
Abstract
Apoptosis plays an essential role in homeostasis and pathogenesis of a variety of human diseases. Endothelial cells are exposed to various environmental and internal stress and endothelial apoptosis is a pathophysiological consequence of these stimuli. Pulmonary endothelial cell apoptosis initiates or contributes to progression of a number of lung diseases. This chapter will focus on the current understanding of the role of pulmonary endothelial cell apoptosis in the development of emphysema and acute lung injury (ALI) and the factors controlling pulmonary endothelial life and death.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Pulmonary
- Endothelial cells
- Apoptosis
- Necrosis
- Necroptosis
- ER stress
- Unfolded protein response
- Autophagy
- Emphysema
- Acute lung injury
- ARDS
- COPD
4.1 Overview of Cell Death
4.1.1 Apoptosis
Apoptosis is a term first used by Kerr et al. in 1972 to describe a genetically determined energy-dependent active form of programmed cellular suicide. Apoptosis is characterized by well-ordered morphologic and molecular features including: cell surface exposure of phosphatidylserine, plasma membrane blebbing, cell shrinkage, cytoskeletal rearrangement, collapse of nuclear membrane, chromatin condensation, DNA fragmentation, and formation of membrane bound fragments known as “apoptotic bodies” (Kerr et al. 1972). Cell surface-exposed phosphatidylserine acts as a chemoattractant for phagocytes to engulf and clear apoptotic bodies (Henson and Tuder 2008). Apoptosis serves to eliminate unwanted, aged, harmful, injured, or infected cells. Due to limited release of intracellular contents, minimal inflammation occurs (Savill et al. 2002). However, if ingestion of apoptotic bodies by monocytes, macrophages, and dendritic cells (efferocytosis) is impaired, inflammation and autoimmunity may be enhanced (Gaipl et al. 2006). Apoptosis plays an essential role in the maintenance of tissue homeostasis and embryonic development. Further, during embryonic development, the timing of apoptosis is genetically determined. Excessive or inadequate apoptosis can, however, contribute to the pathogenesis of a variety of human diseases. Apoptosis is triggered by external stressors (e.g., death ligands, ultraviolet, and γ radiation) and/or internal stimuli (e.g., oxidants, DNA damage, increased Ca2+). Apoptosis is processed by two fundamental signaling pathways: the death receptor-mediated extrinsic pathway and the mitochondria-dependent intrinsic pathway (Olson and Kornbluth 2001; Thorburn 2004). Extrinsic pathway-activated caspase-8 can truncate and activate BID, thus activating the intrinsic pathway (Li et al. 1998). The details on regulation of apoptosis have been reviewed (Harrington et al. 2007; Subramanian and Steer 2010; Ola et al. 2011). Therapies targeting regulators of apoptosis have been used in preclinical and clinical trials for a variety of diseases including the treatment of cancers (Goldar et al. 2015).
4.1.2 Necrosis
Necrosis is a passive and caspase-independent cell death, characterized by cell swelling, mitochondrial degeneration, impaired ATP generation, lysosomal leakage, early rupture of plasma membranes, random fragmentation/degradation of DNA, and leakage of cellular contents into the surrounding environment (Henriquez et al. 2008). Necrosis is usually induced by nonspecific and non-physiological stress. Further, inhibition of caspases leads to necrosis (Henriquez et al. 2008). Due to release of potentially pro-inflammatory and pro-immunogenic cellular contents into surrounding tissues, necrosis often induces inflammation, autoimmune responses, and is often seen concomitant with apoptosis.
4.1.3 Necroptosis
Necroptosis describes a type of active, regulated, and programmed necrosis dependent upon the serine/threonine kinase activity of receptor-interacting protein kinase 1 and 3 (RIPK1/3) (Linkermann and Green 2014). Necroptosis and apoptosis share several upstream signaling elements including death receptors caspase 8 and FLIP. When caspase-8 is inhibited, RIPK1 is activated and forms an intracellular complex with RIPK3 to assemble the necrosome, leading to phosphorylation of mixed lineage kinase domain-like protein (MLKL) and ultimately cell death. Unlike apoptosis, necroptosis promotes harmful innate and adaptive immunologic responses by releasing damage associated molecular patterns (DAMPs). Thus, the reduction of necroptosis might be beneficial by minimizing the release of DAMPs and proinflammatory responses. Necroptosis is, however, a defense mechanism against invading microbes, including viral infections, and promotes the death and removal of virally infected cells. Therefore, blockade of necroptosis may increase susceptibility to viral infections particularly in patients with suppressed immunity. A number of inhibitors of necroptosis, such as necrostatin (specific inhibitor for RIPK1) and necrosulfonamide (specific inhibitor for human MLKL), have been described, providing potential therapeutic tools for treatment. Given the complex role of necroptosis, tissue and cell-specific targeting therapy is needed.
4.1.4 Endoplasmic Reticulum Stress-Induced Apoptosis
The endoplasmic reticulum (ER) is the site of posttranslational modifications and folding of secreted and membrane proteins. A variety of insults, such as ER Ca2+ chelators, reducing agents, glucose starvation, glycosylation antagonists, and protein mutations, can disrupt ER protein folding and lead to an accumulation of unfolded or misfolded proteins in the ER, thus initiating ER stress (Schroder and Kaufman 2005). Cells respond to ER stress by the unfolded protein response (UPR). The UPR includes three arms: pancreatic ER kinase (PKR)-like ER kinase (PERK)/eukaryotic initiation factor 2α (eIF2α), transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1) (Schroder and Kaufman 2005). Through the UPR, cells attempt to restore ER homeostasis in order to maintain cell survival by inhibiting global protein synthesis (to reduce the loading of client protein to the ER for folding), enhancing ER protein folding capacity, and promoting ER-associated degradation of misfolded or unfolded proteins (Schroder and Kaufman 2005).
Prolonged ER stress causes cell death due to simultaneous activation of multiple apoptotic pathways by the UPR (Szegezdi et al. 2006). PERK-induced phosphorylation of eIF2α can lead to apoptosis by induction of pro-apoptotic transcription factor, C/EBP homologous protein (CHOP), which suppresses expression of anti-apoptotic protein, Bcl-2. Activated IRE1 activates c-Jun N-terminal kinase (JNK), which causes apoptosis by phosphorylation and thus inactivation of Bcl-2 and by phosphorylation and thus activation of pro-apoptotic protein, Bim. In addition, increased Ca2+ in the ER activates the death effector, Bax/Bak in the ER membrane, causing movement of Ca2+ from the ER to the mitochondria leading to mitochondrial-dependent apoptosis. ER membrane-localized caspase-12 (rodent) and caspase-4 (human) have also been implicated in ER-stress-induced apoptosis (Szegezdi et al. 2003; Kim et al. 2006). Caspase-12/-4 are cleaved and thus activated by the Ca2+-dependent protease, m-calpain, by ER stress (Groenendyk and Michalak 2005). However, other studies have suggested that ER stress-induced apoptosis depends upon the apoptosome and not caspase-12/-4 (Obeng and Boise 2005; Di Sano et al. 2006).
Cell fate determination is not well understood when both survival (adaptive) and apoptotic pathways are simultaneously activated. It has been proposed that persistent ER stress causes apoptosis due to sustained induction of CHOP and instability of the adaptive pathway (Lin et al. 2007). It has also been suggested that cells survive mild ER stress because of the short half-life of pro-apoptotic proteins, compared to pro-survival proteins (Rutkowski et al. 2006). Robust prolonged ER stress causes apoptosis due to the induction of CHOP excessive to its degradation (Rutkowski et al. 2006).
4.1.5 Autophagy-Associated Cell Death
Autophagy is a dynamic and continuous process by which cells dispose of damaged or unneeded cellular proteins or organelles (mitochondria) by self-digestion to generate intracellular nutrients. During physiological conditions, autophagy is suppressed by mammalian target of rapamycin (mTOR), thus inhibiting the expression of autophagy-related genes (ATGs). Upon external or internal stress: including nutrient starvation, growth factor deprivation, hypoxia, ischemia, or mitochondrial aging, mTOR is inhibited thus initiating autophagy. Autophagy is a multistep sequential process, consisting of the formation of double-membrane vesicles that sequester unwanted cargo (proteins or mitochondria) in autophagosomes, fusion of autophagosomes with endosomes or lysosomes to form amphisomes or autolysosomes, and digestion of cargo by proteases (Hotchkiss et al. 2009; Choi et al. 2013). Autophagy is an evolutionarily conserved housekeeping process that allows recycling of damaged proteins and organelles in order to maintain homeostasis. Impairment in any step of autophagy causes cellular nutrient deficiency and/or accumulation of damaged proteins and organelles leading to cell death (Hotchkiss et al. 2009). Whether autophagy promotes cell survival or death may depend on cell type and setting (Gustafsson and Gottlieb 2008).
