
Abstract
The gastrointestinal (GI) tract is the residence of trillions of microorganisms that include bacteria, archaea, fungi and viruses. The collective genomes of whole microbial communities (microbiota) integrate the gut microbiome. Up to 100 genera and 1000 distinct bacterial species were identified in digestive tube niches. Gut microbiomes exert permanent pivotal functions by promoting food digestion, xenobiotic metabolism and regulation of innate and adaptive immunological processes. Proteins, peptides and metabolites released locally and at distant sites trigger many cell signalling and pathways. This intense crosstalk maintains the host-microbial homeostasis. Diet, age, diet, stress and diseases cause increases or decreases in relative abundance and diversity bacterial specie of GI and other body sites. Studies in animal models and humans have shown that a persistent imbalance of gut’s microbial community, named dysbiosis, relates to inflammatory bowel diseases (IBD), irritable bowel syndrome (IBS), diabetes, obesity, cancer, cardiovascular and central nervous system disorders. Notably specific bacterial communities are promising clinical target to treat inflammatory and infectious diseases. In this context, intestinal microbiota transplantation (IMT) is one optional treatment for IBD, in particular to patients with recurrent Clostridium difficile-induced pseudo-membrane colitis. Here we discuss on recent discoveries linking whole gut microbiome dysbiosis to metabolic and inflammatory diseases and potential prophylactic and therapeutic applications of faecal and phage therapy, probiotic and prebiotic diets.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
The evolution of Homo sapiens is linked to a mutualistic partnership among bacteria, archaea, viruses, protozoans and fungi that cohabit many sites of the human body (Backhed et al. 2012). The number and diversity of the microorganisms are modulated by physiological and environmental factors, such as host genotype, habitat and mainly diet. The total number of bacteria in a standard adult man of 70 kg is estimated to be 3.8 × 1013, which is approximately the number of cells in the human body (Sender et al. 2016). Bacteria are classified morphologically and biochemically based on cell wall type, cell shape, oxygen requirements, endospore production, motility and energy requirements. They are also classified phylogenetically based on the analysis of nucleotide sequences of variable regions of small subunit ribosomal RNA operons or 16S and 18S rRNA genes (Belizario and Napolitano 2015). The next generation sequencing (NGS) metagenomic method has been applied to genomic profiling of known and unknown microbes present in diverse human body sites. The phylogenetic clustering of all bacterial genomes, collectively named microbiomes, into taxonomic groups (domain, kingdom, phyla, class, order, family, genus and species) have generated complex microbiome datasets (Qin et al. 2010; Gevers et al. 2012; Flintoft 2012; Li et al. 2014; The Human Microbiome Project Consortium 2012).
The human microbiomes are divided in four major phyla: Firmicutes (65%), Bacteroidetes (16%), Actinobacteria (9%) and Proteobacteria (5%). Bacteroidetes and Firmicutes represent more than 90% of the relative abundance of gut microbes (Arumugam et al. 2011). The phylum Bacteroidetes is composed of Gram-negative, not spores forming, anaerobic bacteria that tolerate the presence of oxygen but cannot use it for growth. Firmicutes are a diverse phylum of gut microbiota composed mainly of Bacilli and Clostridia classes. They are Gram-positive, anaerobic (Clostridia) and obligate or facultative aerobes (Bacilli) characterized by a low GC content. Clostridium species produce endospores to survive adverse conditions, allowing them to return only when the ecosystem is favourable for their growth. In contrast, Actinobacteria, for example Bifidobacterium, are Gram-positive, multiple branching rods, non-motile, non-spore-forming and anaerobic. Proteobacteria (e.g. Escherichia, Klebsiella, Enterobacter) are aerobic or facultative anaerobic, Gram-negative, non-spore-forming rods, which inhabit the intestinal tract of all vertebrates. While Lactobacillus, Staphylococcus and Escherichia coli are examples of facultative anaerobic bacteria, Bacteroides are obligate anaerobes (Belizário and Napolitano 2015).
Microbiome studies have identified a large number of microbial compositions across a range of people from European, South Americans, Indians and Africans (Qin et al. 2010; Gevers et al. 2012; Flintoft 2012). Phylogenic approaches have been used to compare modern human microbiomes from those of ancestral population (Moeller et al. 2014; Clemente et al. 2013). European children had higher Firmicutes and Proteobacteria and lower levels depletion of Bacteroidetes, which contain the fibre degradation species such as Prevotella and Xylanibacter, in the gut flora (De Filippo et al. 2010). West Africa children had a higher prevalence of Bacteroidetes and absence of Firmicutes (De Filippo et al. 2010). The diversity of bacterial composition, caloric load and nutrient absorption has served to clustering species-level phylotypes into enterotypes (Arumugam et al. 2011; Koren et al. 2013). A personal enterotype is based on presence in the gut of healthy and disease-associated microbial strains/species, and it determines the physiopathological state of each individual. The human skin, lung, oral, genital and gastrointestinal tract microbiomes are influenced by environment and microbe colonization and represent a key step in the dynamic evolution of Homo sapiens (Ley et al. 2008; Qin et al. 2010; Abubucker et al. 2012; Flintoft 2012; Moeller et al. 2014).
The deeper taxonomic resolution (strains and subspecies) and functional analyses of single nucleotide polymorphisms (SNPs) and other variant sequences in microbiome datasets are still being defined (Ley et al. 2008; Qin et al. 2010; Abubucker et al. 2012; Flintoft 2012; Li et al. 2014). At least as important is the analysis of the metabolome, which mirrors the multiple functional activities of the microbiome, along with the corresponding transcriptome and proteome. Human gut tissue-specific genome-scale metabolic models displaying metabolome maps and networks have been developed for this particular purpose (Pornputtapong et al. 2015).
The investigation of such numerous and diverse microbiome datasets is a complex task. It requires biological knowledge and skills on a set of tools to accurately predict the influence of perturbed microbe–microbe interaction network on health and disease outcomes. Here, we will present and discuss studies exploring the role of gut microbiomes on aetiology of gastroenterology and metabolic diseases. We also discuss on emerging nutritional and pharmacological strategies including faecal transplantation, probiotics, prebiotics and phage therapy to modulate species-level phylotypes, as well as general bacterial richness and diversity, of the gut microbiota.
