
Abstract
Inherited ichthyoses are a group of genetic disorders characterized by dry skin, hyperkeratosis, and increased scale desquamation, often associated with erythroderma. The clinical manifestations are due to a series of mutations of genes involved in the development of the stratum corneum and skin barrier formation. The group consists of phenotypic expressions limited only in the skin and its appendages and others with additional extracutaneous involvement. The incidence of ichthyotic conditions in pigmented and black skin ethnic populations is not known. In two reports from the Eastern Province of Saudi Arabia, the incidence of hereditary ichtyosis and non-bullous congenital ichthyosiform erythroderma has been assessed, respectively, 7 per 1000 and 2 per 1000 new patients. These relatively high rates may be attributed to high consanguinity in countries of the Middle East [1, 2]. The commonest type of hereditary ichthyosis was ichthyosis vulgaris followed by non-bullous ichthyosiform erythroderma; 90% of the patients in this group were born with collodion membranes. Also in populations in East Africa, ichthyoses are not rarely seen.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others



Inherited ichthyoses are a group of genetic disorders characterized by dry skin, hyperkeratosis, and increased scale desquamation, often associated with erythroderma. The clinical manifestations are due to a series of mutations of genes involved in the development of the stratum corneum and skin barrier formation. The group consists of phenotypic expressions limited only in the skin and its appendages and others with additional extracutaneous involvement. The incidence of ichthyotic conditions in pigmented and black skin ethnic populations is not known. In two reports from the Eastern Province of Saudi Arabia, the incidence of hereditary ichtyosis and non-bullous congenital ichthyosiform erythroderma has been assessed, respectively, 7 per 1000 and 2 per 1000 new patients. These relatively high rates may be attributed to high consanguinity in countries of the Middle East [1, 2]. The commonest type of hereditary ichthyosis was ichthyosis vulgaris followed by non-bullous ichthyosiform erythroderma; 90% of the patients in this group were born with collodion membranes. Also in populations in East Africa, ichthyoses are not rarely seen.
40.1 Pathogenesis
In the process of epidermal differentiation, intermediate keratin filaments responsible for the structural integrity of keratinocytes are among the first proteins to be expressed in a tightly regulated manner. Keratins 5 and 14 are expressed in the basal layers and keratins 1 and 10 in the suprabasal layers. This overall process of differentiation results in the formation of the stratum corneum as a robust barrier composed of keratinocytes and inter-keratinocyte lipids. Mutations in proteins essential for the formation of this barrier, i.e., keratins and enzymes involved in lipid synthesis, lead to the disruption of its integrity resulting in ichthyotic conditions. Mutations in over 50 genes have been reported to cause ichthyoses, affecting a host of cellular functions including DNA repair, lipid biosynthesis, adhesion, and desquamation, as well as other processes. Based on these findings, a recent consensus conference revised the nomenclature and proposed a new clinical classification that distinguishes 36 different types of ichthyosis and 10 related clinical entities [3]. Major consensus was achieved on the proposal to categorize inherited ichthyosis into two types: Non-syndromic ichthyoses, in which the phenotypic expression of the underlying genetic defect is only seen in the skin, and syndromic ichthyoses, in which the phenotypic expression is also manifested in other organs (Table 40.1).
40.2 Clinical Pictures
Clinical hallmark of all entities is thickening of the cornified layer with visible scaling, often accompanying with features of inflammation presenting as erythroderma. Other symptoms include painful fissuring of the thickened skin, decreased range of motion at joints, decreased tactile sensitivity, hypohidrosis with heat intolerance, and pruritus. In darkly pigmented and black skin, the diagnosis and classification could be challenging because erythroderma is difficult to assess. In addition, patients in tropical countries are faced with challenges of heat intolerance, infections, and social stigma. Families with disfiguring skin diseases usually have difficulties integrating into society, and thus marriage is not encouraged. To safeguard the future of other children, patients with ichthyoses are being isolated and denied further education, thus having no chance for future employment [4].
Non-syndromic ichthyoses include mainly the common ichthyosis vulgaris, a recessive X-linked ichthyosis with delayed onset, the large group of congenital autosomal recessive ichthyotic conditions, and a group of keratinopathic ichthyoses which show flat erosions and bulla formation; the syndromic types are also considered in short.
40.2.1 Non-syndromic Ichthyoses
-
1.
