegeneration measured at 14 days post-TBI, but the therapeutic window for this neuroprotective effect was limited to the initial riluzole dose having to be administered during the first hour after TBI. However, when the infant rats received the LP inhibitor tirilazad at 30 min after TBI, it increased the neuroprotective therapeutic window for riluzole to 4 h after TBI (Smith and Hall 1998). Thus, combination treatments may extend the neuroprotective efficacy window significantly. Accordingly, combination neuroprotective therapy might be able to improve efficacy, reduce variability and improve the therapeutic window for achievement of clinically measurable neuroprotection in TBI patients, although additional work remains to be conducted to determine whether that neuroprotective hypothesis is correct.
References
Althaus JS, Oien TT, Fici GJ, Scherch HM, Sethy VH, VonVoigtlander PF (1994) Structure activity relationships of peroxynitrite scavengers an approach to nitric oxide neurotoxicity. Res Commun Chem Pathol Pharmacol 83(3):243–254
Ates O, Cayli S, Altinoz E, Gurses I, Yucel N, Sener M, Kocak A, Yologlu S (2007) Neuroprotection by resveratrol against traumatic brain injury in rats. Mol Cell Biochem 294(1–2):137–144
Awasthi D, Church DF, Torbati D, Carey ME, Pryor WA (1997) Oxidative stress following traumatic brain injury in rats. Surg Neurol 47(6):575–581
Bains M, Hall ED (2012) Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta 1822(5):675–684
Beit-Yannai E, Zhang R, Trembovler V, Samuni A, Shohami E (1996) Cerebroprotective effect of stable nitroxide radicals in closed head injury in the rat. Brain Res 717(1–2):22–28
Beni SM, Kohen R, Reiter RJ, Tan DX, Shohami E (2004) Melatonin-induced neuroprotection after closed head injury is associated with increased brain antioxidants and attenuated late-phase activation of NF-kappaB and AP-1. FASEB J 18(1):149–151
Bonini MG, Mason RP, Augusto O (2002) The Mechanism by which 4-hydroxy-2,2,6,6-tetramethylpiperidene-1-oxyl (tempol) diverts peroxynitrite decomposition from nitrating to nitrosating species. Chem Res Toxicol 15(4):506–511
Bringold U, Ghafourifar P, Richter C (2000) Peroxynitrite formed by mitochondrial NO synthase promotes mitochondrial Ca2+ release. Free Radic Biol Med 29(3–4):343–348
Carrico KM, Vaishnav R, Hall ED (2009) Temporal and spatial dynamics of peroxynitrite-induced oxidative damage after spinal cord contusion injury. J Neurotrauma 26(8):1369–1378
Carroll RT, Galatsis P, Borosky S, Kopec KK, Kumar V, Althaus JS, Hall ED (2000) 4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol) inhibits peroxynitrite-mediated phenol nitration. Chem Res Toxicol 13(4):294–300
Cebak JE, Singh IN, Hill RL, Wang JA, Hall ED (2017) Phenelzine protects brain mitochondrial function in vitro and in vivo following traumatic brain injury by scavenging the reactive carbonyls 4-hydroxynonenal and acrolein leading to cortical histological protection. J Neurotrauma 34(7):1302–1317
Chan PH, Epstein CJ, Li Y, Huang TT, Carlson E, Kinouchi H, Yang G, Kamii H, Mikawa S, Kondo T et al (1995) Transgenic mice and knockout mutants in the study of oxidative stress in brain injury. J Neurotrauma 12(5):815–824
Chen G, Fang Q, Zhang J, Zhou D, Wang Z (2011) Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J Neurosci Res 89(4):515–523
Chen Z, Park J, Butler B, Acosta G, Alvarez S, Zheng L, Tang J, McCain R, Zhang W, Ouyang Z, Cao P, Shi R (2016) Mitigation of sensory and motor deficits by acrolein scavenger phenelzine in a rat model of spinal cord contusive injury. J Neurochem 138(2):328–338
Cirak B, Rousan N, Kocak A, Palaoglu O, Palaoglu S, Kilic K (1999) Melatonin as a free radical scavenger in experimental head trauma. Pediatr Neurosurg 31(6):298–301
Dash PK, Zhao J, Orsi SA, Zhang M, Moore AN (2009) Sulforaphane improves cognitive function administered following traumatic brain injury. Neurosci Lett 460(2):103–107
Deng-Bryant Y, Singh IN, Carrico KM, Hall ED (2008) Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab 28(6):1114–1126