2 Gut Microbiota Dictates Host’s Immunological Functions
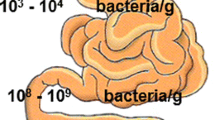
The gut is colonized by 100–400 trillion of microorganisms that live in a close symbiotic relationship (Sender et al. 2016). Most of bacteria are tightly adherent to mucus which forms the outer and inner physical barrier of over 7 m long. Many of these organisms are transmitted at early life to babies, mainly via mother’s milk, which contains higher numbers of Bifidobacteria and Lactobacillus (Palmer et al. 2012). In adults, the majority of the bacteria found in the gut belong to the genera Bacteroides, Parabacteroides (Bacteroidetes) and Clostridium (Firmicutes) (Eckburg et al. 2005; Lepage et al. 2013). The gut is an anaerobic environment and aerobic pathogenic species cannot invade and colonize it; however, anaerobic and facultative pathogenic species can invade it causing disease. Each site of the GI tract has an unique microbiota; bacteria density increases in the jejunum/ileum and in the large intestine in comparison with the stomach and duodenum. The highest cell density is present in the colon, approximately 1012 colony per units/mL (99%). Colon microbiota population is mostly composed of anaerobes such Bacteroides, Porphyromonas, Bifidobacterium, Lactobacillus and Clostridium (genera that belong to the most abundant phyla: Bacteroidetes, Actinobacteria and Firmicutes), with anaerobic bacteria outnumbering aerobic bacteria by a factor of 100 to 1000:1.
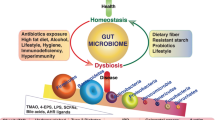
Host and microbial communities have evolved strategies that promote a steady state for their mutual benefit. Dysbiosis refers to an imbalance in microbial species abundance, which is commonly linked to impaired gut barrier function and inflammatory cell activation (Backhed et al. 2012). It is likely that failure to adequately regulate the composition (microbial diversity) is at the onset and chronicity of many diseases, including inflammatory bowel diseases (IBD), irritable bowel syndrome (IBS), diabetes, obesity and cancer (Clemente et al. 2013; Brown et al. 2013a, b; Chang and Lin 2016; Thaiss et al. 2016). The potential association between intestinal dysbiosis and certain human diseases can be assigned, for example, by the presence of anti-inflammatory species, such as Faecalibacterium prausnitzii, which predominate in healthy individuals, or the presence of potentially pro-inflammatory bacteria Bacteroides and Ruminococcus gnavus, which are associated with IBD (Swidsinski et al. 2005). A decreased Bacteroidetes/Firmicutes ratio and low abundance of Bacteroides species have been associated with obesity in animal model studies (Verdam et al. 2013). Dysbiosis is also known in other microbiomes, such as in the skin, vagina, oral cavity, stomach and particularly in the form of small intestine bacterial overgrowth.
The gut microbiota plays a critical role in the development of gut-associated lymphoid tissues including Peyer’s patches, isolated lymphoid follicles and mesenteric lymph nodes, which are responsible for a fully functional immune system (Round and Mazmanian 2009; Brown et al. 2013a, b). At early life, interactions of microbes and their products in the gut (intestine) prepare the immune system to distinguish self from non-self (invaders) (Round and Mazmanian 2009; Hooper et al. 2012; Honda and Littman 2016). The innate immune response is mediated by innate lymphoid cells (ILCs) formed by natural killer (NK) cells, cytotoxic and non-cytotoxic and helper lymphoid cell subsets that include ILC1, ILC2 and ILC3. NK cells and ILC1 produce large amounts of IFN-γ, killer antimicrobial peptides (AMPs), such as granulysins, cathelicins and defensins, and chemokines (Ostaff et al. 2013). Together, they exert critical functions on the regulation of microbial ecology and immune tolerance (Thaiss et al. 2016). Imbalance of either NK activity or AMP production can lead to the development of local inflammatory diseases as well as systemic infectious (Fuchs and Colonna 2011; Ostaff et al. 2013).
The intestinal epithelial cells (IECs) and immune cells express a variety of receptors called pattern recognition receptors (PRR) that mediate the interactions between the immune system and the commensal microbiota. Toll-like receptors (TLRs) and nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) are examples of PRR that recognize unique microbial molecules named pathogen-associated molecular patterns (PAMPs) including lipopolysaccharides, lipid A, peptidoglycans, flagella and microbial RNA/DNA (Sangiuliano et al. 2014). These receptors activate inflammasomes, which in turn activates pro-inflammatory caspases, leading to the production of cytokines interleukins IL-1β and IL-18 (Maynard et al. 2012; Brown et al. 2013a, b; Sangiuliano et al. 2014). The different inflammasomes have redundant roles during infection, and their activation can lead to cell death by pyroptosis, which protects against infection (Jorgensen et al. 2017).
Firmicutes are positively associated with TNF-α and IL-1β production, which are critical cytokines involved in the regulation of immune cells (Maynard et al. 2012; Clemente et al. 2013; Honda and Littman 2016; Webb et al. 2016). Different inflammatory diseases are characterized by mutations or loss of some innate response genes in lymphoid tissues, smaller Peyer’s patches and mesenteric lymph nodes (Hooper et al. 2012; Honda and Littman 2016). The ability to control systemic infection is markedly reduced in NOD1−/− as well as NOD2−/− (Maynard et al. 2012; Brown et al. 2013a, b; Sangiuliano et al. 2014). Experiments with NOD2−/− mice revealed its participation in type 1 diabetes, which is triggered by products from gut bacterial species not yet identified (Costa et al. 2016). Patients’ immunity status and genotypes are directly associated with overgrowth of pathogenic microbes.
Luminal bacteria of gut epithelium induce the synthesis and secretion of approximately 70% of bacterial-specific IgA that prevents invasion of the microorganisms into the gut tissue (Brown et al. 2013a, b). IgA class switching by mucosal B cells is maintained by retinoic acid and transforming growth factor-β (TGF-β), which are critical immune suppressive processes mediated by T helper (Th) 17 cells and regulatory T cells (Treg) (Honda and Littman 2016). The use of germ-free (GF) mice and gnotobiotic mouse models have demonstrated that immunological functions can be restored by the development of bacterial communities after GF are colonizing with microorganisms (Honda and Littman 2016; Webb et al. 2016). GF mice display underdeveloped lymphoid tissues, defective T and B cell function and low numbers of circulating CD4+ T cells and antibody production. The equilibrium between the whole body immune system and immune regulatory functions of gut bacteria appears to be delicate. Loss of a specific species can lead to an overreaction or suppression of the innate immune system (Round and Mazmanian 2009; Webb et al. 2016).