Ichthyosis Vulgaris
Ichthyosis vulgaris is the most common clinical type of ichthyosis, with an overall incidence of 1:250 to 1:1000. It is characterized by xerosis, scaling, pruritus, and eczema, strongly associated with atopic symptoms. The phenotypic manifestations tend to appear from the age of 2 months and often improve in summer. The extensor sides of the lower legs and the back are the most commonly affected location. The chest and the abdomen are often less involved; keratosis pilaris and palmoplantar hyperlinearity are often seen. The discovery of mutations of filaggrin (FLG) as a basic figure of ichthyosis vulgaris led to the insight that the very same loss-of-function mutations confer a strong predisposition to atopic dermatitis [5, 6]. Although the presence of population-specific FLG mutations has been reported in Europeans, Asians, and Africans [7], it is likely that additional factors other than these mutations are involved in African Americans [8] and in Ethiopians [9].
-
2.
Recessive X-Linked Ichthyosis
Recessive X-linked ichthyosis is the second most common ichthyosis with a prevalence of 1:2000 to 1:6000, caused by hereditary deficiency of the steroid sulfatase (STS) enzyme due to mutations of the STS gene. The disease manifests exclusively in males. Cutaneous manifestations are present soon after birth and usually tend not to improve with age. In its clinical expression, X-linked ichthyosis is more severe than ichthyosis vulgaris, as large, dark brown prominent scales form, adhering to the underlying skin and covering the entire body including flexural areas (Fig. 40.1). In mild cases the scaling is less distinguishable but also visible in dark and black skin. Additional features may be present, such as intellectual disability, autism, or attention deficit with hyperactivity, partly influenced by the deletion size of the gene.
Fig. 40.1 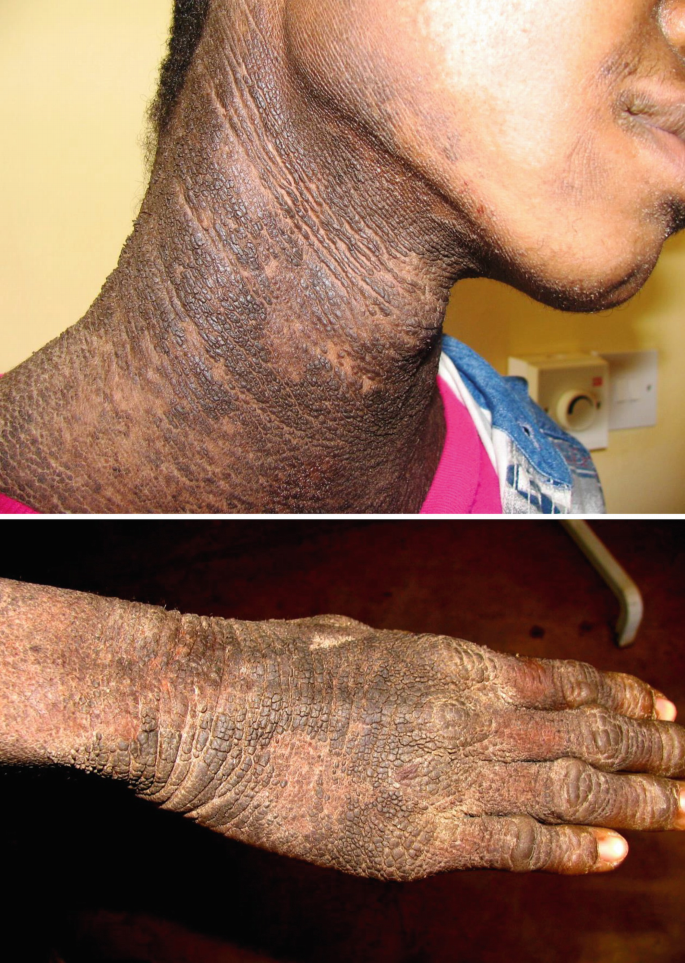
Severe ichthyosis showing hystrix type of hyperkeratoses in joints and hands in a young Tanzanian male (most likely autosomal recessive X-chromosomal type)
-
3.
Autosomal Recessive Congenital Ichthyoses (Non-bullous)
Autosomal recessive congenital ichthyosis (ARCI) is a comprehensive definition term used to represent a generic phenotype of erythrodermic, scaly skin presenting over almost the entire body surface at birth. The ARCI group is clinically divided into three major phenotypes and two subtypes.
