
Abstract
Circulatory shock is accompanied, and likely to be preceded, by profound alterations in autonomic function. In stable conditions, sympathetic and parasympathetic limbs of the autonomic nervous system work in a highly coordinated manner, by virtue of physical and biochemical afferent signals being transduced into coordinated neural activity to maintain homeostasis in multiple organs innervated by specialized autonomic nerves. In this chapter, we assess how normal autonomic activity is regulated and the practical implications of how perturbation of the autonomic nervous system fuels further detrimental changes in shock. We will also highlight how autonomic regulation of cardiovascular and extra-cardiovascular physiology contributes to circulatory shock and define autonomic dysfunction practically in a clinical context.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Circulatory shock is accompanied, and likely to be preceded, by profound alterations in autonomic function. In stable conditions, sympathetic and parasympathetic limbs of the autonomic nervous system work in a highly coordinated manner, by virtue of physical and biochemical afferent signals being transduced into coordinated neural activity to maintain homeostasis in multiple organs innervated by specialized autonomic nerves. In this chapter, we assess how normal autonomic activity is regulated and the practical implications of how perturbation of the autonomic nervous system fuels further detrimental changes in shock. We will also highlight how autonomic regulation of cardiovascular and extra-cardiovascular physiology contributes to circulatory shock and define autonomic dysfunction practically in a clinical context.
1 Introduction
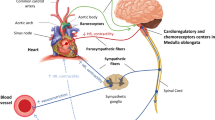
The autonomic system, comprising sympathetic and parasympathetic limbs, plays a crucial role in the homeostatic control of the cardiovascular system. Key neurotransmitters and receptors mediating each limb are summarized in ◘ Fig. 8.1. In healthy subjects, physiological variability in heart rate and blood pressure is controlled by the interplay between the two limbs of autonomic system: parasympathetic and sympathetic system. Contrary to established physiological teaching, advances in autonomic experimental techniques have shed new light on the conventional model of opposing autonomic limbs [1]. Optimal cardiac output requires the simultaneous co-activation of both autonomic limbs, which permits both a longer time for ventricular filling and more efficient contraction of the myocardium [2, 3]. The maintenance of cardiovascular variability in response to routine physiological perturbation is mediated by two key physiological reflexes: the arterial baroreflex and peripheral chemoreflex.


Basic anatomic organization of neurotransmitters of the autonomic nervous system
2 Arterial Baroreflex
The arterial baroreflex mechanism buffers acute fluctuations in blood pressure [4]. The afferent limb of the baroreflex transduces changes in arterial pressure within the aortic arch and carotid sinus into electrical signals, relayed to the brainstem via aortic and glossopharyngeal nerves. These electrical signals are integrated by neurons within the brainstem, principally the nucleus tractus solitarius. Even small increases in blood pressure result in increased parasympathetic activity which, via the vagus nerve, slows the heart. Conversely, falls in arterial blood pressure lead to central neural activation of the sympathetic nervous system and consequent catecholamine release (epinephrine and norepinephrine). Reduced baroreceptor sensitivity is associated with excess morbidity [5] and increased mortality in critical illness [6].
3 Chemoreflex
Respiratory autonomic control is regulated by central and peripheral chemoreceptors, the latter of which are located in the carotid and aortic bodies. Changes in partial pressure of oxygen and carbon dioxide are transduced into neural signals [7]. Through central integration of this information initially within the nucleus tractus solitarii in the brainstem and other higher respiratory centres, changes in ventilation alter efferent autonomic neural signals acting on a diverse array of target organs. Peripheral chemoreceptors influence cardiovascular regulation directly, through hypoxia causing increased heart rate and sympathetic vasoconstrictor nerve activity within skeletal muscle vascular beds, which contributes to the progression of chronic cardiac failure [8]. Acute hypoxia results in resetting of the arterial baroreflex to higher pressures and higher levels of heart rate and muscle sympathetic nerve activity [9]. These effects occur without altering arterial baroreflex sensitivity and are also independent of breathing rate and tidal volume. Thus, autonomic regulation of cardiovascular and respiratory function is very likely to interact during acute shock states. Indeed, peripheral chemoreceptors play a role beyond that of detecting changes in ventilation, since they also sense a broad range of metabolic and inflammatory molecules [10]. Inflammatory mediators robustly increase peripheral chemoreceptor discharge, which is likely to an important reason why respiratory rate is such a strongly predictive clinical parameter for detecting sepsis [11]. Moreover, feedback from the lungs, through hyperventilation driven by hypoxia, acidosis and/or inflammation, also impacts on efferent autonomic activity. Loss of chemoreflex [12] and baroreceptor sensitivity [6] is associated with increased mortality in critical illness [13].