Dimlich RV, Tornheim PA, Kindel RM, Hall ED, Braughler JM, McCall JM (1990) Effects of a 21-aminosteroid (U-74006F) on cerebral metabolites and edema after severe experimental head trauma. Adv Neurol 52:365–375
Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, Clark RS (2004) Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem 279(37):38563–38570
Galvani S, Coatrieux C, Elbaz M, Grazide MH, Thiers JC, Parini A, Uchida K, Kamar N, Rostaing L, Baltas M, Salvayre R, Negre-Salvayre A (2008) Carbonyl scavenger and antiatherogenic effects of hydrazine derivatives. Free Radic Biol Med 45(10):1457–1467
Gladstone DJ, Black SE, Hakim AM (2002) Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke 33(8):2123–2136
Gu Y, Hua Y, Keep RF, Morgenstern LB, Xi G (2009) Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke 40(6):2241–2243
Gutteridge JM (1995) Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 41(12 Pt 2):1819–1828
Hall E (1986) Beneficial effects of acute intravenous ibuprofen on neurological recovery of head injured mice: comparison of cyclooxygenase inhibition of thromboxane A2 synthetase or 5-lipoxygenase. CNS. Trauma 2:75–83
Hall ED, Bosken JM (2009) Measurement of oxygen radicals and lipid peroxidation in neural tissues. Curr Protoc Neurosci Chapter 7:Unit 7 17. 11–51
Hall ED, Yonkers PA, McCall JM, Braughler JM (1988) Effects of the 21-aminosteroid U74006F on experimental head injury in mice. J Neurosurg 68(3):456–461
Hall ED, Yonkers PA, Horan KL, Braughler JM (1989) Correlation between attenuation of posttraumatic spinal cord ischemia and preservation of tissue vitamin E by the 21-aminosteroid U74006F: evidence for an in vivo antioxidant mechanism. J Neurotrauma 6(3):169–176
Hall ED, Braughler JM, Yonkers PA, Smith SL, Linseman KL, Means ED, Scherch HM, Von Voigtlander PF, Lahti RA, Jacobsen EJ (1991) U-78517F: a potent inhibitor of lipid peroxidation with activity in experimental brain injury and ischemia. J Pharmacol Exp Ther 258(2):688–694
Hall ED, Yonkers PA, Andrus PK, Cox JW, Anderson DK (1992) Biochemistry and pharmacology of lipid antioxidants in acute brain and spinal cord injury. J Neurotrauma 9(Suppl 2):S425–S442
Hall ED, Andrus PK, Yonkers PA (1993) Brain hydroxyl radical generation in acute experimental head injury. J Neurochem 60(2):588–594
Hall ED, McCall JM, Means ED (1994) Therapeutic potential of the lazaroids (21-aminosteroids) in acute central nervous system trauma, ischemia and subarachnoid hemorrhage. Adv Pharmacol 28:221–268
Hall ED, Andrus PK, Smith SL, Oostveen JA, Scherch HM, Lutzke BS, Raub TJ, Sawada GA, Palmer JR, Banitt LS, Tustin JM, Belonga KL, Ayer DE, Bundy GL (1995) Neuroprotective efficacy of microvascularly-localized versus brain-penetraiting antioxidants. Acta Neurochir (Suppl) 66:107–113
Hall ED, Andrus PK, Smith SL, Fleck TJ, Scherch HM, Lutzke BS, Sawada GA, Althaus JS, Vonvoigtlander PF, Padbury GE, Larson PG, Palmer JR, Bundy GL (1997) Pyrrolopyrimidines: novel brain-penetrating antioxidants with neuroprotective activity in brain injury and ischemia models. J Pharmacol Exp Ther 281(2):895–904
Hall ED, Kupina NC, Althaus JS (1999) Peroxynitrite scavengers for the acute treatment of traumatic brain injury. Ann N Y Acad Sci 890:462–468
Hall ED, Vaishnav RA, Mustafa AG (2010) Antioxidant therapies for traumatic brain injury. Neurotherapeutics 7(1):51–61
Hall ED, Wang JA, Miller DM (2012) Relationship of nitric oxide synthase induction to peroxynitrite-mediated oxidative damage during the first week after experimental traumatic brain injury. Exp Neurol 238(2):176–182
Halliwell B, Gutteridge J (2008) Free radicals in biology and medicine, 3rd edn. Oxford University Press, New York
Hamann K, Shi R (2009) Acrolein scavenging: a potential novel mechanism of attenuating oxidative stress following spinal cord injury. J Neurochem 111(6):1348–1356
Hamann K, Nehrt G, Ouyang H, Duerstock B, Shi R (2008) Hydralazine inhibits compression and acrolein-mediated injuries in ex vivo spinal cord. J Neurochem 104(3):708–718
