
Abstract
Type 2 diabetes mellitus (T2DM) should be understood as the final common pathway that results from an imbalance between increased insulin resistance (i.e., decreased insulin action) and relative insulinopenia (i.e., impaired insulin secretion). There is consensus that for most patients with T2DM the earliest defect observed is insulin resistance. At early stages, the pancreas is able to compensate for insulin resistance by increasing insulin secretion so that glucose tolerance is preserved. Thus, a state of normal glucose tolerance is maintained at the expense of hyperinsulinemia. Over time, and by mechanisms that are still incompletely understood, pancreatic β-cells fail to maintain this high rate of insulin secretion and impaired glucose tolerance and overt T2DM develop. In the current chapter, the mechanisms leading to insulin resistance and the spectrum of therapeutic approaches for patients with T2DM will be described.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Type 2 diabetes mellitus (T2DM) should be understood as the final common pathway that results from an imbalance between increased insulin resistance (i.e., decreased insulin action) and relative insulinopenia (i.e., impaired insulin secretion) [1].
There is consensus that for most patients with T2DM the earliest defect observed is insulin resistance . For many patients this is genetically determined, but a common acquired factor that worsens insulin resistance is obesity [2]. However, the relative contribution of acquired versus genetic factors is unclear and varies from patient to patient. At early stages, the pancreas is able to compensate for insulin resistance by increasing insulin secretion so that glucose tolerance is preserved. Thus, a state of normal glucose tolerance is maintained at the expense of hyperinsulinemia . Over time, and by mechanisms that are still incompletely understood, pancreatic β-cells fail to maintain this high rate of insulin secretion, and impaired glucose tolerance and overt T2DM develop [3].
In the current chapter, the mechanisms leading to insulin resistance and the spectrum of therapeutic approaches for patients with T2DM will be described.
Causes of Insulin Resistance: Acquired vs. Genetic
The interplay between acquired and genetic defects in the development and promotion of insulin resistance is complex and incompletely understood [4,5,6]. Moreover, the relative contribution of each of these factors varies from case to case, and ranges from monogenic insulin resistance syndromes to fully-acquired cases in the setting of obesity.
Monogenic syndromes of insulin resistance are rare , but they have been consistently reported in the literature in the last few decades [6]. Over 15 different culprit genes have been identified including insulin receptor, peroxisome proliferator-activated receptor-γ (PPAR-γ), pericentrin, perilipin, protein kinase B (Akt2), etc. Of note, most single-gene causes of insulin resistance do not affect insulin signaling pathways, but adipose tissue function, inducing insulin resistance only as a consequence of the d ysfunctional adipose tissue. Other responsible genes identified are involved in DNA repair, but their link to severe insulin resistance has not been fully elucidated [6]. These genetic syndromes range from infantile fatal disease to mild insulin resistance in later life, and have been reviewed in depth elsewhere [7, 8].
As for the acquired defects, obesity has received significant a ttention, as it is commonly associated with insulin resistance [4, 5]. However, teasing out the contribution of genetic factors from that directly attributed to obesity has been difficult. Moreover, the severity of obesity does not always correlate with the severity of insulin resistance, implying that there are other mechanisms that regulate the degree of obesity-related insulin resistance. Accumulating evidence suggests that the initiating event of insulin resistance may not be the presence of obesity itself, but the presence of dysfunctional adipose tissue [4, 5]. This concept helps to explain why non-obese individuals with a family history of T2DM (i.e., with a genetic background) are insulin resistant long before the development of obesity [9]. Based on the same principles, the development of obesity (even in the absence of a genetic background) results in adipose tissue insulin resistance due to a distinctive fat distribution (favoring visceral accumulation, in contrast of subcutaneous adipose tissue), but most importantly, due to the development of ectopic accumulation of fat in insulin sensitive tissues, such as the liver and skeletal muscle [4, 5]. Other common causes of insulin resistance include some medications and are described in Table 3.1 [8, 10].
Physiology of Insulin Resistance
Adipose Tissue
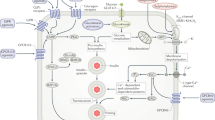
As mentioned above, adipose tissue is likely the tissue where insulin resistance begins [5]. By molecular mechanisms that are beyond the scope of this article and that have been reviewed elsewhere [5, 11, 12], adipose tissue can become insulin resistant in the setting of obesity and/or genetic predisposition. As a consequence of this, hormone-sensitive lipase fails to be inhibited by insulin, which results in increased rates of lipolysis. This, in turn, results in an oversecretion of free fatty acids (FFA) into the circulation, where they can reach other organs promoting lipotoxicity [5]. The term “lipotoxicity ” was originally introduced by Unger [13] to describe the harmful effects of increased FFA levels on β-cell function. However, since its original use, the term lipotoxicity has attained a much broader meaning, and it is now applied to any del eterious effects of FFAs on tissues that would not normally be destined to store large amounts of lipids, such as the liver [14, 15].
In addition to FFA oversecretion, insulin-resistant adipose tissue is also characterized by a pro-inflammatory phenotype [5, 12]. Adipocytokines , such as tumor necrosis factor (TNF)-α and interleukin (IL)-6 are secreted by dysfunctional adipocytes and may contribute to insulin resistance in an autocrine, paracrine, and endo crine fashion. This consolidates a “closed loop” in which insulin resistance promotes inflammation, and inflammation reinforces insulin resistance. Reduced levels of beneficial molecules, such as adiponectin , have also been described in obesity and insulin-resistant states [16]. Moreover, there exists a closed cross-talk between adipocytes and macrophages in the adipose tissue, further expanding the systemi c impact of adipose tissue-derived inflammation [11, 12].