4 Autonomic Variability: An Intrinsic Feature of Health
The successful maintenance of homeostasis, from cells through to the whole organism, requires dynamic interaction between multiple control systems leading to highly complex, variable patterns that are not reflected by static clinical measures such as heart rate and arterial pressure [14]. The maintenance of autonomic variability is a crucial element contributing to this dynamic control of homeostasis, reflecting the ability of afferent, central and efferent autonomic components to detect, and act upon, subtle physiological alterations [15]. While often related to cardiovascular homeostasis, a wealth of recent data shows that the autonomic nervous system also regulates the biological activity of other cell types, including immune cells [16, 17]. Of direct relevance to shock states, both limbs of the autonomic nervous system alter release of inflammatory mediators through the immunomodulatory actions of epinephrine (sympathetic), acetylcholine and vasoactive intestinal peptide (parasympathetic). Although beyond the scope of this chapter, the extra-cardiovascular regulation of inflammation may very well contribute to the magnitude, persistence and/or reversibility of shock states (◘ Fig. 8.2).


Physiologic interaction at the organ level between sympathetic and parasympathetic components of the autonomic nervous system.
5 Key Features of Autonomic Dysfunction in Shock (Circulatory Failure)
Profound changes in autonomic function accompany shock, although the type of shock (cardiogenic, haemorrhagic, septic) may impact upon the precise autonomic phenotype. The autonomic profile of redistributive shock, such as that occurs under general anaesthesia, is further complicated by the peripheral and central actions of pharmacological agents [18]. Human (clinical) studies in humans at the onset of septic or haemorrhagic shock are understandably rare and challenging, so the confounding influence of therapy/sedation needs to be taken into account when understanding the autonomic changes that accompany shock. This means that much of our human physiological understanding of autonomic changes in “pure” shock states is derived from sophisticated physiological experiments where lower body negative pressure is used to produce controlled hypotensive shock [19]. However, it is worth bearing in mind that in patients most susceptible to acquiring infections (e.g. cardiac failure), pre-existing autonomic dysfunction is common and likely to exacerbate the early features of the shock state through cardiovascular and non-cardiovascular mechanisms [20]. Regardless of the model or clinical type of shock, the most ubiquitous autonomic feature of shock is the dramatically heightened activation of the sympathetic nervous system, leading to increased release of catecholamines which spill over into the circulation [21, 22]. Central neural processing of afferent signals further coordinates the emergent neuroendocrine release of vasopressin and angiotensin, amongst other neurohormones, to counteract relative hypovolaemia. Experimental data demonstrate, through direct measurement of neural activity, that sympathetic increases in renal, hepatic, adrenal, splenic and cardiac vascular beds occur during the early phases of shock [23]. Even at very modest levels of hypovolaemia (absolute or redistributive) when heart rate and arterial blood pressure remain unchanged, a baroreflex-mediated increase in muscle sympathetic nerve activity serves to compensate for acute hypovolaemic changes. Non-hypotensive hypovolaemia reduces the diameter of both major arteries containing the stretch-sensitive aortic and carotid arterial baroreceptors, the deactivation of which drives heightened sympathetic drive and parasympathetic withdrawal [24].