4.1.6 Assessments of Cell Death
Based on the unique characteristics of different types of cell death, a variety of assays have been developed to assess the specific types of cell death in vivo and in vitro. Different types of cell death may share common characteristics at different stages of cell death; therefore, it is often necessary to use multiple assays to confirm cell death. The details on the assessments of cell death have been extensively reviewed (Harrington et al. 2007; Henson and Tuder 2008; Lu and Rounds 2009; Klionsky et al. 2016) and will not be discussed in this review.
4.2 Pulmonary Endothelial Cell Apoptosis
Balance of endothelial cell survival and death is crucial for angiogenesis, vessel regression, and barrier function. Due to the unique position of endothelial cells (EC) at the interface of circulating blood and surrounding tissues, EC may be exposed to various environmental stress including: hypoxia, hyperoxia, oxidants, lipopolysaccharide (LPS), and cigarette smoke (CS), or internal stress including: adenosine, ceramide, tumor necrosis factor (TNF)-α, and angiotensin II. Apoptosis is a pathophysiological consequence of these stimuli. However, a variety of biomechanical and biochemical factors are involved in the anti-apoptotic processes. For example, physiological levels of shear stress and cyclic strain, vascular endothelial growth factor (VEGF), focal adhesion kinase (FAK), activated protein C (APC), and sphingosine 1-phosphate (S1P) protect EC against apoptosis. The pro- and anti-apoptotic effects of these mediators have been reviewed (Harrington et al. 2007; Lu and Rounds 2009); therefore, this review will focus on the current understanding of endothelial pro-survival factors (VEGF and FAK) and apoptosis-inducing stress (adenosine, cigarette smoke, and LPS) in the lungs.
4.2.1 Vascular Endothelial Growth Factor
EC express abundant VEGF, which promotes EC survival and maintains normal alveolar structure (Voelkel et al. 2006). Expression of both VEGF and VEGF receptor type 2 (VEGFR2) are decreased in lung tissue of patients with chronic obstructive pulmonary disease (COPD) (Kasahara et al. 2001). This diminished VEGF/VEGFR2 signaling is inversely associated with increased lung EC apoptosis (Kasahara et al. 2001). Lung-targeted inhibition of VEGF or VEGFR2 causes alveolar septal cell apoptosis in mice (Kasahara et al. 2000; Tang et al. 2004). Our group has also shown that blockade of VEGFR2 causes cultured pulmonary artery EC apoptosis in vitro (Lu 2008). These results indicate that VEGF signaling is essential for lung EC survival.
4.2.2 Focal Adhesion Kinase
EC are linked to the basement membrane through binding of cell surface expressed integrins to extracellular matrix (ECM) proteins at focal adhesion complexes (FAC) (Hynes 1992). As anchorage-dependent cells, EC undergo detachment-initiated apoptosis, referred to as anoikis, upon loss of adhesion to underlying basement membrane. FAK, a non-receptor tyrosine kinase and an essential component of FAC, is activated upon integrin engagement of ECM (Guan et al. 1991; Guan and Shalloway 1992; Parsons 2003). FAK provides survival signaling for anchorage-dependent cells such as cultured fibroblasts (Hungerford et al. 1996). Similarly, EC isolated from FAK-null embryos undergo apoptosis (Ilic et al. 1995, 2003). Endothelium-specific deletion of FAK (Cre/FAKflox) is embryonic lethal and causes EC apoptosis (Shen et al. 2005; Braren et al. 2006). Guan and colleagues (Guan et al. 1991; Guan and Shalloway 1992) have demonstrated that FAK tyrosine kinase activity is essential for FAK activity. FAK promotes cell survival by recruiting proteins containing SH2 domain including Src and phosphatidylinositol-3-kinase (PI3K) (Schaller et al. 1994). The activated PI3K recruits and activates Akt (Khwaja et al. 1997), which promotes cell survival via phosphorylation and thus inhibition of pro-apoptotic protein, Bad (Kennedy et al. 1997). FAK also promotes survival by activation of NF-κB and ERK signaling pathways (Huang et al. 2007). Additionally, FAK can translocate to the nucleus and inhibit p53 transcriptional activation and enhance p53 degradation, leading to protection against apoptosis (Ilic et al. 1998).
4.2.3 Adenosine
Adenosine is generated from adenosine-5′-triphosphate (ATP) and adenosine-5′-diphosphate (ADP) by extracellular ecto-5′-nucleotidases, CD39 and CD73, and is metabolized by adenosine deaminase (ADA). Extracellular adenosine exists in low concentrations (40–600 nM) under physiological conditions and is increased due to platelet degranulation, cell necrosis, activation of CD39 and/or CD73, or inhibition of ADA (Thompson et al. 2004; Eltzschig et al. 2006; Volmer et al. 2006; Eckle et al. 2007). Increased extracellular adenosine can interact with cell surface G-protein coupled adenosine receptors (ARs) (Feoktistov et al. 2002; Wyatt et al. 2002; Umapathy et al. 2010). Activation of adenosine receptors, specifically A3-mediated signaling, has been shown to protect against apoptosis and tissue injury (Rivo et al. 2004; Chen et al. 2006; Matot et al. 2006).
However, sustained increased adenosine in ADA-deficient mice enhances alveolar cell apoptosis (Zhou et al. 2009). We have also shown that prolonged exposure to adenosine causes apoptosis of cultured lung EC (Lu et al. 2013). The injurious effect of adenosine is mediated by equilibriative nucleoside transporters. EC predominantly express equilibriative nucleoside transporter 1 (ENT1) and ENT2 (Archer et al. 2004). Upon sustained exposure, adenosine may be taken up into cells by ENTs. Further, similar to other G-protein coupled receptors, prolonged engagement of ARs causes receptor desensitization and internalization (Fredholm et al. 2001). This concept is supported by findings that sustained increased adenosine in ADA-deficient mice enhances alveolar cell apoptosis via a mechanism independent of adenosine receptor, A2BR (Zhou et al. 2009). In addition, sustained exposure to adenosine causes endothelial cell apoptosis; this effect is prevented by inhibition of ENT1/2 however exacerbated by inhibition of either A2AR or A2BR (Lu et al. 2013). These results are consistent with the concept that ENT1/2-facilitated intracellular adenosine uptake and subsequent metabolism mediates adenosine-induced EC apoptosis, whereas AR-mediated signaling limits apoptosis (Simonis et al. 2009).
Once intracellular, adenosine reacts with homocysteine and generates S-adenosyl-l-homocysteine (SAH) by inhibition of SAH hydrolase (SAHH). SAH induces endothelial cell apoptosis independent of homocysteine (Sipkens et al. 2012). SAH is also a product of carboxyl methylation with S-adenosyl-l-methionine (SAM) as a methyl donor. We have demonstrated that exogenous adenosine causes lung EC apoptosis via increased ratio of intracellular SAH to SAM (Rounds et al. 1998). The increased ratio of SAH to SAM suppresses carboxyl methyltransferase activity. Isoprenylcysteine-O-carboxyl methyltransferase (ICMT) is a major methyltransferase for carboxyl methylation of small GTPase, Ras (Clarke 1992), which is a posttranslational modification essential for membrane localization and activation of Ras (Boivin and Beliveau 1995; Fleming et al. 1996; Kranenburg et al. 1997; Michaelson et al. 2001). We have shown that exogenous adenosine causes lung EC apoptosis in part by ICMT inhibition-mediated inhibition of Ras carboxyl methylation and activation (Kramer et al. 2003).
SAM is a precursor to glutathione (GSH) and is synthesized exclusively in the cytosol (Reytor et al. 2009) and also transported into mitochondria (Agrimi et al. 2004). Exogenous SAM has been shown to elevate GSH levels in vivo and prevent alcohol-induced mitochondrial oxidative stress and dysfunction as well as liver and lung injury in animal models (Holguin et al. 1998; Bailey et al. 2006; Cederbaum 2011). p38 is a redox-sensitive protein (Matsuzawa and Ichijo 2008). Reactive oxygen species (ROS)-mediated p38 activation has been implicated in extracellular ATP-induced macrophage apoptosis (Noguchi et al. 2008) and H2O2-induced EC apoptosis (Machino et al. 2003). Activation of p38 has also been implicated in homocysteine-induced apoptosis of endothelial progenitor cells (Bao et al. 2010) and cardiomyocytes (Wang et al. 2011). We have shown that sustained exposure to exogenous adenosine causes mitochondrial defects and endothelial apoptosis via mitochondrial oxidative stress-induced activation of p38 (Lu et al. 2012, 2013). Active p38 causes apoptosis by direct phosphorylation, and thus inhibition of Bcl-2 (De Chiara et al. 2006; Farley et al. 2006) and by increasing mitochondrial translocation of Bax (Capano and Crompton 2006). Future studies are needed to address whether sustained adenosine exposure reduces mitochondrial SAM, thus leading to mitochondrial oxidative stress via increased ratio of SAH to SAM in the cytosol.
In summary, adenosine displays seemingly paradoxical effects on lung EC life and death. Acute exposure protects EC against apoptosis via AR-mediated signaling, whereas prolonged exposure causes EC apoptosis via ENT1/2-mediated intracellular adenosine uptake and subsequent metabolism and mitochondrial oxidative stress.
