
Abstract
Breast cancer affects approximately 1 in 8 women. It is estimated that over 252,710 women in the United States will be diagnosed with breast cancer in 2017. Breast cancer-related deaths have declined over the last two decades as a result of early detection and improved treatment, particularly targeted therapies, such as trastuzumab that targets human epidermal growth factor receptor 2 (HER2), which is frequently overexpressed in breast cancer. However, resistance to trastuzumab, either de novo or acquired resistance, presents a major clinical challenge. Here, we summarize the mechanisms of action and resistance of trastuzumab in breast cancer and discuss potential strategies to overcome resistance.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Receptor tyrosine kinase
- Drug resistance
- Therapeutic antibodies
- Small tyrosine kinase inhibitors
- Breast cancer
3.1 Introduction
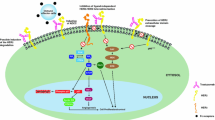
The epidermal growth factor receptor (EGFR) family consists of four members, ERBB1 (EGFR/HER1), ERBB2 (HER2/neu), ERBB3 (HER3), and ERBB4 (HER4), which are cytoplasmic membrane-anchored receptor tyrosine kinases that regulate important biological processes, such as cell growth, differentiation, metabolism, and survival through activation of downstream signaling pathways [1,2,3,4,5,6]. All members of the EGFR family share sequence and structural similarities and contain an extracellular ligand-binding ectodomain, a transmembrane domain, and a cytoplasmic tyrosine kinase domain [1, 7]. Following ligand binding, members of the ERBB family interact to form various combinations of homo- or heterodimers, which then induce autophosphorylation of the tyrosine residues within the kinase domain [1]. Recruitment of adaptor proteins at the phosphotyrosine residues subsequently initiates the downstream signaling cascades, such as phosphatidylinositol 3-kinase (PI3K), Ras, phospholipase Cγ (PLCγ), Janus-activated kinase (JAK) /signal transducer and activator of transcription (STAT) [3, 8].
In contrast to EGFR, ERBB3, and ERBB4, which bind extracellular ligands to trigger downstream signaling, HER2 does not bind to any ligands directly. Rather, HER2 mediates downstream signaling in concert with a ligand-activated coreceptor, e.g., EGFR, ERBB3, or ERBB4 [1, 2, 9]. HER2 can also form homodimers and activate signaling cascades especially at higher concentrations as observed in cancers [2, 9]. The HER2/ERBB3 heterodimer is the most potent activator of two key pathways regulating cell survival and growth, the mitogen-activated protein kinase (MAPK) and the PI3K/Akt signaling cascades, and ERBB3 plays an essential role in HER2-mediated oncogenic signaling [10,11,12]. Activation of HER2 also decreases the protein levels of cell cycle negative regulator p27Kip1 by promoting its mislocation through Jun activation domain-binding protein 1-mediated export into the cytoplasm, and subsequently its degradation via the ubiquitin-proteasome pathway [13].
HER2 is normally expressed at low levels on the cell surface, but in breast cancer, the number of HER2 receptors on the surface of each cell can reach up to 100 times more than a normal cell, which leads to aberrant activation of its downstream signaling cascades and uncontrollable cell growth [14, 15]. HER2 amplification/overexpression is observed in approximately 20% of breast cancer and is associated with poor clinical outcome and disease progression [16,17,18], and HER2 has proved to be one of the most successful targets in breast cancer. In this chapter, we briefly summarize the mechanisms of action and resistance of trastuzumab (Herceptin®; Genentech) as well as treatment strategies to overcome resistance in breast cancer.
3.2 Trastuzumab
3.2.1 Proposed Mechanism of Action
Several HER2-specific mAbs were developed and demonstrated to effectively inhibit tumor growth of HER2-overexpressing cell lines [19]. Among them, the murine mAb 4D5, which was later humanized to become trastuzumab, selectively targets the extracellular domain IV of HER2 with high affinity and prevents ligand-induced dimerization and subsequent activation of downstream pathways [20]. Following the clinical studies which demonstrated that the addition of trastuzumab to chemotherapy, compared with chemotherapy alone, increased the response rates, time to progression, and survival in patients with HER2-positive (HER2+) metastatic breast cancer, trastuzumab received approval by the FDA in 1998 for the treatment of metastatic breast cancer with HER2 overexpression [21, 22].