Liver
The liver receives FFAs from three diff erent sources: adipose tissue lipolysis, diet, and de novo lipogenesis [14, 17]. Of these, adipose tissue lipolysis is the most important one, contributing with approximately ~60% of all FFAs in normal conditions. In the setting of adipose tissue insulin resistance (increased lipolysis, as described above), the flux of FFA to the liver increases. When FFA supply surpasses the metabolic needs of the organ, the liver begins to accumulate them as triglycerides, which results in hepatic steatosis [18]. In addition, increased hepatic FFA oxidation leads to an incomplete oxidation, with the generation of lip id intermediates (e.g., ceramides and diacylglycerols) and reactive oxygen species (ROS) that promote hepatic insulin resistance and inflammation [18].
Hepatic insulin resistance translates into increased rates of hepatic glucose production (HGP) and of very low density lipoprotein (VLDL) secretion, as insulin is u nable to suppress them as under normal conditions [19, 20]. In turn, increased HGP promotes a compensatory hyperinsulinemia in order to maintain normal plasma glucose levels. However, such hyperinsulinemia turns to be deleterious as it increases the rate of intracellular de novo lipogenesis (DNL), further contributing to hepatocyte triglyceride accumulation.
Nonalcoholic fatty liver disease , defined as an intracellular triglyceride accumulation greater than 5.5% in the absence of any secondary cause of steatosis (e.g., alcohol, drugs, viral hepatitis, autoimmune hep atitis, etc.), is increasingly common in patients with obesity and/or T2DM [17, 21,22,23]. It is closely linked to insulin resistance, and in some patients, it can progress to its more severe form known as nonalcoholic steatohepatitis (NASH) , characterized by the presence of hepatic steatosis combined with inflammation, necrosis, and /or fibrosis [21, 22]. In the absence of treatment, this liver condition may progress to cirrhosis and he patocellular carcinoma [21, 22].
Skeletal Muscle
In the setting of this “lipotoxic” environment prom oted by insulin resistance, skeletal muscle insulin resistance also develops, resulting in impaired insulin-stimulated muscle glucose uptake. Several factors contribute to the development of insulin resistance in this tissue. For instance, increased plasma FFA levels promote intramyocellular steatosis and impaired insulin signaling. This is observed among healthy lean individuals, in a dose-dependent manner, when plasma FFA are experimentally increased during an intravenous lipid infusion [24]. This also occurs within 24–48 h after plasma FFA are just slightly increased experimentally to achieve plasma FFA levels typically observed in obesity [25, 26]. However, intramyocellular triglyceride accumulation per se appears not to play a role in the development of insulin resistance. Human studies report that athletes paradoxically have a high triglyceride content despite their normal (or above normal) insulin sensitivity [27]. Thus, the current hypothesis is that insulin resistance-related steatosis is characterized by accumulation of toxic lipid metabolites and pro-inflammatory lipid intermediates that im pair insulin signaling and are responsible for insulin resistance [27].
Chronic hyperinsulinemia , secondary to increased hepatic glucose production, can also promote insulin resistance by downregulating the number of insulin receptors and their downstream signaling steps. An approximately 2 to 3-fold increase in plasma insulin levels causes skeletal muscle insulin resistance after a 72-hour insulin infusion in otherwise insulin-sensitive individuals [28]. The combination of elevated FFA and hyperinsulinemia is the perfect storm for skeletal muscle insulin resistance, which in turn contributes to glucose intolerance.
Pancreatic ß-Cells
In order to keep plasma glucose levels in the normal range in the setting of insulin resistance, a compensatory hyperinsulinemia is required, which results in a demanding burden to the pancreatic ß-cells [29]. While ß-cells can keep up with the workload, glucose tolerance remains within the normal range. However, when this compensation fails, hyperglycemia develops. However, subtle defects in ß-cell function can be detected long before the development of frank hyperglycemia [29]. The underlying mechanisms responsible for this relentless decline in ß-cell function over time are incompletely understood. However, basic and clinical evidence suggests that hyp erglycemia (i.e., glucotoxicity) and chronic elevated plasma FFAs (i.e., lipotoxicity) play key roles in this progression [30,31,32].
In the setting of obesity and insulin resis tance, ß-cells are forced to manage the FFA oversupply. In ideal conditions, this chronic increase of plasma FFA should enhance basal and glucose-stimulated insulin secretion. However, in predisposed patients (e.g., family history of T2DM) increased plasma FFA produces the opposite effect, inducing insulin secretion impairment and favoring the progression to T2DM [31]. These patients appear to have a genetically-determined reduced ß-cell adaptation to excess FFA supply. Once hyperglycemia develops, this generates a positive feedback, where hyperglycemia impairs ß-cell function, further perpetuating the increased plasma glucose levels [30].
Management of T2DM
The management of T2DM requires a multidisciplinary approach, focused on lifestyle modifications, as well as diabetes self-management education and support [33]. At center stage of this approach are nutrition therapy, physical activity, and smoking cessation counseling [33]. Health care providers should focus on how to optimize lifestyle in every patient with T2DM, as modest weight reductions have been shown to improve glycemic control and reduce the need for glucose-lowering medications [34]. Relatively modest reductions of body weight (of approximately 5%) are frequently enough to observe beneficial effects in glycemic control, although weight loss ≥7% are optimal and associated with further benefits on blood pressure, lipids, and NASH [33, 35].