-
(a)
Harlequin ichthyosis
Harlequin ichthyosis is phenotypically the most severe inherited ichthyosis, occasionally fatal. The clinical features include thick, platelike scales with severe ectropion, eclabium, and flattening of the ears. The diagnosis is clinically not difficult, because of its characteristic phenotype.
-
(b)
Lamellar ichthyosis
Lamellar ichthyosis is a major phenotype of autosomal recessive congenital ichthyosis with varying severity (Fig. 40.2a, b), clinically more mild than the harlequin phenotype. The characteristic scales are large, often thickened and dark gray or brown in color, also covering most of the body flexural areas. Palmoplantar keratoderma is frequently seen. A large number of patients who show later in life the classic lamellar phenotype were born as collodion babies (Fig. 40.3). In a Moroccan series, 67% of collodion babies evoluted on lamellar ichthyosis and 21% on congenital ichthyosiform erythroderma [10].
Fig. 40.2 
Lamellar ichthyosis; (a) mild expression of the trunk; (b) severe, generalized involvement
Fig. 40.3 
Collodion babies (a) shortly after birth; (b) persisting ectropion in a 4-year-old child; (c) collodion baby several weeks after birth with severe epidermolytic erythroderma
-
(c)
Congenital ichthyosiform erythroderma
Ichthyosiform erythroderma is another major phenotype of congenital ichthyosis (3b,c); patients are frequently born as collodion babies. After the collodion membrane is dropped, erythroderma and scaling appear. The erythroderma often improves in infancy, but in severe cases the erythrodermic feature becomes persistent. In adults generalized lamellar-type scaling is seen, occasionally with persisting ectropion (Fig. 40.4); in other cases there is prominent dry scaling without erythrodermic features (Fig. 40.5).
Fig. 40.4 
Congenital ichthyosiform erythroderma in an adult, (a) with persisting ectropion (face treated topically with retinoids); (b) severe involvement of the trunk (untreated)
Fig. 40.5 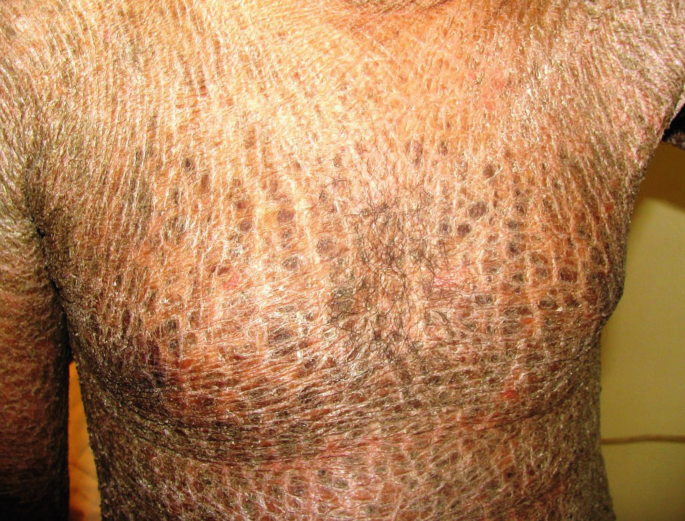
Late stage of congenital ichthyosiform erythroderma; non-bullous, lamellar-type scaling without erythrodermic features
-
(d)
Self-healing collodion baby
Collodion babies present at birth with erythroderma and shiny, tight skin resembling parchment covering the neonate’s body and may cause ectropion, eclabium, flattening of the ears and nostrils, restricted extension of the digits and extremities, and limitation of chest movement. It spares the mucosal surfaces and stops at the edge of natural orifices and at the cutaneous level of the umbilical cord. Hairs usually penetrate the collodion membrane and nails are exceptionally covered. The membrane gradually peels off during the first 4 weeks, but may not completely disappear until the third month of life usually revealing an ichthyotic phenotype. Hypohidrosis is often associated. Less common features include poor sucking, restricted pulmonary ventilation, digital vascular constriction, and edema of the extremities.
-
(e)
Bathing suit ichthyosis
Bathing suit ichthyosis is another minor variant of ARCI, characterized by unique distribution of lesions on the trunk, the most proximal parts of the upper limbs, the scalp, and the neck, but not the central face and the extremities. This unusual phenotype has been first described in a black-skinned population in South Africa [11], and several cases from different ethnic backgrounds were recently reported [12].