Trastuzumab exerts its mechanism of action through several different approaches. First, it disrupts signal transduction pathways, most notably, MAPK and PI3K/Akt signaling, leading to apoptosis and arrest of proliferation [1, 23,24,25]. Trastuzumab produces cytostatic effects associated with downregulation of AKT activity and results in increased G1 growth arrest via enhanced stability of the cell cycle inhibitor p27Kip1 [23, 26, 27]. In addition, trastuzumab can block PI3K signaling by reducing tyrosine phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) and increasing its phosphatase activity and membrane localization [28]. Second, trastuzumab can block proteolytic cleavage of HER2 by the metalloprotease ADAM10 [29], which liberates its extracellular domain (ECD) and produces a truncated, membrane-bound, and kinase active carboxy terminal fragment (CTF) , p95HER2 [30]. Interestingly, HER2 ECD can be detected in the serum of breast cancer patients, and results from clinical studies indicated that a decline in serum HER2 ECD following trastuzumab treatment could predict clinical benefit [31]. Third, trastuzumab exerts an antitumor effect through activation of the antibody-mediated cellular toxicity (ADCC) [32, 33]. Studies have demonstrated in cell lines and xenografts that this immunological effect of trastuzumab is mainly attributed to the binding of the Fc (fragment, crystallizable) region of the antibody to Fc gamma receptor present on natural killer cells [32, 34]. Immunohistochemistry (IHC) analysis of breast tissue samples from patients with HER2-overexpressing advanced breast cancer during a neoadjuvant treatment of trastuzumab and docetaxel in a clinical trial further validated the immune cell-modulated activity of trastuzumab via an increased number of natural killer (NK) cells and cytotoxic proteins, e.g., granzyme B, in tumor infiltrates after trastuzumab treatment [33].
3.2.2 Mechanism of Resistance
Although trastuzumab in combination with chemotherapy has significantly improved the outcome of breast cancer patients, de novo and acquired resistance to trastuzumab pose a major challenge in the clinic [35, 36]. A large proportion of patients with HER2+ breast cancer do not respond to initial trastuzumab treatment (de novo resistance) and those who initially responded eventually experience disease progression (acquired resistance) [37,38,39]. The mechanisms of trastuzumab have been extensively studied and may involve the following: (1) upregulation of downstream signaling due to genetic alterations; (2) hindrance of trastuzumab binding to HER2; and (3) overexpression of ERBB receptors or other tyrosine kinase receptors. Each will be briefly described below.
3.2.3 Upregulation of Downstream Signaling
The constitutive activation of the downstream PI3K/Akt pathway due to mutations in the gene encoding PI3K and/or inactivation or loss of PTEN have been shown to contribute to trastuzumab resistance [28, 40]. PI3K catalyzes the lipid phosphatidylinositol-4,5-bisphosphate (PIP2) to the phosphatidyl-inositol-3,4,5-trisphosphate (PIP3) , which binds to the pleckstrin homology domain of the serine/threonine protein kinase Akt, resulting in the translocation of Akt to the membrane and its subsequent activation to promote cell survival and inhibition of apoptosis [41]. Activating mutations in the PIK3CA gene encoding the catalytic subunit (p110) of PI3K have been reported to induce constitutive activation of the PI3K/Akt pathway. The frequency of PIK3CA activating mutations in HER2+ breast cancers has been reported to be 23–33% [42]. PTEN antagonizes PI3K by dephosphorylating PIP3 and negatively regulates AKT activities [43,44,45,46]. Hence, as PTEN normally blocks PI3K activation, the loss of PTEN results in constitutive activation of PI3K/Akt signaling and subsequently bypassing trastuzumab-mediated growth arrest [46, 47]. Breast cancer patients with PTEN deficiency demonstrated poorer response to trastuzumab compared with those with normal PTEN [28]. Zhang et al. reported that cytoplasmic tyrosine kinase SRC functions as a common mediator of multiple trastuzumab resistance pathways and is regulated via dephosphorylation by PTEN [48]. The increased activation of SRC was observed in both de novo and acquired trastuzumab resistant cells and correlated with trastuzumab resistance in patients [48]. A follow-up clinical trial indicated that patients with HER2-overexpressing metastatic breast cancer with PTEN loss and progressed on trastuzumab-based therapy had decreased overall survival compared with those with normal PTEN [49]. Moreover, studies reported that the combination of trastuzumab with everolimus, an inhibitor against AKT downstream molecule mTOR, provided an objective response rate of 15% and clinical benefit rate of 34% [49]. These findings further validated the role of PTEN deficiency in trastuzumab resistance. In addition, preclinical studies demonstrated that the combination of trastuzumab and the PIK3 inhibitor, GDC-0941, is highly effective against trastuzumab-resistant cells and tumors and can also overcome trastuzumab resistance caused by PTEN loss [24]. The PI3K inhibitors that are currently under clinical investigation for solid tumors harboring PIK3CA or PTEN mutations include buparlisib (BKM120), taselisib (GDC-0032), and GSK2636771 [50].
Akt has also been demonstrated to phosphorylate the tumor suppressor SIRT6 at Ser338, resulting in MDM2 -mediated ubiquitination and subsequent degradation of SIRT6 [51]. The authors further reported a positive correlation between SIRT6 abundance and survival of breast cancer patients. Their findings suggested that stabilization of SIRT6 by preventing its degradation may be a potential therapeutic strategy to overcome trastuzumab resistance.
