
Abstract
Huntington’s disease (HD) is the most common monogenic neurodegenerative disease and the commonest genetic dementia in the developed world. With autosomal dominant inheritance, typically mid-life onset, and unrelenting progressive motor, cognitive and psychiatric symptoms over 15–20 years, its impact on patients and their families is devastating. The causative genetic mutation is an expanded CAG trinucleotide repeat in the gene encoding the Huntingtin protein, which leads to a prolonged polyglutamine stretch at the N-terminus of the protein. Since the discovery of the gene over 20 years ago much progress has been made in HD research, and although there are currently no disease-modifying treatments available, there are a number of exciting potential therapeutic developments in the pipeline. In this chapter we discuss the epidemiology, genetics and pathogenesis of HD as well as the clinical presentation and management of HD, which is currently focused on symptomatic treatment. The principles of genetic testing for HD are also explained. Recent developments in therapeutics research, including gene silencing and targeted small molecule approaches are also discussed, as well as the search for HD biomarkers that will assist the validation of these potentially new treatments.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1.1 Introduction
Huntington’s disease (HD) is one of the most common causes of dominantly inherited neurodegenerative disease [1]. There is typically adult onset, with irreversible progression of motor, psychiatric and cognitive symptoms over 15–20 years, followed by death [2]. The condition was first described in the USA by George Huntingtin in 1872, himself a newly qualified doctor at the time, and his original descriptions of the disease remain true today.
As yet there are still no disease modifying treatments available, but an intensive international research effort is underway with many clinical trials currently on-going. In this chapter we will first cover the epidemiology, genetics , and pathogenesis of HD and then discuss clinical aspects of the disease and the latest developments in HD therapeutics research.
1.2 Epidemiology
The prevalence of HD had been thought to be 4–10/100,000 in Western populations [3], though a more recent study in the UK suggests a figure of 12.3/100,000 [4]. The true prevalence of HD may have been underestimated in the past due to the stigma that has traditionally been attached to the diagnosis; in recent centuries, sufferers have even been accused of witchcraft, pressuring families into hiding the condition [5]. The wider availability of genetic testing may also have contributed to an apparent increase in disease prevalence [6].
The disease is thought to have migrated from North-West Europe to other parts of the world, and there is global variation in prevalence , with naturally low rates in Japan, Hong Kong and Taiwan [7]. One of the highest rates of HD occurs in Venezuela where communities living near the edge of Lake Maracaibo see a prevalence of 700/100,000 people. It was in this population that genetic linkage studies led to the discovery of the causative genetic mutation in HD [8, 9].
1.3 Genetics of Huntington’s Disease
HD is a single-gene disorder with autosomal dominant transmission and so the presence of the mutation on either allele leads to the disease. Therefore an affected parent has a 50% chance of passing it on to their child. The mutation is an expanded CAG triplet repeat near the start of exon 1 of the Huntingtin gene (HTT), which lies on the short arm of chromosome 4. On translation , this leads to the presence of a polyglutamine (polyQ) stretch at the N-terminus of Huntingtin protein (HTT).
The wild-type gene carries 10–35 CAG repeats , with a mean value of 18 CAG repeats across the population (although this mean value is greater in populations with higher disease prevalence ) [7, 10]. The mutation is fully penetrant at 40 or more repeats. Between 36 and 39 repeats there is reduced penetrance; carriers may develop HD symptoms in later life or not at all [11]. HTT with 27–35 CAG repeats is referred to as an “intermediate allele”.
Intermediate alleles in the general population are thought to arise from stepwise expansion of the CAG repeat over many generations. People who inherit intermediate length alleles have long been thought to be unaffected, but a behavioural phenotype has now been identified in this group [12]. During meiosis it is possible for intermediate alleles to expand further, leading to offspring who may carry a disease gene with 36 or more repeats. The risk of this is as high as 21% for those with 35 CAG repeats [13].
This intrinsic instability of the CAG repeat during meiosis especially affects the disease gene, leading to expansions (and sometimes contractions) in repeat length inherited by successive generations [14]. As longer CAG repeat length correlates with earlier age of onset, symptoms may develop at progressively younger ages as the condition is passed down the family tree. This is known as anticipation. Due to differences between spermatogenesis and oogenesis this is more likely to occur when transmission occurs through the paternal bloodline [15]. In rare cases large increases in CAG repeat length above 55 can occur, leading to Juvenile HD (JHD), when the age of onset is less than 20 years old. 90% of JHD cases result from paternal inheritance [16, 17].
As with all genetic conditions a detailed family history is essential to help make a correct diagnosis. However 6–8% of patients with newly diagnosed HD have no family history [18]. As mentioned above, de novo mutations may arise from intermediate length alleles, leading to sporadic cases. Seemingly sporadic cases may occur following non-paternity, misdiagnosis in prior generations, or when deaths of family members occur from other causes before development of neurological symptoms thus masking the presence of the HD gene.
1.3.1 Effect of CAG Repeat Length on Disease Phenotype
Disease onset is defined clinically as the presence of unequivocal extrapyramidal motor signs suggestive of HD. In typical mid-life onset HD with CAG repeat 40–55, the length of the CAG repeat accounts for ~56% of the variability in age at motor onset [19]. A recent genome-wide association (GWA) study has identified genes involved in DNA handling and repair mechanisms, including MLH1, as contributing to further variation in the age of disease onset [20]. Remaining variation is likely determined by other genetic and environmental modifiers [21].
Taking into account the CAG repeat length as well as the number of disease free years already lived, a conditional probability model was developed by Langbehn et al. which is able to estimate the chance of on-going disease free survival over a number of years [22, 23]. However, models based on population data cannot be applied in a one-to-one clinical setting and it is not possible to accurately predict an individual’s age of onset of disease from their CAG repeat length.
Patients often experience psychiatric and cognitive symptoms , as well as very subtle motor disturbances for many years before their official disease onset. CAG repeat length correlates much less strongly with age of onset of psychiatric symptoms , but does show some correlation with rate of disease progression [24, 25]. The duration of disease from diagnosis to death is independent of CAG repeat length.
1.4 Pathogenesis of Huntington’s Disease
Huntingtin is a large 350 kDa protein comprised of multiple repeated units of 50 amino acids, termed HEAT repeats, which assemble into a superhelical structure with a hydrophobic core. Compared to the wild-type protein, the mutant protein contains an expanded polyglutamine stretch starting at residue 18 [26]. This triggers a pathogenic cascade, with both cell autonomous and non-cell autonomous mechanisms involved which are summarised in Table 1.1.
Ultimately there is massive striatal degeneration in the patient brain, as revealed by post-mortem studies. Up to 95% of the GABAergic medium spiny neurons (MSNs) that project to the globus pallidus and substantia nigra are lost, and there is atrophy (though less so) of the cerebral cortex, subcortical white matter, thalamus and hypothalamic nuclei [27].
1.5 Clinical Aspects of Huntington’s Disease
Huntington’s disease is characterized by a triad of progressive motor, cognitive and psychiatric symptoms . The average age of onset is 45 years and the disease is fatal after 15–20 years [62].
1.5.1 Natural History and Disease Progression
As mentioned above, onset of “manifest” HD is said to occur when patients develop definitive motor signs suggestive of HD, which have no other explanation. However prior to this, patients may have a “prodromal” phase of HD for many years, during which subtle motor signs, psychiatric or cognitive symptoms may be present. The TRACK-HD study found differences in speeded tapping (a fine motor task) over 36 months between control and premanifest patients who were predicated to be over 10.8 years from disease onset. During this time a corresponding change in neurobiology, with loss of corticostriatal connectivity and striatal atrophy, is seen (Fig. 1.1).