-
(a)
-
4.
Keratinopathic Ichthyoses (bullous)
The term keratinopathic ichthyosis has been proposed as a comprehensive umbrella for patients with ichthyosis presenting epidermolysis often leading to bulla formation; the group includes patients categorized as epidermolytic ichthyosis or superficial epidermolytic ichthyosis and other rare types described such as annular epidermolytic ichthyosis and ichthyosis type Curth-Macklin.
-
(a)
Epidermolytic ichthyosis (bullous ichthyosiform erythroderma)
This subtype is the most prevalent phenotype of keratinopathic ichthyosis characterized by multiple erosions with erythroderma and widespread formation of blisters. The patients show erythema and blistering at birth, diminishing with age, and generalized epidermolytic hyperkeratosis in adulthood (Fig. 40.6).
Fig. 40.6 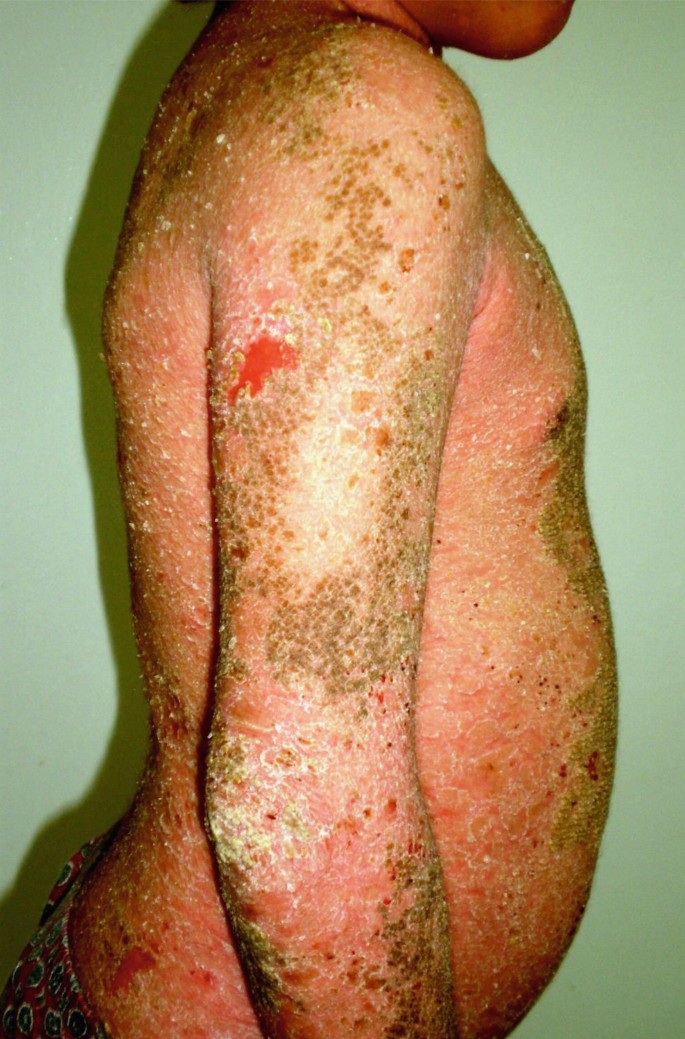
Epidermolytic ichthyosis showing flat erosions and superficial blistering
-
(b)
Superficial epidermolytic ichthyosis (bullous ichthyosis of Siemens)
The term superficial epidermolytic ichthyosis had been proposed to replace the formerly well-defined entity ichthyosis bullosa Siemens, which shows a more superficial pattern of epidermolysis and bulla formation. It is clinically characterized by mild epidermal hyperkeratosis over the flexural areas and the development of denuded hyperkeratotic skin with superficial blistering.
-
(c)
Ichthyosis hystrix , type Curth-Macklin
This is a rare, autosomal dominant disorder characterized by extensive, spiky, or verrucous hyperkeratoses affecting preferably the large joints and the extremities, with or without palmoplantar keratoderma (Fig. 40.7).