3.2.4 Epitope Masking
As indicated above, the ectodomain shedding of HER2 produces a truncated and constitutively active membrane-bound p95HER2 CTF of 95- to 100-kDa. In addition to the ectodomain shedding, the alternative translation initiation of HER2 mRNA can give rise to two other p95HER2 fragments, a membrane-bound 611-CTF (100–115 kDa), which forms constitutively active homodimers, and a soluble 678-CTF (90–95 kDa), which is kinase inactive [52]. Both p95HER2 95–100 kDa and 110–115 kDa fragments lack the epitope for recognition by trastuzumab [53], and circulating HER2 ECD can compete with the full-length membrane-bound HER2 for binding to trastuzumab [54]. Pederson and coworkers found that the 611-CTF regulated genes linked to metastasis, and 611-CTF transgenic mice developed more aggressive and invasive mammary tumors compared with mice with full-length HER2 [52]. Up to 30% of HER2+ breast cancers express p95HER2 and are associated with metastasis and shorter disease-free survival [55, 56]. Retrospective studies indicated that the presence of p95HER2 fragments in tumors is associated with trastuzumab resistance [57, 58]. Interestingly, p95HER2 was shown to preferentially heterodimerize with HER3 to trigger pro-survival signaling [59]. Parra-Palau and coworkers reported that chemotherapy sensitizes p95HER2 (611CTF)-expressing patient derived xenograft from HER2+ breast cancers to trastuzumab [60].
Another mechanism contributing to trastuzumab resistance is the binding of cell surface glycoprotein mucin-4 (MUC4) to the extracellular domain of HER2, which can mask the trastuzumab-binding site on HER2 (epitope masking). Nagy et al. reported that MUC4 expression was correlated negatively with decreased trastuzumab binding, and that knocking down MUC4 reversed trastuzumab resistance in a de novo trastuzumab-resistant JIMT-1 breast cancer cell line [61]. Hyperactivation of the signal transducer and activator of transcription-3 (STAT3) via a positive feedback loop was shown to upregulate MUC4 expression [62]. More recently, Mercogliano et al. reported that TNFα induces elevated expression levels of MUC4 and contributes to trastuzumab resistance in HER2+ breast cancer. The authors further identified MUC4 expression as an independent predictor of poor disease-free survival in HER2+ breast cancer patients and suggested the combination of TNFα-blocking antibodies as a therapeutic option to overcome trastuzumab resistance [63].
3.2.5 Expression of Other Receptor Tyrosine Kinases
HER2 can form heterodimers with other receptor tyrosine kinases to activate downstream signaling cascades to compensate for the inhibition of HER2 signaling by trastuzumab [10, 11, 64,65,66,67]. Ritter et al. demonstrated that trastuzumab-resistant cells exhibited higher levels of EGFR phosphorylation and EGFR/HER2 heterodimers, and the addition of EGFR TKIs, erlotinib, gefitinib, or lapatinib (a dual EGFR/HER2 inhibitor), induced apoptosis in those resistant cells [64]. The HER2-HER3 heterodimer potently activates the PI3K/Akt and MAPK pathways, and trastuzumab is unable to block the ligand-induced HER2/HER3 heterodimer [68]. The HER-2 targeting monoclonal antibody, pertuzumab (see later for more details), was developed to prevent HER2 dimerization with EGFR and HER3 [69].
The receptor tyrosine kinase Eph receptor A2 (EphA2) is overexpressed in many cancer cell lines and human tumor tissue specimens and can form a complex with HER2 and activate signaling promoting cell proliferation and motility [70,71,72]. Eliminating EphA2 expression in ERBB2-driven murine mammary tumor models impaired tumor initiation and metastatic progression [72]. In addition, Zhuang and colleagues found that high levels of EphA2 expression in HER2+ breast cancer patients predict poor prognosis and identified a mechanism by which EphA2 contributes to trastuzumab via EphA2-mediated amplification of the PI3K/Akt and MAPK cascades [65]. Their findings suggested targeting EphA2 as a therapeutic strategy to overcome trastuzumab resistance. Amato et al. demonstrated that the EphA2 kinase inhibitor, ALW-II-41-27, inhibited cell viability of non-small cell lung cancer (NSCLC) cells in vitro and induced tumor regression in a NSCLC xenograft tumor model [73]. Targeting EphA2 was shown to overcome primary and acquired resistance to anti-EGFR therapy, cetuximab, in metastatic colorectal cancer [74]. Whether the addition of ALW-II-41-27 could overcome trastuzumab resistance in breast cancer remains unclear.
The overexpression of the insulin-like growth factor receptor 1 (IGF-1R) and its ligands, insulin growth factor-1 (IGF-1) and IGF-2 , is often observed in breast tumors [75]. Activation of IGF-1R following ligand binding triggers cell survival signals, and overexpression of IGF-1R has been shown to confer resistance to trastuzumab via hyperactivation of SRC [48, 76]. Specifically, ectopic expression of IGF-1R in trastuzumab-sensitive breast cancer cells in the presence of IGF-1 ligand rendered trastuzumab ineffective in reducing cell proliferation, and the addition of IGF-binding protein-3, which suppresses IGF-1R signaling, reversed resistance [76]. The inhibition of SRC renders trastuzumab-resistant IGF-1R breast cancer cells sensitive to trastuzumab [48]. IGF-1R is also reported to form a heterodimeric complex with HER2 and HER3 in breast cancer cells resistant to trastuzumab through enhanced PI3K/Akt signaling and SRC activation [77]. Liu et al. reported that metformin, a type 2 diabetes drug with antitumor effects, reduces HER2 and IGF-1R interactions in trastuzumab-resistant breast cancer cells [78]. Metformin has been shown to activate the adenosine monophosphate (AMP) -activated protein kinase AMPK, which plays a critical role as a regulator of cellular energy homeostasis [79]. Interestingly, metformin inhibits the insulin/IGF signaling by decreasing insulin metabolism in the liver or by reducing IGFR expression. Whether AMPK regulates HER/IGF-1R interaction remains unclear.