While hypocaloric diets (~500–750 kcal/day energy deficit) should be strongly encouraged in patients with T2DM in order to achieve the desired weight reduction, diet composition appears to be of lesser importance [33]. A variety of different diets have proven to be effective, as long as total calorie intake targets are kept in mind. Therefore, diets should be individualized to patients’ preferences and needs. Foods higher in fiber and lower in glycemic load and added sugars should be emphasized (whole grains, vegetables, fruits, legumes, low-fat dairy, nuts, and seeds). Regarding physical activity, the American Diabetes Association recommends for most adults with T2DM at least 150 min of moderate-to-vigorous intensity aerobic physical activity per week. In addition, adults with T2DM should engage in 2–3 sessions/week of resistance exercise on nonconsecutive days [33].
Nevertheless, reaching sufficient weight loss, and especially sustaining such reduction over time is challenging. Even in intensive intervention groups during well-controlled clinical trials (i.e., Look AHEAD) weight loss was only 4.7% after 8 years of follow-up [34]. Weight loss medications (e.g., orlistat, lorcaserin, phentermine/topiramate ER, naltrexone/bupropion, liraglutide 3 mg) can be used as adjunctive therapy in patients with BMI ≥ 27 kg/m2. In addition, metabolic surgery should be considered in patients with a BMI ≥ 40 kg/m2 or ≥35 kg/m2 if hyperglycemia is not adequately controlled despite optimal therapy [33]. Several review articles have addressed the benefits and risks of metabolic surgery in the management of T2DM [36, 37].
Due to the limitations of lifestyle modification to achieve hemoglobin A1c targets (<7% for most adults, but potentially <6.5% or <8% for selected individuals based on risk of hypoglycemia, life expectancy, comorbidities, etc.), pharmacologic treatment should be considered early-on in patients with T2DM [33]. In recent years, we have witnessed an exponential increment in our pharmacological options, with several new drug groups with distinctive mechanisms of action.
Briefly, glucose-lowering agents can be divided based on their mechanisms of action in the following groups: (a) drugs that mainly improve insulin resistance; (b) drugs that mainly improve insulin secretion; (c) drugs with an incretin-mimetic effect; and finally, (d) drugs that are glucose-lowering agents by inducing glycosuria. In this section, we will describe the mechanisms of action, best indications, side effects, and special considerations of each pharmacological agent.
Insulin Sensitizers
Metformin
Metformin is a biguanide that has been available worldwide for the prevention and treatment of T2DM for over 50 years. Its exact mechanism of action remains incompletely understood, although it is clear that it decreases hepatic gluconeogenesis and improves insulin sensitivity at the level of the liver, and to a lesser extent, skeletal muscle [38]. There are several proposed molecular mechanisms for this drug: inhibition of mitochondrial complex I, leading to a reduction in ATP synthesis with the consequent increase in AMP and AMP kinase activation; delayed glucose absorption in the gastrointestinal tract, inhibition of glucagon signaling and gluconeogenic enzymes, glucagon-like peptide (GLP)-1 secretion, and others [38]. Nevertheless, it is debatable whether these actions occur at physiological concentrations or not. A recent study suggested that at the regular doses used in humans, metformin inhibited mitoc hondrial glycerophosphate dehydrogenase in rats, resulting in a reduction in the contribution of both glycerol and lactate to hepatic gluconeogenesis [39].
Given its proven efficacy, overall safety, and cardiovascular benefits, metformin has consolidated over time as the first line of therapy for patients with T2DM and is used at doses that range from 500 to 2000 mg daily [33]. It can be safely used in patients with an eGFR ≥30 mL/min/1.73 m2, but patients should be advised to stop the medication in cases of nausea, vomiting, dehydration, or before a contrast-enhanced computed tomography study. Metformin has also been associated with vitamin B12 deficiency, and therefore, periodic testing of vitamin B12 levels is now recommended [33]. It is usually well tolerated, although a minority may suffer metformin-associated gastrointestinal side effects. Most patients usually develop tolerance to these side effects and slow titration should be always recommended to help to avoid them. Only ~5% of patients are unable to tolerate metformin due to GI side effects. Other side effects, such as lactic acidosis and skin rashes are extremely rare [33].
Thiazolidinediones (TZDs)
Currently, only two available drugs are included in this group: pioglitazone and rosiglitazone. While they both share the same main mechanism of action (i.e., peroxisome proliferator-activated receptor [PPAR]-γ agonism), they have important distinctive effects on lipid metabolism, as well as in the liver and cardiovascular system in humans [5, 17, 40, 41]. By activating PPAR-γ, these molecules improve insulin sensitivity mainly at the level of the adipose tissue, which leads to a reduction in adipocyte triglyceride breakdown (lipolysis) and in plasma FFA levels [42]. This, in turn, results in a reduction of lipotoxicity in other tissues, with the consequent improvements in skeletal muscle and liver insulin sensitivity [43].
In 2007 it was claimed that rosiglitazone could potentially be associated with myocardial infarction and increased cardiovascular disease [44]. However, while the fear among primary care physicians remains, the FDA actually removed that black box warning some years ago, after finding that there was no evidence for that association. Moreover, several studies have shown that pioglitazone actually reduces the progression of cardiovascular disease and cardiovascular events in patients with and without T2DM [45,46,47,48].
Of particular interest, pioglitazone has consistently shown to improve hepatic steatosis and NASH in patients with and without T2DM [43, 49]. This is important as ~70% of patients with T2DM are believed to have nonalcoholic fatty liver disease, of whom ~50% may have the more severe form with inflammation and necrosis (i.e., NASH) [23]. In patients with T2DM and NASH, current guidelines suggest that pioglitazone should be strongly considered early-on after metformin [21, 22]. This has also been embraced by the American Diabetes Association 2018 guidelines for patients with T2DM and NASH. Unlike pioglitazone, rosiglitazone did not show any significant liver histological benefit in patients with NASH [50], suggesting that pioglitazone may have additional mechanisms of action to improve liver histology other than the currently known classical PPAR-γ pathways.