Fig. 40.7 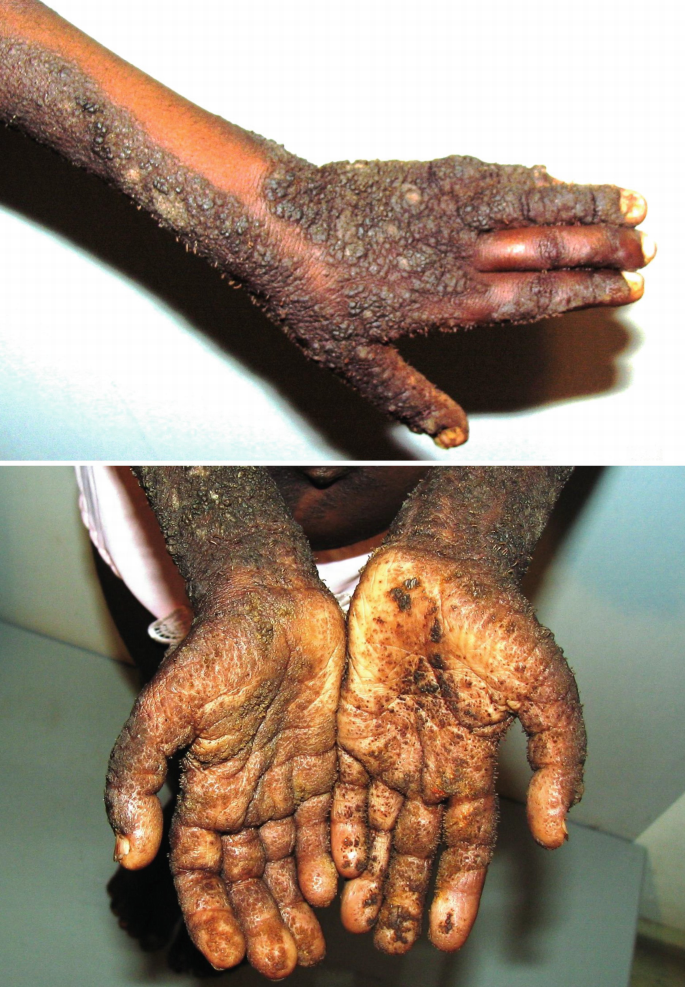
Ichthyosis hystrix, severe type Lambert-Curth-Macklin in an African male (Tanzania), with prominent verrucous, spiky hyperkeratoses on the extremities including the palms and soles (sauriasis)
-
(a)







40.2.2 Syndromic Ichthyoses
-
1.
Refsum Syndrome
Refsum syndrome develops during childhood to adolescence. Most patients show a progressive course, but remission and exacerbation are repeated in some patients. Four of its signs are pigmentous retinitis, peripheral neuritis, cerebellar ataxia, and increased levels of protein in the cerebrospinal fluid. In addition, symptoms, such as anosmia, hemeralopia, vision disorder, cataract, muscular atrophy, cardiomyopathy, bone disorder, progressive myelin degeneration, functional disorder of the nervous system, cochlear hearing loss, ichthyosis, and renal failure, are observed. The skin is dry and cornification is marked, with furfuraceous desquamation. Most scales are brown. Especially in the trunk, cornification is marked. Furthermore, hyperkeratosis is also marked at the olecranon and patella. The diagnosis is verified by the presence of phytanic acid in plasma lipid fractions using gas chromatography.
-
2.
Netherton Syndrome
Netherton syndrome is characterized by three main clinical features: (1) a congenital ichthyotic phenotype of varying expression and severity (ichthyosis linearis circumflexa), (2) hair abnormalities, and (3) atopic predisposition and symptoms. Some patients show symmetrical erythematous lesions in various shapes, such as tortuous and double-edged scaling (Fig. 40.8), whereas in other cases generalized dryness and lichenification predominates. Scalp hairs are short, sparse, dry, and not lustrous; under a light microscope, characteristic nodes of the hair shafts are found, also called bamboo hair, whereby the distal part of the hair shaft invaginates into the proximal (trichorrhexis invaginata). However, bamboo hairs may appear later, and the number and formation of the nodes differ among patients; eyelash or eyebrow defects are also observed in some cases. Chronic hives, bronchial asthma, allergic rhinitis, hay fever, an increase in the serum IgE levels, and eosinophilia have been reported as additional symptoms.
Fig. 40.8 
Netherton syndrome with dry skin and double edge scaling on the extermities (mutation of SPINKS5 and bamboo hairs confirmed)
-
3.