Parametric maps showing regions with statistically significant atrophy in grey and white matter at baseline, 12- and 24 months (from left to right in each frame). PreHD-A and preHD-B are premanifest Huntington’s disease gene carriers with estimated time to clinical disease onset greater than and less than 10.8 years, respectively. HD1 and HD2 are patients with early manifest disease who have no functional impairment and mild functional impairment, respectively. The striatum is affected early on, with more widespread atrophy at later stages. Adapted from Tabrizi et al. [63] from Elsevier publishing group
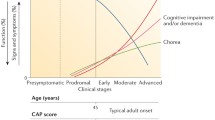
Prior to development of manifest HD, patients who carry the HD gene mutation are referred to as “premanifest”. Around 10–15 years before disease onset, premanifest individuals are indistinguishable from controls. However as disease onset approaches, patients are no longer completely asymptomatic and so the term “perimanifest” disease is used by clinicians to describe patients who are suffering from prodromal HD symptoms , and are thought to likely develop the concrete motor signs of manifest HD in the near future (Fig. 1.2).

Natural history of clinical Huntington’s disease . Adapted from Ross et al. [64] from Nature Publishing Group
1.5.1.1 Rating Scales and Disease Progression
The Unified HD Rating Scale (UHDRS) is comprised of motor, cognitive, behavioural, emotional and functional components and is the most common rating scale for HD [65]. It is widely utilised in HD research, and some of its component parts are also useful in a clinical setting. The total motor score (TMS) subscale is helpful in diagnosing disease onset; based on the score and their overall clinical impression, the clinician assigns a diagnostic confidence score (DCS) between 0 and 4, which reflects their belief that the motor signs are representative of HD.
Diagnosis of disease onset can be a difficult and emotional time for patients and their families, as they are faced with the prospect of inevitable decline, and will have often witnessed other family members suffering the same fate. There are also implications for patients in terms of their employment, insurance and driving, the exact rules of which will vary between countries.
Following disease onset, there is progression through five disease stages, originally described by Shoulson and Fahn (Table 1.2). These stages also correspond to Total Functional Capacity (TFC) subscale score of the UHDRS, which is based on the same functional domains. In a clinical setting, the more general terms of early, moderate and advanced disease are used (Table 1.3).
1.5.2 Clinical Presentation
HD displays much clinical heterogeneity, even within families, in terms of the balance of motor, cognitive and psychiatric features that predominate.
Motor symptoms are comprised of both involuntary movements, seen in early-moderate stage typical adult onset disease, and impaired voluntary movement seen in more advanced stages. Chorea is one of the most striking features, and is defined as short-lived, involuntary, excessive movements, which are semi-purposeful. Chorea progresses from occasional, low amplitude twitches of the face and extremities to constant, large amplitude movements of the entire body. Head turning, eye closure and tongue protrusion occur following activation of facial and neck muscles and back extension results from involvement of axial muscle groups. Chorea may not always bother the patient, even when quite severe. It can however cause problems with writing, eating and maintaining balance.
Other motor features include dystonia , myoclonic jerks and tics. Dystonia is the most common of these and is caused by inappropriate, sustained muscle contractions that lead to abnormal postures such as torticollis (neck turning) and opisthotonos (back arching).
As disease progresses, bradykinesia , akinesia, rigidity and impaired postural reflexes dominate. This can cause particular problems with gait and falls, especially on uneven terrain. Motor symptoms can also rapidly deteriorate in the event of intercurrent infections, stress and anxiety, but with appropriate treatment, this is usually temporary.
Cognitive symptoms are universal in HD. Subtle cognitive impairments may occur years before disease onset [66], and can progress to frank subcortical and frontal dementia in advanced disease. The TRACK-HD study found early deficits in visual attention, psychomotor speed, and visuomotor and spatial integration [25], though patients themselves are often unaware of any problems. Executive dysfunction, with development of concrete thinking is very common. Other cognitive problems include general slowing and impaired short-term memory.
Psychiatric symptoms affect a large proportion of patients; depression is the most common feature, which has a prevalence of 40% in HD patients [67]. Anxiety is the second most common psychiatric condition. Neither depression nor anxiety relate to disease stage and can occur in premanifest HD [68]. Apathy, which is characterised by a general loss of interest, difficulty with initiating activities and passive behaviour, is a common and disabling symptom that is related to disease stage and worsens over time. It has been shown to be a significant predictor of functional decline [25], and is resistant to medication.
Other psychiatric symptoms include irritability and aggression, obsessive, compulsive thoughts and behaviours, and more rarely, psychosis in later stages. Hyper- and hyposexuality can be a problem in early and late HD respectively [69].
Suicide is the second most common cause of death in HD [70]. Risk of this is highest when premanifest patients start to develop manifest disease, and again when loss of independence starts to occur in advanced HD. In a study of 4171 HD patients, 10% had made a previous suicide attempt, and 17.5% had been affected by suicidal thoughts [71]. Risk factors include depression and impulsivity [72].
1.5.2.1 Other Neurological Symptoms
In addition to the triad of characteristic motor, cognitive and psychiatric symptoms described above, patients may have problems with their speech, swallowing and sleep. Speech difficulties arise from a combination of dysarthria (from incoordination of the tongue and orofacial muscles) and word finding difficulties. In advanced disease, some patients may develop complete anarthria. Swallowing problems also result from incoordination of oral and pharyngeal muscles, and can lead to choking episodes and, in severe cases, aspiration pneumonia.
Sleep disturbance may result from primary dysfunction of circadian rhythms, leading to reversal of the sleep-wake cycle [73]. Low mood, anxiety and night-time chorea also contribute to insomnia and can significantly impair quality of life.
1.5.2.2 Peripheral Symptoms
HD is primarily a disease of the central nervous system (CNS) but cells throughout the body express Huntingtin protein, and consequently, a range of systemic symptoms are seen in HD [74]. Severe weight loss, secondary to an underlying catabolic state, is common and may lead to cachexia. This may begin during prodromal HD, and HD patients usually have very high calorie requirement in order to simply maintain their weight. There is an association between higher body mass index at disease onset and slower rate of disease progression [75].
Other peripheral symptoms include osteoporosis and skeletal muscle atrophy. Heart failure occurs in 30% of patients (compared to 2% of age-matched controls) [70]. Endocrine dysfunction in the form of impaired glucose tolerance, hypothyroidism and low male testosterone levels, also occurs. Testicular atrophy with abnormal seminiferous tubules and reduced numbers of germ cells is noted, but does not affect fertility.
1.5.2.3 Juvenile Huntington’s Disease (JHD)
As mentioned previously, JHD is defined by an age of onset less than 20 years old, and generally arises in those who have inherited >55 CAG repeats [76]. The clinical presentation differs from adult onset disease in that rigidity , bradykinesia and akinesia are present from the outset, with fewer hyperkinetic movements. Seizures occur in 30–50% of patients and there may be learning difficulties and behavioural issues whilst at school [76]. This rigid phenotype of HD is also known as the Westphal or akinetic-rigid variant (on rare occasions, adult onset disease may present with this form).
1.5.3 Diagnosis and Investigations
Clinical findings including definite motor signs, combined with a family history suggestive of autosomal dominant transmission are suggestive of the diagnosis, which is confirmed through genetic testing to determine CAG repeat length [77]. As mentioned above, in some cases a family history is absent, so a high index of suspicion is required if the clinical picture is otherwise consistent with HD. MRI and CSF testing do not currently play a role in the diagnosis of HD, other than to rule out alternative diagnoses.