Thiazolidinediones’ benefits (glycemic control, resolution of NASH, reduction of cardiovascular disease, and improvement of polycystic ovarian syndrome) should be weighed against the potential risks of this group of drugs. Fluid retention and peripheral edema may occur in ~7–10% of patients on TZDs, and this percentage increases when combined with insulin [42]. Several mechanisms are likely to contribute to this side effect, being enhanced renal water and sodium reabsorption probably the most important one. Thiazolidinediones can also increase heart failure symptoms in patients with pre-existing disease, and therefore, are contraindicated in patients with known heart failure. They also produce weight gain (in the range of 1–5 kg with chronic use), mild anemia, and a slight increase in bone fractures probably due to a reduction in bone mineral density. In 2011, pioglitazone was associated with bladder cancer [51]. Since then, several studies have tried to replicate those results in retrospective and prospective studies with varying results [52,53,54]. A recent meta-analysis has reported that out of the 23 epidemiological studies published to date, 18 showed no association between bladder cancer and pioglitazone, while those that have reported an association were not confirmed in the same population in subsequent analysis, or either had a significant detection bias or patients on the TZD had significantly more risk factors for bladder cancer than the comparison group [55].
Insulin, Insulin Analogues, and Insulin Secretagogues
Insulin and Insulin Analogs
It is beyond the scope of this review to describe the pharmacodynamic and pharmacokinetic properties of the different insulin preparations, the most appropriate insulin regimen for patients with T2DM, as well as the practical issues to starting or adjusting insulin dosing. We refer the reader to dedicated articles where they have been reviewed in-depth [56, 57].
Insulin analogs can be classified based on their distinctive half-lives: (a) rapid-acting analogs (e.g., aspart, lispro, glulisine, and inhaled insulin); (b) short-acting insulin (e.g., regular); (c) intermediate-acting insulin (e.g., human NPH); (d) long-acting analogs (e.g., glargine, detemir, and degludec); (e) concentrated insulins (e.g., glargine U-300, degludec U-200, NPH U-500) and (f) premixed insulin products (e.g., NPH/regular 70/30, 70/30 aspart mix, 75/25 lispro mix, 50/50 lispro mix). For most patients with T2DM, basal insulin alone is the most convenient initial insulin regimen, beginning with 10–20 IU daily or 0.1–0.3 IU/kg daily. However, many of these patients will require mealtime bolus insulin dosing with disease progression [33]. Rapid-acting analogs are preferred for bolus dosing due to their faster onset of action.
The most common side effect of insulin is hypoglycemia, and special care should be paid to try to avoid them as much as possible, to reduce the risk of cognitive decline and other important deleterious outcomes. Another common side effect of insulin therapy are weight gain and peripheral edema. Less frequent side effects include: self-limited blurred vision (usually at the beginning of therapy, it is likely the result of a disbalance in the osmotic equilibrium between the lens and ocular fluids) or electrolyte disturbances most commonly observed during treatment of an acute decompensation as in diabetic ketoacidosis (e.g., hypokalemia, hypomagnesemia, and/or hypophosphatemia). Dermatologic reactions to insulin can result in lipohypertrophy (as insulin is lipogenic) or lipoatrophy (probably immu nologically mediated). The frequency of lipoatrophy has significantly reduced since the introduction of biosynthetic human insulin, and can also be reduced by alternating the injection site. Less than 1% of patients may present with hypersensitivity a t injection sites, with inflammation and/or subcutaneous nodules [33].
Sulfonylureas
The main mechanism of sulfonylureas is to increase insulin secretion by binding to the sulfonylurea receptor (SUR1). They act by inhibiting the ATP-dependent K+ channel on pancreatic ß-cells [58]. As a consequence of this, ß-cells become depolarized, which produces an influx of calcium into the cytosol and insulin exocytosis. Of note, sulfonylureas induce insulin secretion independently of glucose levels, and they can therefore increase the risk of hypoglycemia [58].
They should be taken 30 min before meals and the typical starting dose should be low and up-titrated every 2 weeks if glycemic control has not been reached [33]. They should be given once or twice per day as they have a prolonged biological effect that lasts longer than their plasma half-life due to receptor interaction and active metabolites. Shorter-duration sulfonylureas, such as glipizide, are preferred due to lower risk of hypoglycemia [33].
While they are usually well tolerated, they still have significant side effects that should be considered. As mentioned above, the most common side effect is hypoglycemia and these episodes can be even more frequent and serious in the elderly, undernourished, in the setting of alcohol abuse, or after exercise or a missed meal. They also produce significant weight gain. Other less common side effects include skin reactions such as erythema multiforme, exfoliative dermatitis, and photosensitivity. Abnormal liver function tests have also been observed. The most important deleterious aspect is their potential for increasing the risk of acute myocardial infarction, stroke, or death, compared with metformin or other agents [58,59,60].
The only advantage of this class of oral agents is their low cost. However, their risk of causing hypoglycemia is high, which combined with their potential to increase cardiovascular disease, makes them the least desirable option for the management of patients with T2DM.