Sjögren-Larsson Syndrome
Primary symptoms include symmetric spastic paralysis of the limbs, particularly marked in their lower part, mental retardation, and congenital ichthyosiform exanthema (Fig. 40.9). Mental retardation is relatively severe, and more than 50% of patients show an IQ of 50 or lower. In most patients, mental developmental retardation is detected during childhood. Gait disorder is often observed. Convulsion is noted in approximately 40% of the patients.
Fig. 40.9 
Associated ichthyosis in Sjögren-Larson syndrome


40.3 Management
There is no definitive treatment for congenital ichthyoses; severe cases usually do not survive the neonatal period. The management of these cases is a challenge for all health services particularly in developing countries [13]. Nevertheless, recent advances in neonatal intensive care and coordinated multidisciplinary management have greatly improved the overall survival [14]. Although some forms may show spontaneous and seasonal variations, most patients require daily management throughout life. The therapeutic outcome is largely limited by nonadherence, as treatments are not only time consuming and involve greasy products but often show disappointing results and patients frequently withdraw from management. The birth of a child with severe congenital ichthyosis represents a shock for the parents. The psychological aspects must be taken in consideration when dealing with these unfortunate patients.
Neonatal care of severe ichthyosis: The collodion baby is born encased in a transparent, parchment-like membrane, which can interfere with respiration and sucking. Over the first 2 weeks of life, the membrane breaks up and desquamates. During this time, management should include careful monitoring of body temperature, hydration, and blood electrolytes. Management should keep the skin soft and pliable to reduce pain from deep skin fissures and to promote desquamation by using a humidified incubator and application of lubricants. The same basic regimen, although more rigorously exercised, also applies to babies suffering from the harlequin phenotype or epidermolytic hyperkeratosis, two forms of ichthyosis which are potentially lethal in the neonatal period.
Bathing and bath additives: Several years ago, patients with ichthyosis were strongly discouraged from bathing to not affect skin surface pH. Today, we know that ichthyosis patients should take a cleansing bath daily and rub their skin gently to mechanically remove some of their scales. In order to improve the barrier function and facilitate desquamation, sodium bicarbonate could be added to the bath water [15].
Hydration and lubrication: This can be accomplished by creams and ointments containing low concentrations of salt, urea (10%), or glycerol, which increase the water-binding capacity of the horny layer. For ichthyoses with thick scaling and markedly increased stratum corneum thickness, addition of one or more keratolytic agents is needed to decrease corneocyte cohesiveness, promote desquamation, and dissolve keratins and lipids. A wide variety of topical keratolytics can be used, including the α-hydroxy acids (e.g., lactic acid and glycolic acid), salicylic acid, high-dose urea, and propylene glycol. Also 2×/day topical application of 10% N-acetylcysteine as a mucolytic agent in combination with 5% urea in a water-in-oil emulsion showed excellent results in lamellar ichthyosis with no significant side effects [16].
Retinoids are particularly helpful for treatment of ichthyotic and hyperkeratotic conditions [17]. Topically they are used in certain anatomic locations including the face, palms and soles, and also in eyelids for treatment of ectropion. Systemic retinoids are widely administered in severe disease but require close follow-up of patients. Isotretinoin or acitretin 0.5–1.0 mg/kg/day should not be exceeded, and the lowest possible therapeutic dose needs to be titrated. Adverse effects may include mucocutaneous toxicities, hair loss, skeletal hyperostoses, and laboratory abnormalities in blood cell counts and serum lipids. Retinoic acid metabolism blocking agents such as liarozole were shown effective with a favorable tolerability profile [18]; in moderate/severe lamellar ichthyosis, 75–150 mg/day oral liarozole improved scaling and life quality index [19].
40.4 Course and Further Monitoring
Most patients require lifelong treatment with daily applications of emollients all over the body. They are potential high consumers of such products which are costly, often not reimbursed by health insurances considered as cosmetics. Topical treatment takes time and often has to be repeated many times per day. In children, one should try to convert the therapeutic sessions into something relaxing and positive.