Hong SC, Goto Y, Lanzino G, Soleau S, Kassell NF, Lee KS (1994) Neuroprotection with a calpain inhibitor in a model of focal cerebral ischemia. Stroke 25(3):663–669
Hummel SG, Fischer AJ, Martin SM, Schafer FQ, Buettner GR (2006) Nitric oxide as a cellular antioxidant: a little goes a long way. Free Radic Biol Med 40(3):501–506
Jin W, Kong J, Wang H, Wu J, Lu T, Jiang J, Ni H, Liang W (2011) Protective effect of tert-butylhydroquinone on cerebral inflammatory response following traumatic brain injury in mice. Injury 42(7):714–718
Kassell NF, Haley EC Jr, Apperson-Hansen C, Alves WM (1996) Randomized, double-blind, vehicle-controlled trial of tirilazad mesylate in patients with aneurysmal subarachnoid hemorrhage: a cooperative study in Europe, Australia, and New Zealand. J Neurosurg 84(2):221–228
Kensler TW, Wakabayashi N, Biswal S (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47:89–116
Kontos HA (1989) Oxygen radicals in CNS damage. Chem Biol Interact 72(3):229–255
Kontos HA, Povlishock JT (1986) Oxygen radicals in brain injury. Cent Nerv Syst Trauma 3(4):257–263
Kontos HA, Wei EP (1986) Superoxide production in experimental brain injury. J Neurosurg 64(5):803–807
Langham J, Goldfrad C, Teasdale G, Shaw D, Rowan K (2000) Calcium channel blockers for acute traumatic brain injury. Cochrane Database Syst Rev 2:CD000565
Lanzino G, Kassell NF (1999) Double-blind, randomized, vehicle-controlled study of high-dose tirilazad mesylate in women with aneurysmal subarachnoid hemorrhage. Part II. A cooperative study in North America. J Neurosurg 90(6):1018–1024
Lewen A, Matz P, Chan PH (2000) Free radical pathways in CNS injury. J Neurotrauma 17(10):871–890
Long DA, Ghosh K, Moore AN, Dixon CE, Dash PK (1996) Deferoxamine improves spatial memory performance following experimental brain injury in rats. Brain Res 717(1–2):109–117
Longoni B, Salgo MG, Pryor WA, Marchiafava PL (1998) Effects of melatonin on lipid peroxidation induced by oxygen radicals. Life Sci 62(10):853–859
Mao L, Wang H, Qiao L, Wang X (2010) Disruption of Nrf2 enhances the upregulation of nuclear factor-kappaB activity, tumor necrosis factor-alpha, and matrix metalloproteinase-9 after spinal cord injury in mice. Mediat Inflamm 2010:238321
Mao L, Wang H, Wang X, Liao H, Zhao X (2011) Transcription factor Nrf2 protects the spinal cord from inflammation produced by spinal cord injury. J Surg Res 170(1):e105–e115
Marklund N, Clausen F, Lewen A, Hovda DA, Olsson Y, Hillered L (2001) Alpha-phenyl-tert-N-butyl nitrone (PBN) improves functional and morphological outcome after cortical contusion injury in the rat. Acta Neurochir 143(1):73–81
Marshall LF, Maas AI, Marshall SB, Bricolo A, Fearnside M, Iannotti F, Klauber MR, Lagarrigue J, Lobato R, Persson L, Pickard JD, Piek J, Servadei F, Wellis GN, Morris GF, Means ED, Musch B (1998) A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg 89(4):519–525
Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED (2008) Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Exp Neurol 209(1):243–253
Mbye LH, Singh IN, Carrico KM, Saatman KE, Hall ED (2009) Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. J Cereb Blood Flow Metab 29(1):87–97
McIntosh TK, Thomas M, Smith D, Banbury M (1992) The novel 21-aminosteroid U74006F attenuates cerebral edema and improves survival after brain injury in the rat. J Neurotrauma 9(1):33–46
Mesenge C, Margaill I, Verrecchia C, Allix M, Boulu RG, Plotkine M (1998) Protective effect of melatonin in a model of traumatic brain injury in mice. J Pineal Res 25(1):41–46
Mikawa S, Kinouchi H, Kamii H, Gobbel GT, Chen SF, Carlson E, Epstein CJ, Chan PH (1996) Attenuation of acute and chronic damage following traumatic brain injury in copper, zinc-superoxide dismutase transgenic mice. J Neurosurg 85(5):885–891
Miller DM, Singh IN, Wang JA, Hall ED (2013) Administration of the Nrf2-ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free Radic Biol Med 57:1–9