Meglitinides
Meglitinides (repaglinide and nateglinide) have a similar mechanism of action as sulfonylureas, but they use a different pancreatic ß-cell receptor. They are also structurally different, and therefore they can be used despite sulfonylurea allergy. They have a rapid onset of action and short half-life. Like sulfonylureas, they should be administered with meals. Side effects include weight gain and hypoglycemia. Repaglinide is mainly metabolized in the liver, therefore it can be used safely in patients with chronic kidney disease [61]. However, the fact that they must be given with each meal (making adherence challenging), and that other agents may be more effective to lower postprandial glucose levels, has largely relegated this class of agents to an infrequent use.
Incretin-Mimetics
Glucagon-Like Peptide (GLP)-1 Receptor Agonists (GLP-1RAs)
The incretin effect is responsible for 50–70% of total insulin secretion after oral glucose administration, but it is absent if intravenous glucose is administered. One of such incretin hormones is GLP-1, which has a short half-life because it is quickly inactivated by dipeptidyl peptidase (DDP)-4. In addition to increasing insulin secretion in a glucose-dependent manner, GLP-1RAs also suppress elevated glucagon levels, delay gastric emptying, suppress appetite, and induce weight loss [33]. Beyond their beneficial effects in glycemic controls, some members of this class, such as liraglutide [62] and semaglutide [63], have shown to reduce cardiovascular events, cardiovascular mortality, and even overall mortality. Similar results have not been reported with other GLP-1 agonists, such as lixisenatide [64] or long-acting once weekly exenatide [65], and therefore it remains unclear whether differences are attributable to the different populations studied or really unique properties of some GLP-1RAs in the class.
Glucagon-like peptide-1 agonists have become widely used in patients with T2DM and their metabolic effects have been quite consistent across studies. They are administered by subcutaneous injections and can be classified based on their half-lives in short-acting (exenatide twice daily [BID], lixisenatide once daily [QD]) or long-acting (liraglutide QD, albiglutide once weekly [QW], dulaglutide QW, semaglutide QW, and exenatide long-acting release QW) [33]. Among their side effects, nausea, vomiting, and diarrhea are probably the most common ones, but patients usually develop tolerance to them with chronic use. However, they are contraindicated in patients with gastroparesis. The risk of hypoglycemia is low as insulin secretion is stimulated only if the plasma glucose levels are elevated [62]. These medications have been found to slightly increase heart rate, most likely as a reflex secondary to blood pressure reduction [33]. Injection site reactions, such as rash, erythema, or pruritus are frequent with these drugs. Patients may develop antibodies against the GLP-1 a gonists, but the clinical significance of such antibodies is unclear (although more injection site reactions are observed in patients with positive antibodies). In most cases, they appear not to affect efficacy of the drugs [33]. There has been some concern regarding the association of this group of drugs with pancreatitis, pancreatic cancer, and medullar thyroid carcinoma. However, studies have shown conflicting results regarding these associations, and therefore more research is needed before final conclusions can be drawn. A recent meta-analysis that included 113 randomized controlled trials did not find an increased incidence of pancreatitis or of pancreatic cancer with GLP1-RA therapy versus comparator arms, but there was a small but significant greater risk of cholelithiasis (OR [95% CI] 1.30 [1.01–1.68], P = 0.041) [66]. Of note, there have also been reports of GLP-1RAs resulting in significant improvements in patients with psoriasis [67]. While this awaits further confirmation in larger studies, a potential explanation for this effect comes from studies showing an overexpression of GLP-1 receptors in psoriatic plaques likely due to infiltration with immune cells [68].
Dipeptidyl Peptidase (DDP)-4 Inhibitors
In the United States, this class of drugs includes saxagliptin, sitagliptin, linagliptin, and alogliptin. They inhibit DPP-4, the enzyme responsible for the degradation of GLP-1 (among other peptides), potentiating endogenous GLP-1 action on pancreatic β-cells. However, they are likely to have a broader spectrum of effects that are just now being better understood, as reviewed elsewhere [69]. While they share the incretin effect with GLP-1 agonists, they have several important differences with that drug group.
This class of oral medications leads to a modest improvement in hemoglobin A1c (~0.7% compared to decreases between 1.0 and 1.5% with other classes of oral agents or GLP-1RA) [33]. As DPP-4 is an enzyme expressed on most cell types and deactivates many different bioactive peptides in addition to GLP-1, its inhibition probably affects plasma glucose by different mechanisms [69]. They only show a modest effect on plasma GLP-1 levels when compared to GLP-1RAs. Unlike GLP-1RAs , they do not produce significant weight loss, and although they have proven to be safe from a cardiovascular standpoint, they have not shown any cardiovascular improvement like some GLP-1RAs [70, 71]. Moreover, in a large randomized controlled trial with saxagliptin, there was a small but significant increase in hospitalizations for heart failure [71]. This has not been found with other members of this class and it remains unclear if it is a real effect or not, as some observational studies have failed to observe such effect on heart failure with saxagliptin [33, 69].
This group of drugs have been usually very well tolerated and they have a low risk of hypoglycemia. They are popular among clinicians given their good safety profile, relative low-cost and that they all have been combined in a tablet formulation with metformin to improve patient adherence. The most commonly reported side effects are headache, nasopharyngitis, and upper respiratory tract infection [33]. While acute pancreatitis has been reported with these drugs, and there is some concern for pancreatic cancer, it is still unknown whether there is a causal relationship [33, 69, 72]. There have been several reports of skin lesions with these drugs, including hypersensitivity reactions, angioedema, and blistering skin conditions (e.g., Stevens–Johnson syndrome). Of note, use of a DPP-4 inhibitor in combination with an ACE inhibitor may further increase the risk of angioedema due to prolongation of bradykinin and substance P half-lives [33]. Linagliptin is the only member of the group that is primarily eliminated via the enterohepatic system, and therefore, it does not require dose adjustment for chronic kidney disease as the other drugs of the group.