Around 1% of patients who are thought to have HD, subsequently test negative for the HD gene mutation. These patients may have different underlying genetic mutations that are collectively known as “HD phenocopies” [78]. The most common cause is the C9orf72 hexanucleotide repeat expansion mutation, which is also a major cause of sporadic and familial frontotemporal lobar degeneration and amyotrophic lateral sclerosis [79]. Known causes of HD phenocopies are shown in Table 1.4. In the majority of cases however, the diagnosis remains unknown.
1.5.4 Management of HD
A multidisciplinary approach involving neurologists, psychiatrists, general practitioners, physiotherapists, occupational therapists, speech and language therapists, dieticians and nurses, is required for the management of HD [80]. This is best achieved through specialist HD clinics, where representatives from local HD support groups can also attend to give help with practical, emotional and social matters. Such clinics are also useful for recruiting patients into research studies or clinical trials if appropriate; taking part in research can be of great psychological benefit to patients and their carers.
1.5.4.1 Drug Treatments
Although there are no disease-modifying treatments available currently, there are medications to alleviate symptoms [81, 82].
Chorea may not always require treatment as it often does not bother patients, but when function is impaired as a result then medication should be considered. Tetrabenazine (TBZ) is a dopamine-depleting agent that has proven efficacy at reducing chorea . It can trigger or worsen depression and should be avoided in those with a history of severe depression or current low mood [83]. Patients must be warned of this side effect and monitored. TBZ must not be prescribed with cytochrome P 450 2D6 inhibitors (such as fluoxetine and paroxetine) as this is required for its clearance [84].
Although a Cochrane review concluded that only TBZ showed clear benefit for chorea , in practice other medications are also used [85]. Atypical neuroleptics such as olanzapine, risperidone and quetiapine are very useful, especially if there is coexistent irritability, agitation or anxiety. Older typical neuroleptics such as sulpiride and haloperidol are also used, but there are greater side effects associated. All neuroleptic use warrants an ECG review prior to starting the prescription, and regular monitoring of blood glucose and cholesterol whilst on treatment. Clonazepam is useful if chorea is combined with dystonia , rigidity or myoclonus. Sodium valproate or levetiracetam can be useful if there is significant myoclonus alone.
In later disease, anti-chorea medications may need to be weaned and stopped as rigidity and spasticity develop. Tizanidine and baclofen may be helpful at this stage and botulinum toxin injections are used for targeted muscle spasm. In JHD, a trial of levodopa may be helpful.
In terms of psychiatric symptoms , depression may be treated with selective serotonin reuptake inhibitors (SSRIs) such as citalopram. Mirtazapine, a sedating antidepressant, is useful when insomnia is also a problem. A specific risk assessment with respect to suicide should be carried out when evaluating psychiatric symptoms .
SSRIs may also be useful for the management of anxiety. Irritability and aggression may be treated with atypical neuroleptics (as for chorea ). Higher doses may occasionally be needed for the treatment of psychosis. For sleep disturbances, a short-term course of zopiclone could be tried, or melatonin can also be useful to address the reversal of the sleep-wake cycle.
1.5.4.2 Non-Drug Treatments
Physiotherapy is useful to optimise gait and balance for as long as possible, and in later stages physiotherapists can offer walking aids if needed. Occupational therapists adapt the home environment in order to prolong independent functioning and increase safety.
Speech and language therapists can improve communication, both verbally up to a point, and later with communication aids. They can also assess swallowing and advise on appropriate diet. In advanced HD, enteral nutrition via a gastrostomy may be needed to maintain adequate nutrition. The feeding tube may be inserted as a percutaneous endoscopic gastrostomy (PEG) or through radiological guidance (RIG) if endoscopy cannot be tolerated. Dieticians can then set up enteral feeding regimens, and also give more general advice on calorie supplementation.
It is essential to liaise with local community mental health teams that provide psychiatric care and regular follow up or monitoring close to home. Cognitive behavioural therapy (CBT) can be administered by psychologists to appropriately selected patients, and can help with anxiety, depression and OCD symptoms .
Social workers are able to arrange home care when patients are no longer fully able to care for themselves, and if necessary to co-ordinate moving into a residential or nursing home. HD support organisations, e.g. the Huntington’s disease association (HDA) in the UK, are invaluable with helping patients and carers with a range of issues from legal matters, employment issues, and claiming any entitlements, to organising local discussion/support groups.
One important issue that it is essential to address with patients is that of driving. By law, patients must notify the driving authorities once they have a diagnosis of manifest HD, and it is inevitable that at some point during the disease course, their driving licence will be revoked. It can cause patients a great deal of distress when this happens, particularly if they do not recognise that their driving ability is impaired.
1.5.4.3 End of Life Care
Inevitably as HD is a progressive condition, end of life care is important for all patients. Though discussion around this subject can be difficult, it is worth addressing these issues whilst patients retain capacity to make decisions, and before significant cognitive or psychiatric deterioration takes place.
As swallowing difficulties progress, oral feeding may become very risky. Whilst some patients are keen to establish enteral feeding under these circumstances, others do not want this under any circumstances. In the event of potentially terminal medical deterioration, patients also differ in the levels of care they would want in terms of antibiotics, hospital admission, resuscitation etc.
In the UK patients can draw up an advanced directive, which outlines the care they would want under particular circumstances should they lose capacity in the future. Alternatively patients may nominate an individual to have lasting power of attorney (LPA), who can make decisions on their behalf if it becomes necessary. In either case, these decisions must be made whilst the patient still has capacity to do so.
In advanced disease, home care may no longer be possible and a residential or nursing home may be necessary. At the very end of life, some patients may not want to go to hospital, preferring instead to be cared for at home or in a hospice setting. Community palliative care teams, district nurses and GPs can help provide symptomatic relief in these circumstances.
1.5.5 Genetic Testing
There is a readily available genetic test that determines the CAG repeat length in each allele of the HTT gene, which can be carried out on a standard blood sample. The relative ease of performing the actual test belies the complexity of the issues surrounding the genetic testing process [77].
1.5.5.1 Diagnostic Testing
Diagnostic genetic tests are requested by neurologists in order to confirm the suspected diagnosis in those patients who are already displaying motor signs of HD. Informed consent from the patient must be obtained prior to testing. The implications of a positive result for the patient and their family must be clearly outlined. Both the nature of HD itself and its autosomal dominant inheritance must be explained. It should be made clear that if they test positive for HD, any of their children would have a 50% chance of inheriting it. Their siblings would also be at risk with a 50% chance of having inherited the mutation themselves (and some may already be developing signs of HD).
Test results must be given in person at a follow up appointment, and in the case of a positive result, referral to a specialist HD clinic should be offered. Details of HD support groups should be given, and family members should be offered referral for genetic counselling if they wish.
It is worth noting that patients who have psychiatric or cognitive symptoms in the context of a positive family history of HD, but who do not have motor signs, must be referred to a clinical geneticist for testing as this would qualify as predictive rather than diagnostic testing.
1.5.5.2 Predictive Testing
Predictive testing refers to genetic testing that is carried out in asymptomatic individuals, who are at known to be at risk of inheriting the mutation due to a positive family history. There are international guidelines for predictive testing in HD [86,87,88]; testing must take place in specialist centres by a clinical geneticist. There must be at least one session of pre-test counselling, followed by a period of reflection, and a second counselling session. Written consent must be given, and as always, strict confidentiality observed. Results must be given face-to-face with further post-test counselling available.