The hepatocyte growth factor (HGF) receptor (also known as c-Met), which regulates important biological processes, including morphogenesis, cell proliferation, survival, differentiation, and anti-apoptosis, is also implicated in the progression and metastasis of many human cancers [80]. Overexpression of c-Met is observed in 20–30% of breast cancers and it has been reported to be an independent prognostic of poor prognosis for breast cancer patients [81,82,83]. Shattuck et al. reported the co-expression of c-Met and HER2 in HER2-overexpressing breast cancer cells and HER+ breast cancer tumor tissues [67]. Moreover, the inhibition of c-Met sensitized HER2-overexpressing breast cancer cells to trastuzumab, suggesting that c-Met contributes to trastuzumab resistance [67]. High risk of trastuzumab treatment failure in breast cancer patients has been reported to associate with high MET and HGF gene copy numbers [84].
3.3 Treatment Strategies to Overcome Resistance
Below we describe some strategies to overcome trastuzumab resistance.
3.3.1 Pertuzumab
The humanized monoclonal antibody pertuzumab (Perjeta®; Genentech) binds to domain II (trastuzumab binds to domain IV) of HER2 and blocks ligand-dependent HER2 heterodimerization with EGFR, HER3, or HER4, but most potently targets the heregulin-mediated HER2/HER3 signaling heterodimer [69, 85]. Inhibiting the formation of the HER2/HER3 heterodimer prevents the activation of downstream signaling, e.g., PI3K and MAPK , that regulate cell survival and growth [1, 11]. Similar to trastuzumab, pertuzumab also triggers ADCC [86]. Clinical studies of patients with advanced HER2+ breast cancer after trastuzumab treatment demonstrated that pertuzumab in combination with trastuzumab was more efficacious than pertuzumab alone [87]. In 2012, the FDA approved pertuzumab in combination with trastuzumab and docetaxel for the treatment of HER2+ metastatic breast cancer patients who have not received prior anti-HER2 therapy or chemotherapy for metastatic disease based on a phase III multicenter randomized clinical trial [88]. More recently, follow-up data extended the results of previous analyses demonstrating the efficacy of the pertuzumab plus trastuzumab and docetaxel combination [89]). Pertuzumab was later approved in 2013 for use in combination with trastuzumab and docetaxel as neoadjuvant treatment of patients with HER2+, locally advanced, inflammatory, or early stage breast cancer [90]. Interestingly, tumor gene expression analyses indicated high expression of the programmed death ligand 1 (PD-L1), an immune checkpoint protein that facilitates cancer immunesurveillance escape, is associated with resistance after neoadjuvant treatment with regimens containing HER2-targeted treatments [91]. A phase I trial is currently underway to evaluate the safety and pharmacokinetics of the PD-L1 monoclonal antibody, atezolizumab , in combination with trastuzumab and pertuzumab in HER2+ breast cancer (NCT02605915).
3.3.2 Lapatinib
Lapatinib (Tykerb®; Novartis) is a reversible ATP-competitive small molecule tyrosine kinase inhibitor (TKI) which binds to the intracellular ATP binding domain of EGFR and HER2 and inhibits the activation of downstream signaling [92,93,94]. The combination of lapatinib and capecitabine in a randomized phase III trial was found to be superior than capecitabine alone in patients with metastatic breast cancer who progressed after treatment with regimens that included an anthracycline, a taxane, and trastuzumab [95, 96]. On the basis of the phase III data, the FDA approved lapatinib in combination with capecitabine for the treatment of patients with advanced or metastatic HER2+ breast cancer and who have received prior therapy including an anthracycline, a taxane, and trastuzumab. Synergistic growth inhibition was observed in trastuzumab-treated HER2+ breast cancer cell lines for the lapatinib and trastuzumab combination [93]. Lapatinib combined with trastuzumab was later evaluated in a phase III clinical trial with results showing a significant overall survival advantage of the combination compared with lapatinib alone in HER2+ metastaic breast cancer patients whose disease progressed during trastuzumab treatment [97]. These findings further supported the dual blockage of HER2 as an approach to overcome resistance.