3 Gut Microbiota Control the Host’s Metabolic States
Microorganisms perform their functions largely through enzyme pathways to digest complex dietary carbohydrates and proteins. The synthesis of a wide range of low-molecular weight signalling molecules, including methane, hydrogen sulfide and metabolites, play either harmful or beneficial effects to the host (Wikoff et al. 2009; Tremaroli and Bäckhed 2012). Those products turn on or off both host genes and genes from other microbes. Thousands of microbiota-derived metabolites have been identified as components of the human metabolome (Overbeek et al. 2005). The maintenance of a stable, fermentative gut microbiota requires diets rich in whole plant foods particularly high in dietary fibre and polyphenols, which are processed by the enzymes from intestinal microbiota such as glycoside hydrolases and polysaccharide lyases. Under anaerobic conditions, species belonging to the Bacteroides family, and to the Clostridiaceae and Lactobacillaceae families, particularly Citrobacter and Serratia strains, produce short-chain fatty acids (SCFAs). SCFAs produced by microbiota are acetate (with two carbons), propionate (with three carbons) and butyrate (with four carbons). Butyrate provides energy for cellular metabolism. Acetate and propionate pass into the bloodstream, and they are taken by liver and peripheral organs, where they can act as substrates for gluconeogenesis and lipogenesis, which, in turn, control body weight gain. SCFAs are preferred source of energy used by colonocytes (colon) (Meijer et al. 2010). SCFAs also bind to the G-protein-coupled receptors (GPRs), among them GPR41 and 43, which are present in a variety of tissues, including adipose, gut enteroendocrine cells and inflammatory cells. Acting in the gut, SCFAs induces the secretion of glucagon-like peptide (GLP-1) and peptide YY (PYY). PYY inhibits nutrient absorption from the intestinal lumen and appetite, whereas GLP1 stimulate β-cells of the pancreas to produce insulin. The gut microbiota contributes to fat deposition through the regulation of the farnesoid X receptor (FXR), the bile acid receptor that is responsible for the regulation of bile acid synthesis and hepatic triglyceride accumulation. Moreover, microbiota converts choline to trimethylamine and thus regulates directly the bioavailability of choline and indirectly the accumulation of triglycerides in the liver. Therefore, targeting FXR signalling by bile acid derivatives could help control the gut bacterial community.
SCFAs play a key role in controlling mucosal proliferation, differentiation and maintenance of mucosal integrity. Impairment in the production of SCFAs, tryptophan metabolites, GABA, noradrenaline, dopamine, acetylcholine and 5-HT (5-hydroxytryptamine or serotonine), all related to microbial metabolism, is involved in GI abnormalities, metabolic diseases and neuropsychiatric derangements (Round and Mazmanian 2009; Tremaroli and Bäckhed 2012; Gilbert et al. 2016). Indeed, several of these molecules are major neurotransmitters. Serotonin, in particular, has been vigorously investigated in connection with its central roles in appetite, sexuality, substance addiction, emotions and stress response. There are reasons to believe that most of the brain serotonin either originates from the gut microbiome or has its synthesis modulated by the brain–gut axis (O’Mahony et al. 2015).
Furthermore, the microbiota produces large quantities of epigenetically active metabolites, such as folate and vitamin A. Therefore, changes in gut microbiota (dysbiosis) can result in the epigenome changes not only directly in adjacent intestinal cells, but also distant cell populations, such as hepatocytes and adipocytes (Honda and Littman 2016). SCFA generated by microbial fermentation are critical for generation of Treg. Finally, bacteria can inhibit the growth of their competitors by specific microbial communication, cell signalling through cell-to-cell contact, metabolites and quorum sensing peptides (Miller and Bassler 2001; Ostaff et al. 2013). These phenomena are relevant for the prevention of intestinal infection and colonization by pathobionts (deleterious microorganisms).
4 Gut Dysbiosis Induces Inflammatory and Metabolic Diseases
Elevated intestinal barrier permeability and translocation of bacteria or endotoxin are associated with GI diseases. Certain strains are sometimes mentioned such as E. coli, Klebsiella, Proteus, Enterobacter, Shigella, Salmonella and Serratia. However, it is still not clear whether these changes are a contributing factor or a consequence of the disease (Harley and Karp 2012). Microbiota composition varies between different locations in the gastrointestinal tract (Eckburg et al. 2005; Arumugam et al. 2011; Cucchiara et al. 2012; Lepage et al. 2013). However, most microbiome studies reported in the literature have focused on only distal large intestine or faecal microbiota. The faecal samples contain over 1000 bacterial species, and up to 50% are uncultivable and thus uncharacterized molecularly (Qin et al. 2010; Zhou et al. 2014; Li et al. 2014). 16S rRNA gene sequencing was used to precisely identify 28 clinically relevant species and genera in stool samples of 897 healthy individuals (Almonacid et al. 2017). This curated database was used to define a 99% healthy confidence interval for each target (14 species and 14 genera). Then, the authors used this healthy reference range as a clinical tool to evaluate for positive and negative associations in people who have one of various chronic disease and medical conditions, including diarrhoea, constipation, indigestion, inflammatory bowel syndrome, ulcerative colitis, Crohn’s disease, obesity, type II diabetes and other conditions. Table 13.1 presents the taxon associated and inversely associated with one or more condition.
The ratio of potentially pathogenic to beneficial commensal microbes seems to be more crucial for disease development (Baumler and Sparandio 2016). Clostridium difficile infection is one of the most common infectious that cause of nosocomial diarrhoea and colitis, associated with antibiotic use (Antharam et al. 2013). The vast majority of Firmicutes sequences (70%) are assigned into the Clostridia class, which is divided into 19 clusters on the basis of growth, metabolic and morphological parameters (Antharam et al. 2013). Clostridium difficile overgrowth is associated with the depletion of Ruminococcaceae, Lachnospiraceae and butyrogenic bacteria belonging to Clostridium cluster IV or XIVa.