As with diagnostic testing, it is essential to explain the dominant inheritance of HD and its potential impact on the whole family. A positive test result means that an individual will get HD at some point in the future, but it is not possible to predict their specific age of onset or rate of progression [22]. Patients who have CAG repeat lengths of reduced penetrance (36–40) or intermediate repeat lengths (27–35) should have a full explanation of the implications of this [89] (see Sect. 5.3. Genetics of Huntington’s disease ).
The personal consequences of either a positive or a negative result should be explored with individuals prior to testing. Following a positive result, patients will know that they are going to develop an incurable neurodegenerative disease in the future, and they may have already witnessed first-hand the devastating impact of this on their own relatives. A negative test result can also cause enormous feelings of guilt when other family members are affected, and lead to strain within family relationships. Difficult situations can also arise if certain family members do not wish to know their gene status. For example the grandchild of an affected individual may wish to undergo predictive testing, when their own parent does not. In this case a positive test result in the grandchild would necessarily mean that their at-risk parent was in fact carrying the mutation also.
It is not legal to carry out predictive testing in children under the age of 18. Children cannot give informed consent, and no one can consent on their behalf because they have a right not to know their gene status as an adult. Growing up with the knowledge of a positive test result would in any case be very damaging for the psychological development of most children.
5–20% of at-risk patients proceed with predictive genetic testing [90]. Common reasons for proceeding include planning for the future, especially in terms of careers or deciding to have children.
1.5.5.3 Reproductive Options
Pre-implantation genetic diagnosis (PGD) is available for HD gene carriers in many countries, including the UK. A number of embryos are created using in vitro fertilisation (IVF) techniques, and then screened for the mutation. Negative embryos are then selected for implantation into the uterus. Multiple cycles of IVF are sometimes required before a successful pregnancy results, and the process can therefore be emotionally and physically demanding. There are also high costs associated with PGD, although in the UK a certain number of cycles are usually funded by the national healthcare system.
It is possible for at-risk patients to undergo PGD without finding out their own gene status. Linkage analysis to look for genetic markers linked to the HTT genes of the parents of the at-risk patient, can identify embryos that have a 50% chance or <1% chance of carrying the mutation. Some private clinics also offer non-disclosure testing when the at-risk prospective parent is tested for HD in order to make a decision on embryo selection, but then the same IVF procedure is followed and the genetic test results are not disclosed to the patient.
Another option is antenatal testing during pregnancy. Chorionic villous sampling (CVS) can be carried out at 11–13 weeks of gestation, or amniocentesis after 16 weeks, to obtain foetal tissue for genetic testing. Detailed counselling must be given prior to this, as parents need to be certain that they would terminate a pregnancy in which the foetus was positive for the mutation. If the pregnancy is continued in the face of a positive test result than the resulting child would effectively have had a positive predictive test to which they never consented. Other reproductive options include using a sperm or egg donor, or adoption.
It is worth discussing with patients who wish to become parents that although they may well be symptom free or many years (or lifelong if they are in fact gene negative), there is a chance that they may develop symptoms whilst their children are still young, or even during pregnancy. Patients may wish to see a neurologist for examination before making their decision to proceed with having children, which is of course a highly personal decision. Indeed some patients who have had positive predictive testing in the past, or who know they are at risk, may simply choose to start a family without seeking further medical help or any intervention at all.
1.6 Huntington’s Disease Therapeutics Research
1.6.1 Disease Modifying Therapies
There is an urgent and as yet unmet need for a treatment which can slow, prevent or even reverse the symptoms of HD in mutation carriers. A number of promising approaches, including the reduction of Huntingtin expression and targeted small molecule therapeutics are currently under investigation in research studies and clinical trials (Fig. 1.3).

Therapeutic targets under investigation in Huntington’s disease . Reproduced from Ross et al. [64], Nature Publishing Group
1.6.1.1 “Gene Suppression” or Reduction of Huntingtin Expression
Mutant HTT primarily exerts a dominant toxic effect, and so a reduction in its expression should reduce any downstream pathology. Strategies to lower HTT levels include both post-transcriptional inhibition such as RNA interference (RNAi) and anti-sense oligonucleotides (ASOs), and genome editing techniques such as zinc finger proteins (ZFPs) and CRISPR/Cas9.
Administration of complementary small interfering RNA molecules (siRNAs) results in Argonaut-2 mediated cleavage and degradation of mature, spliced HTT mRNA in the cytosol. ASOs have a more upstream site of action and recruit RNaseH1, an endogenous enzyme that recognises RNA/DNA duplexes and degrades HTT pre-mRNA [91].
siRNAs [92, 93] and ASOs [94] designed to lower total HTT levels have both been shown to improve disease symptoms in rodents. In humans there is potential concern with regards to the long-term effect of reducing wild-type HTT, although reduction of endogenous HTT has been shown to be well tolerated in non-human primates [95, 96].
It has been challenging to achieve allele-specific lowering of mutant HTT. However ASOs complementary to the expanded CAG have recently been produced which have selectivity for mHTT over wild-type HTT, and have led to phenotypic improvement in rodent models [97]. siRNAs designed to target single nucleotide polymorphisms (SNPs) that reside only on the mutant HTT allele could also be used to achieve allele specific suppression [98, 99]. ASOs targeting mutant-allele linked SNPs have also been designed that lead to even greater (up to 50-fold) selectivity of the mHTT allele over the normal allele [100, 101]. Such compounds will soon proceed to clinical trials, and involve screening potential trial subjects for the presence of specific SNPs prior to drug administration.
Reduction of Huntingtin expression can also be achieved at the transcriptional level, using zinc finger proteins (ZFPs). These contain a zinc finger domain that can be synthetically manipulated to bind HTT DNA, fused to a functional domain such as a nuclease. Drug delivery is through the use of viral vectors, and this approach will also be proceeding into clinical trials in the near future.
Another recently discovered genome editing technique is through manipulation of the endogenous clustered regulatory interspaced short palindromic repeat (CRISPR)/Cas9 system which recognises and destroys foreign DNA in prokaryotic cells. The mutant allele is inactivated by a synthetic guide RNA (gRNA) strand that targets a particular DNA location for cutting, followed by the insertion of a desired DNA sequence (for example stop codons) [102]. The technique has been successfully demonstrated in HD patient-derived fibroblasts, leading to the dramatic reduction of mutant HTT RNA and mHTT protein [103].
A seminal Phase 1/2a clinical trial (IONIS-HTTRx) is currently being carried out by Ionis Pharmaceuticals to test the safety and tolerability of an ASO targeting human HTT, delivered by lumbar intrathecal bolus administration to early stage HD patients [104]. Encouragingly, a recent clinical trial of intrathecal infusion of an ASO targeting SOD1 mRNA in patients with amyotrophic lateral sclerosis (ALS) has demonstrated the safety and tolerability of this general approach [105]. Furthermore, patients with spinal muscular atrophy (SMA) Type 1 (infantile onset) have been treated with lumbar intrathecal bolus injections of an ASO (Nusinersen) that alters mRNA splicing, and have shown dramatic improvement [106].
1.6.1.2 Targeted Small Molecule Approaches
Mutant Huntingtin has been shown to exert its toxic effect through many different pathological pathways. Therefore a number of potential targets for disease modification exist that have been or are currently being tested in animal models and in some cases, in clinical trials .