6 Autonomic Dysfunction and Cardiovascular Collapse
After the onset of shock, in the absence or presence of clinical intervention, circulatory collapse may develop. This most likely occurs as a result of the acute impairment of arterial baroreflex control, rather than loss of sympathetic vasomotor activity. Once coherence between arterial blood pressure and sympathetic nerve activity is lost, profound vasodilation and decreased systemic vascular resistance occur [24]. Other physiological stressors that are common features of acute critical illness, including pain, anxiety and sympatholytic anaesthetic/analgesic agents disrupt autonomic coherence in clinically unpredictable ways. A similar phenomenon is attributable to circulatory collapse following pathological cardiovagal reflex activity [25], where the sudden attenuation of baroreflex function occurs before haemodynamic decompensation. The often unpredictable, sudden circulatory collapse that ensues once shock is established may be underpinned by individual differences in autonomic function. Multiple experimental studies demonstrate highly variable individual differences in the ability of healthy humans to tolerate central hypovolemia. This observation is likely to be even more pertinent in patients at risk of shock who frequently have pre-existing comorbidity associated with autonomic impairment [5, 26]. Genetic variability has been linked to toleration of shock [27], as well as differences in the release of vasoactive hormones [28], baroreflex gain of sympathetic nerve activation [29] and speed of onset of increased sympathetic neural activity [30]. Myocardial cell injury is readily induced by excessive sympathetic activity during shock [31], and thus persistent sympathetic activation may limit cardiac output through this secondary mode of insult.
7 Autonomic Dysfunction as a Feature of Persistent Shock
The traditional notion of autonomic impairment is exemplified by abnormal responses to the Valsalva manoeuvre and neurological syndromes characterized by paradoxical hypotension or hypertension. Contrary to these clinically defined phenotypes, a far more sophisticated, biologically relevant model has developed with the understanding of how adrenoreceptor expression is regulated. Signalling mechanisms following G-protein-coupled receptor activation (e.g. beta-adrenoreceptor) require dynamic regulatory mechanisms that enable rapid adaptation to meet cellular demands. Within minutes of agonist activation, the process of desensitization of GPCRs begins. In the heart, catecholamines bind to β1 and β2- GPCRs leading to conformational changes in GPCR structure that trigger the dissociation of heterotrimeric G-proteins into α and βγ subunits. In turn, this activates signalling via various downstream proteins. Deactivation of GPCR-elicited signalling is essential for efficient receptor-mediated signalling, requiring GRKs to desensitize the receptor to agonist stimulation. GRKs (chiefly GRK2 AND 5 in the heart) firstly phosphorylate the active receptor to enable binding of β-arrestin, which subsequently may lead to clathrin-induced endocytosis, reactivation or degradation of the receptor. GTPases promote G-protein trimer reformation, which reprimes the GPCR in readiness for further agonist stimulation. When the balance between activation and deactivation is disrupted, cardiac dysfunction develops. In clinical practice, this phenomenon is manifest by acutely raised sympathetic drive (as seen at the onset of septic shock [32]) leading to “cardiac uncoupling” where there is a functional disconnection between sympathetic autonomic function and the physiological response of the cardiac myocyte [33]. Moreover, experimental models, and translational studies, demonstrate that baroreflex dysfunction is associated with reduced cardiac contractility. Reduced baroreceptor sensitivity leads to unrestrained release of (higher) angiotensin. High levels of plasma angiotensin generate injurious release of reactive oxygen species through activation of nicotinamide adenine dinucleotide phosphate oxidase subunit 2 leading to reduced cardiac contractility [26]. This cardiac dysfunction is associated with the upregulation of G-protein-coupled receptor kinase expression in cardiomyocytes [26]. Persistent exposure to elevated sympathetic activity, and hence endogenous catecholamine release, is a core feature of critical illness following shock even after apparently successful resuscitation. For example, prolonged bed rest is likely to contribute to persistent autonomic baroreflex dysfunction [34].