IBD is a chronic relapsing inflammatory disorder affecting the GI tract, and its two main clinical phenotypes are Crohn’s disease (CD) and ulcerative colitis (UC). IBDs are characterized by the infiltration of neutrophils, monocytes and lymphocytes into the intestinal lamina propria causing tissue injury, loss of goblet cells, fibrosis, erosions and ulcerations. These diseases are associated with Firmicutes and Bacteroidetes, and increases in Proteobacteria, Actinobacteria, in particular, within the families Pasteurellaceae, Veillonellaceae, Fusobacteriaceae, Enterobacteriaceae, and the adherent-invasive E. coli strains. IBS is a heterogeneous clinical condition diagnosed by abdominal pain, bowel movement irregularity and bloating. Patient’s faecal samples have decreased Bifidobacterium Lactobacillus, Bacteroidetes and Actinobacteria and increased Firmicutes and Proteobacteria. The methane production, which is sometimes elevated, can be measured by breath testing. This is caused by the archaebacteria Methanobrevibacter smithii.
Obesity, insulin resistance and the metabolic syndromes result from a complex interaction of genetic, immune and microbial factors produced by associated gut microbiota (Ley et al. 2006; Cani et al. 2007; Harley and Karp 2012). Metabolic syndrome is a chronic inflammatory disease characterized by translocation of bacteria into blood causing metabolic endotoxemia. The clinical manifestations of metabolic syndrome include cardiovascular disease, insulin resistance and type 2 diabetes, hypertension and fatty liver disease. Individuals colonized by bacteria of genera Faecalibacterium, Bifidobacterium, Lactobacillus, Coprococcus and Methanobrevibacter have significantly less tendency to develop metabolic disturbances and inflammation that lead to type-2-diabetes and ischemic cardiovascular disorders (Ley et al. 2006; Chatelier et al. 2013). These species are characterized by their high production of SCFAs as well as hydrogen peroxides, which are known to inhibit biofilm formation and activity of pathogenic species including Staphylococcus aureus (Chatelier et al. 2013). Genetic and diet-induced mouse models of obesity have shown that Firmicutes/Bacteroidetes ratio is increased in the obese animals as compared to non-obese animals (Ley et al. 2006; De Filippo et al. 2010; Chatelier et al. 2013; Verdam et al. 2013). However, conflicting results exist regarding the overall gut microbiome taxonomic differences (Harley and Karp 2012; Debelius et al. 2016; Almonacid et al. 2017). Metabolic disturbances in human and animal models of obesity may be caused by increased intestinal permeability and diffusion of LPS through circulation, which in turn promotes low-grade inflammation and insulin resistance (Cani et al. 2007; Cani and Delzenne 2011; Kootte et al. 2012). Recently, one study described the effects of high-fed diet on acetate produced by intestinal microbiota using a rat model (Perry et al. 2016). Through its effects on both digestive tract and parasympathetic system, acetate plasma levels may be responsible to increased glucose-stimulated insulin secretion, increased ghrelin secretion, hyperphagia and obesity (Perry et al. 2016). As stated by the authors, variation of acetate levels could explain why consumption of calorically concentrated foods in abundance promotes obesity and insulin resistance.
The gut–brain axis may be crucial for controlling the animal metabolic states and metabolic diseases (Seeley et al. 2015). Gastric bypass surgery named Roux-en-Y Gastric Bypass (RYGB) was applied to study food preference in rats (Hankir et al. 2017). Experimentally, RYGB induces anatomic changes, and the enteroendocrine adaptations modify intestinal gene-encoding hormone expression. Normally, a fatty-acid derivative named oleoylethanolamide (OEA) is produced when fat is ingested and it directly activates PPAR-α receptors, which is responsible to promote satiety via vague nerve that controls dopamine release in the brain (Hankir et al. 2017). Dopamine-suppressed obese animals have low levels of OEA and dopamine. By means of surgically increasing the levels of OEA and dopamine 1 receptor (D1R) expression, a shift in GI–brain axis signalling is speculated to altering animal behaviour and leading them to prefer low fat food. An ongoing human pan-omics protocol is exploring RYGB-induced genetic and enteroendocrine changes in the GI mucosa and the impact of gut microbiome metabolites in humans (Sala et al. 2016). There has been a lot of expectation that dysbiosis associated with gastrointestinal diseases, obesity and diabetes could be treated by targeted modulation of some bacteria species and/or phyla in intestinal tract.
5 Targeting Gut Microbiota Dysbiosis
5.1 Faecal Microbial Transplant (FMT)
Antibiotics have been used to treat infectious diseases over the past century. However, it is clear that antibiotic treatment can change gut microbial community significantly (Dethlefsen et al. 2008; Forslund et al. 2013). High doses and frequent use of antibiotics particularly against anaerobes, such as vancomycin, can disrupt and destabilize the normal bowel microbiome (Andersson and Hughes 2014). One of the consequences could be Clostridium difficile colitis. The chronic recurrent nosocomial pattern of this colitis is associated with two toxins, TcdA and TcdB, produced by C. difficile (Baumler and Sparandio 2016). Antibiotic resistant infections, which are not uncommon, are associated with pathogens named “ESKAPE” which include Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanii, Pseudomonas aeruginosa and Enterobacter species (Baumler and Sparandio 2016).
Faecal bacteriotherapy refers to transference of microbiota of unprocessed stool, via the upper or lower GI tract. FMT has been used as a clinical procedure, in which a liquid suspension of stool from a human (a family member, or a disease-free, screened donor) is inoculated, in order to treat refractory cases of C. difficile colorectal infection (Gough et al. 2011; Rupnik 2015). The faecal preparation contains from 3.0% to 10% of viable and dead bacteria and colonic cells.
It is a matter of discussion whether bacteria and cell’s components affect transplant outcomes, by enhancing ILCs immunostimulatory functions (Bojanova and Bordenstein 2016). Among over 150 on-going clinical trials involving FMT, most of them– in the USA, only few target kidney- and liver-transplanted patients, however, with antibiotic-resistant bacterial colonization or infection (https://clinicaltrials.gov).
About 90% of the patients that receive microbiota transplantation for C. difficile infection are cured, versus 31% of those receiving vancomycin (Van Nood et al. 2013). A few species, including Lactobacillus, Enterococcus, Bifidobacterium and Bacteroides species, have inhibitory activities against C. difficile. Some problems remain to be solved before FMT becomes widespread (Brandt and Reddy 2011). It is known that a variety of viruses, archaea, fungi, parasitic species and metabolites are transferred together with commensal bacteria (Bojanova and Bordenstein 2016). One of the pending questions is their role, potentially both positive and negative, in intestinal immunity and restoration of microbiome normality.