4.2.4 Cigarette Smoke
Lung EC apoptosis is significantly elevated in human smokers with emphysema (Kasahara et al. 2001) and mice with mild emphysema caused by CS exposure (Sakhatskyy et al. 2014). We (Sakhatskyy et al. 2014) and others (Tuder et al. 2000; Damico et al. 2011) have shown that CS extract (CSE) causes cultured lung macro- and microvascular EC apoptosis in vitro. The mechanisms underlying CS-induced lung EC apoptosis are rather complicated and involve FAK, p53, UPR, and autophagy.
FAK is a survival signal for anchorage-dependent cells (Hungerford et al. 1996). Tyrosine 397 phosphorylation of FAK is essential for its activation (Schaller et al. 1994). CSE decreases FAK phosphorylation at tyrosine-397 in an oxidative stress-dependent manner (Lu et al. 2011)—essential in CSE-induced EC apoptosis (Sakhatskyy et al. 2014). FAK also promotes cell survival via suppression of p53 (Ilic et al. 1998). Further, activation of p53 has contributed to CSE-induced pulmonary EC apoptosis (Damico et al. 2011). Thus, we speculate that CSE causes lung EC apoptosis via oxidative stress-mediated inhibition of FAK and subsequent activation of p53.
The UPR is an important mechanism of the elimination of ER stress and enhanced cell survival (Schroder and Kaufman 2005). The UPR is activated in lung tissue of smokers who do not have emphysema (Kelsen et al. 2008). The UPR is also activated by CSE in cultured human bronchial epithelial cells and 3T3 fibroblasts (Hengstermann and Müller 2008; Jorgensen et al. 2008) and cultured pulmonary EC (Sakhatskyy et al. 2014). Using mouse models of CS exposure, we have demonstrated a strong link between impairment of eIF2α signaling with lung EC apoptosis (Sakhatskyy et al. 2014). Future studies are necessary to determine if impaired eIF2α signaling contributes to lung EC apoptosis.
Autophagy is increased in response to deficiencies in extracellular and intracellular nutrients. Enhanced autophagy is observed in the lung tissue of smokers with emphysema (Chen et al. 2008). Autophagy is also activated by CSE exposure in lung epithelial cells and fibroblasts (Kim et al. 2008) as well as lung EC (Sakhatskyy et al. 2014). Increased autophagy has contributed to CS-induced alveolar epithelial cell apoptosis in mice (Chen et al. 2010). In contrast, increased autophagy has also been shown to protect against pulmonary endothelial cell apoptosis induced by cadmium, a component of cigarette smoke (Surolia et al. 2015). We have reported that autophagy was not altered in the lung tissue of a mouse strain susceptible to CS-induced lung EC apoptosis and emphysema (Sakhatskyy et al. 2014). The role of autophagy in CS-induced apoptosis may be dependent on cell types and stimuli.
Due to open structure and limited repair capacity, mitochondrial DNA is 50 times more sensitive to oxidative damage than nuclear DNA (Yakes and Van Houten 1997). Oxidative stress-induced mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis of lung EC (Ruchko et al. 2005). The role of mitochondrial DNA damage in CS-induced lung EC apoptosis remains to be studied.
4.2.5 Lipopolysaccharide
LPS, also known as lipoglycans or endotoxin, is a component of the outer envelope of gram-negative bacteria and elicits pro-inflammatory responses. It is well established that LPS-induced EC activation, dysfunction, and apoptosis play an important role in bacterial sepsis and endotoxemia. In the blood circulation, LPS binds to soluble CD14 via LPS-binding protein (LBP), followed by engagement of toll-like receptor (TLR)-4. This engagement results in the recruitment of adaptor, myeloid differentiation factor 88 (MyD88), and subsequent activation of interleukin (IL)-1 receptor associated kinase (IRAK)-1, TNF receptor associated (TRAF)-6, NF-kB, and MAPK pathways (Desch et al. 1989; Wang et al. 2001; Bannerman and Goldblum 2003).
NF-kB has been shown to transcriptionally upregulate anti-apoptotic genes such as IAP-1, IAP-2, and FLIP (LaCasse et al. 1998; Bannerman et al. 2004). However, suppression of NF-kB has minimal effect on LPS-induced EC apoptosis (Zen et al. 1999). This is due to FADD/MyD88-dependent negative regulation of LPS-induced NF-kB activation (Martin et al. 2005; Zhande et al. 2007); Fas is no longer able to activate MyD88, thus stimulating LPS/TLR4/NF-kB signaling (Martin et al. 2005). LPS also stimulates MyD88-independent signaling of endothelial apoptosis (Dauphinee and Karsan 2006). Heterotrimeric Gi/Go proteins play a role in LPS-induced TLR signaling independent of the MyD88-dependent pathway, leading to MAPK, Akt, and IFN activation of endothelial cells (Dauphinee et al. 2011). Whether LPS-induced stimulation of heterotrimeric G coupled proteins plays a role in EC apoptosis is unknown. LPS can activate the BID-dependent intrinsic pathway of apoptosis in lung EC (Wang et al. 2007). Conversely, LPS has been shown to upregulate mRNA of anti-apoptotic molecules, thus preventing EC apoptosis (Hu et al. 1998). LPS-induced intrinsic apoptosis and cytoprotection in disease states are not well understood and require further study.
4.3 Pulmonary EC Apoptosis in Lung Diseases
Apoptosis has been shown to ameliorate or exacerbate lung injury. Pulmonary EC apoptosis plays an important role in physiological processes including vasculogenesis and angiogenesis during lung development. Pulmonary EC apoptosis may also initiate or contribute to the progression of a number of lung diseases, as reviewed elsewhere (Harrington et al. 2007; Lu and Rounds 2009). In this review, we will focus on the role of pulmonary EC apoptosis in development of emphysema and Acute Lung Injury (ALI).
4.3.1 Emphysema
Chronic obstructive pulmonary disease (COPD), a progressive respiratory condition consisting of emphysema and chronic bronchitis, is the fourth leading cause of death worldwide and may become the third leading cause of death by 2030 based on prediction by the World Health Organization (Khaltaev 2005). The prevalence of COPD in the United States in 2013 was estimated to be 6.4% (15.7 million adults) (Wheaton et al. 2015). COPD is also an important contributor of mortality and disability in the United States (Murray et al. 2013). Further, COPD-related medical costs were estimated at $32 billion in the USA in 2010 with an additional $4 billion in costs due to absence from work (Ford et al. 2015). α1-antitrypsin (AAT) deficiency and other genetic predispositions contribute to the development of COPD (Sandford et al. 1997). However, tobacco smoke remains the leading cause of this devastating disease. Indoor air pollution (such as biomass fuel used for cooking and heating), outdoor air pollution, and occupational dusts and chemicals also increase the risk of COPD (Diette et al. 2012). Although the pathology of COPD has been well defined, the pathogenesis of the disease initiation and progression is not understood. Currently, there is no specific treatment available to reverse COPD.
Emphysema, a common and debilitating manifestation of COPD, is characterized by alveolar airspace enlargement, loss of alveolar capillary septa, and resultant impaired gas exchange. Several hypotheses have been proposed to explain alveolar wall damage in emphysema. Protease/anti-protease imbalance has been accepted as a major mechanism for emphysematous lung destruction (Shapiro 1995, 1999; Shapiro et al. 2003; Taraseviciene-Stewart and Voelkel 2008). It is believed that neutrophil elastase and macrophage matrix metalloproteinases enzymatically degrade elastin in alveolar septa, leading to emphysema (Taraseviciene-Stewart and Voelkel 2008). This notion is supported by findings that patients with genetic deficiency of the anti-protease, AAT, develop emphysema (No Authors 1997). Additionally, intra-tracheal instillation of proteases causes an emphysema phenotype in rats (Pastor et al. 2006). However, less than 5% of emphysema patients have AAT deficiency. Inflammatory cell infiltration is also seen in human emphysema. However, lung inflammation in pneumonia or acute lung injury does not usually result in emphysema. This suggests that inflammation may not be sufficient by itself for the development of emphysema. Oxidant stress and immunological injury also play a role in the pathogenesis of emphysema (Taraseviciene-Stewart and Voelkel 2008). Emerging evidence has highlighted a role of apoptosis, particularly EC apoptosis, in the initiation and progression of emphysema (Kasahara et al. 2000, 2001; Giordano et al. 2008).