Miller D, Wang J, Buchanan A, Hall E (2014) Temporal and spatial dynamics of Nrf2-ARE-mediated gene targets in cortex and hippocampus following controlled cortical impact traumatic brain injury in mice. J Neurotrauma 31:1194–1201
Miller DM, Singh IN, Wang JA, Hall ED (2015) Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp Neurol 264:103–110
Monyer H, Hartley DM, Choi DW (1990) 21-Aminosteroids attenuate excitotoxic neuronal injury in cortical cell cultures. Neuron 5(2):121–126
Mori T, Kawamata T, Katayama Y, Maeda T, Aoyama N, Kikuchi T, Uwahodo Y (1998) Antioxidant, OPC-14117, attenuates edema formation, and subsequent tissue damage following cortical contusion in rats. Acta Neurochir Suppl (Wien) 71:120–122
Muizelaar JP, Kupiec JW, Rapp LA (1995) PEG-SOD after head injury. J Neurosurg 83(5):942
Mustafa AG, Singh IN, Carrico KM, Hall ED (2010) Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem 114(1):271–280
Mustafa AG, Wang JA, Carrico KM, Hall ED (2011) Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J Neurochem 117(3):579–588
Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, Bullock MR, Choi SC, Clifton GL, Contant CF, Coplin WM, Dietrich WD, Ghajar J, Grady SM, Grossman RG, Hall ED, Heetderks W, Hovda DA, Jallo J, Katz RL, Knoller N, Kochanek PM, Maas AI, Majde J, Marion DW, Marmarou A, Marshall LF, McIntosh TK, Miller E, Mohberg N, Muizelaar JP, Pitts LH, Quinn P, Riesenfeld G, Robertson CS, Strauss KI, Teasdale G, Temkin N, Tuma R, Wade C, Walker MD, Weinrich M, Whyte J, Wilberger J, Young AB, Yurkewicz L (2002) Clinical trials in head injury. J Neurotrauma 19(5):503–557
Ozdemir D, Tugyan K, Uysal N, Sonmez U, Sonmez A, Acikgoz O, Ozdemir N, Duman M, Ozkan H (2005a) Protective effect of melatonin against head trauma-induced hippocampal damage and spatial memory deficits in immature rats. Neurosci Lett 385(3):234–239
Ozdemir D, Uysal N, Gonenc S, Acikgoz O, Sonmez A, Topcu A, Ozdemir N, Duman M, Semin I, Ozkan H (2005b) Effect of melatonin on brain oxidative damage induced by traumatic brain injury in immature rats. Physiol Res 54(6):631–637
Panter SS, Braughler JM, Hall ED (1992) Dextran-coupled deferoxamine improves outcome in a murine model of head injury. J Neurotrauma 9:47–53
Pellegrini-Giampietro DE, Cherici G, Alesiani M, Carla V, Moroni F (1990) Excitatory amino acid release and free radical formation may cooperate in the genesis of ischemia-induced neuronal damage. J Neurosci 10(3):1035–1041
Readnower RD, Pandya JD, McEwen ML, Pauly JR, Springer JE, Sullivan PG (2011) Post-injury administration of the mitochondrial permeability transition pore inhibitor, NIM811, is neuroprotective and improves cognition after traumatic brain injury in rats. J Neurotrauma 28(9):1845–1853
Rohn TT, Hinds TR, Vincenzi FF (1993) Ion transport ATPases as targets for free radical damage. Protection by an aminosteroid of the Ca2+ pump ATPase and Na+/K+ pump ATPase of human red blood cell membranes. Biochem Pharmacol 46(3):525–534
Rohn TT, Hinds TR, Vincenzi FF (1996) Inhibition of Ca2+-pump ATPase and the Na+/K+-pump ATPase by iron-generated free radicals. Protection by 6,7-dimethyl-2,4-DI-1- pyrrolidinyl-7H-pyrrolo[2,3-d] pyrimidine sulfate (U-89843D), a potent, novel, antioxidant/free radical scavenger. Biochem Pharmacol 51(4):471–476
Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J, Kamins J, Okamoto S, Izumi M, Shirasawa T, Lipton SA (2008) Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem 104(4):1116–1131
Sharma S, Zhuang Y, Ying Z, Wu A, Gomez-Pinilla F (2009) Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience 161(4):1037–1044
Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH (2003) Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 23(8):3394–3406
Singh IN, Sullivan PG, Hall ED (2007) Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J Neurosci Res 85(10):2216–2223
Singh IN, Gilmer LK, Miller DM, Cebak JE, Wang JA, Hall ED (2013) Phenelzine mitochondrial functional preservation and neuroprotection after traumatic brain injury related to scavenging of the lipid peroxidation-derived aldehyde 4-hydroxy-2-nonenal. J Cereb Blood Flow Metab 33(4):593–599