Pediatric Crohn’s disease patients were treated with FMT from suitable donors (Suskind et al. 2015). In this study, 31 species in the faecal microbiome following FMT at two weeks were highlighted, including Ruminococcus torques, Odoribacter splanchnicus, Bacteroides caccae, Alistipes shahii, A. putredinis and Parabacteroides merdae. E. coli expansion contributed to disease outcome and may be a helpful predictor of efficacy of faecal therapy (Suskind et al. 2015).
In animals, the gut microbiome plays a crucial role in weight gain and obesity (Ley et al. 2006; Harley and Karp 2012; Tremaroli and Bäckhed 2012). In ob/ob mice, there is a 50% reduction in Bacteroidetes, with proportional increases in Firmicutes and Archaea in obese mice (Turnbaugh et al. 2006). Transferring the microbiota from fat mice to germ-free hosts induces greater weight gain than in those receiving the microbiota from lean donors (Ley et al. 2006). High-fat intake increases intestinal permeability. In contrast, the administration of Bifidobaterium infantis reduced the production of proinflammatory cytokines and white adipose tissue gain in mice (Cani et al. 2007).
Results in human metabolic disorders have not been conclusive. Transfer of intestinal microbiota from lean donors mildly increased insulin sensitivity in subjects with metabolic syndrome (Vrieze et al. 2012). So far, however, these results have not been confirmed in larger protocols. In 75 obese and prediabetic volunteers that underwent an 8-day antibiotic treatment (amoxicillin and vancomycin), dysbiosis did not alter gut permeability (Reijnders et al. 2016). Neither did LPS blood levels, used as biomarker of systemic inflammation, nor enzymes involved in the regulation of hepatic metabolism of lipids and cholesterol, despite tremendous alterations in gut microbiota (Reijnders et al. 2016).
The use of microbiome-modulating therapies to treat obesity is still a big challenge (Debelius et al. 2016). Novel preventive and therapeutic approaches to obesity depend on better experimental techniques. In this way, recently scientific communities have initiated global efforts to standardization, which has the support of the Unified Microbiome Initiative consortium and the Microbiome Quality Control Project (Alivisatos et al. 2015, http://www.mbqc.org/).
5.2 Probiotics and Prebiotics
Probiotics are defined as live microorganisms that can survive and temporarily colonize the gut. The gastric juice pH ranges from 3 to 5, and this is the first major obstacle that one probiotic must overcome in clinical practice to recover the gut microbiomic dysbiosis. Lactobacillus plantaraum and Bifidobacterium are probiotic bacteria capable of modulating negative effects of high fat diets and even to manage immunological reactions mediated by inflammatory diseases (Maynard et al. 2012; Hooper et al. 2012). Non-pathogenic lactic acid producer such as bioengineered E. coli strains named Nissle 1917 (Ukena et al. 2007) act also as probiotics. Lactobacillus rhamnosus and gasseri and Bifidobacterium lactis may reduce adiposity, body weight, and weight gain in humans (Mekkes et al. 2014). Furthermore, these probiotics can prevent or ameliorate clinical symptoms of irritable bowels syndrome, inflammatory and necrotizing enterocolitis and acute diarrhoea (Whelan and Quigley 2013).
Dietary prebiotics are selectively fermented ingredients that can change the composition and activity of the gastrointestinal microbiota in beneficial ways (Roberfroid 2007). Dietary prebiotic components include poorly digestible carbohydrates, such as non-starch polysaccharides, resistant starch, non-digestible oligosaccharides and polyphenols. Colonic bacteria produce carbohydrate hydrolysing enzymes and via fermentation of prebiotics produce hydrogen, methane, carbon dioxide and SCFAs, which can affect host energy levels and gut hormone regulation. Mixtures of probiotic and prebiotic ingredients can selectively stimulate growth or activity of health promoting bacteria (Roberfroid 2007). Large-scale metagenomics studies have confirmed that synergic association of pro- and prebiotics really change the gut microbial specie dynamics at the genomic level.
5.3 Phage Therapy and CRISPR/Cas9 System
Phages have been used as antimicrobials in human medicine in Eastern Europe as replacement of antibiotics for over 90 years (Abedon 2014). Phage therapy consists of using bacterial viruses (known as phages) as vector for controlling microbial populations (Sulakvelidze et al. 2001; Abedon 2014). Bacteriophages attach to specific receptors in the host membrane and then inject their genetic material into bacteria. Phage infection can result in lysis, lysogeny or resistance. Lytic bacteriophages induce host cell death. Lysogenic (or temperate) phages insert their genome into the host DNA bacteria as a prophage that can transfer it to another during excision-infection process. Temperate and lytic bacteriophages can be programmed to sensitize and kill antibiotic-resistant bacteria.
The use of phages as antibacterial therapeutics has gained new frontier with discoveries of single targeted bacterial specific phage therapies to Pseudomonas aeruginosa, Salmonella, Klebsiella pneumonia, Shigela dysenteriae, Vibrio cholerae, Vancomycin-resistant Enterococcus, methicillin-resistant Staphylococcus aureus (MRSA) strains and Escherichia coli referred to as extended-spectrum beta-lactamase producing E. coli resistant to several classes of antibiotics (Rhoads et al. 2009; Kutter et al. 2015). Clinical studies have confirmed the effectiveness of large spectrum commercial bacteriophage cocktails, such as Enko-phage, SES-bacteriophage, Pyo-bacteriophage and Intesti-bacteriophage. However, the uses of these generic phage cocktails have been approved only in few Eastern European countries (Pelfrene et al. 2016).
Bacteria and archaea have evolved various mechanisms of defence against phage infections (Sampson et al. 2010). Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR associated proteins (Cas) system is an RNA-mediated DNA interference mechanism of defence identified in both Archaea and Bacteria domain (Garneau et al. 2010; Zhang et al. 2013; Haeussler and Concordet 2016). Cas genes encode a heterogeneous family of nucleases, helicases, polymerases, and polynucleotide-binding proteins. This adaptive immune system is triggered upon phage invasion. Bacteria integrate in its genome short fragments of the foreign DNA, called protospacers. These sequences are integrated into specific regions of the host chromosome known as “CRISPR arrays” that are rich in short palindromic invariable repeats. The protospacer becomes a spacer sequence, which is flanked by the repeated regions. The spacer and the invariable repeats are then transcribed into a CRISPR-RNA (crRNA), which guides specific endonuclease Cas9 to a target DNA containing regions complementary to the spacer fragment of the crRNA. Upon recognition, Cas9 cleaves invasive DNA preventing phage infection.