3.3.3 Neratinib
Neratinib (Puma Biotechnology) is an irreversible ATP-competitive small molecule TKI that blocks the intracellular ATP-binding site of EGFR, HER2, and HER4 [92,93,94]. The results from preclinical studies demonstrated that neratinib inhibits proliferation of HER2-overexpressing human breast cancer cell lines in vitro as well as an EGFR-overexpressing epidermal carcinoma cell line [92]. Phase II studies showed that neratinib was well tolerated among advanced HER2+ breast cancer patients with or without prior treatment with trastuzumab [98]. In a phase III (ExteNET) study, neratinib treatment significantly improved the 2-year disease-free survival of HER2-positive breast cancer patients after chemotherapy and trastuzumab-based adjuvant therapy [99]. Currently, neratinib is being evaluated in a number of clinical trials as a neoadjuvant therapy for patients with HER2+ breast cancer and as a treatment for patients with metastatic HER2+ breast cancer (clinicaltrials.gov). A new drug application for neratinib for extended adjuvant treatment of HER2+ early stage breast cancer has been accepted by the FDA and awaiting approval.
3.3.4 Ado-Trastuzumab Emtansine (T-DM1)
T-DM1 is an antibody-drug conjugate (ADC) containing trastuzumab covalently linked to the cytotoxic microtubule inhibitor, emtansine (DM1), via a thioester linker MCC (4-[N-maleimidomethyl] cyclohexane-1-carboxylate). T-DM1 contains about 3.5 molecules of DM1 per antibody and is internalized following binding of trastuzumab to HER2 on the cell surface [100, 101]. After binding of T-DM1 to HER2 on the cell surface, the HER2-T-DM1 complex is internalized via receptor-mediated endocytosis followed by lysosomal degradation, resulting in the release of intracellular DM-1-containing catabolites that bind to and inhibit microtubule polymerization, and subsequently induce cell cycle arrest and cell death [101]. In retaining the activity of trastuzumab, T-DM1 also disrupts the PI3K/Akt signaling cascade, inhibits the HER2 ectodomain shedding, and induces ADCC [100, 101]. Preclinical studies of T-DM1 indicated greater activity compared with trastuzumab with retained selectivity toward HER2 [100]. Favorable results from clinical studies led to the approval of T-DM1 in second-line therapy by the FDA in 2013 for patients whose advanced HER2+ breast cancer progressed after trastuzumab treatment [102,103,104]. A phase III study (MARIANNE) evaluating T-DM1 for first-line treatment of HER2-positive, advanced breast cancer indicated that T-DM1 and T-DM1 plus pertuzumab did not achieve superiority compared with trastuzumab plus a taxane [105]. The acquired resistance to T-DM1 has been reported, and factors contributing to T-DM1 resistance include poor internalization and defective intracellular trafficking of the T-DM1-HER2 complexes, inefficient lysosomal degradation of T-DM1, expression of drug efflux proteins, and altered tubulins in addition to those mechanisms known to induce trastuzumab resistance [106]. To circumvent resistance, clinical studies to evaluate T-DM1 in combination with other targeted therapies, for example, immunotherapy (pembrolizumab or atezolizumab), HER/HER3 antibody (pertuzumab), TKIs (lapatinib or neratinib), and cyclin D kinase 4/6 inhibitor (palbociclib or ribociclib) as well as in triple combination (chemotherapy and TKI), for metastatic breast cancer are currently underway. Studies on T-DM1 combined with PI3K inhibitors (taselisib) are also ongoing (clinicaltrials.gov).
3.4 Conclusion
Trastuzumab has demonstrated remarkable clinical success and increased patient outcome. However, acquired and de novo resistance via multiple mechanisms remain a clinical challenge. Furthering our understanding of the resistance mechanisms has led to the development of therapeutic strategies to overcome this resistance and improve patient outcome. HER2 somatic mutations have been reported in breast cancer, but these mutations occur almost always in the absence of HER2 gene amplification [107, 108]. HER2 mutations were functionally characterized in breast cancer without HER2 amplification [109]. While many of the identified mutations were found to be sensitive to HER2-targeted therapies in cell lines, those harboring the L755_T759 deletion mutation were resistant to lapatinib [109]. How this mutation affects patients treated with the combination therapy containing lapatinib should be further evaluated. Antibodies against immune checkpoints, e.g., PD-L1, PD-1, and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), to unleash T cell-mediated anti-tumor activity have demonstrated success as a cancer treatment in recent years [110]. Preclinical studies indicated that PD-1 antibodies significantly improved the therapeutic activity of trastuzumab [111]. A phase Ib/II clinical trial is currently underway to evaluate the efficacy of PD-1 antibody (MK-3475) and trastuzumab in patients with trastuzumab-resistant, HER2+ metastatic breast cancers (NCT02129556). As more combination therapies are being evaluated, optimizing patient selection and predictive biomarkers are required to maximize clinical efficacy.