Smith SL, Hall ED (1998) Tirilazad widens the therapeutic window for riluzole-induced attenuation of progressive cortical degeneration in an infant rat model of the shaken baby syndrome. J Neurotrauma 15(9):707–719
Smith SL, Andrus PK, Zhang JR, Hall ED (1994) Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J Neurotrauma 11(4):393–404
Sonmez U, Sonmez A, Erbil G, Tekmen I, Baykara B (2007) Neuroprotective effects of resveratrol against traumatic brain injury in immature rats. Neurosci Lett 420(2):133–137
Sullivan PG, Thompson MB, Scheff SW (1999) Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol 160(1):226–234
Sullivan PG, Krishnamurthy S, Patel SP, Pandya JD, Rabchevsky AG (2007) Temporal characterization of mitochondrial bioenergetics after spinal cord injury. J Neurotrauma 24(6):991–999
Toklu HZ, Hakan T, Biber N, Solakoglu S, Ogunc AV, Sener G (2009) The protective effect of alpha lipoic acid against traumatic brain injury in rats. Free Radic Res 43(7):658–667
Wang X, de Rivero Vaccari JP, Wang H, Diaz P, German R, Marcillo AE, Keane RW (2012) Activation of the nuclear factor E2-related factor 2/antioxidant response element pathway is neuroprotective after spinal cord injury. J Neurotrauma 29(5):936–945
Wood PL, Khan MA, Moskal JR, Todd KG, Tanay VA, Baker G (2006) Aldehyde load in ischemia-reperfusion brain injury: neuroprotection by neutralization of reactive aldehydes with phenelzine. Brain Res 1122(1):184–190
Wood PL, Khan MA, Moskal JR (2008) Mechanism of action of the disease-modifying anti-arthritic thiol agents D-penicillamine and sodium aurothiomalate: restoration of cellular free thiols and sequestration of reactive aldehydes. Eur J Pharmacol 580(1–2):48–54
Wu A, Ying Z, Gomez-Pinilla F (2006) Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Exp Neurol 197(2):309–317
Xiong Y, Peterson PL, Muizelaar JP, Lee CP (1997) Amelioration of mitochondrial function by a novel antioxidant U-101033E following traumatic brain injury in rats. J Neurotrauma 14(12):907–917
Xiong Y, Peterson PL, Verweij BH, Vinas FC, Muizelaar JP, Lee CP (1998) Mitochondrial dysfunction after experimental traumatic brain injury: combined efficacy of SNX-111 and U-101033E. J Neurotrauma 15(7):531–544
Xiong Y, Shie FS, Zhang J, Lee CP, Ho YS (2005) Prevention of mitochondrial dysfunction in post-traumatic mouse brain by superoxide dismutase. J Neurochem 95(3):732–744
Yan W, Wang HD, Hu ZG, Wang QF, Yin HX (2008) Activation of Nrf2-ARE pathway in brain after traumatic brain injury. Neurosci Lett 431(2):150–154
Zhang DD (2006) Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev 38(4):769–789
Zhang JR, Scherch HM, Hall ED (1996) Direct measurement of lipid hydroperoxides in iron-dependent spinal neuronal injury. J Neurochem 66(1):355–361
Zhang R, Shohami E, Beit-Yannai E, Bass R, Trembovler V, Samuni A (1998) Mechanism of brain protection by nitroxide radicals in experimental model of closed-head injury. Free Radic Biol Med 24(2):332–340
Zhang H, Squadrito GL, Uppu R, Pryor WA (1999) Reaction of peroxynitrite with melatonin: a mechanistic study. Chem Res Toxicol 12(6):526–534
Acknowledgements
Portions of the work reviewed in this chapter were supported by funding from 5R01 NS046566, 5P30 NS051220, and 5P01 NS58484 and currently by 5R01 NS083405, 5R01 NS084857 and 1R01 NS100093 and from the Kentucky Spinal Cord & Head Injury Research Trust.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Hall, E.D., Singh, I.N., Cebak, J.E. (2018). Oxidative Damage Mechanisms in Traumatic Brain Injury and Antioxidant Neuroprotective Approaches. In: Fujikawa, D. (eds) Acute Neuronal Injury. Springer, Cham. https://doi.org/10.1007/978-3-319-77495-4_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-77495-4_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-77494-7
Online ISBN: 978-3-319-77495-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)