Practical Implications
Although the measurement of autonomic function may have a lot of potential applications, there are several inherent challenges in capturing processing and interpretation these data. Furthermore, pre-existing autonomic impairment is common in surgical patients with a similar clinical profile as those who develop critical illness for different reasons. This suggests that a more detailed understanding of these patients’ autonomic physiology may help us understand rational, targeted treatments either to prevent, or reverse, shock. However, there is still very little evidence about the clinical impact of this information on the management of critically ill patients. For example, heart rate variability has been explored in the critical care setting (including in the early stages of septic shock and traumatic hypovolaemia), but has not been widely adopted because of technical limitations and lack of outcome data. Dynamic tests of beta-adrenoreceptor responsivity appear to hold most promise [35,36,37,38], where the cardiometabolic response to catecholamine infusion appears to identify a relationship between beta-adrenoreceptor signalling/physiology and outcome. Across several studies, the failure of patients to respond to β-1 adrenoreceptor agonists is strongly predictive of outcome. Even where many patients required treatment with vasopressors for persistent circulatory shock, graded dobutamine challenge revealed that preserved cardiac responsivity to dobutamine stimulation was more frequently present in survivors. Early studies, using an intravenous infusion of dobutamine at 10 microg/kg/min for 1 h after resuscitation, demonstrated that survivors were far more likely to increase oxygen consumption by more than 15%. In a study where the majority of patients continued to require pressor support, dobutamine not only increased oxygen delivery and consumption in responders but also exerted a significant metabolic effect as reflected by a greater temperature increase. Thus, an intact cardiometabolic response following a dobutamine “stress test” is consistent with the idea that disruption of beta-adrenoreceptor physiology is pivotal in determining outcome from shock. Further evidence is provided by experimental models of baroreflex dysfunction and clinical measurement of spontaneous baroreflex sensitivity, which further support the hypothesis that disruption of beta-adrenoreceptor recycling underlies the development, and persistence, of circulatory shock.
8 Clinical Interventions
The developing interest in critical care of controlling heart rate with beta-blockers [39], novel sedative agents [40] and early mobilization [41] is likely to exert profound effects on autonomic control. For the reasons outlined previously, the benefits of many of these apparently unconnected interventions may centre on reversing the detrimental effects of prolonged sympathetic activation on receptor recycling mechanisms.
Take-Home Messages
-
Multiple drugs used in the ICU may mask or provoke features of autonomic dysfunction. Cardiovascular therapy needs to take this into account the whole clinical picture and not just focus on numerical targets.
-
Static measures of heart rate and blood pressure do not reflect underlying autonomic compromise. Rapid changes in patient position, or painful/stimulatory interventions, may evoke unpredictable, exaggerated or even ablated changes in heart rate/blood pressure.
-
Tachycardia may reflect a number of underlying pathophysiological features that are typical of critical illness. The clinical exclusion of triggers for tachycardia, such as pain, hypovolaemia and adequate sedation, does not mean that persistently high heart rates are benign.
-
In established critical illness, recurrent episodes of postural hypotension, intermittent pressor requirement, dysrhythmias (including atrial fibrillation) and/or persistent tachycardia should prompt consideration of more in-depth cardiovascular interrogation. Transthoracic echocardiography offers a rapid evaluation of cardiorespiratory physiology that may assist in ruling out primary autonomic abnormalities if pathological alterations are evident.
Conclusion
Shock and critical illness induce profound alterations in autonomic function. Several core features of shock may largely be explained by the pre-existence, or rapid development, of autonomic impairment. The longer-term implications of this are likely to mirror outcomes in cardiovascular disease, where extremes of autonomic impairment are independently predictive of survival.
References
Paton JF, Boscan P, Pickering AE, Nalivaiko E. The yin and yang of cardiac autonomic control: Vago-sympathetic interactions revisited. Brain Res Brain Res Rev. 2005;49(3):555–65.
Machhada A, Marina N, Korsak A, Stuckey DJ, Lythgoe MF, Gourine AV. Origins of the vagal drive controlling left ventricular contractility. J Physiol. 2016;594(14):4017–30.
Machhada A, Trapp S, Marina N, Stephens RCM, Whittle J, Lythgoe MF, et al. Vagal determinants of exercise capacity. Nat Commun. 2017;8:15097.
Wehrwein EA, Joyner MJ. Regulation of blood pressure by the arterial baroreflex and autonomic nervous system. Handb Clin Neurol. 2013;117:89–102.