Lung tissue from patients with emphysema displays increased apoptosis of both epithelial and endothelial cells in the alveolar septa (Kasahara et al. 2001; Imai et al. 2005). Bcl-2 single-nucleotide polymorphisms have been associated with severity of human emphysema (Sata et al. 2007). We have shown that lung EC apoptosis is elevated in a mouse model of emphysema induced by CS exposure (Sakhatskyy et al. 2014). Interestingly, induction of alveolar cell apoptosis by intratracheal instillation of the active caspase-3 causes emphysema in rats (Aoshiba et al. 2003). Additionally, inhibition of VEGF signaling causes alveolar septal cell apoptosis and emphysema in mice (Kasahara et al. 2000; Tang et al. 2004). Similarly, intra-tracheal instillation of C12 ceramide triggers alveolar endothelial and epithelial cell apoptosis and emphysema-like changes in mice (Petrache et al. 2005). Further, lung EC-targeted induction of apoptosis led to emphysema and enhanced oxidative stress and lung inflammation (Giordano et al. 2008). More importantly, inhibition of apoptosis using pan-caspase inhibitors prevented the emphysematous changes induced by either ceramide (Petrache et al. 2005) or blockage of VEGF signaling (Kasahara et al. 2000; Tang et al. 2004). These results support a central role of lung EC apoptosis in the development of emphysema. Anti-protease, AAT, inhibits CSE-induced pulmonary EC apoptosis in vitro by direct interaction with caspase-3 (Aldonyte et al. 2008). Overexpression of AAT also inhibits lung endothelial apoptosis and attenuates emphysema caused by either active caspase-3 or blockade of VEGF signaling (Petrache et al. 2006). These studies suggest that lung EC apoptosis is a critical step in the pathogenesis of emphysema.
Inhibition of FAK causes emphysema-like change in rat lungs (Mizuno et al. 2012). We have shown that CS exposure for 3 weeks enhanced pulmonary EC apoptosis and decreased FAK activity in mice susceptible to CS-induced emphysema (Sakhatskyy et al. 2014). Further studies are necessary to address whether reduced FAK activity contributes to CS-induced lung EC apoptosis and emphysema in humans in vivo. We have shown that CS exposure increases lung tissue adenosine levels in mice, an effect associated with lung EC apoptosis and early emphysema (Lu et al. 2013). Sustained increased adenosine in ADA-deficient mice also enhances alveolar cell apoptosis and causes emphysema in mice (Zhou et al. 2009). ADA expression and activity are reduced in the lung of smokers with COPD (Zhou et al. 2010). Whether chronically elevated adenosine contributes to CS-induced lung endothelial cell apoptosis and development of emphysema remains to be investigated.
Ceramide is upregulated in emphysematous lungs of patients and animal models, as well as in cultured pulmonary EC exposed to CSE (Petrache et al. 2005). This increase in ceramide is associated with enhanced alveolar cell apoptosis (Petrache et al. 2005). Interestingly, intratracheal instillation of C12 ceramide triggers airspace enlargement and apoptosis of alveolar EC and type II epithelial cells (Petrache et al. 2005). Further, inhibition of de novo ceramide synthesis significantly attenuated lung cell apoptosis and emphysema induced by VEGFR2 blockade (Petrache et al. 2005). These results suggest that ceramide is also an important mediator of alveolar cell apoptosis and emphysema (Petrache et al. 2005).
Only 10–15% of smokers develop emphysema. The mechanism underlying increased susceptibility to emphysema remains unclear. The UPR is elevated in the lungs of smokers without evidence of emphysema (Kelsen et al. 2008). Nrf2, a redox-sensitive, antioxidant transcription factor, is activated by eIF2α, a branch of UPR (Digaleh et al. 2013). Nrf2 knockout mice demonstrate enhanced susceptibility to cigarette smoke-induced emphysema in comparison to wild-type mice (Iizuka et al. 2005). We have shown that active eIF2α was significantly reduced in the lungs of AKR mice with mild emphysema induced by CS (Sakhatskyy et al. 2014). Future studies are needed to address whether Nrf2 is reduced in the lungs and whether inadequate induction of Nrf2 contributes to development of emphysema.
Autophagy is significantly increased in lung tissue of patients with COPD; the degree of autophagy positively correlates with the clinical severity of disease (Chen et al. 2008). Increased autophagy has contributed to CS-induced alveolar epithelial cell apoptosis and emphysema in mice (Chen et al. 2010; Mizumura et al. 2014). In contrast, increased autophagy protects against pulmonary endothelial cell apoptosis and emphysema induced by cadmium, a component of cigarette smoke (Surolia et al. 2015). We have reported that autophagy was not altered in lung tissue of a mouse strain with increased lung EC apoptosis and mild emphysema induced by CS (Sakhatskyy et al. 2014). Thus, the role of autophagy in regulating lung EC apoptosis and early onset of CS-induced emphysema needs further study.
4.3.2 Acute Lung Injury
ALI and its more severe form, acute respiratory distress syndrome (ARDS), are life-threatening disorders clinically characterized by severe hypoxemia and pulmonary bilateral infiltrates. In the United States, ARDS affects approximately 190,000 patients annually (Rubenfeld et al. 2005). ARDS accounts for 3.6 million associated hospital days (Rubenfeld et al. 2005; Adhikari et al. 2010). The global impact of ARDS has been difficult to assess due to varying definitions of the broad clinical phenotypes and limited data. Thus, ARDS remains an underreported disease of treated incidence, as opposed to actual incidence, in the undeveloped world (Buregeya et al. 2014). Although the mortality rate of ARDS has decreased to around 30–40% due to lung protective ventilation strategies (Amato et al. 1998; Villar et al. 2006), ARDS remains a deadly syndrome without a specific cure. Currently, there are no pharmacological interventions available to reduce the mortality of ARDS.
Sepsis, bacterial and viral pneumonia, and trauma remain the leading risk factors for the development of ARDS. Emerging evidence from epidemiologic studies, animal models, and cultured cell models have suggested that both active and passive cigarette smoke exposure modifies the susceptibility for development of ALI and ARDS (Iribarren et al. 2000; Calfee et al. 2011; Lu et al. 2011, 2013; Hsieh et al. 2014; Borgas et al. 2016).
The pathophysiology of ARDS is characterized by increased permeability of the alveolar-capillary barrier, influx of protein and inflammatory cell-rich fluid into the alveolar space, attenuated gas exchange between alveolar-capillary barrier, and dysregulated inflammation. Increased permeability of the microvascular endothelium and alveolar epithelium promotes edema formation, and this concept has been accepted as an important mechanism for the initiation of ARDS (Matthay et al. 2012). It is well established that polymorphonuclear cells (PMN) and immunological injury also play a significant role in the pathogenesis of ARDS (Perl et al. 2011). PMN accumulation is observed in the broncheoalveolar lavage fluid (BALF) (Pittet et al. 1997) and lung biopsies of early ARDS patients (Bachofen and Weibel 1977, 1982). Further, neutrophilia has been correlated with exacerbation of sepsis-induced ALI (Steinberg et al. 1994). However, ARDS may also develop in neutropenic patients, and neutrophil activation and migration may be observed in human lungs without injury (Martin et al. 1989; Downey et al. 1999). This suggests that inflammation may not be sufficient by itself for the development of ARDS.
Emerging evidence has suggested a role of pulmonary cell apoptosis in the initiation and progression of ARDS. The death receptor, Fas, and its ligand, FasL system, is an important death receptor-mediated extrinsic pathway of apoptosis. FasL is expressed and released by inflammatory cells, including neutrophils and lymphocytes, whereas Fas is expressed on the surface of lung EC, alveolar and bronchial epithelial cells, Clara cells, and alveolar macrophages. Fas and FasL are increased in pulmonary edema fluid and in lung tissue of patients with ARDS (Albertine et al. 2002). Silencing of Fas/FasL reduces lung cell apoptosis and ALI in a mouse model of sepsis (Perl et al. 2005, 2007). Soluble FasL (sFasL) is a cleaved form of FasL by metalloproteinases and is increased in BAL fluid of patients with ARDS (Matute-Bello et al. 1999). sFasL released from inflammatory cells is capable of inducing lung epithelial cell apoptosis (Matute-Bello et al. 1999). The role of Fas/FasL in lung EC apoptosis is not yet clear. Robust pulmonary endothelial cell apoptosis has been observed in patients with severe ARDS (Abadie et al. 2005) and in mice with ALI induced by LPS (Fujita et al. 1998). Sepsis-induced ARDS in mice indicates evidence for pulmonary microvascular endothelial cell death as a cause of barrier dysfunction and edema (Gill et al. 2014, 2015). Inhibition of apoptosis using a broad-spectrum caspase inhibitor prolonged survival of mice exposed to LPS (Kawasaki et al. 2000). Since apoptosis of alveolar endothelial, epithelial, and interstitial inflammatory cells occurs during ALI, future studies are needed to address the role of apoptosis of specific cells in initiation of ALI/ARDS.
Apoptosis has been thought of to be a non-inflammatory means of removing injurious cells, thus facilitating lung repair. However, there is increasing evidence indicating that Fas/FasL-mediated lung epithelial apoptosis results in release of pro-inflammatory cytokines (such as TNF-α and TGF-β1), leading to inflammation and progression from ARDS to fibrosis (Chapman 1999). Whether pulmonary endothelial cell apoptosis occurs during initiation or progression of pulmonary fibrosis is unknown.
The role of necroptosis in development of ARDS is yet to be determined. Of interest, a recent study of blood transfusion-related acute lung injury indicates that banked red blood cell (RBC) transfusion enhances susceptibility to lung inflammation and ARDS in critically ill transfused patients and mice through necroptosis of lung EC and subsequent release of DAMPs (Qing et al. 2014).