CRISPR/Cas9 system has served as tool for many genetic studies, particularly genomic editing (Jiang et al. 2013; Haeussler and Concordet 2016). One of its enormous potential is the manipulation of bacterial genome (Garneau et al. 2010; Vercoe et al. 2013; Selle and Barrangou 2015; Yosef et al. 2015; Marraffini and Sontheimer 2008). Engineering of commensal bacteria with CRISPR/Cas9 system can be used to introduce specific mutations into essential, antibiotic resistance, virulence genes encode resistance genes and lytic phages. CRISPR/Cas system may constitute an effective vaccination tool in public health for prevention of diseases. However, much work is needed to be done in order to anticipate undesired off-target effects and efficiency of delivery system.
A healthy human gut contains viral communities—the human gut “virome” (over 1012 per person) that infect host cells and other types of microorganisms that inhabit us (Virgin 2014). The viral communities, many of which were unknown so far, may affect human health and disease as do bacteria and archaea. Metagenomic DNA sequencing remains the unique way to identify the growing number of bacteriophages in commensal and pathogenic bacteria present in human body. Lytic phages have also been identified in environmental reservoirs such as zoo composting and human and domestic animal faecal sewages (Amgarten et al. 2017). These are promising tools for the development of more adequate phage-based therapies.
Finally, the functional exploration of pathogen-specific bacteriophages and gene therapy depends on development of in vitro microbiome-mediated disease models that are not so easy to standardize (Fritz et al. 2013). Next, proof-of-concept for phage therapy approaches needs to be addressed in well-designed human clinical trials.
6 Conclusions and Perspectives
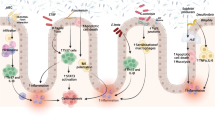
Metagenomic DNA sequencing of human microbiomes has revealed the high complexity and diversity of microbial communities living in various sites of the human body. These human–microbiota ecosystems are critical in health-promoting processes, and thus clinical studies have highlighted the unexpected and wide-ranging consequences of killing bacteria with antibiotics. Therefore, bacteriotherapy has been explored as a strategy to the steady-state control of bacterial community in disease-causing processes, although yet to be fully determined. The annotation and analyses of many human microbiomes have shown that absence of specific microbial species facilitate the breakdown of epithelial-immune cell barrier and dissemination of pathogenic species into the distant sites. Cultivated and uncultivated microbes may contribute to aetiology of human disorders. Emerging new biomarkers for better discriminatory phyla and species will contribute to diagnosis of type II diabetes, obesity, metabolic disorders, inflammatory bowel diseases and even certain cancers (Fig. 13.1).

The ecological community of commensal, symbiotic and pathogenic microorganisms play a central role in host metabolism, energy homeostasis and the innate and adaptive immune response. A shift in the gut microbial ecosystem (dysbiosis) results from a decrease in symbiont and/or an increase in pathobiont bacteria. The complex interaction between diet, age, host genetic and environmental factors (antibiotics and infections) and products released by gut microbiota such as SCFA, tyrosine, tryptophan and phenylalanine and their derivatives affect host signalling pathways and thus host metabolism and immunity. Increases in gut permeability and in the systemic levels of bacterial products such as LPS, a condition called metabolic endotoxemia, cause systemic inflammation. Low grade inflammation leads to impaired insulin action, insulin resistance, fat deposition and obesity and the metabolic syndrome. Pharmaceutical and nutraceutical agents targeting specific bacterial species or phylum/class may help to re-establish homeostasis and microbiome balance. SCFA, short-chain fatty acid, PAMP (Pathogen-Associated Molecular Patterns), DAMP (danger-associated molecular patterns and danger signals), AMPs (antimicrobial peptides)
The use of antibiotics compromises genome defence and increases bacteria’s ability to acquire antibiotic resistance. Prebiotics, probiotics, synbiotics, phage therapy and CRISPR/cas systems are emerging as tools to eradicate and modulate pathogenic bacterial species in microbial communities. Genetic engineering and synthetic biology offer alternative and complementary approaches to the production of quorum sensing natural products and their analogues. Engineering of multidrug resistant bacteria-specific bacteriophages will be possible and fully explored in near future. The use of novel pharmaceuticals and nutraceuticals to manage microbial colonization and development of healthy gut microbial community at early childhood will help the adult human body functions and prevent the occurrence of several pathologies.
Funding Statement
This work was supported by grants from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES/PNPD proc 2011/188-37/2011), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, proc 2011/486048) and Fundação de Amparo a Pesquisa do Estado de Sao Paulo (2012/02497-7, 2014/20847-0).