Abbreviations
- ADAM10:
-
A disintegrin and metalloproteinase domain-containing protein 10
- ADC:
-
Antibody-drug conjugate
- ADCC:
-
Antibody-mediated cellular toxicity
- AMPK:
-
Adenosine monophosphate (AMP)-activated protein kinase
- CTF:
-
Carboxy terminal fragment
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- ECD:
-
Extracellular domain
- EGFR:
-
Epidermal growth factor receptor
- EphA2:
-
Ephrin receptor A2
- HER2:
-
Human epidermal growth factor receptor 2
- HGF:
-
Hepatocyte growth factor
- IGF-1:
-
Insulin growth factor-1
- IGF-1R:
-
Insulin-like growth factor receptor 1
- IGF-2:
-
Insulin growth factor-2
- IHC:
-
Immunohistochemistry
- JAK:
-
Janus-activated kinase
- MAPK:
-
Mitogen-activated protein kinase
- MDM2:
-
Murine double minute 2
- MUC4:
-
Glycoprotein mucin-4
- NK:
-
Natural killer
- NSCLC:
-
Non-small cell lung cancer
- PD-1:
-
Programmed death-1
- PD-L1:
-
Programmed death ligand-1
- PI3K:
-
Phosphatidylinositol 3-kinase
- PIP2 :
-
Phosphatidylinositol-4,5-bisphosphate
- PIP3 :
-
Phosphatidyl-inositol-3,4,5-trisphosphate
- PLCγ:
-
Phospholipase Cγ
- PTEN:
-
Phosphatase and tensin homologue
- STAT:
-
Signal transducer and activator of transcription
- T-DM1:
-
Ado-trastuzumab emtansine
- TKI:
-
Tyrosine kinase inhibitor
- TNFα:
-
Tumor necrosis factor α
References
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–37.
Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7(7):505–16.
Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol. 2009;218(3):460–6.
Yarden Y, Shilo BZ. SnapShot: EGFR signaling pathway. Cell. 2007;131(5):1018.
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–34.
Chen MK, Hung MC. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015;282(19):3693–721.
Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer. 2001;8(1):11–31.
Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21(2):177–84.
Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19(53):6102–14.
Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008;68(14):5878–87.
Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9(7):463–75.
Garrett JT, Sutton CR, Kurupi R, Bialucha CU, Ettenberg SA, Collins SD, et al. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110alpha inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer Res. 2013;73(19):6013–23.
Yang HY, Zhou BP, Hung MC, Lee MH. Oncogenic signals of HER-2/neu in regulating the stability of the cyclin-dependent kinase inhibitor p27. J Biol Chem. 2000;275(32):24735–9.
Kallioniemi OP, Kallioniemi A, Kurisu W, Thor A, Chen LC, Smith HS, et al. ERBB2 amplification in breast cancer analyzed by fluorescence in situ hybridization. Proc Natl Acad Sci U S A. 1992;89(12):5321–5.
Iqbal N, Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int. 2014;2014:852748.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82.
Pegram M, Slamon D. Biological rationale for HER2/neu (c-erbB2) as a target for monoclonal antibody therapy. Semin Oncol. 2000;27(5 Suppl 9):13–9.
Owens MA, Horten BC, Da Silva MM. HER2 amplification ratios by fluorescence in situ hybridization and correlation with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clin Breast Cancer. 2004;5(1):63–9.
Klapper LN, Vaisman N, Hurwitz E, Pinkas-Kramarski R, Yarden Y, Sela M. A subclass of tumor-inhibitory monoclonal antibodies to ErbB-2/HER2 blocks crosstalk with growth factor receptors. Oncogene. 1997;14(17):2099–109.
Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89(10):4285–9.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–92.
Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(3):719–26.
Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002;62(14):4132–41.
Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15(5):429–40.
Mohsin SK, Weiss HL, Gutierrez MC, Chamness GC, Schiff R, Digiovanna MP, et al. Neoadjuvant trastuzumab induces apoptosis in primary breast cancers. J Clin Oncol. 2005;23(11):2460–8.
Lane HA, Beuvink I, Motoyama AB, Daly JM, Neve RM, Hynes NE. ErbB2 potentiates breast tumor proliferation through modulation of p27(Kip1)-Cdk2 complex formation: receptor overexpression does not determine growth dependency. Mol Cell Biol. 2000;20(9):3210–23.
Le XF, Claret FX, Lammayot A, Tian L, Deshpande D, LaPushin R, et al. The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-induced G1 cell cycle arrest and tumor growth inhibition. J Biol Chem. 2003;278(26):23441–50.
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–27.
Liu PC, Liu X, Li Y, Covington M, Wynn R, Huber R, et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol Ther. 2006;5(6):657–64.
Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61(12):4744–9.
Kostler WJ, Schwab B, Singer CF, Neumann R, Rucklinger E, Brodowicz T, et al. Monitoring of serum Her-2/neu predicts response and progression-free survival to trastuzumab-based treatment in patients with metastatic breast cancer. Clin Cancer Res. 2004;10(5):1618–24.
Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–6.
Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. 2006;94(2):259–67.
Lewis GD, Figari I, Fendly B, Wong WL, Carter P, Gorman C, et al. Differential responses of human tumor cell lines to anti-p185HER2 monoclonal antibodies. Cancer Immunol Immunother. 1993;37(4):255–63.
Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011;9(1):16–32.
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659–72.
Lan KH, CH L, Mechanisms YD. of trastuzumab resistance and their clinical implications. Ann N Y Acad Sci. 2005;1059:70–5.