4.4 Conclusions and Perspectives
Cell life and death are tightly regulated by survival signaling and death inducing programs. Pulmonary EC apoptosis significantly contributes to the development of emphysema and ALI/ARDS, as depicted in Fig. 4.1. Pan-caspase inhibitors have been used to inhibit lung cell apoptosis and prevent emphysema and ALI in animal models. However, use of such drugs to treat apoptosis-associated lung diseases may be problematic due to breakdown of tissue homeostasis and activation of necroptosis (Linkermann and Green 2014). The therapeutic potential of drugs that modulate cell death is dependent upon cell type-specific, tissue-specific, and vascular bed-specific actions. Thus, drugs acting locally and with cell type specificity are needed. Areas where research is needed include: (1) apoptosis susceptibility of different EC (conduit artery versus microvascular versus progenitor); (2) role of apoptosis of specific lung cells in initiation and/or progression of lung diseases; (3) role of necrosis and necroptosis in development of lung diseases, such as emphysema and ALI.

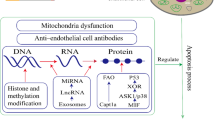
Signaling pathways to CS-induced pulmonary endothelial cell apoptosis. Multiple signaling pathways are involved in CS-induced pulmonary endothelial cell apoptosis. (1) CS reduces VEGF/VEGFR2 signaling, leading to induction of ceramide and consequent apoptosis; (2) CS reduces FAK activation, leading to activation of p53 and inhibition of PI3K/Akt signaling, which results in apoptosis; (3) CS causes mitochondrial oxidative stress and mitochondrial dysfunction, leading to apoptosis; (4) CS elevates adenosine levels, leading to inactivation of Ras and mitochondrial oxidative stress, resulting in apoptosis; (5) CS impairs unfolded protein response, leading to apoptosis
Abbreviations
- AAT:
-
Alpha1-anti-trypsin
- ADA:
-
Adenosine deaminase
- ADP:
-
Adenosine-5′-diphosphate
- APC:
-
Activated protein C
- ARDS:
-
Acute respiratory distress syndrome
- ARs:
-
Adenosine receptors
- ATF6:
-
Transcription factor 6
- ATGs:
-
Autophagy-related genes
- ATP:
-
Adenosine-5′-triphosphate
- BALF:
-
Broncheoalveolar lavage fluid
- CD39:
-
ecto-5′-nucleotidase
- CD73:
-
ecto-5′-nucleotidase
- CHOP:
-
C/EBP homologous protein
- COPD:
-
Chronic obstructive pulmonary disease
- CS:
-
Cigarette smoke
- CSE:
-
Cigarette smoke extract
- DAMPs:
-
Damage associated molecular patterns
- EC:
-
Endothelial cells
- ECM:
-
Extracellular matrix
- eIF2α:
-
Eukaryotic initiation factor 2α
- ENT1/2:
-
Equilibrative nucleoside transporter 1/2
- ER:
-
Endoplasmic reticulum
- FAC:
-
Focal adhesion complexes
- FAK:
-
Focal adhesion kinase
- GSH:
-
Glutathione
- ICMT:
-
Isoprenylcysteine-O-carboxyl methyltransferase
- IRAK-1:
-
Interleukin (IL)-1 receptor associated kinase
- IRE1:
-
Inositol-requiring enzyme 1
- JNK:
-
c-Jun N-terminal kinase
- LPS:
-
Lipopolysaccharide
- MLKL:
-
Mixed lineage kinase domain-like protein
- mTOR:
-
Mammalian target of rapamycin
- MyD88:
-
Myeloid differentiation factor 88
- PERK:
-
Pancreatic ER kinase like ER kinase
- RBC:
-
Red blood cells
- RIPK1/3:
-
Receptor-interacting protein kinase 1 and 3
- ROS:
-
Reactive oxygen species
- S1P:
-
Sphingosine 1-phosphate
- SAH:
-
S-adenosyl-l-homocysteine
- SAHH:
-
S-adenosyl-l-homocysteine hydrolase
- SAM:
-
S-Adenosyl-l-Methionine
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor-alpha
- TRAF-6:
-
TNF receptor associated factor-6
- UPR:
-
Unfolded protein response
- VEGF:
-
Vascular endothelial growth factor
- VEGFR2:
-
VEGF receptor type 2
References
Abadie Y, Bregeon F, Papazian L, Lange F, Chailley-Heu B, Thomas P, Duvaldestin P, Adnot S, Maitre B, Delclaux C (2005) Decreased VEGF concentration in lung tissue and vascular injury during ARDS. Eur Respir J 25(1):139–146
Adhikari NK, Fowler RA, Bhagwanjee S, Rubenfeld GD (2010) Critical care and the global burden of critical illness in adults. Lancet 376(9749):1339–1346
Agrimi G, Di Noia MA, Marobbio CM, Fiermonte G, Lasorsa FM, Palmieri F (2004) Identification of the human mitochondrial S-adenosylmethionine transporter: bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem J 379(Pt 1):183–190
Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, Matthay MA, Ware LB (2002) Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol 161(5):1783–1796
Aldonyte R, Hutchinson ET, Jin B, Brantly M, Block E, Patel J, Zhang J (2008) Endothelial alpha-1-antitrypsin attenuates cigarette smoke induced apoptosis in vitro. COPD 5:153–162
Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY, Carvalho CR (1998) Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 338(6):347–354
Aoshiba K, Yokohori N, Nagai A (2003) Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol 28:555–562
Archer RG, Pitelka V, Hammond JR (2004) Nucleoside transporter subtype expression and function in rat skeletal muscle microvascular endothelial cells. Br J Pharmacol 143(1):202–214
Bachofen M, Weibel ER (1977) Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis 116(4):589–615
Bachofen M, Weibel ER (1982) Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med 3(1):35–56
Bailey SM, Robinson G, Pinner A, Chamlee L, Ulasova E, Pompilius M, Page GP, Chhieng D, Jhala N, Landar A, Kharbanda KK, Ballinger S, Darley-Usmar V (2006) S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am J Physiol Gastrointest Liver Physiol 291(5):G857–G867
Bannerman DD, Goldblum SE (2003) Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol 284(6):L899–L914
Bannerman DD, Eiting KT, Winn RK, Harlan JM (2004) FLICE-like inhibitory protein (FLIP) protects against apoptosis and suppresses NF-kappaB activation induced by bacterial lipopolysaccharide. Am J Pathol 165(4):1423–1431
Bao XM, Wu CF, Lu GP (2010) Atorvastatin inhibits homocysteine-induced oxidative stress and apoptosis in endothelial progenitor cells involving Nox4 and p38MAPK. Atherosclerosis 210(1):114–121
Boivin D, Beliveau R (1995) Subcellular distribution and membrane association of Rho-related small GTP-binding proteins in kidney cortex. Am J Phys 269:F180–F189
Borgas D, Chambers E, Newton J, Ko J, Rivera S, Rounds S, Lu Q (2016) Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am J Respir Cell Mol Biol 54(5):683–696
Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R (2006) Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol 172(1):151–162
Buregeya E, Fowler RA, Talmor DS, Twagirumugabe T, Kiviri W, Riviello ED (2014) Acute respiratory distress syndrome in the global context. Glob Heart 9(3):289–295
Calfee CS, Matthay MA, Eisner MD, Benowitz N, Call M, Pittet JF, Cohen MJ (2011) Active and passive cigarette smoking and acute lung injury after severe blunt trauma. Am J Respir Crit Care Med 183(12):1660–1665
Capano M, Crompton M (2006) Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem J 395(1):57–64
Cederbaum AI (2011) Hepatoprotective effects of S-adenosyl-L-methionine against alcohol- and cytochrome P450 2E1-induced liver injury. World J Gastroenterol 16(11):1366–1376
Chapman HA (1999) A Fas pathway to pulmonary fibrosis. J Clin Invest 104:1–2
Chen GJ, Harvey BK, Shen H, Chou J, Victor A, Wang Y (2006) Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J Neurosci Res 84(8):1848–1855
Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, Nakahira K, Pilewski JM, Lee JS, Zhang Y, Ryter SW, Choi AM (2008) Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 3(10):e3316
Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, Choi AM (2010) Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A 107(44):18880–18885
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368(7):651–662
Clarke S (1992) Protein isoprenylation and methylation at carboxyl terminal cysteine residues. Annu Rev Biochem 61:355–386
Damico R, Simms T, Kim BS, Tekeste Z, Amankwan H, Damarla M, Hassoun PM (2011) p53 mediates cigarette smoke-induced apoptosis of pulmonary endothelial cells: inhibitory effects of macrophage migration inhibitor factor. Am J Respir Cell Mol Biol 44(3):323–332
Dauphinee SM, Karsan A (2006) Lipopolysaccharide signaling in endothelial cells. Lab Investig 86(1):9–22
Dauphinee SM, Voelcker V, Tebaykina Z, Wong F, Karsan A (2011) Heterotrimeric Gi/Go proteins modulate endothelial TLR signaling independent of the MyD88-dependent pathway. Am J Physiol Heart Circ Physiol 301(6):H2246–H2253