Toner A, Jenkins N, Ackland GL, POM-O Study Investigators. Baroreflex impairment and morbidity after major surgery. Br J Anaesth. 2016;117(3):324–31.
Sharshar T, Gray F, de la Grandmaison GL, Hopklnson NS, Ross E, Dorandeu A, et al. Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet. 2003;362(9398):1799–805.
Kara T, Narkiewicz K, Somers VK. Chemoreflexes – physiology and clinical implications. Acta Physiol Scand. 2003;177(3):377–84.
Toledo C, Andrade DC, Lucero C, Schultz HD, Marcus N, Retamal M, et al. Contribution of peripheral and central chemoreceptors to sympatho-excitation in heart failure. J Physiol. 2017;595(1):43–51.
Querido JS, Wehrwein EA, Hart EC, Charkoudian N, Henderson WR, Sheel AW. Baroreflex control of muscle sympathetic nerve activity as a mechanism for persistent sympathoexcitation following acute hypoxia in humans. Am J Physiol Regul Integr Comp Physiol. 2011;301(6):R1779–85.
Ackland GL, Kazymov V, Marina N, Singer M, Gourine AV. Peripheral neural detection of danger-associated and pathogen-associated molecular patterns. Crit Care Med. 2013;41(6):e85–92.
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–10.
Schmidt H, Muller-Werdan U, Nuding S, Hoffmann T, Francis DP, Hoyer D, et al. Impaired chemoreflex sensitivity in adult patients with multiple organ dysfunction syndrome – the potential role of disease severity. Intensive Care Med. 2004;30(4):665–72.
Schmidt H, Müller-Werdan U, Hoffmann T, Francis DP, Piepoli MF, Rauchhaus M, et al. Autonomic dysfunction predicts mortality in patients with multiple organ dysfunction syndrome of different age groups*. Crit Care Med. 2005;33(9):1994–2002.
Ernst G. Heart-rate variability-more than heart beats? Front Public Health. 2017;5:240.
Thayer JF, Lane RD. The role of vagal function in the risk for cardiovascular disease and mortality. Biol Psychol. 2007;74(2):224–42.
Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve – an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52(4):595–638.
Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol. 2012;30:313–35.
Neukirchen M, Kienbaum P. Sympathetic nervous system: evaluation and importance for clinical general anesthesia. Anesthesiology. 2008;109(6):1113–31.
Wolthuis RA, Bergman SA, Nicogossian AE. Physiological effects of locally applied reduced pressure in man. Physiol Rev. 1974;54(3):566–95.
van de Borne P, Montano N, Pagani M, Oren R, Somers VK. Absence of low-frequency variability of sympathetic nerve activity in severe heart failure. Circulation. 1997;95(6):1449–54.
Chan JY, Ou CC, Wang LL, Chan SH. Heat shock protein 70 confers cardiovascular protection during endotoxemia via inhibition of nuclear factor-kappaB activation and inducible nitric oxide synthase expression in the rostral ventrolateral medulla. Circulation. 2004;110(23):3560–6.
de Montmollin E, Aboab J, Mansart A, Annane D. Bench-to-bedside review: Beta-adrenergic modulation in sepsis. Crit Care. 2009;13(5):230.
Ninomiya I, Nisimaru N, Irisawa H. Sympathetic nerve activity to the spleen, kidney, and heart in response to baroceptor input. Am J Phys. 1971;221(5):1346–51.
Floras JS, Butler GC, Ando SI, Brooks SC, Pollard MJ, Picton P. Differential sympathetic nerve and heart rate spectral effects of nonhypotensive lower body negative pressure. Am J Physiol Regul Integr Comp Physiol. 2001;281(2):R468–75.
Ocon AJ, Medow MS, Taneja I, Stewart JM. Respiration drives phase synchronization between blood pressure and RR interval following loss of cardiovagal baroreflex during vasovagal syncope. Am J Physiol Heart Circ Physiol. 2011;300(2):H527–40.
Ackland GL, Whittle J, Toner A, Machhada A, Del Arroyo AG, Sciuso A, et al. Molecular mechanisms linking autonomic dysfunction and impaired cardiac contractility in critical illness. Crit Care Med. 2016;44(8):e614–24.