40.5 Prenatal Diagnosis and Genetic Counseling
Parents of children with ichthyosis should be offered appropriate genetic counseling to explain the nature of the disorder, its mode of inheritance, and the probability of future manifestations in the family. In some developed countries, prenatal testing is offered for individuals or couples at risk of having children with severe type of congenital ichthyoses, performed by chorionic villus sampling at 10–12 weeks of gestation. The ethical situation may differ for other types such as moderate lamellar ichthyosis. Helpful contact details can be downloaded from the Foundation for Ichthyosis and Related Skin Types (F.I.R.S.T.) and the Network for Ichthyoses and Related Keratinization disorders (NIRK). Unfortunately, patient organizations and foundations supporting patients with ichthyosis are almost limited to Western countries.
References
Al-Zayir AA, Al-Amro Al-Alakloby OM. Clinico-epidemiological features of primary hereditary ichthyoses in the eastern province of Saudi Arabia. Int J Dermatol. 2006;45:257–64.
Al-Amro Al-Akloby OM, Al-Zayir AA. Clinico-epidemiological features of congenital nonbullous ichthyosiform erythroderma in the eastern province of Saudi Arabia. J Eur Acad Dermatol Venereol. 2004;18:659–64.
Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the first Ichthyosis consensus conference in Soreze 2009. J Am Acad Dermatol. 2010;63:607–41.
Ibekwe PU, Ogunbiyi AO, Ogun OG, et al. Social stigmatization of two sisters with lamella ichthyosis in Ibadan, Nigeria. Int J Dermatol. 2012;51:67–8.
Palmer CN, Irvine AD, Terron-Kwiatkowski A, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–6.
Sandilands A, Terron-Kwiatkowski A, Hull PR, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39:650–4.
Taylan F, Nilsson D, Asad S, et al. Whole-exome sequencing of Ethiopian patients with ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol. 2015;136:507–9.e519.
Polcari I, Becker L, Stein SL, et al. Filaggrin gene mutations in African Americans with both ichthyosis vulgaris and atopic dermatitis. Pediatr Dermatol. 2014;31:489–92.
Taylan F, Nilsson D, Asad S, et al. Whole-exome sequencing of Ethiopian patients with ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol. 2015;136:507–9.e19.
Khadir K, Benharbit B, Habibeddine S, et al. Treatment outcome for collodion babies: the experience of the dermatology Department of Ibn Rochd Teaching Hospital. Casablanca Ann Dermatol Venereol. 2009;136:731–2.
Jacyk WK. Bathing-suit ichthyosis. A peculiar phenotype of lamellar ichthyosis in South African blacks. Eur J Dermatol. 2005;15:433–6.
Benmously-Mlika R, Zaouak A, Mrad R, et al. Bathing suit ichthyosis caused by a TGM1 mutation in a Tunisian child. Int J Dermatol. 2014;53:1478–80.
Migowa AN, Murungi CW, Gatinu BW, et al. Harlequin ichthyosis in an African child: case report. East Afr Med J. 2010;87:389–92.
Glick JB, Craiglow BG, Choate KA, et al. Improved management of harlequin ichthyosis with advances in neonatal intensive care. Pediatrics. 2017;139(1). https://doi.org/10.1542/peds.2016-1003.
Traupe H, Burgdorf WHC. Treatment of ichthyosis—there is always something you can do! In memoriam: Wolfgang Küster. J Am Acad Dermatol. 2007;57:542–7.
Bassotti A, Moreno S, Criado E. Successful treatment with topical N-acetylcysteine in urea in five children with congenital lamellar ichthyosis. Pediat Dermatol. 2011;28:451–5.
Digiovanna JJ, Mauro T, Milstone LM, et al. Systemic retinoids in the management of ichthyoses and related skin types. Dermatol Ther. 2013;26:26–38.
van Steensel MA. Emerging drugs for ichthyosis. Expert Opin Emerg Drugs. 2007;12:647–56.
Vahlquist A, Blockhuys S, Steijlen P, et al. Oral liarozole in the treatment of patients with moderate/severe lamellar ichthyosis: results of a randomized, double-blind, multinational, placebo-controlled phase II/III trial. Br J Dermatol. 2014;170:173–81.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Mokni, M. (2018). Inherited Ichthyoses. In: Orfanos, C., Zouboulis, C., Assaf, C. (eds) Pigmented Ethnic Skin and Imported Dermatoses. Springer, Cham. https://doi.org/10.1007/978-3-319-69422-1_40
Download citation
DOI: https://doi.org/10.1007/978-3-319-69422-1_40
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-69421-4
Online ISBN: 978-3-319-69422-1
eBook Packages: MedicineMedicine (R0)