De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G, Armirotti A, Amodei S, Palamara AT, Russo T, Garaci E, Cozzolino F (2006) Bcl-2 phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem 281(30):21353–21361
Desch CE, O’Hara P, Harlan JM (1989) Antilipopolysaccharide factor from horseshoe crab, Tachypleus tridentatus, inhibits lipopolysaccharide activation of cultured human endothelial cells. Infect Immun 57(5):1612–1614
Di Sano F, Ferraro E, Tufi R, Achsel T, Piacentini M, Cecconi F (2006) Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism. J Biol Chem 281(5):2693–2700
Diette GB, Accinelli RA, Balmes JR, Buist AS, Checkley W, Garbe P, Hansel NN, Kapil V, Gordon S, Lagat DK, Yip F, Mortimer K, Perez-Padilla R, Roth C, Schwaninger JM, Punturieri A, Kiley J (2012) Obstructive lung disease and exposure to burning biomass fuel in the indoor environment. Glob Heart 7(3):265–270
Digaleh H, Kiaei M, Khodagholi F (2013) Nrf2 and Nrf1 signaling and ER stress crosstalk: implication for proteasomal degradation and autophagy. Cell Mol Life Sci 70(24):4681–4694
Downey GP, Dong Q, Kruger J, Dedhar S, Cherapanov V (1999) Regulation of neutrophil activation in acute lung injury. Chest 116(1 Suppl):46S–54S
Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, Rosenberger P, Eltzschig HK (2007) Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol 178(12):8127–8137
Eltzschig HK, Faigle M, Knapp S, Karhausen J, Ibla J, Rosenberger P, Odegard KC, Laussen PC, Thompson LF, Colgan SP (2006) Endothelial catabolism of extracellular adenosine during hypoxia: the role of surface adenosine deaminase and CD26. Blood 108(5):1602–1610
Farley N, Pedraza-Alva G, Serrano-Gomez D, Nagaleekar V, Aronshtam A, Krahl T, Thornton T, Rincon M (2006) p38 mitogen-activated protein kinase mediates the Fas-induced mitochondrial death pathway in CD8+ T cells. Mol Cell Biol 26(6):2118–2129
Feoktistov I, Goldstein AE, Ryzhov S, Zeng D, Belardinelli L, Voyno-Yasenetskaya T, Biaggioni I (2002) Differential expression of adenosine receptors in human endothelial cells: role of A2B receptors in angiogenic factor regulation. Circ Res 90(5):531–538
Fleming IN, Elliott CM, Exton JH (1996) Differential translocation of Rho family GTPases by lysophosphatidic acid, endothelin-1, and platelet-derived growth factor. J Biol Chem 271:33067–33073
Ford ES, Murphy LB, Khavjou O, Giles WH, Holt JB, Croft JB (2015) Total and state-specific medical and absenteeism costs of COPD among adults aged ≥18 years in the United States for 2010 and projections through 2020. Chest 147(1):31–45
Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53(4):527–552
Fujita M, Kuwano K, Kunitake R, Hagimoto N, Miyazaki H, Kaneko Y, Kawasaki M, Maeyama T, Hara N (1998) Endothelial cell apoptosis in lipopolysaccharide-induced lung injury in mice. Int Arch Allergy Immunol 117(3):202–208
Gaipl US, Kuhn A, Sheriff A, Munoz LE, Franz S, Voll RE, Kalden JR, Herrmann M (2006) Clearance of apoptotic cells in human SLE. Curr Dir Autoimmun 9:173–187
Gill SE, Taneja R, Rohan M, Wang L, Mehta S (2014) Pulmonary microvascular albumin leak is associated with endothelial cell death in murine sepsis-induced lung injury in vivo. PLoS One 9(2):e88501
Gill SE, Rohan M, Mehta S (2015) Role of pulmonary microvascular endothelial cell apoptosis in murine sepsis-induced lung injury in vivo. Respir Res 16:109
Giordano RJ, Lahdenranta J, Zhen L, Chukwueke U, Petrache I, Langley RR, Fidler IJ, Pasqualini R, Tuder RM, Arap W (2008) Targeted induction of lung endothelial cell apoptosis causes emphysema-like changes in the mouse. J Biol Chem 283(43):29447–29460
Goldar S, Khaniani MS, Derakhshan SM, Baradaran B (2015) Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac J Cancer Prev 16(6):2129–2144
Groenendyk J, Michalak M (2005) Endoplasmic reticulum quality control and apoptosis. Acta Biochim Pol 52:381–395
Guan JL, Shalloway D (1992) Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature 358(6388):690–692
Guan JL, Trevithick JE, Hynes RO (1991) Fibronectin/integrin interaction induces tyrosine phosphorylation of a 120-kDa protein. Cell Regul 2(11):951–964
Gustafsson AB, Gottlieb RA (2008) Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol 44:654–661
Harrington EO, Lu Q, Rounds S (2007) Endothelial cell apoptosis. In: Endothelial biomedicine. Cambridge Press, Cambridge
Hengstermann A, Müller T (2008) Endoplasmic reticulum stress induced by aqueous extracts of cigarette smoke in 3T3 cells activates the unfolded-protein-response-dependent PERK pathway of cell survival. Free Radic Biol Med 44:1097–1107
Henriquez M, Armisén R, Stutzin A, Quest AF (2008) Cell death by necrosis, a regulated way to go. Curr Mol Med 8(3):187–206
Henson PM, Tuder RM (2008) Apoptosis in the lung: induction, clearance and detection. Am J Physiol Lung Cell Mol Physiol 294:L601–L611
Holguin F, Moss I, Brown LA, Guidot DM (1998) Chronic ethanol ingestion impairs alveolar type II cell glutathione homeostasis and function and predisposes to endotoxin-mediated acute edematous lung injury in rats. J Clin Invest 101(4):761–768
Hotchkiss RS, Strasser A, McDunn JE, Swanson PE (2009) Cell death. N Engl J Med 361(16):1570–1583
Hsieh SJ, Zhuo H, Benowitz NL, Thompson BT, Liu KD, Matthay MA, Calfee CS, L. National Heart, N. Blood Institute Acute Respiratory Distress Syndrome (2014) Prevalence and impact of active and passive cigarette smoking in acute respiratory distress syndrome. Crit Care Med 42(9):2058–2068
Hu X, Yee E, Harlan JM, Wong F, Karsan A (1998) Lipopolysaccharide induces the antiapoptotic molecules, A1 and A20, in microvascular endothelial cells. Blood 92(8):2759–2765
Huang D, Khoe M, Befekadu M, Chung S, Takata Y, Ilic D, Bryer-Ash M (2007) Focal adhesion kinase mediates cell survival via NF-kappaB and ERK signaling pathways. Am J Physiol Cell Physiol 292(4):C1339–C1352
Hungerford JE, Compton MT, Matter ML, Hoffstrom BG, Otey CA (1996) Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J Cell Biol 135(5):1383–1390
Hynes RO (1992) Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69(1):11–25
Iizuka T, Ishii Y, Itoh K, Kiwamoto T, Kimura T, Matsuno Y, Morishima Y, Yamamoto M, Sekizawa K (2005) Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 10(12):1113–1125
Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377(6549):539–544
Ilic D, Almeida EA, Schlaepfer DD, Dazin P, Aizawa S, Damsky CH (1998) Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol 143(2):547–560
Ilic D, Kovacic B, McDonagh S, Jin F, Baumbusch C, Gardner DG, Damsky CH (2003) Focal adhesion kinase is required for blood vessel morphogenesis. Circ Res 92(3):300–307
Imai K, Mercer BA, Schulman LL, Sonett JR, D’Armiento JM (2005) Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J 25(2):250–258
Iribarren C, Jacobs DR Jr, Sidney S, Gross MD, Eisner MD (2000) Cigarette smoking, alcohol consumption, and risk of ARDS: a 15-year cohort study in a managed care setting. Chest 117(1):163–168
Jorgensen E, Stinson A, Shan L, Yang J, Gietl D, Albino AP (2008) Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 8:229
Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF (2000) Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106:1311–1319
Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF (2001) Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 163:737–744
Kawasaki M, Kuwano K, Hagimoto N, Matsuba T, Kunitake R, Tanaka T, Maeyama T, Hara N (2000) Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am J Pathol 157(2):597–603
Kelsen SG, Duan X, Ji R, Perez O, Liu C, Merali S (2008) Cigarette smoke induces an unfolded protein response in the human lung: a proteomic approach. Am J Respir Cell Mol Biol 38:541–550
Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N (1997) The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev 11(6):701–713
Kerr J, Wyllie A, Currie A (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Khaltaev N (2005) WHO strategy for prevention and control of chronic obstructive pulmonary disease. Exp Lung Res 31(Suppl 1):55–56
Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J (1997) Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J 16(10):2783–2793