Sodium-Glucose Cotransporter (SGLT)-2 Inhibitors
This group of drugs is composed of canagliflozin, dapagliflozin, and empagliflozin [33]. Their mechanism of action is inhibition of SGLT-2 transporters in the renal proximal tubule, promoting urinary glucose excretion. Their glucose-lowering effect therefore depends on the filtered glucose load (i.e., baseline hyperglycemia) [33].
In addition to their glucose-lowering effects, these drugs have shown to have a number of other beneficial metabolic effects. Both empagliflozin and canagliflozin have shown to reduce the composite primary outcome of death from cardiovascular causes, nonfatal myocardial infarction, or nonfatal stroke in large randomized controlled trials [73, 74]. Moreover, they both significantly decreased heart failure hospitalization compared to placebo. While empagliflozin [73], but not canagliflozin [74], significantly reduced overall cardiovascular mortality, both had similar mean reductions despite different confident intervals [73, 74]. Additional benefits of these drugs are to reduce blood pressure, delay the progression of microvascular disease, and produce modest weight loss. These benefits occur despite a small increase in plasma LDL-C [73, 74].
As these drugs produce an osmotic diuresis, it is important to pay attention to the volume status of patients when starting these drugs and during follow-up [33]. They have been associated with orthostatic hypotension, dehydration, and/or acute kidney injury. This is even more frequent in elderly patients or those concomitantly taking diuretics, ACE inhibitors or angiotensin receptor blockers (ARB) [75]. Potential hyperkalemia has also been observed. Because of this, use in elderly populations should be done with care [33]. Patients taking these medications are at increased risk of urinary tract infections (especially in women), acute balanitis or balanoposthitis, and vulvovaginal candida infections [75]. Among other less frequent, but potentially more serious adverse events, there may be an increase risk of bone fractures, especially in patients taking canagliflozin [75]. While the mechanism remains elusive, it is suspected that increased falls (due to volume depletion) may play an important role in this increase. However, reduced bone mineral density has also been reported [76]. Euglycemic diabetes ketoacidosis has also been reported in patients with T2DM on SGLT-2 inhibitors. This occurs mainly in patients with type 1 diabetes mellitus or T2DM concomitantly using insulin, after decreasing insulin doses due to better control when a SGLT-2 inhibitor is added or after prolonged fasting [77]. Canagliflozin has also been associated with increased amputation risk [74]. Patients on canagliflozin with a previous amputation, peripheral vascular disease, and/or neuropathy were at highest risk for amputation. At the current time, it is unclear whether other members of this drug class can also lead to amputations. Dapagliflozin has been associated with bladder cancer, but it is unknown whether this is a causal association [78].
Conclusions
Hyperglycemia in T2DM develops as pancreatic β-cells fail to meet the demands of chronic insulin resistance and acquired insults, such as obesity, in genetically predisposed subjects. As more drugs become available for the treatment of T2DM, health care providers are more frequently faced with the burden of making decisions regarding the most appropriate medication for each patient. Due to its safety profile, efficacy, low cost, cardiovascular benefits and long-term clinical experience, metformin has consolidated as the first-line therapy for T2DM. The question remains what the best second line of therapy is. The results from recent large randomized controlled trials of diabetes medications showing cardiovascular risk reduction will make some GLP-1RAs (liraglutide, semaglutide) and SGLT2 inhibitors (empagliflozin, canagliflozin) second-line therapy for patients with T2DM and proven cardiovascular disease. In patients with T2DM and NASH, pioglitazone will become the agent of choice given its benefit in this population. Ultimately, physicians must individualize care to the unique medical and social situation of each patient. However, optimizing therapy is a challenge when we do not routinely measure insulin resistance or insulin secretion in our patients when we choose treatment. Doctors would probably benefit from understanding in each patient what is their predominant metabolic defect leading to hyperglycemia. Unfortunately, we basically choose treatment based on their cost, safety profile, and physicians’/patients’ personal preferences. A more targeted treatment approach will likely be beneficial in the future. The choices mentioned above based on liver or cardiovascular disease are a beginning. Clearly, more work is needed to better tailor treatment in the future.
References
DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers. 2015;1:15019.
Cusi K. The epidemic of type 2 diabetes mellitus: its links to obesity, insulin resistance, and lipotoxicity. In: Regensteiner JG, Reusch JEB, Stewart KJ, Veves A, editors. Diabetes and exercise. 1st ed. New York: Humana Press; 2009. p. 3–54.
Cusi K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr Diab Rep. 2010;10:306–15.
Caprio S, Perry R, Kursawe R. Adolescent obesity and insulin resistance: roles of ectopic fat accumulation and adipose inflammation. Gastroenterology. 2017;152:1638–46.
Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142:711–25. e716
Parker VE, Semple RK. Genetics in endocrinology: genetic forms of severe insulin resistance: what endocrinologists should know. Eur J Endocrinol. 2013;169:R71–80.
Semple RK, Savage DB, Cochran EK, Gorden P, O’Rahilly S. Genetic syndromes of severe insulin resistance. Endocr Rev. 2011;32:498–514.
Semple RK. EJE PRIZE 2015: how does insulin resistance arise, and how does it cause disease? Human genetic lessons. Eur J Endocrinol. 2016;174:R209–23.
Cusi K. Lessons learned from studying families genetically predisposed to type 2 diabetes mellitus. Curr Diab Rep. 2009;9:200–7.
Repaske DR. Medication-induced diabetes mellitus. Pediatr Diabetes. 2016;17:392–7.