One approach is to target the post-translational modifications of mHTT, for example by increasing phosphorylation at certain neuroprotective residues. Administration of the ganglioside GM1 causes reversal of disease phenotype in HD mice, which is thought to occur by increasing beneficial phosphorylation [107]. Acetylation of mHTT promotes its clearance by autophagy , and the inhibition of the deacetylase sirtuin 1 by selisistat has been shown to have benefit in various HD models and more recently, has been found to be safe and tolerable to human patients [108].
Inhibition of phosphodiesterase (PDE) 10A (which is a major modulator of striatal synaptic biology regulating cAMP and cGMP signalling and synaptic plasticity) led to multiple phenotypic improvements in HD mice [109]. However, the recently completed Pfizer “Amaryllis” trial of PDE10A inhibition in patients has not found any beneficial effect on motor function, or other HD symptoms that were tested [110].
Potentiation of the neurotrophin BDNF (which is depleted in HD), through agonism of the tyrosine receptor kinase B (TrkB), was initially thought to be a promising approach [111]. Agonism by monocloncal antibodies is currently under investigation [112]. However cysteamine, which is also thought to act through increasing BDNF levels, has not demonstrated efficacy in a recent clinical trial (CYST-HD) [113].
Inhibition of kynurenine 3-monooxygenase (KMO) improves the balance of neuroprotective over excitotoxic tryptophan metabolites produced by microglia. The peripherally administered KMO inhibitor JM6 has shown benefit in HD mouse models [114]. However more recently this has not been replicated using the novel KMO inhibitor CHDI-340246, although electrophysiological alterations were restored [115].
There is hyperactivity of the innate immune system in HD. The immune modulator laquinimod, which has been shown to reduce NFkB activation in astrocytes [116] and has demonstrated potential in the treatment of multiple sclerosis [117], is currently being tested in LEGATO-HD [118]. Its effect on motor function and brain imaging in early HD is being assessed.
Cellular metabolism is known to be affected in HD; the PPARγ agonist rosiglitazone has been shown to reduce mHTT-induced toxicity in striatal cells and improve motor function in HD mice [119]. The mitogen-activated protein kinase (MAPK) signalling pathway is deranged in HD and presents many therapeutic targets for potential amelioration, however this area is complex and not yet fully understood [120]. Counteracting excitotoxicity caused by mHTT has been tested using ceftriaxone, which upregulates the expression of the EAAT2 glutamate transporter, and has been shown to be beneficial in mouse models [121].
Other therapeutic targets that have shown potential success include the upregulation of chaperone proteins to reduce mHTT misfolding and aggregation [122, 123]. Inhibition of histone deacetylases (HDACs) in an effort to reduce the transcriptional dysregulation caused by mHTT has also been shown to be of benefit in HD models, although the mechanism by which it exerts its effects were different to that expected [124].
1.6.1.3 Stem Cell Therapy
There have been a number of small trials of foetal striatal transplantation in HD patients over the last 15 years in the UK [124−126], France [127, 128], Germany [129], Italy [130] and the USA [131], but the outcomes form these trials have been mixed. Improvement or stabilisation of motor, functional and neuropsychiatric symptoms has been reported by some [128], but not all the groups. Variability in results may be accounted for by the small sample size of patients at each centre and the heterogeneity between studies in terms of patient selection. There were also differences with respect to the type of tissue transplanted (cells from whole ganglionic eminence versus lateral ganglionic eminence, and the foetal gestation), as well as the preparation of the donor tissue and numbers of cells transplanted [125].
Post-mortem studies have shown poor long-term graft survival, possibly due to allograft immunoreactivity, excitotoxicity or microglial responses directed against donor tissue [132]. More recently, aggregates of mHTT have been found in the grafted donor cells, implying the spread of mutant protein between neurons, and calling into question the feasibility of foetal striatal transplantation therapy as a strategy to treat HD [44].
1.6.1.4 Other Current and Recent HD Clinical Trials
Thus far there have been no completed Phase 3 clinical trials of potentially disease modifying therapies with a successful outcome. Trials of vitamin E, idebenone, baclofen, lamotrigine, creatine, coenzyme Q10 + remacemide, ethyl-eicosapentanoic acid and riluzole have all had negative results [133].
Trials aimed at improving overall function for people with HD include 2CARE and CREST-E, but both were recently terminated prematurely on the grounds of futility [134, 135]. 2CARE evaluated coenzyme Q10 at a dose of 2400 mg/day for a planned duration of 5 years, and CREST-E evaluated creatine at doses of up to 40 g/day for a planned duration of 3 years. Assessment of overall function was based on the Total Functional Capacity (TFC) score, but this may not have been a sensitive enough tool to detect any potential impact on disease progression [62].
Pridopidine, which is thought to act as a dopamine stabiliser in the CNS, failed to reach its primary endpoint in two clinical trials : HART [136] and MermaiHD [137]. However, both trials showed an improvement in motor symptoms as evaluated by the Total Motor Score (TMS) and so this drug was re-evaluated for its effect on TMS in the PRIDE-HD trial. Unfortunately, once again the drug did not differentiate from placebo, although there was some beneficial effect on TFC at lower doses [138]. This highlights the need for sensitive markers of clinical progression when designing clinical trials .
Deutetrabenazine (SD-809), designed for the treatment of chorea , has been shown in the FIRST-HD trial to result in improved motor signs over a period of 12 weeks when compared to placebo [139]. The ongoing, open-label extension of this study (ARC-HD) has shown that overnight conversion from tetrabenazine to deutetrabenazine is safe and effectively maintained chorea control [140]. A trial of deep brain stimulation for treatment of motor symptoms , HD-DBS [141], is also underway.
Non-motor features of HD have also been a target of clinical trials. The Reach2HD trial tested the drug PBT2 (thought to reduce metal-induced aggregation of mutant Huntingtin ) in patients with early and moderate HD. The drug was found to be safe and well tolerated in patients, and a potential benefit on cognition (executive function) was observed, which needs to be followed up with a larger study [142]. Other trials targeting non-motor features of HD are Action-HD, which has recently reported no significant effect of the drug bupropion on apathy (though there was a large placebo/participation effect in this study), and the ETON-Study [143], which is looking at the effect of epigallocatechin gallate on cognitive function. Finally, NEUROHD [144] is comparing the effect of olanzapine, tetrabenazine and tiapiride on overall function as assessed by the independence scale of the UHDRS.
1.6.2 Biomarkers
Objective biomarkers that accurately track disease progression are essential in order to detect any beneficial effects of potential therapies in a trial setting. Commonly used clinical scales such as the UHDRS might not be sensitive enough to detect subtle changes, and are susceptible to inter- and intra-rater variability. Furthermore, once effective therapies exist, preventing or delaying neurodegeneration in premanifest patients would be the goal; biomarkers are key in helping to decide when to time such interventions.
Longitudinal observational studies, including TRACK-HD [25] and PREDICT-HD [66] led to a more detailed understanding of the natural progression of HD from premanifest through to manifest disease. Structural MRI has demonstrated significantly faster rates of decline in striatal volume in premanifest and manifest individuals compared with age-matched controls, even in those estimated to be >15 years from estimated disease onset [23].
Other imaging modalities that have potential as biomarkers include diffusion tensor imaging (DTI) which has shown abnormalities in neuronal fibre orientation and integrity in white matter and subcortical grey matter structures in both premanifest and manifest HD, functional MRI techniques, and [18] F-fluorodeoxyglucose (FDG)-PET [145]. Magnetic resonance spectroscopy (MRS) also demonstrates abnormal metabolic activity, indicative of neuronal health and could be used to assess response to a therapeutic intervention [146].