Kim SJ, Zhang Z, Hitomi E, Lee YC, Mukherjee AB (2006) Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum Mol Genet 15(11):1826–1834
Kim HP, Wang X, Chen ZH, Lee SJ, Huang MH, Wang Y, Ryter SW, Choi AM (2008) Autophagic proteins regulate cigarette smoke-induced apoptosis: protective role of heme oxygenase-1. Autophagy 4(7):887–895
Klionsky DJ et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 12(1):1–222
Kramer K, Harrington EO, Lu Q, Bellas R, Newton J, Sheahan KL, Rounds S (2003) Isoprenylcysteine carboxyl methyltransferase activity modulates endothelial cell apoptosis. Mol Biol Cell 14(3):848–857
Kranenburg O, Poland M, Gebbink M, Oomen L, Moolenaar WH (1997) Dissociation of LPA-induced cytoskeletal contraction from stress fiber formation by differential localization of RhoA. J Cell Sci 110:2417–2427
LaCasse EC, Baird S, Korneluk RG, MacKenzie AE (1998) The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 17(25):3247–3259
Li H, Zhu H, Xu C, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas-pathway of apoptosis. Cell 94:491–501
Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318(5852):944–949
Linkermann A, Green DR (2014) Necroptosis. N Engl J Med 370(5):455–465
Lu Q (2008) Transforming growth factor-beta1 protects against pulmonary artery endothelial cell apoptosis via ALK5. Am J Physiol Lung Cell Mol Physiol 295(1):L123–L133
Lu Q, Rounds S (2009) Pulmonary endothelial cell death: implications for lung disease pathogenesis. In: Voelkel NF, Rounds S (eds) The pulmonary endothelium: function in health and disease. Wiley, Chichester, pp 243–260
Lu Q, Sakhatskyy P, Grinnell K, Newton J, Ortiz M, Wang Y, Sanchez-Esteban J, Harrington EO, Rounds S (2011) Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol 301(6):L847–L857
Lu Q, Newton J, Hsiao V, Shamirian P, Blackburn MR, Pedroza M (2012) Sustained adenosine exposure causes lung endothelial barrier dysfunction via nucleoside transporter-mediated signaling. Am J Respir Cell Mol Biol 47(5):604–613
Lu Q, Sakhatskyy P, Newton J, Shamirian P, Hsiao V, Curren S, Gabino Miranda GA, Pedroza M, Blackburn MR, Rounds S (2013) Sustained adenosine exposure causes lung endothelial apoptosis: a possible contributor to cigarette smoke-induced endothelial apoptosis and lung injury. Am J Physiol Lung Cell Mol Physiol 304(5):L361–L370
Machino T, Hashimoto S, Maruoka S, Gon Y, Hayashi S, Mizumura K, Nishitoh H, Ichijo H, Horie T (2003) Apoptosis signal-regulating kinase 1-mediated signaling pathway regulates hydrogen peroxide-induced apoptosis in human pulmonary vascular endothelial cells. Crit Care Med 31(12):2776–2781
Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA (1989) Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest 84(5):1609–1619
Martin TR, Hagimoto N, Nakamura M, Matute-Bello G (2005) Apoptosis and epithelial injury in the lungs. Proc Am Thorac Soc 2(3):214–220
Matot I, Weiniger CF, Zeira E, Galun E, Joshi BV, Jacobson KA (2006) A3 adenosine receptors and mitogen-activated protein kinases in lung injury following in vivo reperfusion. Crit Care 10(2):R65
Matsuzawa A, Ichijo H (2008) Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta 1780(11):1325–1336
Matthay MA, Ware LB, Zimmerman GA (2012) The acute respiratory distress syndrome. J Clin Invest 122(8):2731–2740
Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR (1999) Soluble Fas ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 163(4):2217–2225
Michaelson D, Silletti J, Murphy G, Peter DE, Rush M, Philips MR (2001) Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol 152:111–126
Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM (2014) Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124(9):3987–4003
Mizuno S, Yasuo M, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, Farkas D, Farkas L, Voelkel NF (2012) Copper deficiency induced emphysema is associated with focal adhesion kinase inactivation. PLoS One 7(1):e30678
Murray CJ, Atkinson C, Bhalla K, Birbeck G, Burstein R, Chou D, Dellavalle R, Danaei G, Ezzati M, Fahimi A, Flaxman D, Foreman S, Gabriel E, Gakidou N, Kassebaum S, Khatibzadeh S, Lim S, Lipshultz E, London S, Lopez AD, MacIntyre MF, Mokdad AH, Moran A, Moran AE, Mozaffarian D, Murphy T, Naghavi M, Pope C, Roberts T, Salomon J, Schwebel DC, Shahraz S, Sleet DA, Murray CJ, Abraham J, Ali MK, Atkinson C, Bartels DH, Bhalla K, Birbeck G, Burstein R, Chen H, Criqui MH, Dahodwala N, Jarlais DC, Ding EL, Dorsey ER, Ebel BE, Ezzati M, Fahami SF, Flaxman AD, Gonzalez-Medina D, Grant B, Hagan H, Hoffman H, Kassebaum N, Khatibzadeh S, Leasher JL, Lin J, Lipshultz SE, Lozano R, Lu Y, Mallinger L, McDermott MM, Micha R, Miller TR, Mokdad AA, Mokdad AH, Mozaffarian D, Naghavi M, Narayan KM, Omer SB, Pelizzari PM, Phillips D, Ranganathan D, Rivara FP, Roberts T, Sampson U, Sanman E, Sapkota A, Schwebel DC, Sharaz S, Shivakoti R, Singh GM, Singh D, Tavakkoli M, Towbin JA, Wilkinson JD, Zabetian A, Murray CJ, Abraham J, Ali MK, Alvardo M, Atkinson C, Baddour LM, Benjamin EJ, Bhalla K, Birbeck G, Bolliger I, Burstein R, Carnahan E, Chou D, Chugh SS, Cohen A, Colson KE, Cooper LT, Couser W, Criqui MH, Dabhadkar KC, Dellavalle RP, Jarlais DC, Dicker D, Dorsey ER, Duber H, Ebel BE, Engell RE, Ezzati M, Felson DT, Finucane MM, Flaxman S, Flaxman AD, Fleming T, Foreman K, Forouzanfar MH, Freedman G, Freeman MK, Gakidou E, Gillum RF, Gonzalez-Medina D, Gosselin R, Gutierrez HR, Hagan H, Havmoeller R, Hoffman H, Jacobsen KH, James SL, Jasrasaria R, Jayarman S, Johns N, Kassebaum N, Khatibzadeh S, Lan Q, Leasher JL, Lim S, Lipshultz SE, London S, Lopez AD, Lozano R, Lu Y, Mallinger L, Meltzer M, Mensah GA, Michaud C, Miller TR, Mock C, Moffitt TE, Mokdad AA, Mokdad AH, Moran A, Naghavi M, Narayan KM, Nelson RG, Olives C, Omer SB, Ortblad K, Ostro B, Pelizzari PM, Phillips D, Raju M, Razavi H, Ritz B, Roberts T, Sacco RL, Salomon J, Sampson U, Schwebel DC, Shahraz S, Shibuya K, Silberberg D, Singh JA, Steenland K, Taylor JA, Thurston GD, Vavilala MS, Vos T, Wagner GR, Weinstock MA, Weisskopf MG, Wulf S, Murray CJ, U. S. B. o. D. Collaborators (2013) The state of US health, 1990–2010: burden of diseases, injuries, and risk factors. JAMA 310(6):591–608
No Authors (1997) Alpha 1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ 75(5):397–415
Noguchi T, Ishii K, Fukutomi H, Naguro I, Matsuzawa A, Takeda K, Ichijo H (2008) Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J Biol Chem 283(12):7657–7665
Obeng EA, Boise LH (2005) Caspase-12 and caspase-4 are not required for caspase-dependent endoplasmic reticulum stress-induced apoptosis. J Biol Chem 280(33):29578–29587
Ola MS, Nawaz M, Ahsan H (2011) Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem 351(1–2):41–58
Olson M, Kornbluth S (2001) Mitochondria in apoptosis and human disease. Curr Mol Med 1(1):91–122
Parsons JT (2003) Focal adhesion kinase: the first ten years. J Cell Sci 116(Pt 8):1409–1416
Pastor LM, Sánchez-Gascón F, Girona JC, Bernal-Mañas CM, Morales E, Beltrán-Frutos E, Canteras M (2006) Morphogenesis of rat experimental pulmonary emphysema induced by intratracheally administered papain: changes in elastic fibres. Histol Histopathol 21(12):1309–1319
Perl M, Chung CS, Lomas-Neira J, Rachel TM, Biffl WL, Cioffi WG, Ayala A (2005) Silencing of Fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am J Pathol 167(6):1545–1559
Perl M, Chung CS, Perl U, Lomas-Neira J, de Paepe M, Cioffi WG, Ayala A (2007) Fas-induced pulmonary apoptosis and inflammation during indirect acute lung injury. Am J Respir Crit Care Med 176(6):591–601
Perl M, Lomas-Neira J, Venet F, Chung CS, Ayala A (2011) Pathogenesis of indirect (secondary) acute lung injury. Expert Rev Respir Med 5(1):115–126
Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM (2005) Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 11:491–498
Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L, Petrache HI, Flotte TR, Tuder RM (2006) Alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol 169:1155–1166
Pittet JF, Mackersie RC, Martin TR, Matthay MA (1997) Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med 155(4):1187–1205
Qing DY, Conegliano D, Shashaty MG, Seo J, Reilly JP, Worthen GS, Huh D, Meyer NJ, Mangalmurti NS (2014) Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am J Respir Crit Care Med 190(11):1243–1254