Narayan M, Wilken JA, Harris LN, Baron AT, Kimbler KD, Maihle NJ. Trastuzumab-induced HER reprogramming in “resistant” breast carcinoma cells. Cancer Res. 2009;69(6):2191–4.
Piccart M. Circumventing de novo and acquired resistance to trastuzumab: new hope for the care of ErbB2-positive breast cancer. Clin Breast Cancer. 2008;8(Suppl 3):S100–13.
Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402.
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–7.
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224.
Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96(8):4240–5.
Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100(4):387–90.
Leslie NR, Downes CPPTEN. The down side of PI 3-kinase signalling. Cell Signal. 2002;14(4):285–95.
Parsons R, Simpson L. PTEN and cancer. Methods Mol Biol. 2003;222:147–66.
Pandolfi PP. Breast cancer—loss of PTEN predicts resistance to treatment. N Engl J Med. 2004;351(22):2337–8.
Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17(4):461–9.
Morrow PK, Wulf GM, Ensor J, Booser DJ, Moore JA, Flores PR, et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol. 2011;29(23):3126–32.
Balko J, Mayer I, Levy M, Arteaga C. PIK3CA in Breast Cancer. My Cancer Genome. 2017. https://www.mycancergenome.org/content/disease/breast-cancer/pik3ca/.
Thirumurthi U, Shen J, Xia W, LaBaff AM, Wei Y, Li CW, et al. MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Sci Signal. 2014;7(336):ra71.
Pedersen K, Angelini PD, Laos S, Bach-Faig A, Cunningham MP, Ferrer-Ramon C, et al. A naturally occurring HER2 carboxy-terminal fragment promotes mammary tumor growth and metastasis. Mol Cell Biol. 2009;29(12):3319–31.
Arribas J, Baselga J, Pedersen K, Parra-Palau JL. p95HER2 and breast cancer. Cancer Res. 2011;71(5):1515–9.
Tortora G. Mechanisms of resistance to HER2 target therapy. J Natl Cancer Inst Monogr. 2011;2011(43):95–8.
Saez R, Molina MA, Ramsey EE, Rojo F, Keenan EJ, Albanell J, et al. p95HER-2 predicts worse outcome in patients with HER-2-positive breast cancer. Clin Cancer Res. 2006;12(2):424–31.
Scaltriti M, Chandarlapaty S, Prudkin L, Aura C, Jimenez J, Angelini PD, et al. Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin Cancer Res. 2010;16(9):2688–95.
Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99(8):628–38.
Sperinde J, Jin X, Banerjee J, Penuel E, Saha A, Diedrich G, et al. Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clin Cancer Res. 2010;16(16):4226–35.
Xia W, Liu LH, Ho P, Spector NL. Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW572016. Oncogene. 2004;23(3):646–53.
Parra-Palau JL, Morancho B, Peg V, Escorihuela M, Scaltriti M, Vicario R, et al. Effect of p95HER2/611CTF on the response to trastuzumab and chemotherapy. J Natl Cancer Inst. 2014;106(11)
Nagy P, Friedlander E, Tanner M, Kapanen AI, Carraway KL, Isola J, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005;65(2):473–82.
Li G, Zhao L, Li W, Fan K, Qian W, Hou S, et al. Feedback activation of STAT3 mediates trastuzumab resistance via upregulation of MUC1 and MUC4 expression. Oncotarget. 2014;5(18):8317–29.
Mercogliano MF, De Martino M, Venturutti L, Rivas MA, Proietti CJ, Inurrigarro G, et al. TNFalpha-induced mucin 4 expression elicits trastuzumab resistance in HER2-positive breast cancer. Clin Cancer Res. 2017;23(3):636–48.
Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13(16):4909–19.
Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S, et al. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. 2010;70(1):299–308.
Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–28.
Shattuck DL, Miller JK, Carraway KL 3rd, Sweeney C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008;68(5):1471–7.
Wehrman TS, Raab WJ, Casipit CL, Doyonnas R, Pomerantz JH, Blau HM. A system for quantifying dynamic protein interactions defines a role for Herceptin in modulating ErbB2 interactions. Proc Natl Acad Sci U S A. 2006;103(50):19063–8.
Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5(4):317–28.
Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6(12):1795–806.
Boyd AW, Bartlett PF, Lackmann M. Therapeutic targeting of EPH receptors and their ligands. Nat Rev Drug Discov. 2014;13(1):39–62.
Brantley-Sieders DM, Zhuang G, Hicks D, Fang WB, Hwang Y, Cates JM, et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J Clin Invest. 2008;118(1):64–78.
Amato KR, Wang S, Hastings AK, Youngblood VM, Santapuram PR, Chen H, et al. Genetic and pharmacologic inhibition of EPHA2 promotes apoptosis in NSCLC. J Clin Invest. 2014;124(5):2037–49.
Martini G, Belli V, Vitiello PP, Troiani T, Cardone C, Napolitano S, et al. EPHA2 blockade overcomes primary and acquired resistance to anti-epidermal growth factor receptor (EGFR) therapy in metastatic colorectal cancer (mCRC). Ann Oncol. 2016;27(suppl 6):10.