Cognitive biomarkers are often limited by floor and ceiling effects, and confounded by educational level and mood. One potential measure is the Huntington’s disease cognitive assessment battery (HD-CAB) [147]. Deterioration in emotion recognition is found in premanifest HD, and in early HD, performance on the Stroop test and indirect circle tracing also tracks clinical decline. Quantitative motor tasks such as the speeding tapping task (which is affected in premanifest HD), grip force variability and tongue force measures may also be of use in HD drug trials [25].
An ultrasensitive single-molecule counting (SMC) mHTT immunoassay has been developed that can quantify very low levels (in the femtomolar range) of mHTT in the cerebrospinal fluid (CSF). The level of mHTT detected was associated with proximity to disease onset and diminished cognitive and motor function, and is a new biomarker being taken forward into clinical trials [148]. Levels of tau in the CSF also show promise as a biomarker in HD [149]. Recently the level of neurofilament light protein (NfL) in plasma, has demonstrated potential as a prognostic blood biomarker of disease onset and progression in Huntington’s disease [150].
1.7 Conclusion
Thanks to intensive, collaborative, international research efforts spanning the last few decades, we now know a great deal about the genetics , pathogenesis and natural history of HD. Clinical presentation is with progressive motor, cognitive and psychiatric features for which there are currently only symptomatic treatments. Management is best achieved through specialist multidisciplinary clinics, which are linked to a wider research team that patients can access if they wish to. There are currently many exciting therapeutic developments in the pipeline, giving hope for a potentially disease modifying treatment in the near future.
References
Fisher ER, Hayden MR (2014) Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord 29:105–114
Ross CA, Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98
Harper P (2002) The epidemiology of Huntington’s disease. Huntington’s disease. Oxford Medical Publications, Oxford
Evans SJ, Douglas I, Rawlins MD, Wexler NS, Tabrizi SJ, Smeeth L (2013) Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry
Rawlins M (2010) Huntington’s disease out of the closet? Lancet 376:1372–1373
Morrison PJ (2012) Prevalence estimates of Huntington disease in Caucasian populations are gross underestimates. Mov Disord 27:1707–1708. (author reply 8–9)
Kay C, Fisher E, Michael H (2014) Epidemiology. In: Tabrizi SJ, Jones L (eds) Bates G. Oxford University Press, Huntington’s disease, pp 131–164
Gusella JF, Wexler NS, Conneally PM et al (1983) A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306:234–238
MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G et al (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72:971–983
Snell RG, MacMillan JC, Cheadle JP et al (1993) Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat Genet 4:393–397
Rubinsztein DC, Leggo J, Coles R et al (1996) Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am J Hum Genet 59:16–22
Killoran A, Biglan KM, Jankovic J, et al (2013) Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS. Neurology
Semaka A, Kay C, Doty C et al (2013) CAG size-specific risk estimates for intermediate allele repeat instability in Huntington disease. J Med Genet 50:696–703
Zühlke C, Riess O, Bockel B, Lange H, Thies U (1993) Mitotic stability and meiotic variability of the (CAG)n repeat in the Huntington disease gene. Hum Mol Genet 2:2063–2067
Kremer B, Almqvist E, Theilmann J et al (1995) Sex-dependent mechanisms for expansions and contractions of the CAG repeat on affected Huntington disease chromosomes. Am J Hum Genet 57:343–350
Barbeau A (1970) Parental ascent in the juvenile form of Huntington’s chorea. Lancet 2:937
Kremer B (2002) Clinical Neurology of Huntington’s disease. In: Harper P, (ed) Huntington’s disease. Oxford Medical Publications, Oxford
Siesling S, Vegter-van de Vlis M, Losekoot M et al (2000) Family history and DNA analysis in patients with suspected Huntington’s disease. J Neurol Neurosurg Psychiatry 69:54–59
Gusella JF, MacDonald ME, Lee JM (2014) Genetic modifiers of Huntington’s disease. Mov Disord 29:1359–1365
Consortium GMoHsDG-H (2015) Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 162:516–26
Wexler NS, Lorimer J, Porter J et al (2004) Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A 101:3498–3503
Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR (2004) Group IHsDC. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet 65:267–277
Aylward EH, Nopoulos PC, Ross CA et al (2011) Longitudinal change in regional brain volumes in prodromal Huntington disease. J Neurol Neurosurg Psychiatry 82:405–410
Rosenblatt A, Kumar BV, Mo A, Welsh CS, Margolis RL, Ross CA (2012) Age, CAG repeat length, and clinical progression in Huntington’s disease. Mov Disord 27:272–276
Tabrizi SJ, Scahill RI, Owen G, et al (2013) Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol
Zuccato C, Valenza M, Cattaneo E (2010) Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev 90:905–981
Halliday GM, McRitchie DA, Macdonald V, Double KL, Trent RJ, McCusker E (1998) Regional specificity of brain atrophy in Huntington’s disease. Exp Neurol 154:663–672
Hughes A, Jones L (2014) Pathogenic mechanisms in Huntington’s disease. In: Bates GP, Tabrizi SJ, Jones L (eds) Huntington’s Disease, 4th edn. Oxford University Press
Sathasivam K, Neueder A, Gipson TA et al (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A 110:2366–2370
Mangiarini L, Sathasivam K, Seller M et al (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87:493–506
Barbaro BA, Lukacsovich T, Agrawal N et al (2015) Comparative study of naturally occurring huntingtin fragments in Drosophila points to exon 1 as the most pathogenic species in Huntington’s disease. Hum Mol Genet 24:913–925
Sun CS, Lee CC, Li YN et al (2015) Conformational switch of polyglutamine-expanded huntingtin into benign aggregates leads to neuroprotective effect. Sci Rep 5:14992
Arrasate M, Finkbeiner S (2012) Protein aggregates in Huntington’s disease. Exp Neurol 238:1–11
Zuccato C, Marullo M, Conforti P, MacDonald ME, Tartari M, Cattaneo E (2008) Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol 18:225–238
Steffan JS, Kazantsev A, Spasic-Boskovic O et al (2000) The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A 97:6763–6768
Zuccato C, Tartari M, Crotti A et al (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35:76–83
Hodges A, Strand AD, Aragaki AK et al (2006) Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet 15:965–977
Butler R, Bates GP (2006) Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci 7:784–796
Pavese N, Gerhard A, Tai YF et al (2006) Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology 66:1638–1643
Tai YF, Pavese N, Gerhard A et al (2007) Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 130:1759–1766
Crotti A, Benner C, Kerman BE et al (2014) Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci 17:513–521
Björkqvist M, Wild EJ, Thiele J et al (2008) A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med 205:1869–1877
Pecho-Vrieseling E, Rieker C, Fuchs S et al (2014) Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat Neurosci 17:1064–1072
Cicchetti F, Lacroix S, Cisbani G et al (2014) Mutant huntingtin is present in neuronal grafts in huntington disease patients. Ann Neurol 76:31–42
Pardo R, Molina-Calavita M, Poizat G, Keryer G, Humbert S, Saudou F (2010) pARIS-htt: an optimised expression platform to study huntingtin reveals functional domains required for vesicular trafficking. Mol Brain 3:17
Gauthier LR, Charrin BC, Borrell-Pagès M et al (2004) Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127–138