Reytor E, Perez-Miguelsanz J, Alvarez L, Perez-Sala D, Pajares MA (2009) Conformational signals in the C-terminal domain of methionine adenosyltransferase I/III determine its nucleocytoplasmic distribution. FASEB J 23(10):3347–3360
Rivo J, Zeira E, Galun E, Matot I (2004) Activation of A3 adenosine receptor provides lung protection against ischemia-reperfusion injury associated with reduction in apoptosis. Am J Transplant 4(12):1941–1948
Rounds S, Yee WL, Dawicki DD, Harrington E, Parks N, Cutaia MV (1998) Mechanism of extracellular ATP- and adenosine-induced apoptosis of cultured pulmonary artery endothelial cells. Am J Phys 275(2 Pt 1):L379–L388
Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD (2005) Incidence and outcomes of acute lung injury. N Engl J Med 353(16):1685–1693
Ruchko M, Gorodnya O, LeDoux SP, Alexeyev MF, Al-Mehdi AB, Gillespie MN (2005) Mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis in oxidant-challenged lung endothelial cells. Am J Physiol Lung Cell Mol Physiol 288(3):L530–L535
Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ (2006) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol 4(11):e374
Sakhatskyy P, Gabino Miranda GA, Newton J, Lee CG, Choudhary G, Vang A, Rounds S, Lu Q (2014) Cigarette smoke-induced lung endothelial apoptosis and emphysema are associated with impairment of FAK and eIF2alpha. Microvasc Res 94:80–89
Sandford AJ, Weir TD, Pare PD (1997) Genetic risk factors for chronic obstructive pulmonary disease. Eur Respir J 10(6):1380–1391
Sata M, Takabatake N, Inoue S, Shibata Y, Abe S, Machiya J, Wada T, Ji G, Kido T, Matsuura T, Muramatsu MA, Kubota I (2007) Intronic single-nucleotide polymorphisms in Bcl-2 are associated with chronic obstructive pulmonary disease severity. Respirology 12:34–41
Savill J, Dransfield I, Gregory C, Haslett C (2002) A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2(12):965–975
Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT (1994) Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol 14(3):1680–1688
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789
Shapiro SD (1995) The pathogenesis of emphysema: the elastase:antielastase hypothesis 30 years later. Proc Assoc Am Physicians 107(3):346–352
Shapiro SD (1999) The macrophage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 160(5 Pt 2):S29–S32
Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A (2003) Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol 163(6):2329–2335
Shen TL, Park AY, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan JL (2005) Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol 169(6):941–952
Simonis G, Wiedemann S, Joachim D, Weinbrenner C, Marquetant R, Strasser RH (2009) Stimulation of adenosine A2b receptors blocks apoptosis in the non-infarcted myocardium even when administered after the onset of infarction. Mol Cell Biochem 328(1-2):119–126
Sipkens JA, Hahn NE, Blom HJ, Lougheed SM, Stehouwer CD, Rauwerda JA, Krijnen PA, van Hinsbergh VW, Niessen HW (2012) S-Adenosylhomocysteine induces apoptosis and phosphatidylserine exposure in endothelial cells independent of homocysteine. Atherosclerosis 221(1):48–54
Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD (1994) Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med 150(1):113–122
Subramanian S, Steer CJ (2010) MicroRNAs as gatekeepers of apoptosis. J Cell Physiol 223(2):289–298
Surolia R, Karki S, Kim H, Yu Z, Kulkarni T, Mirov SB, Carter AB, Rowe SM, Matalon S, Thannickal VJ, Agarwal A, Antony VB (2015) Heme oxygenase-1-mediated autophagy protects against pulmonary endothelial cell death and development of emphysema in cadmium-treated mice. Am J Physiol Lung Cell Mol Physiol 309(3):L280–L292
Szegezdi E, Fitzgerald U, Samali A (2003) Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci 1010:186–194
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7(9):880–885
Tang K, Rossiter HB, Wagner PD, Breen EC (2004) Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J Appl Physiol 97:1559–1566
Taraseviciene-Stewart L, Voelkel NF (2008) Molecular pathogenesis of emphysema. J Clin Invest 118(2):394–402
Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP (2004) Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med 200(11):1395–1405
Thorburn A (2004) Death receptor-induced cell killing. Cell Signal 16(2):139–144
Tuder RM, Wood K, Taraseviciene L, Flores SC, Voekel NF (2000) Cigarette smoke extract decreases the expression of vascular endothelial growth factor by cultured cells and triggers apoptosis of pulmonary endothelial cells. Chest 117(5 Suppl 1):241S–242S
Umapathy NS, Fan Z, Zemskov EA, Alieva IB, Black SM, Verin AD (2010) Molecular mechanisms involved in adenosine-induced endothelial cell barrier enhancement. Vasc Pharmacol 52(5–6):199–206
Villar J, Kacmarek RM, Perez-Mendez L, Aguirre-Jaime A (2006) A high positive end-expiratory pressure, low tidal volume ventilatory strategy improves outcome in persistent acute respiratory distress syndrome: a randomized, controlled trial. Crit Care Med 34(5):1311–1318
Voelkel NF, Vandivier RW, Tuder RM (2006) Vascular endothelial growth factor in the lung. Am J Physiol Lung Cell Mol Physiol 290:L209–L221
Volmer JB, Thompson LF, Blackburn MR (2006) Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol 176(7):4449–4458
Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412(6844):346–351
Wang HL, Akinci IO, Baker CM, Urich D, Bellmeyer A, Jain M, Chandel NS, Mutlu GM, Budinger GR (2007) The intrinsic apoptotic pathway is required for lipopolysaccharide-induced lung endothelial cell death. J Immunol 179(3):1834–1841
Wang X, Cui L, Joseph J, Jiang B, Pimental D, Handy DE, Liao R, Loscalzo J (2011) Homocysteine induces cardiomyocyte dysfunction and apoptosis through p38 MAPK-mediated increase in oxidant stress. J Mol Cell Cardiol 52(3):753–760
Wheaton AG, Cunningham TJ, Ford ES, Croft JB, C. Centers for Disease and Prevention (2015) Employment and activity limitations among adults with chronic obstructive pulmonary disease–United States, 2013. MMWR Morb Mortal Wkly Rep 64(11):289–295
Wyatt AW, Steinert JR, Wheeler-Jones CP, Morgan AJ, Sugden D, Pearson JD, Sobrevia L, Mann GE (2002) Early activation of the p42/p44MAPK pathway mediates adenosine-induced nitric oxide production in human endothelial cells: a novel calcium-insensitive mechanism. FASEB J 16(12):1584–1594
Yakes FM, Van Houten B (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 94(2):514–519
Zen K, Karsan A, Stempien-Otero A, Yee E, Tupper J, Li X, Eunson T, Kay MA, Wilson CB, Winn RK, Harlan JM (1999) NF-kappaB activation is required for human endothelial survival during exposure to tumor necrosis factor-alpha but not to interleukin-1beta or lipopolysaccharide. J Biol Chem 274(40):28808–28815
Zhande R, Dauphinee SM, Thomas JA, Yamamoto M, Akira S, Karsan A (2007) FADD negatively regulates lipopolysaccharide signaling by impairing interleukin-1 receptor-associated kinase 1-MyD88 interaction. Mol Cell Biol 27(21):7394–7404
Zhou Y, Mohsenin A, Morschl E, Young HW, Molina JG, Ma W, Sun CX, Martinez-Valdez H, Blackburn MR (2009) Enhanced airway inflammation and remodeling in adenosine deaminase-deficient mice lacking the A2B adenosine receptor. J Immunol 182(12):8037–8046
Zhou Y, Murthy JN, Zeng D, Belardinelli L, Blackburn MR (2010) Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS One 5(2):e9224
Acknowledgments
This work was supported with the use of facilities at the Providence VA Medical Center and by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103652 (Lu, project 1), P20GM103652 (Rounds), VA Merit Review (Rounds), NIH Ro1 HL130230 (Lu).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Chambers, E., Rounds, S., Lu, Q. (2018). Pulmonary Endothelial Cell Apoptosis in Emphysema and Acute Lung Injury. In: Parthasarathi, K. (eds) Molecular and Functional Insights Into the Pulmonary Vasculature. Advances in Anatomy, Embryology and Cell Biology, vol 228. Springer, Cham. https://doi.org/10.1007/978-3-319-68483-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-68483-3_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-68482-6
Online ISBN: 978-3-319-68483-3
eBook Packages: MedicineMedicine (R0)