Asghar A, Sheikh N. Role of immune cells in obesity induced low grade inflammation and insulin resistance. Cell Immunol. 2017;315:18–26.
Sell H, Habich C, Eckel J. Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol. 2012;8:709–16.
Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes. 1995;44:863–70.
Bril F, Lomonaco R, Cusi K. The challenge of managing dyslipidemia in patients with nonalcoholic fatty liver disease. Clin Lipidol. 2012;7:471–81.
Lomonaco R, Ortiz-Lopez C, Orsak B, Webb A, Hardies J, Darland C, et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology. 2012;55:1389–97.
Unger RH, Scherer PE, Holland WL. Dichotomous roles of leptin and adiponectin as enforcers against lipotoxicity during feast and famine. Mol Biol Cell. 2013;24:3011–5.
Bril F, Cusi K. Nonalcoholic fatty liver disease: the new complication of type 2 diabetes mellitus. Endocrinol Metab Clin N Am. 2016;45:765–81.
Sunny NE, Bril F, Cusi K. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol Metab. 2017;28:250–60.
Bril F, Barb D, Portillo-Sanchez P, Biernacki D, Lomonaco R, Suman A, et al. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology. 2017;65:1132–44.
Bril F, Sninsky JJ, Baca AM, Superko HR, Portillo Sanchez P, Biernacki D, et al. Hepatic steatosis and insulin resistance, but not steatohepatitis, promote atherogenic dyslipidemia in NAFLD. J Clin Endocrinol Metab. 2016;101:644–52.
Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2017. https://doi.org/10.1002/hep.29367.
European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. Diabetologia. 2016;59:1121–40.
Bril F, Cusi K. Management of nonalcoholic fatty liver disease in patients with type 2 diabetes: a call to action. Diabetes Care. 2017;40:419–30.
Belfort R, Mandarino L, Kashyap S, Wirfel K, Pratipanawatr T, Berria R, et al. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes. 2005;54:1640–8.
Kashyap SR, Belfort R, Berria R, Suraamornkul S, Pratipranawatr T, Finlayson J, et al. Discordant effects of a chronic physiological increase in plasma FFA on insulin signaling in healthy subjects with or without a family history of type 2 diabetes. Am J Physiol Endocrinol Metab. 2004;287:E537–46.
Kashyap S, Belfort R, Cersosimo E, Lee S, Cusi K. Chronic low-dose lipid infusion in healthy subjects induces markers of endothelial activation independent of its metabolic effects. J Cardiometab Syndr. 2008;3:141–6.
Amati F, Dube JJ, Alvarez-Carnero E, Edreira MM, Chomentowski P, Coen PM, et al. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes. 2011;60:2588–97.
Iozzo P, Pratipanawatr T, Pijl H, Vogt C, Kumar V, Pipek R, et al. Physiological hyperinsulinemia impairs insulin-stimulated glycogen synthase activity and glycogen synthesis. Am J Physiol Endocrinol Metab. 2001;280:E712–9.
Abdul-Ghani MA, Tripathy D, DeFronzo RA. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 2006;29:1130–9.
Del Prato S, Leonetti F, Simonson DC, Sheehan P, Matsuda M, DeFronzo RA. Effect of sustained physiologic hyperinsulinaemia and hyperglycaemia on insulin secretion and insulin sensitivity in man. Diabetologia. 1994;37:1025–35.
Kashyap S, Belfort R, Gastaldelli A, Pratipanawatr T, Berria R, Pratipanawatr W, et al. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52:2461–74.
Solomon TP, Knudsen SH, Karstoft K, Winding K, Holst JJ, Pedersen BK. Examining the effects of hyperglycemia on pancreatic endocrine function in humans: evidence for in vivo glucotoxicity. J Clin Endocrinol Metab. 2012;97:4682–91.
American Diabetes Association. Standards of medical care in diabetes-2017: summary of revisions. Diabetes Care. 2017;40:S4–5.
Look AHEAD Research Group, Wing RR, Bolin P, Brancati FL, Bray GA, Clark JM, et al. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med. 2013;369:145–54.
Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149:367–78.
Keidar A. Bariatric surgery for type 2 diabetes reversal: the risks. Diabetes Care. 2011;34(Suppl 2):S361–266.
Adams TD, Davidson LE, Litwin SE, Kim J, Kolotkin RL, Nanjee MN, Gutierrez JM, Frogley SJ, Ibele AR, Brinton EA, Hopkins PN, McKinlay R, Simper SC, Hunt SC. Weight and metabolic outcomes 12 years after gastric bypass. N Engl J Med. 2017;377:1143–55.
Rena G, hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–85.
Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–6.
Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, et al. A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care. 2005;28:1547–54.
Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20:573–91.
Yau H, Rivera K, Lomonaco R, Cusi K. The future of thiazolidinedione therapy in the management of type 2 diabetes mellitus. Curr Diab Rep. 2013;13:329–41.
Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. 2016;165:305–15.
Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71.
Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D’Agostino RB Sr, et al. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: a randomized trial. JAMA. 2006;296:2572–81.
Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A, et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA. 2008;299:1561–73.
Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive study (PROspective pioglitAzone clinical trial in macroVascular events): a randomised controlled trial. Lancet. 2005;366:1279–89.
Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med. 2016;374:1321–31.
Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–307.
Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann-Heurtier A, Serfaty L, et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled fatty liver improvement with rosiglitazone therapy (FLIRT) trial. Gastroenterology. 2008;135:100–10.
Lewis JD, Ferrara A, Peng T, Hedderson M, Bilker WB, Quesenberry CP Jr, et al. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care. 2011;34:916–22.