Roux JC, Zala D, Panayotis N, Borges-Correia A, Saudou F, Villard L (2012) Modification of Mecp2 dosage alters axonal transport through the Huntingtin/Hap1 pathway. Neurobiol Dis 45:786–795
Li H, Wyman T, Yu ZX, Li SH, Li XJ (2003) Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet 12:2021–2030
Jin YN, Johnson GV (2010) The interrelationship between mitochondrial dysfunction and transcriptional dysregulation in Huntington disease. J Bioenerg Biomembr 42:199–205
Siddiqui A, Rivera-Sánchez S, MeR Castro et al (2012) Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic Biol Med 53:1478–1488
Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB (1986) Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 321:168–171
Joshi PR, Wu NP, André VM et al (2009) Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci 29:2414–2427
Benn CL, Slow EJ, Farrell LA et al (2007) Glutamate receptor abnormalities in the YAC128 transgenic mouse model of Huntington’s disease. Neuroscience 147:354–372
Huang SS, He J, Zhao DM, Xu XY, Tan HP, Li H (2010) Effects of mutant huntingtin on mGluR5-mediated dual signaling pathways: implications for therapeutic interventions. Cell Mol Neurobiol 30:1107–1115
Tong X, Ao Y, Faas GC et al (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci 17:694–703
Bennett EJ, Shaler TA, Woodman B et al (2007) Global changes to the ubiquitin system in Huntington’s disease. Nature 448:704–708
Martinez-Vicente M, Talloczy Z, Wong E et al (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 13:567–576
Qi L, Zhang XD, Wu JC et al (2012) The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE 7:e46834
Kennedy L, Evans E, Chen CM et al (2003) Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet 12:3359–3367
Swami M, Hendricks AE, Gillis T et al (2009) Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet 18:3039–3047
Dragileva E, Hendricks A, Teed A et al (2009) Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis 33:37–47
Bates GP, Dorsey R, Gusella JF et al (2015) Huntington disease. Nat Rev Dis Primers 1:15005
Tabrizi SJ, Reilmann R, Roos RA et al (2012) Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol 11:42–53
Ross CA, Aylward EH, Wild EJ et al (2014) Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 10:204–216
Kieburtz K, Penney JB, Corno P, Ranen N, Shoulson I, Feigin A, Abwender D et al (2001) Unified Huntington’s disease rating scale: reliability and consistency. Huntington Study Group. Mov Disord 11:136–142
Paulsen JS, Langbehn DR, Stout JC et al (2008) Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 79:874–880
van Duijn E, Kingma EM, van der Mast RC (2007) Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci 19:441–448
Craufurd D, Snowden J (2002) Neuropsychological and neuropsychiatric aspects of Huntington’s disease, in Huntington’s disease. In: PS H (ed) Huntington’s disease. Oxford Medical Publications, Oxford
Novak MJ, Tabrizi SJ (2010) Huntington’s disease. BMJ 340:c3109
Lanska DJ, Lanska MJ, Lavine L, Schoenberg BS (1988) Conditions associated with Huntington’s disease at death. A case-control study. Arch Neurol 45:878–880
Paulsen JS, Hoth KF, Nehl C, Stierman L (2005) Critical periods of suicide risk in Huntington’s disease. Am J Psychiatry 162:725–731
Lipe H, Schultz A, Bird TD (1993) Risk factors for suicide in Huntingtons disease: a retrospective case controlled study. Am J Med Genet 48:231–233
Videnovic A, Leurgans S, Fan W, Jaglin J, Shannon KM (2009) Daytime somnolence and nocturnal sleep disturbances in Huntington disease. Parkinsonism. Relat Disord 15:471–474
van der Burg JM, Björkqvist M, Brundin P (2009) Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol 8:765–774
Myers RH, Sax DS, Koroshetz WJ et al (1991) Factors associated with slow progression in Huntington’s disease. Arch Neurol 48:800–804
Andrew SE, Goldberg YP, Kremer B et al (1993) The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat Genet 4:398–403
Craufurd D, MacLeod R, Frontali M, et al (2014) Diagnostic genetic testing for Huntington’s disease. Pract Neurol
Wild EJ, Mudanohwo EE, Sweeney MG et al (2008) Huntington’s disease phenocopies are clinically and genetically heterogeneous. Mov Disord 23:716–720
Hensman Moss DJ, Poulter M, Beck J et al (2014) C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 82:292–299
Novak MJ, Tabrizi SJ (2011) Huntington’s disease: clinical presentation and treatment. Int Rev Neurobiol 98:297–323
Bonelli RM, Hofmann P (2007) A systematic review of the treatment studies in Huntington’s disease since 1990. Expert Opin Pharmacother 8:141–153
Frank S (2014) Treatment of Huntington’s disease. Neurotherapeutics 11:153–160
Group HS (2006) Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology 66:366–372
Guay DR (2010) Tetrabenazine, a monoamine-depleting drug used in the treatment of hyperkinetic movement disorders. Am J Geriatr Pharmacother 8:331–373
Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C (2009) Therapeutic interventions for symptomatic treatment in Huntington’s disease. Cochrane Database Syst Rev CD006456
Craufurd D, Tyler A (1992) Predictive testing for Huntington’s disease: protocol of the UK Huntington’s Prediction Consortium. J Med Genet 29:915–918
Went L (1990) Ethical issues policy statement on Huntington’s disease molecular genetics predictive test. International Huntington Association. World Federation of Neurology. J Med Genet 27:34–38
Guidelines for the molecular genetics predictive test in Huntington’s disease. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington’s Chorea. Neurology 1994;44:1533–6
Semaka A, Hayden MR (2014) Evidence-based genetic counselling implications for Huntington disease intermediate allele predictive test results. Clin Genet 85:303–311
Harper PS, Lim C, Craufurd D (2000) Ten years of presymptomatic testing for Huntington’s disease: the experience of the UK Huntington’s Disease Prediction Consortium. J Med Genet 37:567–571
Keiser MS, Kordasiewicz HB, McBride JL (2016) Gene suppression strategies for dominantly inherited neurodegenerative diseases: lessons from Huntington’s disease and spinocerebellar ataxia. Hum Mol Genet 25:53–64
Harper SQ, Staber PD, He X et al (2005) RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A 102:5820–5825
Stanek LM, Sardi SP, Mastis B et al (2014) Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum Gene Ther 25:461–474
Kordasiewicz HB, Stanek LM, Wancewicz EV et al (2012) Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 74:1031–1044
McBride JL, Pitzer MR, Boudreau RL et al (2011) Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther 19:2152–2162
Grondin R, Kaytor MD, Ai Y et al (2012) Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain 135:1197–1209
Datson NA, González-Barriga A, Kourkouta E et al (2017) The expanded CAG repeat in the huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain. PLoS ONE 12:e0171127
Pfister EL, Kennington L, Straubhaar J et al (2009) Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol 19:774–778
Miller JRC, Pfister EL, Liu W et al (2017) Allele-selective suppression of mutant Huntingtin in primary human blood cells. Sci Rep 7:46740
Southwell AL, Skotte NH, Kordasiewicz HB et al (2014) In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther 22:2093–2106
Carroll JB, Warby SC, Southwell AL et al (2011) Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther 19:2178–2185
Cox DB, Platt RJ, Zhang F (2015) Therapeutic genome editing: prospects and challenges. Nat Med 21:121–131
Shin JW, Kim KH, Chao MJ et al (2016) Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet 25:4566–4576
Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of IONIS-HTTRx in Patients With Early Manifest Huntington’s Disease. NCT02519036. Aug 1st 2015
Miller TM, Pestronk A, David W et al (2013) An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 12:435–442