Jerome L, Shiry L, Leyland-Jones B. Deregulation of the IGF axis in cancer: epidemiological evidence and potential therapeutic interventions. Endocr Relat Cancer. 2003;10(4):561–78.
Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst. 2001;93(24):1852–7.
Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, et al. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 2010;70(3):1204–14.
Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle. 2011;10(17):2959–66.
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–23.
Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–48.
Ghoussoub RA, Dillon DA, D'Aquila T, Rimm EB, Fearon ER, Rimm DL. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 1998;82(8):1513–20.
Lengyel E, Prechtel D, Resau JH, Gauger K, Welk A, Lindemann K, et al. C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int J Cancer. 2005;113(4):678–82.
Lee WY, Chen HH, Chow NH, WC S, Lin PW, Guo HR. Prognostic significance of co-expression of RON and MET receptors in node-negative breast cancer patients. Clin Cancer Res. 2005;11(6):2222–8.
Minuti G, Cappuzzo F, Duchnowska R, Jassem J, Fabi A, O'Brien T, et al. Increased MET and HGF gene copy numbers are associated with trastuzumab failure in HER2-positive metastatic breast cancer. Br J Cancer. 2012;107(5):793–9.
Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2(2):127–37.
Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009;69(24):9330–6.
Cortes J, Fumoleau P, Bianchi GV, Petrella TM, Gelmon K, Pivot X, et al. Pertuzumab monotherapy after trastuzumab-based treatment and subsequent reintroduction of trastuzumab: activity and tolerability in patients with advanced human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol. 2012;30(14):1594–600.
Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366(2):109–19.
Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372(8):724–34.
Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(1):25–32.
Bianchini G, Gianni L. The immune system and response to HER2-targeted treatment in breast cancer. Lancet Oncol. 2014;15(2):e58–68.
Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64(11):3958–65.
Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66(3):1630–9.
ClinicalTrials.gov. Available from: https://clinicaltrials.gov.
Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–43.
Opdam FL, Guchelaar HJ, Beijnen JH, Schellens JH. Lapatinib for advanced or metastatic breast cancer. Oncologist. 2012;17(4):536–42.
Blackwell KL, Burstein HJ, Storniolo AM, Rugo HS, Sledge G, Aktan G, et al. Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 Study. J Clin Oncol. 2012;30(21):2585–92.
Burstein HJ, Sun Y, Dirix LY, Jiang Z, Paridaens R, Tan AR, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol. 2010;28(8):1301–7.
Chan A, Delaloge S, Holmes FA, Moy B, Iwata H, Harvey VJ, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17(3):367–77.
Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–90.
Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128(2):347–56.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–91.
Giordano SH, Temin S, Kirshner JJ, Chandarlapaty S, Crews JR, Davidson NE, et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2-positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(19):2078–99.
Krop IE, Kim SB, Gonzalez-Martin A, LoRusso PM, Ferrero JM, Smitt M, et al. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(7):689–99.
Perez EA, Barrios C, Eiermann W, Toi M, Im YH, Conte P, et al. Trastuzumab emtansine with or without pertuzumab versus trastuzumab plus taxane for human epidermal growth factor receptor 2-positive, advanced breast cancer: primary results from the phase III MARIANNE Study. J Clin Oncol. 2017;35(2):141–8.
Barok M, Joensuu H, Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014;16(2):209.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70.
Balko JM, Mayer AI, Levy M, Arteaga CL. HER2 (ERBB2) in Breast Cancer. My Cancer Genome. 2013. https://www.mycancergenome.org/content/disease/breast-cancer/erbb2/ (Updated 10 April 2013).
Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3(2):224–37.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
Stagg J, Loi S, Divisekera U, Ngiow SF, Duret H, Yagita H, et al. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci U S A. 2011;108(17):7142–7.
Acknowledgments
The work was supported by the following grants: National Institutes of Health (CCSG CA016672); Cancer Prevention & Research Institutes of Texas (RP160710 and RP150245); National Breast Cancer Foundation, Inc.; Breast Cancer Research Foundation; Patel Memorial Breast Cancer Endowment Fund; The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund; Ministry of Science and Technology, International Research-intensive Centers of Excellence in Taiwan (I-RiCE; MOST 106-2911-I-002-302); Ministry of Health and Welfare, China Medical University Hospital Cancer Research Center of Excellence (MOHW106-TDU-B-212-144003); and Center for Biological Pathways.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Hsu, J.L., Hung, MC. (2018). Mechanisms of Action and Resistance of Trastuzumab in Breast Cancer. In: Yarden, Y., Elkabets, M. (eds) Resistance to Anti-Cancer Therapeutics Targeting Receptor Tyrosine Kinases and Downstream Pathways. Resistance to Targeted Anti-Cancer Therapeutics, vol 15. Springer, Cham. https://doi.org/10.1007/978-3-319-67932-7_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-67932-7_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-67930-3
Online ISBN: 978-3-319-67932-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)