Lewis JD, Habel LA, Quesenberry CP, Strom BL, Peng T, Hedderson MM, et al. Pioglitazone use and risk of bladder cancer and other common cancers in persons with diabetes. JAMA. 2015;314:265–77.
Tuccori M, Filion KB, Yin H, Yu OH, Platt RW, Azoulay L. Pioglitazone use and risk of bladder cancer: population based cohort study. BMJ. 2016;352:i1541.
Korhonen P, Heintjes EM, Williams R, Hoti F, Christopher S, Majak M, et al. Pioglitazone use and risk of bladder cancer in patients with type 2 diabetes: retrospective cohort study using datasets from four European countries. BMJ. 2016;354:i3903.
Levin D, Bell S, Sund R, Hartikainen SA, Tuomilehto J, Pukkala E, et al. Pioglitazone and bladder cancer risk: a multipopulation pooled, cumulative exposure analysis. Diabetologia. 2015;58:493–504.
Lasserson DS, Glasziou P, Perera R, Holman RR, Farmer AJ. Optimal insulin regimens in type 2 diabetes mellitus: systematic review and meta-analyses. Diabetologia. 2009;52:1990–2000.
Fonseca VA, Haggar MA. Achieving glycaemic targets with basal insulin in T2DM by individualizing treatment. Nat Rev Endocrinol. 2014;10:276–81.
Roumie CL, Hung AM, Greevy RA, et al. Comparative effectiveness of sulfonylurea and metformin monotherapy on cardiovascular events in type 2 diabetes mellitus: a cohort study. Ann Intern Med. 2012;157:601–10.
Pladevall M, Riera-Guardia N, Margulis AV, Varas-Lorenzo C, Calingaert B, Perez-Gutthann S. Cardiovascular risk associated with the use of glitazones, metformin and sufonylureas: meta-analysis of published observational studies. BMC Cardiovasc Disord. 2016;16:14.
Azoulay L, Suissa S. Sulfonylureas and the risks of cardiovascular events and death: a methodological meta-regression analysis of the observational studies. Diabetes Care. 2017;40:706–14.
Guardado-Mendoza R, Prioletta A, Jimenez-Ceja LM, Sosale A, Folli F. The role of nateglinide and repaglinide, derivatives of meglitinide, in the treatment of type 2 diabetes mellitus. Arch Med Sci. 2013;9:936–43.
Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311–22.
Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–44.
Pfeffer MA, Claggett B, Diaz R, Dickstein K, Gerstein HC, Kober LV, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247–57.
Holman RR, Bethel MA, Mentz RJ, Thompson VP, Lokhnygina Y, Buse JB, Chan JC, Choi J, Gustavson SM, Iqbal N, Maggioni AP, Marso SP, Öhman P, Pagidipati NJ, Poulter N, Ramachandran A, Zinman B, Hernandez AF, EXSCEL Study Group. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2017;377:1228–39.
Monami M, Nreu B, Scatena A, Cresci B, Andreozzi F, Sesti G, Mannucci E. Safety issues with glucagon-like peptide-1 receptor agonists (pancreatitis, pancreatic cancer and cholelithiasis): data from randomized controlled trials. Diabetes Obes Metab. 2017;19:1233–41.
Drucker DJ, Rosen CF. Glucagon-like peptide-1 (GLP-1) receptor agonists, obesity and psoriasis: diabetes meets dermatology. Diabetologia. 2011;54:2741–4.
Faurschou A, Pedersen J, Gyldenlove M, Poulsen SS, Holst JJ, Thyssen JP, et al. Increased expression of glucagon-like peptide-1 receptors in psoriasis plaques. Exp Dermatol. 2013;22:150–2.
Andersen ES, Deacon CF, Holst JJ. Do we know the true mechanism of action of the DPP-4 inhibitors? Diabetes Obes Metab. 2017. https://doi.org/10.1111/dom.13018. [Epub ahead of print].
Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232–42.
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–26.
Gokhale M, Buse JB, Gray CL, Pate V, Marquis MA, Sturmer T. Dipeptidyl-peptidase-4 inhibitors and pancreatic cancer: a cohort study. Diabetes Obes Metab. 2014;16:1247–56.
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–28.
Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644–57.
DeFronzo RA, Norton L, Abdul-Ghani M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat Rev Nephrol. 2017;13:11–26.
Bilezikian JP, Watts NB, Usiskin K, Polidori D, Fung A, Sullivan D, et al. Evaluation of bone mineral density and bone biomarkers in patients with type 2 diabetes treated with canagliflozin. J Clin Endocrinol Metab. 2016;101:44–51.
Peters AL, Buschur EO, Buse JB, Cohan P, Diner JC, Hirsch IB. Euglycemic diabetic ketoacidosis: a potential complication of treatment with sodium-glucose cotransporter 2 inhibition. Diabetes Care. 2015;38:1687–93.
Jabbour S, Seufert J, Scheen A, Bailey CJ, Karup C, Langkilde AM. Dapagliflozin in patients with type 2 diabetes mellitus: a pooled analysis of safety data from phase 2b/3 clinical trials. Diabetes Obes Metab. 2017. https://doi.org/10.1111/dom.13124.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Bril, F., Cusi, K. (2018). Basic Concepts in Insulin Resistance and Diabetes Treatment. In: Cohen Sabban, E., Puchulu, F., Cusi, K. (eds) Dermatology and Diabetes. Springer, Cham. https://doi.org/10.1007/978-3-319-72475-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-72475-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-72474-4
Online ISBN: 978-3-319-72475-1
eBook Packages: MedicineMedicine (R0)