References
Abedon ST (2014) Phage therapy: eco-physiological pharmacology. Scientifica 214:581639
Abubucker S, Segata N, Goll J, Schubert AM, Izard J et al (2012) Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8(6):e1002358
Alivisatos AP, Blaser MJ, Brodie EL, Chun M, Dangl JL, Donohue TJ, Dorrestein PC, Gilbert JA, Green JL, Jansson JK, Knight R, Maxon ME, McFall-Ngai MJ, Miller MF, Pollard KS, Ruby EG, Taha SA, Unified Microbiome Initiative Consortium (2015) A unified initiative to harness Earth’s microbiomes. Science 350(6260):507–508
Almonacid DE, Kraal L, Ossandon FJ, Budovskaya YV, Cardenas JP, Bik EM, Goddard AD, Richman J, Zachary S, Apte ZS (2017) 16S rRNA gene sequencing and healthy reference ranges for 28 clinically relevant microbial taxa from the human gut microbiome. PLoS One 12(5):e0176555
Amgarten D, Martins LG, Lombardi KC, Antunes LP, Souza APS et al (2017) Three novel Pseudomonas phages isolated from composting provide insights into the evolution and diversity of tailed phages. BMC Genomics 18:346
Andersson DI, Hughes D (2014) Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol 12:465–478
Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, Wang GP (2013) Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol 51(9):2884–2892
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T et al (2011) Enterotypes of the human gut microbiome. Nature 473(7346):174–180
Backhed F, Fraser CM, Ringel Y, Sanders ME, Sartor B, Sherman PM, Versalovic J, Young V, Finlay BB (2012) Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe 12:611–622
Baumler AJ, Sparandio V (2016) Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535(7610):85–93
Belizario JE, Napolitano M (2015) Human microbiomes and their role in dysbiosis, common diseases and novel therapeutic approaches. Front Microbiol 6:1050
Bojanova DP, Bordenstein SR (2016) Faecal transplants: what is being transferred? PLoS Biol 13(7):e1002503
Brandt LJ, Reddy SS (2011) Faecal microbiota transplantation for recurrent Clostridium difficile infection. J Clin Gastroenterol 45(suppl):S159–S167
Brown EM, Sadarangani M, Finlay BB (2013a) The role of the immune system in governing host–microbe interactions in the intestine. Nat Immunol 14(2013):660–667
Brown CT, Sharon I, Thomas BC, Castelle CJ, Morowitz MJ, Banfield JF (2013b) Genome resolved analysis of a premature infant gut microbial community reveals a Varibaculum cambriense genome and a shift towards fermentation-based metabolism during the third week of life. Microbiome 1(1):30. https://doi.org/10.1186/2049-2618-1-30
Cani PD, Delzenne NM (2011) The gut microbiome as therapeutic target. Pharmacol Ther 130:202–212
Cani DP, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR, Delzenne NM (2007) Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50:2374–2383
Chang C, Lin H (2016) Dysbiosis in gastrointestinal disorders. Best Pract Res Clin Gastroenterol 30:3–15
Chatelier EL, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M et al (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500:541–546
Clemente JC, Ursell LK, Parfrey LW, Knight R (2013) The impact of the gut microbiota on human health: an integrative view. Cell 148(6):1258–1270
Costa FRC, Françozo MCS, Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, Ramos SG, Câmara NO, Zoete MR, Palm NW, Flavell RA, Silva JS, Carlos D (2016) Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med 213:1223–1239
Cucchiara S, Stronati L, Aloi M (2012) Interactions between intestinal microbiota and innate immune system in pediatric inflammatory bowel disease. J Clin Gastroenterol 46(Suppl):S64–S66
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA 107:14691–14696
Debelius J, Song SJ, Vazquez-Baeza Y, Xu ZZ, Gonzalez A, Knight R (2016) Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol 17:217
Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6:e280
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L et al (2005) Diversity of the human intestinal microbial flora. Science 308:1635–1638
Flintoft L (2012) Disease genomics: associations go metagenome-wide. Nat Rev Genet 13 (11):756–757. https://doi.org/10.1038/nrg3347
Forslund K, Sunagawa S, Kultima JR, Mende DR, Arumugam M, Typas A, Bork P (2013) Country-specific antibiotic use practices impact the human gut resistome. Genome Res 23(7):31–39
Fritz JV, Desai MS, Shah P, Schneider JG, Wilmes P (2013) From meta-omics to causality: experimental models for microbiome research. Microbiome 1:14
Fuchs A, Colonna A (2011) Natural killer (NK) and NK-like cells at mucosal epithelia: mediators of anti-microbial defense and maintenance of tissue integrity. Eur J Microbiol Immunol 1:257–266
Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R et al (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71
Gevers D, Po M, Schloss PD, Huttenhower C (2012) Bioinformatics for the human microbiome project. PLoS Comput Biol 8(11):e1002779
Gilbert JA, Quinn RA, Debelius J, Xu ZZ, Morton J, Garg N, Jansson JK, Dorrestein PC, Knight R (2016) Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 535:94–103
Gough N, Shaikh H, Manges AR (2011) Systematic review of intestinal microbiota transplantation (Faecal Bacteriotherapy) for recurrent Clostridium difficile infection. Clin Infect Dis 53(10):994–1002
Haeussler M, Concordet J-P (2016) Genome editing with CRISPR-Cas9: Can it get any better? J Genet Genomic 43:239–250
Hankir MK, Seyfried F, Hintschich CA, Diep TA, Kleberg K, Kranz M, Deuther-Conrad W et al (2017) Gastric bypass surgery recruits a gut PPAR-α-striatal D1R pathway to reduce fat appetite in obese rats. Cell Metab 25(2):335–344
Harley ITW, Karp CL (2012) Obesity and the gut microbiome: striving for causality. Mol Metab 1(1–2):21–31
Honda K, Littman DR (2016) The microbiota in adaptive immune homeostasis and disease. Nature 535:75–84
Hooper LV, Littman DR, Macpherson AJ (2012) Interactions between the microbiota and the immune system. Science 336:1268–1273
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA (2013) RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotech 31:233–239
Jorgensen I, Rayamajhi M, Miao EA (2017) Programmed cell death as a defence against infection. Nat Rev Immunol 17:151–164
Kootte RS, Vrieze A, Holleman F, Dallinga-Thie GM, Zoetendal EG, de Vos WM et al (2012) The therapeutic potential of manipulating gut microbiota in obesity and type2 diabetes mellitus. Diabetes Obes Metab 14:112–120
Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, Huttenhower C, Ley RE (2013) A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput Biol 9(1):e1002863
Kutter EM, Kuhl SJ, Abedon ST (2015) Re-establishing a place for phage therapy in western medicine. Future Microbiol 10:685–688
Lepage P, Leclerc MC, Joossens M, Mondot S, Blottière HM, Raes J, Ehrlich D, Doré J (2013) A metagenomic insight into our gut's microbiome. Gut 62(1):146–158
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023
Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI (2008) Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6(10):776–788
Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S et al (2014) An integrated catalogue of reference genes in the human gut microbiome. Nat Biotechnol 32:834–841
Marraffini LA, Sontheimer EJ (2008) CRISPR interference limits horizontal gene transfer in 623 Staphylococci by targeting DNA. Science 322(5909):1843–184
Maynard CL, Elson CO, Hatton RD, Weaver CT (2012) Reciprocal interactions of the intestinal microbiota and immune system. Nature 489:231–241
Meijer K, de Vos P, Priebe MG (2010) Butyrate and other short-chain fatty acids as modulators of immunity: what relevance for health? Curr Opin Clin Nutr Metab Care 13:715–721
Mekkes MC, Weenen TC, Brummer RJ, Claassen E (2014) The development of probiotic treatment in obesity: a review. Benef Microbes 5(1):19–28
Miller MB, Bassler BL (2001) Quorum sensing in bacteria. Annu Rev Microbiol 55:165–199
Moeller AH, Li Y, Mpoudi Ngole E, Ahuka-Mundeke S, Lonsdorf EV, Pusey AE, Peeters M, Hahn BH, Ochman H (2014) Rapid changes in the gut microbiome during human evolution. Proc Natl Acad Sci USA 111(46):16431–16435
O’Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF (2015) Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res 277:32–48
Ostaff MJ, Stange EF, Wehkamp J (2013) Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol Med 5:1465–1483
Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crécy-Lagard V et al (2005) The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33(17):5691–5702
Palmer DJ, Metcalfe J, Prescott SL (2012) Preventing disease in the 21st century: the importance of maternal and early infant diet and nutrition. J Allergy Clin Immunol 130(3):733–734
Pelfrene E, Willebrand E, Sanches AC, Sebris Z, Cavaleri M (2016) Bacteriophage therapy: a regulatory perspective. J Antimicrob Chemother 71:2071–2207
Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, Shulman GI (2016) Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 534(7606):213–217
Pornputtapong N, Nookaew I, Nielsen J (2015) Human metabolic atlas: an online resource for human metabolism. Database (Oxford) 2015:bav068. https://doi.org/10.1093/database/bav068
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65
Reijnders D, Goossens GH, Hermes GDA, Neis EPJG, van der Beek CM, Most J et al (2016) Effects of gut microbiota manipulation by antibiotics on host metabolism in obese humans: a randomized double-blind placebo-controlled trial. Cell Metabol 24:63–74
Rhoads DD, Wolcott RD, Kuskowski MA, Wolcott BM, Ward LS, Sulakvelidze A (2009) Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. J Wound Care 18(6):237–244
Roberfroid MB (2007) Prebiotics: the concept revisited. J Nutr 137(3):830s–837s
Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9(5):313–323
Rupnik M (2015) Toward a true bacteriotherapy for Clostridium difficile infection. N Engl J Med 372:1566–1568
Sala P, Belarmino G, Machado NM, Cardinell CS et al (2016) The SURMetaGIT study: design and rationale for a prospective pan-omics examination of the gastrointestional response to Roux-em-Y gastric bypass surgery. J Intl Med Res 44(6):1359–1375
Sampson TR, Saroj SD, Llewellyn AC, Tzeng YL, Weiss DS (2010) A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 479:254–257
Sangiuliano B, Perez M, Moreira D, Belizário J (2014) Cell death associated molecular-pattern molecules: inflammatory signalling and control. Mediators Inflamm 2014:249784
Seeley RJ, Chambers AP, Sandoval DA (2015) The role of gut adaptation in the potent effects of multiple bariatric surgeries on obesity and diabetes. Cell Metab 21:369–378
Selle K, Barrangou R (2015) Harnessing CRISPR-Cas systems for bacterial genome editing. Trends Microbiol 23:225–232
Sender R, Fuchs S, Milo R (2016) Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14(8):e1002533
Sulakvelidze A, Alavidze Z, Morris JC Jr (2001) Bacteriophage therapy. Antimicrob Agents Chemother 45(3):649–659
Suskind DL, Brittnacher MJ, Wahbeh G, Shaffer ML, Hayden HS, Qin X, Singh N, Damman CJ, Hager KR, Nielson H, Miller SI (2015) Faecal microbial transplant effect on clinical outcomes and faecal microbiome in active Crohn’s disease. Inflamm Bowel Dis 21(3):556–563
Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H (2005) Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol 43:3380–3389
Thaiss CA, Zmora N, Levy M, Elinav E (2016) The microbiome and innate immunity. Nature 535:65–74
The Human Microbiome Project Consortium, Hutternhower C, Gevers D, Knight R, Abubucker S, Badger JH et al (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214
Tremaroli V, Bäckhed F (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031
Ukena SN, Singh A, Dringenberg U, Engelhardt R, Seidler U et al (2007) Probiotic Escherichia coli Nissle 1917 inhibits leaky gut by enhancing mucosal integrity. PLoS One 2(12):e1308
Van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JB, Tijssen JG, Speelman P, Dijkgraaf MG, Keller JJ (2013) Duodenal infusion of donor faeces for recurrent Clostridium difficile. N Engl J Med 368:407–415
Vercoe RB, Chang JT, Dy RL, Taylor C, Gristwood T, Clulow JS et al (2013) Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet 9:e1003454
Verdam FJ, Fuentes S, de Jonge C, Zoetendal EG, Erbil R, Greve JW, Buurman WA, de Vos WM, Rensen SS (2013) Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) 21(12):E607–E615
Virgin HW (2014) The virome in mammalian physiology and disease. Cell 157(1):142–150
Vrieze A, van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in subjects with metabolic syndrome. Gastroenterology (4):913–916
Webb CR, Koboziev I, Furr KL, Grisham MB (2016) Protective and pro-inflammatory roles of intestinal bacteria. Pathophysiology 23:67–80
Whelan K, Quigley EM (2013) Probiotics in the management of irritable bowel syndrome and inflammatory bowel disease. Curr Opin Gastroenterol 29(2):184–189
Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdaka G (2009) Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 106(10):3698–3703
Yosef MM, Kiro R, Qimron U (2015) Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc Natl Acad Sci USA 112(23):7267–7272
Zhang Q, Rho M, Tang H, Doak TG, Ye Y (2013) CRISPR-Cas systems target a diverse collection of invasive mobile genetic elements in human microbiomes. Genome Biol 14:R40
Zhou Y, Mihindukulasuriya KA, Gao H, La Rosa P, Wylie KM, Martin JC et al (2014) Exploration of bacterial community classes in major human habitats. Genome Biol 15(5):R66
Acknowledgements
We thank Aline Maria da Silva, João Carlos Setubal, Dan Waitzberg and colleagues of University of Sao Paulo and Clinical Hospital of Medical School for insights and productive discussions.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Belizário, J.E., Faintuch, J. (2018). Microbiome and Gut Dysbiosis. In: Silvestre, R., Torrado, E. (eds) Metabolic Interaction in Infection. Experientia Supplementum, vol 109. Springer, Cham. https://doi.org/10.1007/978-3-319-74932-7_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-74932-7_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-74931-0
Online ISBN: 978-3-319-74932-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)