Finkel RS, Chiriboga CA, Vajsar J et al (2016) Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388:3017–3026
Di Pardo A, Maglione V, Alpaugh M et al (2012) Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc Natl Acad Sci U S A 109:3528–3533
Süssmuth SD, Haider S, Landwehrmeyer GB et al (2015) An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br J Clin Pharmacol 79:465–476
Giampà C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR (2010) Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS ONE 5:e13417
Health NIo. A Phase 2, Double-Blind Randomized, Sequential Treatment Group, Placebo-Controlled Study To Evaluate The Safety, Tolerability And Brain Cortico-Striatal Function Of 2 Doses Of PF-02545920 In Subjects With Early Huntington’s Disease. Pfizer; 2014
Simmons DA, Belichenko NP, Yang T et al (2013) A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J Neurosci 33:18712–18727
Todd D, Gowers I, Dowler SJ et al (2014) A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington’s disease. PLoS ONE 9:e87923
Verny C, Bachoud-Lévi AC, Durr A et al (2017) A randomized, double-blind, placebo-controlled trial evaluating cysteamine in Huntington’s disease. Mov Disord 32:932–936
Zwilling D, Huang SY, Sathyasaikumar KV et al (2011) Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 145:863–874
Beaumont V, Mrzljak L, Dijkman U et al (2016) The novel KMO inhibitor CHDI-340246 leads to a restoration of electrophysiological alterations in mouse models of Huntington’s disease. Exp Neurol 282:99–118
Brück W, Pförtner R, Pham T et al (2012) Reduced astrocytic NF-κB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol 124:411–424
Comi G, Jeffery D, Kappos L et al (2012) Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med 366:1000–1009
A Clinical Study in Subjects With Huntington’s Disease to Assess the Efficacy and Safety of Three Oral Doses of Laquinimod (LEGATO-HD). Teva Branded Pharmaceutical Products, R&D Inc. 2016 ongoing
Jin J, Albertz J, Guo Z et al (2013) Neuroprotective effects of PPAR-γ agonist rosiglitazone in N171-82Q mouse model of Huntington’s disease. J Neurochem 125:410–419
Wild EJ, Tabrizi SJ (2014) Targets for future clinical trials in Huntington’s disease: What’s in the pipeline? Mov Disord 29:1434–1445
Miller BR, Dorner JL, Shou M et al (2008) Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience 153:329–337
Sontag EM, Joachimiak LA, Tan Z et al (2013) Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates mutant Huntingtin cellular phenotypes. Proc Natl Acad Sci U S A 110:3077–3082
Labbadia J, Novoselov SS, Bett JS et al (2012) Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain 135:1180–1196
Mielcarek M, Landles C, Weiss A et al (2013) HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol 11:e1001717
Barker RA, Mason SL, Harrower TP et al (2013) The long-term safety and efficacy of bilateral transplantation of human fetal striatal tissue in patients with mild to moderate Huntington’s disease. J Neurol Neurosurg Psychiatry 84:657–665
Reuter I, Tai YF, Pavese N et al (2008) Long-term clinical and positron emission tomography outcome of fetal striatal transplantation in Huntington’s disease. J Neurol Neurosurg Psychiatry 79:948–951
Bachoud-Lévi AC, Rémy P, Nguyen JP et al (2000) Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet 356:1975–1979
Bachoud-Lévi AC, Gaura V, Brugières P et al (2006) Effect of fetal neural transplants in patients with Huntington’s disease 6 years after surgery: a long-term follow-up study. Lancet Neurol 5:303–309
Capetian P, Knoth R, Maciaczyk J et al (2009) Histological findings on fetal striatal grafts in a Huntington’s disease patient early after transplantation. Neuroscience 160:661–675
Gallina P, Paganini M, Lombardini L et al (2010) Human striatal neuroblasts develop and build a striatal-like structure into the brain of Huntington’s disease patients after transplantation. Exp Neurol 222:30–41
Hauser RA, Furtado S, Cimino CR et al (2002) Bilateral human fetal striatal transplantation in Huntington’s disease. Neurology 58:687–695
Wijeyekoon R, Barker RA (2011) The current status of neural grafting in the treatment of Huntington’s disease. A Review. Front Integr Neurosci 5:78
Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C (2009) Therapeutic interventions for disease progression in Huntington’s disease. Cochrane Database Syst Rev CD006455
Announcement of 2CARE Early Study Closure. http://huntingtonstudygroup.org/tag/2care/2014
Group HS nnouncement of CREST-E Early Study Closure. http://huntingtonstudygroup.org/tag/crest-e/2014
Investigators HSGH (2013) A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington’s disease. Mov Disord 28:1407–1415
de Yebenes JG, Landwehrmeyer B, Squitieri F et al (2011) Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol 10:1049–1057
Reilmann R, AM, Landwehrmeyer G, Kieburtz K, Grachev I, Eyal E, Savola J, Borowsky B, Papapetropoulos S, Hayden M (2017) Efficacy, safety, and tolerability of pridopidine in Huntington disease (HD): results from the phase ii dose-ranging study, pride-HD. In: 21st international congress of parkinson’s disease and movement disorders, 2017, Vancouver, BC, Mov Disord. 32(suppl 2), http://www.mdsabstracts.org/abstract/efficacy-safety-and-tolerability-of-pridopidine-in-huntington-disease-hd-results-from-the-phase-ii-dose-ranging-study-pride-hd/
Frank S, Testa CM, Stamler D et al (2016) Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA 316:40–50
Frank S, Stamler D, Kayson E et al (2017) Safety of converting from tetrabenazine to deutetrabenazine for the treatment of chorea. JAMA Neurol 74:977–982
Deep Brain Stimulation (DBS) of the Globus Pallidus (GP) in Huntington’s Disease (HD) (HD-DBS). 2015, ongoing
Investigators HSGRH. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2015;14:39–47
Effects of EGCG (Epigallocatechin Gallate) in Huntington’s Disease (ETON-Study) (ETON)
Neuroleptic and Huntington Disease Comparison of: Olanzapine, la Tetrabenazine and Tiapride (NEUROHD
Tang CC, Feigin A, Ma Y et al (2013) Metabolic network as a progression biomarker of premanifest Huntington’s disease. J Clin Invest 123:4076–4088
Sturrock A, Laule C, Decolongon J et al (2010) Magnetic resonance spectroscopy biomarkers in premanifest and early Huntington disease. Neurology 75:1702–1710
Stout JC, Queller S, Baker KN et al (2014) HD-CAB: a cognitive assessment battery for clinical trials in Huntington’s disease 1,2,3. Mov Disord 29:1281–1288
Wild EJ, Boggio R, Langbehn D et al (2015) Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J Clin Invest 125:1979–1986
Rodrigues FB, Byrne L, McColgan P et al (2016) Cerebrospinal fluid total tau concentration predicts clinical phenotype in Huntington’s disease. J Neurochem 139:22–25
Byrne LM, Rodrigues FB, Blennow K et al (2017) Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: a retrospective cohort analysis. Lancet Neurol 16:601–609
Acknowledgements
Dr. Rhia Ghosh is funded entirely by a Medical Research Council UK Clinical Research Fellowship.
Professor Sarah J Tabrizi receives grant funding for her research from the EU FP7 health call, Medical Research Council UK, CHDI Foundation, Huntington Disease Association of the UK, Dementiaand Neurodegenerative Disease Network UK, European Huntington’s Disease Network, the Wellcome Trust, the UCL/UCLH Biomedical Research Centre and BBSRC.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Ghosh, R., Tabrizi, S.J. (2018). Clinical Features of Huntington’s Disease. In: Nóbrega, C., Pereira de Almeida, L. (eds) Polyglutamine Disorders. Advances in Experimental Medicine and Biology, vol 1049. Springer, Cham. https://doi.org/10.1007/978-3-319-71779-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-71779-1_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-71778-4
Online ISBN: 978-3-319-71779-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)