Klemcke HG, Joe B, Rose R, Ryan KL. Life or death? A physiogenomic approach to understand individual variation in responses to hemorrhagic shock. Curr Genomics. 2011;12(6):428–42.
Convertino VA, Sather TM. Vasoactive neuroendocrine responses associated with tolerance to lower body negative pressure in humans. Clin Physiol. 2000;20(3):177–84.
Wijeysundera DN, Butler GC, Ando S, Pollard M, Picton P, Floras JS. Attenuated cardiac baroreflex in men with presyncope evoked by lower body negative pressure. Clin Sci (Lond). 2001;100(3):303–9.
Convertino VA, Rickards CA, Ryan KL. Autonomic mechanisms associated with heart rate and vasoconstrictor reserves. Clin Auton Res. 2012;22(3):123–30.
Ellison GM, Torella D, Karakikes I, Purushothaman S, Curcio A, Gasparri C, et al. Acute beta-adrenergic overload produces myocyte damage through calcium leakage from the ryanodine receptor 2 but spares cardiac stem cells. J Biol Chem. 2007;282(15):11397–409.
Annane D, Trabold F, Sharshar T, Jarrin I, Blanc AS, Raphael JC, et al. Inappropriate sympathetic activation at onset of septic shock: a spectral analysis approach. Am J Respir Crit Care Med. 1999;160(2):458–65.
Norris PR, Ozdas A, Cao H, Williams AE, Harrell FE, Jenkins JM, et al. Cardiac uncoupling and heart rate variability stratify ICU patients by mortality: a study of 2088 trauma patients. Ann Surg. 2006;243(6):804–12; discussion 812–4.
Hughson RL, Shoemaker JK. Autonomic responses to exercise: deconditioning/inactivity. Auton Neurosci. 2015;188:32–5.
Kumar A, Schupp E, Bunnell E, Ali A, Milcarek B, Parrillo JE. Cardiovascular response to dobutamine stress predicts outcome in severe sepsis and septic shock. Crit Care (London, England). 2008;12(2):R35.
Vallet B, Chopin C, Curtis SE, Dupuis BA, Fourrier F, Mehdaoui H, et al. Prognostic value of the dobutamine test in patients with sepsis syndrome and normal lactate values: a prospective, multicenter study. Crit Care Med. 1993;21(12):1868–75.
Rhodes A, Lamb FJ, Malagon I, Newman PJ, Grounds RM, Bennett ED. A prospective study of the use of a dobutamine stress test to identify outcome in patients with sepsis, severe sepsis, or septic shock. Crit Care Med. 1999;27(11):2361–6.
Jellema WT, Groeneveld AB, Wesseling KH, Thijs LG, Westerhof N, van Lieshout JJ. Heterogeneity and prediction of hemodynamic responses to dobutamine in patients with septic shock. Crit Care Med. 2006;34(9):2392–8.
Morelli A, Ertmer C, Westphal M, Rehberg S, Kampmeier T, Ligges S, et al. Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: a randomized clinical trial. JAMA. 2013;310(16):1683–91.
Cruickshank M, Henderson L, MacLennan G, Fraser C, Campbell M, Blackwood B, et al. Alpha-2 agonists for sedation of mechanically ventilated adults in intensive care units: a systematic review. Health Technol Assess. 2016;20(25):v–xx, 1–117
Schweickert WD, Pohlman MC, Pohlman AS, Nigos C, Pawlik AJ, Esbrook CL, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet. 2009;373(9678):1874–82.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 European Society of Intensive Care Medicine
About this chapter
Cite this chapter
Ackland, G.L. (2019). Autonomic Dysfunction in Shock. In: Pinsky, M.R., Teboul, JL., Vincent, JL. (eds) Hemodynamic Monitoring. Lessons from the ICU. Springer, Cham. https://doi.org/10.1007/978-3-319-69269-2_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-69269-2_8
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-69268-5
Online ISBN: 978-3-319-69269-2
eBook Packages: MedicineMedicine (R0)